

Abstract
Non-small cell lung cancer (NSCLC) is a frequent type of cancer, which is mainly characterized clinically by high aggressiveness and high mortality. KRAS oncoprotein is the most common molecular protein detected in NSCLC, accounting for 25% of all oncogenic mutations. Constitutive activation of the KRAS oncoprotein triggers an intracellular cascade in cancer cells, leading to uncontrolled cell proliferation of cancer cells and aberrant cell survival states. The results of multiple clinical trials have shown that different KRAS mutation subtypes exhibit different sensitivities to different chemotherapy regimens. Meanwhile, anti-angiogenic drugs have shown differential efficacy for different subtypes of KRAS mutated lung cancer. It was explored to find if the specificity of the KRAS mutation subtype would affect PD-L1 expression, so immunotherapy would be of potential clinical value for the treatment of some types of KRAS mutations. It was discovered that the specificity of the KRAS mutation affected PD-L1, which opened up immunotherapy as a potential clinical treatment option. After several breakthrough studies, the preliminary test data of many early clinical trials showed that it is possible to directly inhibit KRAS G12C mutation, which has been proved to be a targeted treatment that is suitable for about 10%–12% of patients with advanced NSCLC, having a significant impact on the prolongation of their survival and the improvement of their quality of life. This article reviews the latest progress of treatments for NSCLC with KRAS mutation, in order to gain insight into the biological diversity of lung cancer cells and their potential clinical implications, thereby enabling individualized treatment for patients with KRAS-mutant NSCLC.
Keywords: KRAS, NSCLC, chemotherapy, immunotherapy, targeted therapy
Background
Lung cancer is one of the leading causes of cancer death in the world with 1.8 million deaths every year. The 5-year survival rate for patients with lung cancer is approximately 20% (1). Non-small cell lung cancer (NSCLC) accounts for 80%–85% of the total number of lung cancer cases (2). Through recent research, there has been great advancement in the treatment of NSCLC patients with epidermal growth factor receptor (EGFR) mutation and anaplastic lymphoma receptor tyrosine kinase (ALK) rearrangement (3–6). However, effective treatments for kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have not been developed. KRAS mutations are found in 25%–50% of Caucasian NSCLC patients and 5%–10% of Asian NSCLC patients (7–10). In patients with stage IV NSCLC, the results of platinum-based chemotherapy as the first form of treatment are very poor. It is obviously necessary to improve the treatment methods and provide individualized treatment for each patient (11). The NSCLC molecular spectrum is the key factor in treatment decision-making. There are many emerging carcinogenic targets and active targeted drugs. Somatic mutation of EGFR and rearrangement of ALK, proto-oncogene tyrosine protein kinase (ROS1), and proto-oncogene (RET) are supposed to be dependable biomarkers and effective drug targets for NSCLC (12, 13). However, the rat sarcoma viral oncogene homolog (RAS) family is the most common mutated oncogene, yet this oncogene has been defined as untreatable. Despite more than 40 years of basic and clinical research, there is still no effective anti-RAS therapy in the actual clinical diagnosis and treatment process. In recent years, targeted therapy and immunotherapy have been booming. At the same time, direct KRAS targeting and KRAS-related immunotherapy have also made great progress (14, 15). This paper will look to review the biological basis of KRAS mutations in NSCLC and discuss the potential causes of previous failures. Additionally, this paper will analyze the therapeutic effects of chemotherapy, targeted therapy, and immunotherapy in clinical practice and look to provide individualized treatment strategies for patients with KRAS mutations in lung cancer.
KRAS Biology
KRAS Function
KRAS is a member of the RAS oncogene family and encodes a small membrane-bound GTPase that toggles between a bound state of active guanosine triphosphate (GTP) and a bound state of inactive guanosine diphosphate (GDP) (16–18). RAS proteins act like cellular switches that are controlled by stimuli, and when stimulated, in the GTP-bound form, these proteins activate diverse signaling pathways that regulate elemental cellular processes (19, 20).
The activation of RAS signaling is strictly controlled by the regulatory factors that promote GDP–GTP exchange (guanine nucleotide exchange factors (GEFs)) or affect GTPase activity (GTPase-activating proteins (GAPs)). GEFs and GAPs are capable of binding to one or two pockets on RAS proteins, termed switch I and switch II regions, respectively. The former enhances the GDP release from RAS and stimulates its replacement by GTP, leading to RAS activation; the latter increases the inherent GTPase activity of RAS, leading to the rapid active–inactive transition of RAS state (21, 22). The main functional difference between mutant RAS oncoproteins and normal RAS oncoproteins is that mutant oncogene weakens the ability of RAS proteins to hydrolyze GTP (23–25). The RAS mutant oncoprotein is locked in a state of constitutive GTP-bound activity, leading to uncontrolled cell proliferation and survival ( Figure 1 ). Therefore, RAS proteins are one of the mutant cellular proteins that were proven to be the driving force in human cancer. However, RAS proteins have not succumbed to any kind of targeted therapy and have even been known as “undruggable” for many years. This is because RAS proteins do not seem to provide suitable pockets that allow drug binding, except for their GDP/GTP binding sites. Unfortunately, what binds RAS proteins to these nucleotides is picomolar affinity, with very slow off-rates. In addition, GTPase signaling is mediated by protein–protein interactions (PPIs) involving extended and shallow surfaces. The tight binding and the high intracellular concentration of GTP make the identification of competitive nucleotide analogs seem almost impossible for a long time (26–28). Cysteine 12-modifying KRAS inhibitors that impair RAF binding and downstream signals (29); quinazoline-based compounds and guanosine mimetic inhibitors that suppress GTP loading of KRAS G12C and cell proliferation (30, 31); and allele-specific inhibitors that inhibit mutant KRAS-driven signaling by binding to the GDP-bound oncoprotein and preventing activation (32) are all recent discoveries that have started to shift the common perception that RAS proteins are undruggable.
Figure 1.

KRAS function and its main downstream pathways. This diagram is a summary of KRAS oncogenic mutations that impair its activity to hydrolyze GTP, thereby activating three major signaling pathways that mediate basic cellular processes. KRAS, kirsten rat sarcoma viral oncogene homolog; Gh2, growth hormone 2; SOS, Son of Sevenless; GTP, guanosine triphosphate; GDP, guanosine diphosphate; GAPs, GTPase-activating proteins; PI3K, phosphoinositide 3-kinase; Akt/PKB, protein kinase B; BAD, BCL-2/BCL-XL-associated death promoter; CASP9, Recombinant Caspase 9; PIP3, phosphatidyl inositol triphosphate; PDK1, 3-phosphoinositide dependent kinase-1; RASSF1, Ras association domain family 1; MST1, human macrophage stimulation 1; Raf, rat fibrosarcoma; MEK, mitogen-activated protein kinase kinase; ERK, extracellular regulated kinase; NORE1A, Ras-association domain family 5.
Downstream Effector Pathways
In addition to binding to GTP, RAS proteins must also establish a connection with the cell membrane to interact with GEF and other upstream regulators, such as EGFR, fibroblast growth factor receptor (FGFR), and human EGFR 2-4 (HER2-4/ERBB2-4). This happens so that extracellular signals can be transmitted to downstream signaling pathways (17, 18).
The biological effects of RAS depend on the signaling network it regulates. In this way, it is pivotal to understand not only the activation mode of RAS but also the mechanism of its downstream molecular effectors (33). There are more than ten reported RAS effectors implicated in multiple signaling cascades, including the canonical rat fibrosarcoma/mitogen-activated protein kinase/extracellular regulated kinase (Raf–MEK–ERK) pathway, a common overactivated pathway in cancer, which causes abnormal proliferation of cells by regulating the cell cycle. RAS also activates phosphoinositide 3-kinase/protein kinase B (PI3K–Akt/PKB) signaling, which plays a pivotal role in RAS protein-mediated antiapoptosis (34, 35). The RAS association domain family 1 (RASSF1) pathway is another RAS downstream effector pathway that is required for RAS-dependent apoptosis reduction and proliferation (36) ( Figure 1 ).
In brief, RAS proteins play important roles in regulating cell proliferation, differentiation, and apoptosis by regulating signal transduction through different effectors that control diverse cellular functions. Constitutive activation of RAS oncoproteins initiates intracellular cascade reactions in the absence of extracellular signaling. This can lead to unlimited cell proliferation and aberrant cell survival. The deregulation of these cellular functions gives rise to hallmarks of cancer formation of various specificities (37).
KRAS Mutations
KRAS oncogenes are mainly mutations in exons 2, 3, and 4, which cause constitutive activation of the mitogen-activated protein kinase (MAPK) pathway. Approximately 90% of KRAS mutations occur at codon 12 (exon 2). This is especially noticeable in patients with NSCLC. The most common allele variants are G12C (GGT–TGT) and G12V (GGT–GTT), which are caused by classical smoking transformed from a G:C–T:A (38). The bioactive function of KRAS is related to the protein structure that depends on the bound state to GTP. Notably, KRAS mutations are heterogeneous and primarily involve substitutions in codons 12, 13, or 61 (39). In particular, G12 is situated on the p-ring and is involved in assisting nucleotide stability during activation, resulting in changes in intrinsic hydrolysis and gap-induced hydrolysis without changing the rate of nucleotide exchange (40). KRAS G12C and G12D are the most common types of mutations in lung cancer patients, accounting for 33.6% and 23.9% of total KRAS mutations, respectively. Other types of KRAS mutations are G12V (22.1%), G12A (7.1%), Q61H (5.3%), G13D (3.5%), Q13C (1.8%), G12S (1.8%), and G61R (0.9%) (41) ( Figure 2 ).
Figure 2.

Percentage of various mutant subtypes of KRAS.
Specific KRAS mutations have unique biological characteristics. For example, although substitutions of KRAS G12, G13, and Q61 attenuate GTP hydrolysis capacity, other mutations such as KRAS A146T maintained hydrolysis levels similar to wild-type (WT) KRAS. The A146T substitution likely promotes KRAS-GTP formation in the form of increased nucleotide exchange, thereby reducing this isoform’s oncogenic capacity (42). Different types of KRAS mutations also cause differences in downstream signaling pathways. Basic experimental analysis revealed that lung cancer cell lines harboring KRAS G12C or KRAS G12V mutations exhibited increased Ras-related protein (RAL) A/B signaling but decreased PI3K/Akt signaling compared with other KRAS mutant isoforms or WT cell lines (43). Contrarily, cell lines containing KRAS G12D were more likely to activate the PI3K–Akt pathway (31, 44–47) ( Figure 3 ).
Figure 3.
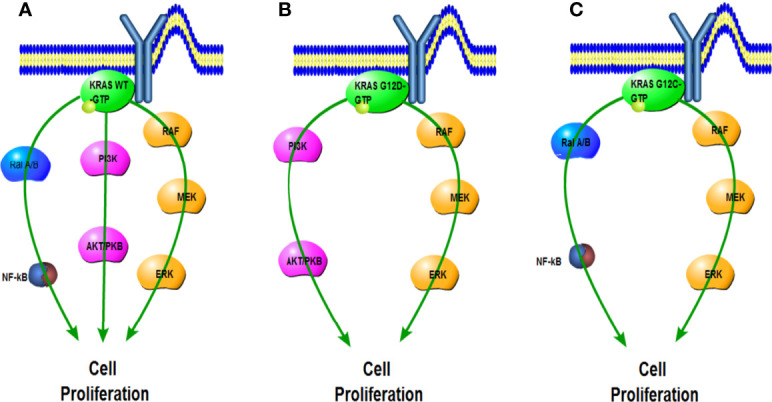
Impact of G12C and G12D mutations on KRAS downstream pathways compared with wild type. (A) Activated KRAS wild-type signals the Ral A/B, PI3K, and RAF pathways. (B) KRAS G12D preferentially activates the PI3K and RAF pathways. (C) KRAS G12C preferentially activates the Ral A/B and RAF pathways.
Current Approaches of KRAS-Mutant Non-Small Cell Lung Cancer and Their Efficacy in Different Subtypes
Although KRAS is one of the earliest oncogenic driver genes detected to date, no therapies have been found that effectively target KRAS mutations. Numerous therapeutic strategies have been developed including but not limited to chemotherapy, anti-angiogenic therapy, immunotherapy, blockage of downstream, and direct targeting of KRAS (18) ( Figure 4 ). However, the vast majority of treatments have not been studied for individual KRAS mutation subtypes. In all subtypes of KRAS-mutant NSCLC, mutations occur primarily at codon 12 (>80%) and 13 (15%). Additionally, KRAS-G12C mutation accounts for approximately 39% of all KRAS mutants. Other frequently occurring mutations involve KRAS G12V (18%–21%) and KRAS G12D (17%–18%) variants (17). Further efforts are dedicated to elucidating the impact of different KRAS mutation subtypes in lung cancer patients on treatment efficacy. Different signaling and drug sensitivity patterns among these subtypes have been determined by preclinical studies, which suggested that differences may occur at the level of amino acid substitution (47, 48). Therefore, we reviewed the efficacy of the above treatments in different subtypes, aiming to provide ideas for personalized therapies of KRAS-mutant NSCLC ( Table 1 ).
Figure 4.

Current approaches of KRAS-mutant NSCLC. NSCLC, non-small cell lung cancer; KRAS, kirsten rat sarcoma viral oncogene homolog; PD-1, programmed cell death protein 1; PD-L1, programmed cell death-ligand 1; CTLA4, cytotoxic T-lymphocyte-associated protein 4; Raf, rat fibrosarcoma; MEK, mitogen-activated protein kinase kinase; PI3K, phosphoinositide 3-kinase; Akt/PKB, protein kinase B.
Table 1.
Summary of clinical trials investigating the outcome of different KRAS mutation subtypes.
| Study | Pts | KRAS status | Treatment | Endpoint | KRAS status | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Jia et al. (49) | 170 | WT | G12C | G12V | G12D | Rare | First-line chemotherapy | WT | G12C | G12V | G12D | Rare | |||
| 59% (100) | 14% (23) | 11% (18) | 5% (9) | 11% (20) | ORR (%) | 19.0 | 26.1 | 22.2 | 11.1 | 20.0 | |||||
| PFS (months) | 6.4 | 4.4 | 2.9 | 7.0 | 4.7 | ||||||||||
| DCR (%) | 86 | 78.3 | 55.6 | 66.7 | 60 | ||||||||||
| Cserepes et al. (50) | 494 | WT | G12C | G12V | G12D | Rare | COD13 MUT | First-line chemotherapy | WT | G12C | G12V | G12D | Rare | COD13 MUT | |
| 68% (338) | 12% (61) | 6% (29) | 6% (27) | 4% (19) | 4% (20) | PFS (days) | 211 | 191 | 233 | 150 | 198 | 157 | |||
| OS (days) | 479 | 561 | 470 | 325 | 559 | 330 | |||||||||
| Sun et al. (51) | 304 | WT | G12C | G12V | G12D | Rare | First-line chemotherapy | WT | G12C | G12V | G12D | Rare | |||
| 87% (265) | 3% (9) | 3% (10) | 4% (13) | 3% (7) | OS (months) | 15 | 7.7 | 9.6 | 8.1 | 5.5 | |||||
| Ghimessy et al. (52) | WT | G12C | G12V | G12D | Rare | BEV/CHT | WT | G12/13X | G12D | ||||||
| 213 | 61% (130) | 16% (35) | 10% (20) | 9% (19) | 4% (9) | PFS (months) | 11.70 | 8.27 | 3.70 | ||||||
| OS (months) | 21.0 | 16.1 | 7.2 | ||||||||||||
| G12C | Sotorasib | G12C | |||||||||||||
| Skoulidis et al. (53) | 124 | 124 | ORR (%) | 37.1 | |||||||||||
| PFS (months) | 6.8 | ||||||||||||||
| Jeanson et al. (54) | G12A | G12C | G12D | G12V | G13C | ICIs | G12A | G12C | G12D | G12V | G13C | ||||
| 144 | 10% (15) | 48% (69) | 17% (25) | 17% (24) | 8% (11) | ORR (%) | 13.3 | 18.5 | 20.0 | 18.2 | 18.2 | ||||
| PFS (months) | 2.66 | 3.09 | 3.91 | 2.69 | 4.60 | ||||||||||
| Jänne et al. (55) | G12C/V | Non-G12C/V | Selumetinib + docetaxel | G12C/V | Non-G12C/V | ||||||||||
| 83 | 57% (47) | 43% (36) | PFS (months) | 5.7 | 4.9 | ||||||||||
| OS (months) | 9.6 | 8.6 | |||||||||||||
| Placebo + docetaxel | PFS (months) | 1.4 | 2.6 | ||||||||||||
| OS (months) | 4.4 | 7.1 | |||||||||||||
KRAS, kirsten rat sarcoma viral oncogene homolog; Pts, patients; ORR, objective response rate; PFS, progression-free survival; DCR, disease control rate; OS, overall survival; BEV, bevacizumab; CHT, chemotherapy; ICIs, immune checkpoint inhibitors.
Status of Chemotherapy in Patients With Different KRAS Mutation Subtypes of Non-Small Cell Lung Cancer
In recent advances in the treatment of NSCLC, most patients with the advanced-stage disease are still treated with platinum-based chemotherapy regimens. The predictive value of KRAS mutations in NSCLC was investigated in patients receiving definitive chemotherapy (56), postoperative radiotherapy adjuvant chemotherapy (57), or the phase III TRIBUTE trial comparing first-line carboplatin/paclitaxel with erlotinib or placebo for advanced NSCLC (58). In the above settings, KRAS mutation has not been found to be a predictor of response rate, progression-free survival (PFS), or overall survival (OS) in patients with lung cancer. The JBR10 trial showed a significant positive effect with chemotherapy in only KRAS WT. Yet the difference was not shown to be statistically significant (p = 0.29) (59). In addition, an Asian cohort study analyzed the prognosis of lung cancer patients who received different chemotherapy regimens according to KRAS mutation status. OS was markedly worse in KRAS mutant patients treated with pemetrexed or gemcitabine (p = 0.12). Meanwhile, among KRAS mutated lung cancer patients, OS was longer and statistically significant for adenocarcinoma patients compared with squamous carcinoma patients (22.7 vs. 11.5 months; p = 0.051) (51). It is worth noting that clinical studies show that PFS and OS are remarkably shortened in patients with KRAS codon 13 mutation, which indicates that the KRAS codon 13 mutation has a potential negative impact on chemotherapy (60). Additionally, analysis of clinical data from platinum-based chemotherapy according to KRAS mutation status demonstrated that patients with mutations at codon 13 experienced a shorter PFS and OS compared when compared with patients with mutations at codon 12 (61). According to a retrospective study that involved 2,183 Chinese cases that exhibited KRAS mutations, patients with KRAS G12V mutations seemed to have a poorer response to chemotherapy than others. In this study, a shorter and statistically significant PFS was observed when the G12V mutant was compared with WT patients (2.9 vs. 6.4 months, respectively; p = 0.001). Patients with KRAS G12V mutations were less sensitive to chemotherapy and had worse PFS than non-KRAS G12V mutated patients (median PFS (mPFS), 2.9 vs. 4.7 months; p = 0.046). There was no difference in PFS for other KRAS subtypes that were compared. In addition, PFS may be shorter in patients with KRAS mutated adenocarcinoma histology (4.3 vs. 6.7 months; p = 0.051). However, KRAS WT patients had a significantly higher disease control rate (DCR) to platinum-based chemotherapy (86.0% vs. 65.7%, p = 0.002). Although G12V had the lowest DCR of 55.6%, the response profile to platinum-based chemotherapy did not appear to be statistically significant between mutational subtypes (p < 0.05) (49). However, in a recent study, patients with KRAS G12V mutant lung adenocarcinoma (LUAD) not only tended to have a better response to platinum-based chemotherapy (p = 0.077) but also, although there was no significant difference, were more likely to have longer PFS than patients with other codon 12 mutations (p = 0.145) (50). However, these differences were not statistically significant. In these results, it can be found that the KRAS 13 codon is less sensitive to chemotherapy than the KRAS 12 codon. G12V showed poor efficacy in first-line chemotherapy, but it showed strong sensitivity to platinum-based chemotherapy. However, the KRAS mutant is less sensitive to chemotherapy than the KRAS WT.
Therefore, we propose a clinically significant hypothesis, namely, that the different types of KRAS mutations can produce different reactions to chemotherapy. This was seen in a study of cell lines with KRAS mutations by Garassino et al. In comparison with the WT clones, the G12C mutation was associated with a reduced response to cisplatin but increased sensitivity to taxol and pemetrexed, whereas G12V mutation showed a strong sensitivity to cisplatin but less sensitivity to pemetrexed. For cell lines with G12D mutations, taxol had minimal effects, but sorafenib had sound results (48).
Status of Anti-Angiogenic Therapy in Patients With Different KRAS Mutation Subtypes of Non-Small Cell Lung Cancer
In addition to initiating tumor formation by stimulating proliferation, oncogenic RAS ensures tumor progression by promoting tumor angiogenesis (62, 63). Different downstream pathways of oncogenic RAS are ultimately involved in promoting tumor angiogenesis through the upregulation of vascular endothelial growth factor (VEGF) and CXC chemokine interleukin-8 (IL-8) (62, 64–67). Angiogenesis inhibition is one of the most important strategies against solid tumors. Cutting off the blood supply to a tumor micro area leads to a lack of oxygen to the solid tumor; this results in extensive tissue necrosis within the tumor organization. The difference between normal and tumor tissue angiogenesis activation makes the process of antitumor drug discovery an attractive strategy target (68). Over the past decades, the VEGF signaling pathway has been identified as a central axis in the process of tumor angiogenesis. The advent of recombinant antibody technology has facilitated the development of bevacizumab (BEV), a humanized antibody that targets VEGF and is the current leading clinical treatment to inhibit tumor angiogenesis (69).
However, although it has been proved that VEGF plays an indispensable role in tumor angiogenesis mediated by RAS, seldom do we have studies involving the relationship between KRAS mutations and antiangiogenic therapy efficacy (70–72). A phase II trial assessing the efficacy of BEV in chemotherapy found that all KRAS mutated patients had no pathological response to neoadjuvant BEV combined with chemotherapy, whereas 35% of patients with WT KRAS showed a significant pathological response (73). In a recent clinical retrospective study by Ghimessy et al., patients with KRAS mutations, and especially patients with KRAS G12D mutant lung cancer, had a significantly shorter OS than those with KRAS WT or other KRAS mutant tumors (p = 0.0223 and p = 0.0144, respectively). At the same time, the KRAS WT or all other codon 12/13 (G12/13x) KRAS mutations other than KRAS G12D mutation had significant adverse effects on PFS (p = 0.0032). Thus, G12D mutations may define a subset of KRAS types for which LUAD patients with such mutations are not eligible for treatments with BEV-based antiangiogenic drugs (52).
Status of KRAS Targeted Therapy in Patients With Different KRAS Mutation Subtypes of Non-Small Cell Lung Cancer
Although KRAS was discovered decades ago, none of the studies targeting KRAS therapy have achieved significant results until recent years. Several studies have shown that specific mutant KRAS may cause differential sensitivity to EGFR tyrosine kinase inhibitors (EGFR-TKIs). One such study demonstrated that patients with the KRAS codon 13 mutation experienced worse outcomes when compared with patients with KRAS codon 12 mutations and KRAS WT patients (p < 0.0001 and p = 0.01 for PFS and OS, respectively) (74). Another study proved the potential OS benefit of EGFR-TKIs in patients with KRAS G12D/G12S mutations (HR = 0.49, p = 0.05). It was also observed that EGFR-TKIs tended to reduce survival in patients with G12C/G12V mutations (HR = 1.41, p = 0.07), which was more significant in the adenocarcinoma subgroup (HR = 1.73, p = 0.01), while the harmful effects of G12V mutation alone were more prominent (HR = 1.96, p = 0.04) (75). Contrarily, Fiala et al. reported that EGFR-TKIs improved PFS in patients with non-G12C KRAS mutant tumors when compared with the G12C group (76). However, the poor outcomes of EGFR WT/KRAS-mutant NSCLC patients indicate that the KRAS mutation is neither prognostic nor predictive of benefit from EGFR-TKIs (77). Recent advancement in RAS targeted drugs is the development of allele-specific inhibitors. The locations of KRAS oncogenic mutations are mainly clustered at several hotspot residues, especially in G12 (78). KRAS G12C mutants have cysteine residues that have been used to design covalent inhibitors with preclinical activity recently, which makes the inability to drug KRAS a thing of the past (29, 32, 79). Mutation-selective KRAS inhibitors utilize reactivity and the nucleophilic cysteine at No. 12. Thus, modified by disulfide bonds, these covalent conjugates can be incorporated into allosteric isomers and allosterically inhibit KRAS oncoprotein activity, or bind to the orthosteric substrate site and compete with GDP/GTP to inhibit protein activation. Ostrem et al. found compounds 6 and 12 and identified their corresponding new allosteric site switch II pocket (S-IIP), which opened the way for the development of allosteric KRAS G12C covalent inhibitors (29).
AMG510 is a small-molecule compound that irreversibly and specifically binds to G12C and functions to lock KRAS in an inactive state with GDP. This covalent inhibitor slowly converts the KRAS active state to the KRAS-GDP state with a 30-min half-life. In a recently concluded phase II clinical trial (NCT 03600883) in which 124 patients were evaluated, 37.1% patients with NSCLC had a confirmed objective response (4 had a complete response and 42 had a partial response; 95% CI, 28.6 to 46.2), and 80.6% had a disease control response (95% CI, 72.6 to 87.2); the mPFS was 6.8 months (95% CI, 5.1 to 8.2) (53). Another ongoing phase I/II trial targeting KRAS G12C (NCT 03785249) considers MRTX849, a similar small-molecule direct inhibitor with a half-life of 20 h, irreversibly binds to cysteine 12 in the switch II pocket induced by KRAS G12C and locks the KRAS protein in an inactive GDP bound state, resulting in the inhibition of the RAS/MAPK signaling pathway (80). A phase I trial (NCT04006301) conducted by Janssen evaluating JNJ-74699157 has just begun recruitment. The drug is an investigational, orally available small molecule that is designed to potently and selectively inhibit KRAS G12C (81). Eli Lilly drug LY3499446, a new compound under development as KRAS G12C inhibitors (NCT #04165031), will be evaluated as a single agent or in combination with other agents such as abemaciclib, cetuximab, and erlotinib in advanced solid tumors including NSCLC (81). We can conclude that NSCLC patients with KRAS G12D/G12V/G13C mutations are better candidates for immunotherapy than patients with KRAS G12A/G12C mutations.
Status of Immunotherapy in Patients With Different KRAS Mutation Subtypes of Non-Small Cell Lung Cancer
It is widely indicated that the degree of programmed cell death protein 1 (PD-1) expression is tightly correlated with the KRAS subtype status, and KRAS mutation is, to some extent, considered a possible biomarker for immune checkpoint inhibitors (ICIs) (82). Furthermore, clinical benefit from the application of PD-1 inhibitors in patients with KRAS mutations was reported in a comprehensive analysis (83). Increased expression of PD-1 has been affirmed in KRAS mutant cells, accompanied by the demonstration that ERK activation mediates upregulation of programmed cell death-ligand 1 (PD-L1) through KRAS mutations (84). A study based on the intrinsic link between the degree of PD-L1 expression on tumor cells and the type of KRAS mutation found that, as a PD-1 inhibitor, pembrolizumab, or an ERK inhibitor can restore the body’s antitumor immunity and prevent apoptosis of CD3+ T cells by preventing the immune escape of tumor cells (85). At the same time, a large number of studies have confirmed that PD-L1 expression has a close relationship with circulating tumor cells (CTCs). Reduced CTC numbers are strongly associated with a good response to immunotherapy and longer OS and PFS (86, 87). Additionally, other available data indicate that high CTC values before treatment are associated with an increased risk of patient death and progression (88). The results of Wang et al. showed that CTCs can also be used to detect the dynamic changes of PD-L1 during radiotherapy in lung cancer patients (89). Nicolazzo et al. summarized that stage IV patients with NSCLC who received ICI nivolumab therapy could have their resistance to immunotherapy measures through the persistence of PD-L1-positive CTCs (90). In addition, there is evidence for variability in the biological behavior of different KRAS mutation subtypes due to the high heterogeneity in the presentation of KRAS mutations. Therefore, attention needs to be paid to heterogeneity in the efficacy of immunosuppressive agents in lung cancer patients with KRAS mutations when immunotherapy is administered. Meanwhile, differences in the tumor microenvironment (TME) of different lung cancer patients affect the efficacy of immunotherapy. There is evidence that the above TME differences can affect the sensitivity of lung cancer patients to immunotherapy. Some of these differences are the status of neutrophils, the number of NK cell counts, the activity of dendritic cells (DCs), the expression of PD-L1 on macrophages, Foxp3+ Ti/S ratio, and CD8+ Ti/S ratio, and chromosomal stability (91–96). It should also be mentioned that gut microbiota can shape TME by modulating the immune and hormonal factors throughout the host (97, 98). The metabolites of the gut microbiota also have implications for the TME and tumor immunosuppressive therapy (99–101). Modulation of the gut microbiota has been reported to enhance the effects of cancer immunotherapy (101). Therefore, when administering immunotherapy to patients with KRAS mutated lung cancer, attention needs to be paid to the differences in the TME of patients with lung cancer while paying attention to the heterogeneity in the efficacy of immunosuppressive agents. This is all with respect to the expectation of achieving the individualization of immunotherapy for patients with lung cancer.
In a retrospective study, Jeanson et al. analyzed the extent of PD-1 expression in 128 patients with advanced NSCLC (all histological subgroups, predominantly LUAD) treated with ICIs (anti-programmed death 1, anti-PD-L1, or anti-cytotoxic T-lymphocyte-associated protein 4 antibodies). Although no significant differences were observed when comparing the efficacy and toxicity of ICIs between different subtypes of KRAS mutations, there were statistically significant differences in PD-L1 expression: a higher proportion of patients with KRAS G12D, G12V, or G13C mutations had PD-L1 positive tumors, and a higher proportion of PD-L1-negative tumors had G12A and G12C mutations. Interestingly, KRAS-mutant NSCLC was investigated according to the degree of PD-L1 expression; they found that a better objective response rate (ORR) and longer PFS were observed for PD-L1-positive tumors. Meanwhile, in patients with KRAS G12A and G12V mutant cancers, the degree of PD-L1 expression was similar to the ORR and PFS in patients treated with ICIs (54). We may conclude that NSCLC patients with KRAS G12D/G12V/G13C mutations are better candidates for immunotherapy, whereas patients with KRAS G12A/G12C mutations are not.
Downstream Pathway Inhibitors Vary Between Patients With Different KRAS Mutation Subtypes of Non-Small Cell Lung Cancer
The KRAS signaling pathway is highly complex and dynamically changing, and the downstream pathways involve multiple effectors. The representative ones are the Raf–MEK–ERK and PI3K–Akt signaling networks (102, 103). Due to the slightly different biochemical characteristics of each allele, the downstream pathways involved may vary in quantity and quality (104). The best existing example is the varying sensitivity of CRC cell lines expressing different KRAS alleles to MEK1/2 inhibition. Cell lines expressing A146T were sensitive to a single MEK1/2 inhibitor, but not to other KRAS-activated mutations (105). Also, the effects of different KRAS mutation subtypes on downstream signaling pathways such as PI3K may result in differential response to therapy (47). Therefore, it is important to understand whether the metabolic levels of cells with different KRAS mutation statuses are affected by these inhibitors. This will help to inform new combination regimens that have the potential to form targeted therapies for WT and mutant cancer cells to help patients receive tailored treatment.
Caiola et al. studied KRAS WT and G12C mutated NSCLC clones to determine the response of both to PI3K–Akt inhibitors (BEZ235 and BKM120). Metabolomic analysis revealed that although the final effects of both mutation types on cell growth, cell cycle distribution, and caspase activation were similar, glutamine metabolism in KRAS G12C and serine metabolism in KRAS WT were impaired after PI3K signaling pathway blockade by inhibitors. PI3K inhibitors cause autophagy in KRAS WT, but not KRAS G12C. At the same time, there was significantly reduced KRAS G12C ammonia production, possibly as a result of impaired glutamine metabolism (106). A randomized phase II trial of selumetinib plus docetaxel in KRAS mutant advanced NSCLC suggested the impact of KRAS codon subtypes. Patients with KRAS G12V mutation had longer PFS and ORR than other subtypes (p = 0.24 and p = 0.189, respectively), while KRAS G12C mutation may have longer OS than other mutation types (p = 0.48). Further analysis at week 6 suggested tumors harboring KRAS G12V may have had a better response: G12V (n = 9) for 62%; reduction across all codons (n = 81) for 18% (55).
Impact of KRAS Concurrent Pathogenic Mutations on Outcomes of Therapy
KRAS-mutant NSCLC has been proven to be a genetically heterogeneous disease. In addition to having different types of point mutations, it is often associated with other co-mutations in lung cancer, which has been reported in various papers in recent years (107, 108). Approximately 50% of NSCLC with KRAS mutations have additional co-accompanied mutations that are critical in tumorigenesis, such as TP53, STK11/LKB1, KEAP1, and SAMARCA4—which are the most commonly reported mutations (109). We summarized the clinical trials that are investigating the outcomes of different KRAS co-mutations below, in order to provide references for the personalized treatments of relevant patients ( Table 2 ). Each of these co-mutational partners is a key contributor to Ras signaling and the TME in lung tumor cells and has resulted in more prominent molecular and clinical heterogeneity of KRAS-driven NSCLC (38).
Table 2.
Summary of clinical trials investigating the outcomes of different KRAS co-mutations.
| Study | Pts | KRAS status | Treatment | Endpoint | KRAS status | ||||
|---|---|---|---|---|---|---|---|---|---|
| Skoulidis et al. (110) | K-only | KP | KL | ICIs | K-only | KP | KL | ||
| 174 | 37% (64) | 32% (56) | 31% (54) | ORR (%) | 28.6 | 35.7 | 7.4 | ||
| PFS (months) | 2.7 | 3.0 | 1.8 | ||||||
| OS (months) | 16.1 | 16.0 | 6.4 | ||||||
| Skoulidis et al. (53) | KRAS MUT | KRAS-KEAP1 MUT | Sotorasib | KRAS MUT | KRAS-KEAP1 MUT | ||||
| 104 | 81% (84) | 19% (20) | ORR (%) | 44 | 20 | ||||
| Alessi et al. (111) | K-only | KS | ICIs | KS | K-only | ||||
| 176 | 90% (159) | 10% (17) | ORR (%) | 0 | 22.0 | ||||
| PFS (months) | 1.4 | 4.1 | |||||||
| OS (months) | 3.0 | 15.1 | |||||||
| Liu et al. (112) | K-only | KP | KS | Non-immunotherapy | K-only | KP | KS | ||
| 155 | 61% (94) | 33% (52) | 6% (9) | DFS (months) | 18.0 | 16.31 | 10.97 | ||
| OS (months) | 20.11 | 18.48 | 15.37 | ||||||
| ICIs | |||||||||
| 77 | 56% (43) | 32% (25) | 12% (9) | PFS (months) | 2.77 | 4.63 | 1.73 | ||
KRAS, kirsten rat sarcoma viral oncogene homolog; Pts, patients; KL, KRAS-STK11/LKB1 co-mutant; KP, KRAS-TP53 co-mutant; ORR, objective response rate; PFS, progression-free survival; OS, overall survival; MUT, mutation; KEAP1, Kelch-like ECH-associated protein 1; ICIs, immune checkpoint inhibitors; KS, KRAS-SMARCA4 co-mutant; DFS, disease-free survival.
KRAS Co-Mutated With TP53 and STK11/LKB1
KRAS mutations in NSCLC patients frequently occur together with mutations in tumor protein 53 (TP53) and serine–threonine kinase 11/liver kinase B1 (STK11/LKB1). Genomic alterations co-occurred in the TP53 and STK11/LKB1 tumor suppressor genes, which define the unique biology, therapeutic sensitivities, and immune conditions of different subgroups of NSCLC with KRAS mutations (113). STK11 / LKB1 encodes a serine threonine kinase, which plays a role in cell metabolism ,energy homeostasis, growth and polarity regulation through the phosphorylation of adenosine monophosphate activated protein kinase (AMPK) and 12 AMPK related kinases. (114). Inactivation of STK11 (or its protein product LKB1) through mutational or non-mutational mechanisms has been associated with an inert or “cold” TME. It leads to the accumulation of neutrophils with T cell-suppressive effects, accompanied by a corresponding increase in the expression of T-cell exhaustion markers and tumor-promoting cytokines. In human tumors and genetically engineered mouse models, the density of invasive cytotoxic CD8+ T lymphocytes was decreased, along with the reduced expression of PD-L1 (113, 115, 116). In contrast, extensive infiltration of cytotoxic CD8+ Th1 tumor-infiltrating lymphocytes (TILs), as well as high expression of interferon (IFN)-dependent genes and IFN-induced PD-L1, was predominantly observed in KRAS-TP53 co-mutated tumors (110).
In the study of Skoulidis et al., LUAD patients were divided into three groups according to whether TP53 or STK11/LKB1 gene mutations occurred. The majority of KRAS-STK11/LKB1 co-mutated (KL) tumors were shown to be significantly more resistant to PD-1 inhibitors, with lower response rates observed for this subtype in three independent databases [9.1% for MDACC (MD Anderson Cancer Center), 9.1% for MSKCC (Memorial Sloan Kettering Cancer Center), and 4.8% for DFCI/MGH (Dana-Farber Cancer Institute/Massachusetts General Hospital)]. On the other hand, the KRAS-TP53 co-mutant (KP) group showed greater sensitivity to PD-1 inhibitors. When it comes to PFS, the KL group showed significantly shorter PFS than either K-only (hazard ratio (HR) 1.98, 95% CI, 1.33 to 2.94; p < 0.001) or KP (HR 1.77, 95% CI, 1.16 to 2.69; p = 0.0072) groups in pairwise comparisons, while the latter two groups had similar PFS. Meanwhile, this significant difference in OS was also observed among the three subgroups in the SU2C cohort and was statistically significant (p = 0.0045). Median OS was 6.4 months in KL compared with 16.0 months in KP and 16.1 months in K-only LUAD (110). In the study by La Fleur et al., worse OS was observed for LUAD patients with a mutation in either TP53 or STK11/LKB1. In the LUAD KRAS mutation group, poor survival appeared to be related to TP53 or STK11/LKB1 co-mutations instead of a single KRAS aberration. This result was also found in the open data analysis of cBioPortal (117).
Co-mutations in KRAS and TP53 suggest that in lung cancer, tumors carrying these mutations may be more sensitive to immune checkpoint suppression (83). Conversely, tumors with both KRAS and STK11 mutations may be associated with an immunosuppressive microenvironment (110, 118). In addition, in the presence of oncogenic KRAS mutations, STK11/LKB1 deficiency promoted the synthesis of interleukin-6 (IL-6), which predominantly recruited large numbers of neutrophils but suppressed T-cell infiltration, and it had higher criteria for markers of T-cell exhaustion (mainly PD-1). Moreover, PD-L1 expression was also suppressed in cancer cells, indicating that STK11-deficient KRAS mutations lead to anti-PD-1/PD-L1 resistance in cancer cells (110, 115).
In summary, KRAS-TP53 co-mutant NSCLC patients are more suitable for treatment with ICIs, while those with both KRAS and STK11/LKB1 mutations demonstrated resistance.
KRAS Co-Mutated With KEAP1
Kelch-like ECH-associated protein 1 (KEAP1), a principal repressor of nuclear factor erythroid 2-like 2 (NFE2L2; hereafter NRF2), functions primarily as a transcriptional regulator during the cellular oxidative stress response and is one of the most frequent co-mutations in KRAS mutated tumors that co-occur with genomic changes that affect tumor biology and response to systemic therapy (92, 104, 105, 119). According to a pan-cancer analysis, the amount of KEAP1 mutations in 40,167 patients with distinct cancer types was 2.7%; patients with NSCLC had the highest levels of KEAP1 mutations (15.8%) (120). Nearly 20% of KRAS mutant lung cancers harbor concurrent loss of function (LOF) mutations in KEAP1 (121–123).
The results demonstrate that KEAP1 mutations activate the NRF2 antioxidant program and promote LUAD progression in concert with mutant KRAS (124), demonstrating that cancer cells can overcome oxidative stress barriers during tumorigenesis (125–131). The metabolic requirement for glutaminolysis may also similarly manifest as a therapeutic vulnerability in other cancers with genetic (132–137), epigenetic (138–140), or post transcriptional (141) alterations in the KEPA1/NRF2 signaling pathway, a hypothesis that illustrates the importance of kinase-targeted therapeutic strategies for KRAS-KEAP1 mutant lung cancer (142). Furthermore, in KRAS-mutant LUAD, tumors with LKB1 loss are highly enriched for concurrent KEAP1 mutations, which activate the KEAP1/NRF2 pathway. A recent study investigated the biological consequences of these co-occurring alterations and explored whether they conferred specific therapeutic vulnerabilities. The results collectively found that in kallikrein-related peptidases (KLK) tumor cells, activation of the KEAP1/NRF2 pathway limits metabolic flexibility and promotes glutamine-addictive metabolism to maintain the tricarboxylic acid (TCA) cycle in addition to redox homeostasis, rendering these tumor cells selectively vulnerable to glutaminase inhibitors (143).
In an exploratory analysis by Skoulidis et al., the activity of sotorasib was observed across a spectrum of prevalent co-occurring mutations, including STK11 and KEAP1, both of which are related to inferior treatment outcomes and a poor prognosis in patients with NSCLC (110, 117, 120, 144–147). Among the 104 patients (KRAS-mutant NSCLC is mainly adenocarcinoma, accounting for 95.2%) who were assessed for co-occurring genomic alterations, efficacy was seen in the subgroups with mutated STK11, KEAP1, or TP53. After total genomic changes were assessed in 104 patients, efficacy was significantly improved in the STK11, KEAP1, or TP53 mutated subgroups. Fifty percent (95% CI, 28 to 72) of patients in the STK11 mutant subgroup and WT KEAP1 subgroup responded, and 39% (95% CI, 30 to 49) of evaluable patients responded. Among patients with KEAP1 mutations, 23% of patients in the STK11 and KEAP1 subgroups (95% CI, 5 to 54) responded, compared with 14% of patients in the WT STK11 and KEAP1 subgroups (95% CI, 0 to 58) (53). In total, KEAP1 co-mutation is an adverse factor for NSCLC patients with KRAS mutations who receive sotorasib therapy.
KRAS Co-Mutated With SMARCA4
The SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complexes control DNA accessibility to transcriptional factors and regulate transcriptional programming (148, 149). The genes encoding SWI/SNF complex subunits are among the most highly mutated in cancer. Among various kinds of cancer, SWI/SNF multi-subunit protein complex composition of genomic changes has taken place. It is estimated that at least 20% of malignancies have SWI/SNF complex subunit mutations (150, 151). SWI/SNF related, matrix associated, actin-dependent regulator of chromatin, subfamily A, member 4 (SMARCA4) encodes brahma-related gene1 (BRG1), one of two mutually exclusive ATPase subunits of the SWI/SNF complex. Mutations in the SMARCA4 gene are found in a variety of cancers and tended to co-occur with KRAS mutations frequently (10%) (122, 152–155). Studies have shown that inactivation of SMARCA4 promotes the invasion of NSCLC by altering chromatin organization (156), while decreased expression of SMARCA4 results in a poor prognosis of lung cancer (157–159).
Lissanu et al. showed that SMARCA4 through synergies with lack of p53 and KRAS activation plays a role of tumor suppressor, and these SMARCA4 mutations in the tumor were highly sensitive to the inhibition of oxidative phosphorylation (160). Another study indicated that decreased expression of SMARCA4 resulted in a poor prognosis of lung cancer. Besides, the presence of SMARCA4 co-mutations in KRAS mutated NSCLC patients was found to contribute to poor immunotherapy outcomes (157). Besides, the presence of SMARCA4 co-mutations in KRAS mutated NSCLC patients was found to contribute to poor immunotherapy outcomes (111, 161). In the study of Alessi et al., compared with K-only subgroup, ORR (22% vs. 0%, p = 0.03), mPFS (4.1 vs. 1.4 months, HR = 0.25, 95% CI, 0.14 to 0.42, p < 0.001) and median OS (15.1 vs. 3.0 months, HR = 0.29, 95% CI, 0.17 to 0.50, p < 0.001) in KRAS-SMARCA4 co-mutant (KS) subgroup were significantly shortened (149). To make the conclusions more comprehensive, the analysis by Liu et al. concluded that genomic changes in SMARCA4 are one of the reasons for the poor prognosis of KRAS mutant LUAD patients regardless of whether they received non-immunotherapy or immunotherapy. Among patients receiving non-immunotherapy, the KS subgroup had a significantly shorter DFS than the KP (HR 4.47, 95% CI, 1.52 to 13.22, p = 0.003) and K-only (HR 2.43, 95% CI, 1.46 to 4.05, p = 1.2E−4) two subgroups. A retrospective review of LUAD patients treated with immunotherapy yielded that the KS co-mutated group had shorter PFS than the other subtypes in various subgroup analyses (HR = 2.82, 95% CI, 1.17 to 6.81, p = 0.016) (112).
NSCLC patients with concurrent KRAS and SMARCA4 mutations require another targeted therapeutic strategy. Cisplatin-based chemotherapy was shown to be beneficial to patients with NSCLC with low SMARCA4 expression in a clinical study (157). CDK4 inhibitors including palbociclib may also be a potential alternative (162). In addition, a recent study showed that SMARCA4-deficient lung cells and xenograft tumors suppressed oxidative phosphorylation evidently (160). All observations suggest that therapeutic strategies are encouraged, but further clinical trials are needed.
Conclusion
As of today, differences in the effectiveness of chemotherapy, antiangiogenic therapy, targeted therapy, or immunotherapy among lung cancer patients with different KRAS mutant subtypes are not known. However, the above research results show that G12C/V is effective for platinum-based chemotherapy, while G12D is more sensitive to first-line chemotherapy. EGFR inhibition has a poor effect on KRAS mutation, but codon 12 mutations are more sensitive than codon 13 mutations. Meanwhile, patients with KRAS G12C lung cancer are likely to find success with covalent inhibitors such as AMG 510 and MRTX849, an anti-endogenous protein degradation molecule. Codon 13 mutations are more sensitive to ICIS than codon 12 mutations. KRAS co-mutated with STK11/LKB1 is insensitive to ICIs, while TP53 co-mutation is the opposite. KRAS combined with SMARCA4 mutant LUAD has a poor response to non-immunotherapy and immunotherapy, and SMARCA4 mutation may be a genetic factor contributing to its poor response ( Figure 5 ). The reasons for the poor efficacy of patients with KRAS-mutant NSCLC and the large interpatient variability may relate to oncogenic mechanisms and not to the function of the target itself. The key point is that there is a high degree of heterogeneity among the subtypes of KRAS mutations. Coexisting genetic events and differences in KRAS allele mutations determine different metabolic profiles and TME, both of which will produce significant differential drug sensitivities in seemingly similar tumors. Therefore, with individualized treatments for different KRAS mutant subtypes, we may eventually change the process of fatal NSCLC. In conclusion, the innovation of traditional treatment strategies and the emergence of new promising drugs may change the treatment pattern of KRAS mutant lung cancer. Yet the therapeutic strategy of KRAS gene mutation remains to be further explored.
Figure 5.
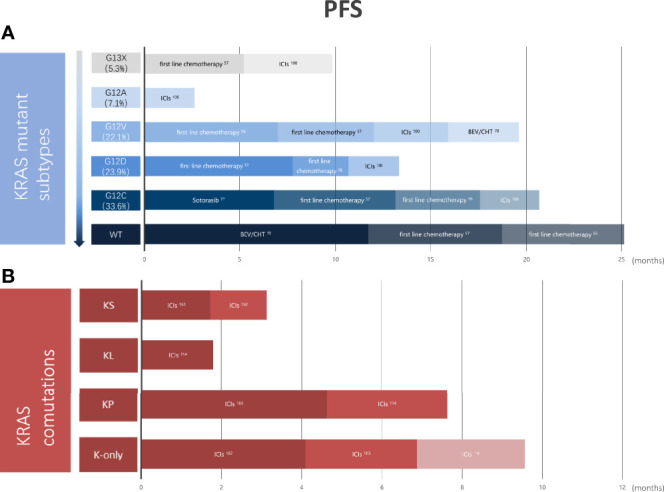
Outcomes vary in primary treatments in different KRAS mutation subtypes or different KRAS co-mutations. (A) Pooled PFS of NSCLC patients with KRAS wild-type and different KRAS mutations from clinical trials. (B) Pooled PFS of K-only and KRAS co-mutated patients with NSCLC from clinical trials. KRAS, kirten rat sarcoma viral oncogene homolog; COD, codon; WT, wild type; ICIs, immune checkpoint inhibitors; BEV, bevacizumab; CHT, chemotherapy; KS, KRAS-SAMARCA4 co-mutant; KL, KRAS-STK11/LKB1 co-mutant; KP, KRAS-KEAP1 co-mutant; PFS, progression-free survival; NSCLC, non-small cell lung cancer.
Author Contributions
HY provided the initial idea for this review. MS and RQ were in charge of data acquisition and drafting of the article. JR and DL revised the article. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (no. 81874221), Basic Public Welfare Research Project of Zhejiang Province (LGF21H160027), and Science and Technology Agency of Taizhou City (20ywa09).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors gratefully acknowledge Dr. Feng-Ming (Spring) Kong, from Case Western Reserve University, for the editorial assistance in the abstract.
Glossary
- NSCLC
non-small cell lung cancer
- EGFR
epidermal growth factor receptor
- ALK
anaplastic lymphoma receptor tyrosine kinase
- KRAS
kirsten rat sarcoma viral oncogene homolog
- ROS1
proto-oncogene tyrosine-protein kinase
- RET
rearranged during transfection proto-oncogene
- RAS
rat sarcoma viral oncogene homolog
- GTP
guanosine triphosphate
- GDP
guanosine diphosphate
- GEFs
guanine nucleotide exchange factors
- GAPs
GTPase-activating proteins
- FGFR
fibroblast growth factor receptor
- HER2–4/ERBB2–4
human epidermal growth factor receptors 2–4
- Raf
rat fibrosarcoma
- MEK
mitogen-activated protein kinase kinase
- ERK
extracellular regulated kinase
- PI3K
phosphoinositide 3-kinase
- Akt/PKB
protein kinase B
- RASSF1
Ras association domain family 1
- MAPK
mitogen-activated protein kinase
- RAL
RAS-associated protein
- PFS
progression-free survival
- OS
overall survival
- WT
wild type
- DCR
disease control rate
- LUAD
lung adenocarcinoma
- VEGF
vascular endothelial growth factor
- IL-8
CXC chemokine interleukin-8
- BEV
bevacizumab
- EGFR-TKIs
EGFR tyrosine kinase inhibitors
- S-IIP
switch II pocket
- mPFS
median progression-free survival
- PD-1
programmed cell death protein 1
- PD-L1
programmed cell death-ligand 1
- ICIs
immune checkpoint inhibitors
- ORR
objective response rate
- TME
tumor microenvironment
- TP53
tumor protein 53
- STK11
serine-threonine kinase 11
- LKB1
liver kinase B1
- KL
KRAS-STK11/LKB1 co-mutant
- KP
KRAS-TP53 co-mutant
- AMPK
activating phosphorylation of protein kinase
- TILs
tumor-infiltrating lymphocytes
- IFN
interferon
- IL-6
interleukin-6
- KEAP1
Kelch-like ECH-associated protein 1
- NFE2L2/NRF2
negative regulator of nuclear factor erythroid 2-like 2
- LOF
loss of function
- KLK
kallikrein-related peptidases
- TCA
tricarboxylic acid
- SWI/SNF
SWItch/Sucrose Non-Fermentable
- SMARCA4
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4
- BRG1
brahma-related gene1
- KS
KRAS-SMARCA4 co-mutant
References
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- 2. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong K-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat Rev Cancer (2014) 14:535–46. doi: 10.1038/nrc3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou C, Wu Y-L, Chen G, Feng J, Liu X-Q, Wang C, et al. Erlotinib Versus Chemotherapy as First-Line Treatment for Patients With Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol (2011) 12:735–42. doi: 10.1016/S1470-2045(11)70184-X [DOI] [PubMed] [Google Scholar]
- 4. Gao B, Sun Y, Zhang J, Ren Y, Fang R, Han X, et al. Spectrum of LKB1, EGFR, and KRAS Mutations in Chinese Lung Adenocarcinomas. J Thorac Oncol (2010) 5:1130–5. doi: 10.1097/JTO.0b013e3181e05016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK Inhibitor Ceritinib Overcomes Crizotinib Resistance in Non-Small Cell Lung Cancer. Cancer Discov (2014) 4:662–73. doi: 10.1158/2159-8290.CD-13-0846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Solomon BJ, Mok T, Kim DW, Wu Y-L, Nakagawa K, Mekhail T, et al. First-Line Crizotinib Versus Chemotherapy in ALK-Positive Lung Cancer. N Engl J Med (2014) 371:2167–77. doi: 10.1056/NEJMoa1408440 [DOI] [PubMed] [Google Scholar]
- 7. Riely GJ, Marks J, Pao W. KRAS Mutations in Non-Small Cell Lung Cancer. Proc Am Thorac Soc (2009) 6:201–5. doi: 10.1513/pats.200809-107LC [DOI] [PubMed] [Google Scholar]
- 8. Piva S, Ganzinelli M, Garassino MC, Caiola E, Farina G, Broggini M, et al. Across the Universe of K-RAS Mutations in Non-Small-Cell-Lung Cancer. Curr Pharm Des (2014) 20:3933–43. doi: 10.2174/13816128113196660761 [DOI] [PubMed] [Google Scholar]
- 9. Xia N, An J, Jiang QQ, Li M, Tan J, Hu C-p. Analysis of EGFR, EML4-ALK, KRAS, and C-MET Mutations in Chinese Lung Adenocarcinoma Patients. Exp Lung Res (2013) 39:328–35. doi: 10.3109/01902148.2013.819535 [DOI] [PubMed] [Google Scholar]
- 10. Miyanaga A, Shimizu K, Noro R, Seike M, Kitamura K, Kosaihira S, et al. Activity of EGFR-Tyrosine Kinase and ALK Inhibitors for EML4-ALK-Rearranged Non-Small-Cell Lung Cancer Harbored Coexisting EGFR Mutation. BMC Cancer (2013) 13:262. doi: 10.1186/1471-2407-13-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dogan S, Shen R, Ang DC, Johnson ML, D'Angelo SP, Paik PK, et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin Cancer Res (2012) 18:6169–77. doi: 10.1158/1078-0432.CCR-11-3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahmadzada T, Kao S, Reid G, Boyer M, Mahar A, Cooper WA, et al. An Update on Predictive Biomarkers for Treatment Selection in Non-Small Cell Lung Cancer. J Clin Med (2018) 7(6):153. doi: 10.3390/jcm7060153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Furugaki K, Mochizuki M, Kohno M, Shu S, Harada N, Yoshimura Y, et al. Expression of C-Terminal ALK, RET, or ROS1 in Lung Cancer Cells With or Without Fusion. BMC Cancer (2019) 19:301. doi: 10.1186/s12885-019-5527-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune Checkpoint Inhibitors for Patients With Advanced Lung Cancer and Oncogenic Driver Alterations: Results From the IMMUNOTARGET Registry. Ann Oncol (2019) 30:1321–8. doi: 10.1093/annonc/mdz167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the Undruggable RAS: Mission Possible? Nat Rev Drug Discov (2014) 13:828–51. doi: 10.1038/nrd4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kranenburg O. The KRAS Oncogene: Past, Present, and Future. Biochim Biophys Acta (2005) 1756:81–2. doi: 10.1016/j.bbcan.2005.10.001 [DOI] [PubMed] [Google Scholar]
- 17. Ferrer I, Zugazagoitia J, Herbertz S, John W, Paz-Ares L, Schmid-Bindert G. KRAS-Mutant Non-Small Cell Lung Cancer: From Biology to Therapy. Int J Mol Sci (2020) 21(12):4325. doi: 10.3390/ijms21124325 [DOI] [PubMed] [Google Scholar]
- 18. Uras IZ, Moll HP, Casanova E. Targeting KRAS Mutant Non-Small-Cell Lung Cancer: Past, Present and Future. Int J Mol Sci (2020) 21(12):4325. doi: 10.3390/ijms21124325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barbacid M. Ras Genes. Annu Rev Biochem (1987) 56:779–827. doi: 10.1146/annurev.bi.56.070187.004023 [DOI] [PubMed] [Google Scholar]
- 20. Malumbres M, Barbacid M. RAS Oncogenes: The First 30 Years. Nat Rev Cancer (2003) 3:459–65. doi: 10.1038/nrc1097 [DOI] [PubMed] [Google Scholar]
- 21. Karnoub AE, Weinberg RA. Ras Oncogenes: Split Personalities. Nat Rev Mol Cell Biol (2008) 9:517–31. doi: 10.1038/nrm2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cox AD, Der CJ. Ras History: The Saga Continues. Small GTPases (2010) 1:2–27. doi: 10.4161/sgtp.1.1.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gibbs JB, Sigal IS, Poe M, Scolnick EM. Intrinsic GTPase Activity Distinguishes Normal and Oncogenic Ras P21 Molecules. Proc Natl Acad Sci USA (1984) 81:5704–8 .doi: 10.1073/pnas.81.18.5704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McGrath JP, Capon DJ, Goeddel DV, Levinson AD. Comparative Biochemical Properties of Normal and Activated Human Ras P21 Protein. Nature (1984) 310:644–9. doi: 10.1038/310644a0 [DOI] [PubMed] [Google Scholar]
- 25. Sweet RW, Yokoyama S, Kamata T, Feramisco JR, Rosenberg M, Gross M. The Product of Ras Is a GTPase and the T24 Oncogenic Mutant Is Deficient in This Activity. Nature (1984) 311:273–5. doi: 10.1038/311273a0 [DOI] [PubMed] [Google Scholar]
- 26. McCormick F. KRAS as a Therapeutic Target. Clin Cancer Res (2015) 21:1797–801. doi: 10.1158/1078-0432.CCR-14-2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging Ras Back in the Ring. Cancer Cell (2014) 25:272–81. doi: 10.1016/j.ccr.2014.02.017 [DOI] [PubMed] [Google Scholar]
- 28. Cromm PM, Spiegel J, Grossmann TN, Waldmann H. Direct Modulation of Small GTPase Activity and Function. Angew Chem Int Ed Engl (2015) 54:13516–37. doi: 10.1002/anie.201504357 [DOI] [PubMed] [Google Scholar]
- 29. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature (2013) 503:548–51. doi: 10.1038/nature12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong Y, Lu J, Hunter J, Li L, Scott D, Choi HG, et al. Covalent Guanosine Mimetic Inhibitors of G12C KRAS. ACS Med Chem Lett (2017) 8:61–6. doi: 10.1021/acsmedchemlett.6b00373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, et al. Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C. Cell Chem Biol (2017) 24:1005–1016.e3. doi: 10.1016/j.chembiol.2017.06.017 [DOI] [PubMed] [Google Scholar]
- 32. Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective Inhibition of Oncogenic KRAS Output With Small Molecules Targeting the Inactive State. Cancer Discov (2016) 6:316–29. doi: 10.1158/2159-8290.CD-15-1105 [DOI] [PubMed] [Google Scholar]
- 33. Roberts PJ, Stinchcombe TE. KRAS Mutation: Should We Test for It, and Does It Matter? J Clin Oncol (2013) 31:1112–21. doi: 10.1200/JCO.2012.43.0454 [DOI] [PubMed] [Google Scholar]
- 34. Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of Ras to Phosphoinositide 3-Kinase P110alpha Is Required for Ras-Driven Tumorigenesis in Mice. Cell (2007) 129:957–68. doi: 10.1016/j.cell.2007.03.051 [DOI] [PubMed] [Google Scholar]
- 35. Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, et al. Role of Phosphoinositide 3-OH Kinase in Cell Transformation and Control of the Actin Cytoskeleton by Ras. Cell (1997) 89:457–67. doi: 10.1016/S0092-8674(00)80226-3 [DOI] [PubMed] [Google Scholar]
- 36. van der Weyden L, Adams DJ. The Ras-Association Domain Family (RASSF) Members and Their Role in Human Tumourigenesis. Biochim Biophys Acta (2007) 1776:58–85. doi: 10.1016/j.bbcan.2007.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lambert JM, Lambert QT, Reuther GW, Malliri A, Siderovski DP, Sondek J, et al. Tiam1 Mediates Ras Activation of Rac by a PI(3)K-Independent Mechanism. Nat Cell Biol (2002) 4:621–5. doi: 10.1038/ncb833 [DOI] [PubMed] [Google Scholar]
- 38. Passiglia F, Malapelle U, Del Re M, Righi L, Pagni F, Furlan D, et al. KRAS Inhibition in Non-Small Cell Lung Cancer: Past Failures, New Findings and Upcoming Challenges. Eur J Cancer (2020) 137:57–68. doi: 10.1016/j.ejca.2020.06.023 [DOI] [PubMed] [Google Scholar]
- 39. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res (2019) 47:941–7. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karachaliou N, Mayo C, Costa C, Magrí I, Gimenez-Capitan A, Molina-Vila MA, et al. KRAS Mutations in Lung Cancer. Clin Lung Cancer (2013) 14:205–14. doi: 10.1016/j.cllc.2012.09.007 [DOI] [PubMed] [Google Scholar]
- 41. Liu Y, Li H, Zhu J, Zhang Y, Liu X, Li R, et al. The Prevalence and Concurrent Pathogenic Mutations of KRAS (G12C) in Northeast Chinese Non-Small-Cell Lung Cancer Patients. Cancer Manag Res (2021) 13:2447–54. doi: 10.2147/CMAR.S282617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poulin EJ, Bera AK, Lu J, Lin Y-J, Strasser SD, Paulo JA, et al. Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov (2019) 9:738–55. doi: 10.1158/2159-8290.CD-18-1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nadal E, Chen G, Prensner JR, Shiratsuchi H, Sam C, Zhao L, et al. KRAS-G12C Mutation Is Associated With Poor Outcome in Surgically Resected Lung Adenocarcinoma. J Thorac Oncol (2014) 9:1513–22. doi: 10.1097/JTO.0000000000000305 [DOI] [PubMed] [Google Scholar]
- 44. Xu S, Long BN, Boris GH, Chen A, Ni S, Kennedy MA, et al. Structural Insight Into the Rearrangement of the Switch I Region in GTP-Bound G12A K-Ras. Acta Crystallogr D Struct Biol (2017) 73:970–84. doi: 10.1107/S2059798317015418 [DOI] [PubMed] [Google Scholar]
- 45. Cruz-Migoni A, Canning P, Quevedo CE, Bataille CJR, Bery N, Miller A, et al. Structure-Based Development of New RAS-Effector Inhibitors From a Combination of Active and Inactive RAS-Binding Compounds. Proc Natl Acad Sci USA (2019) 116:2545–50. doi: 10.1073/pnas.1811360116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–A Visualization System for Exploratory Research and Analysis. J Comput Chem (2004) 25:1605–12. doi: 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 47. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. J Natl Cancer Inst (2012) 104:228–39. doi: 10.1093/jnci/djr523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garassino MC, Marabese M, Rusconi P, Rulli E, Martelli O, Farina G, et al. Different Types of K-Ras Mutations Could Affect Drug Sensitivity and Tumour Behaviour in Non-Small-Cell Lung Cancer. Ann Oncol (2011) 22:235–7. doi: 10.1093/annonc/mdq680 [DOI] [PubMed] [Google Scholar]
- 49. Jia Y, Jiang T, Li X, Zhao C, Zhang L, Zhao S, et al. Characterization of Distinct Types of KRAS Mutation and Its Impact on First-Line Platinum-Based Chemotherapy in Chinese Patients With Advanced Non-Small Cell Lung Cancer. Oncol Lett (2017) 14:6525–32. doi: 10.3892/ol.2017.7016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cserepes M, Ostoros G, Lohinai Z, Raso E, Barbai T, Timar J, et al. Subtype-Specific KRAS Mutations in Advanced Lung Adenocarcinoma: A Retrospective Study of Patients Treated With Platinum-Based Chemotherapy. Eur J Cancer (2014) 50:1819–28. doi: 10.1016/j.ejca.2014.04.001 [DOI] [PubMed] [Google Scholar]
- 51. Sun JM, Hwang DW, Ahn JS, Ahn M-J, Park K. Prognostic and Predictive Value of KRAS Mutations in Advanced Non-Small Cell Lung Cancer. PloS One (2013) 8:e64816. doi: 10.1371/journal.pone.0064816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ghimessy AK, Gellert A, Schlegl E, Hegedus B, Raso E, Barbai T, et al. KRAS Mutations Predict Response and Outcome in Advanced Lung Adenocarcinoma Patients Receiving First-Line Bevacizumab and Platinum-Based Chemotherapy. Cancers (Basel) (2019) 11(10):1514. doi: 10.3390/cancers11101514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers With KRAS P.G12C Mutation. N Engl J Med (2021) 384:2371–81. doi: 10.1056/NEJMoa2103695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jeanson A, Tomasini P, Souquet-Bressand M, Brandone N, Boucekine M, Grangeon M, et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J Thorac Oncol (2019) 14:1095–101. doi: 10.1016/j.jtho.2019.01.011 [DOI] [PubMed] [Google Scholar]
- 55. Janne PA, Smith I, McWalter G, Mann H, Dougherty B, Walker J, et al. Impact of KRAS Codon Subtypes From a Randomised Phase II Trial of Selumetinib Plus Docetaxel in KRAS Mutant Advanced Non-Small-Cell Lung Cancer. Br J Cancer (2015) 113:199–203. doi: 10.1038/bjc.2015.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rodenhuis S, Boerrigter L, Top B, Slebos RJ, Mooi WJ, Veer Lv, et al. Mutational Activation of the K-Ras Oncogene and the Effect of Chemotherapy in Advanced Adenocarcinoma of the Lung: A Prospective Study. J Clin Oncol (1997) 15:285–91. doi: 10.1200/JCO.1997.15.1.285 [DOI] [PubMed] [Google Scholar]
- 57. Schiller JH, Adak S, Feins RH, Keller SM, Fry WA, Livingston RB, et al. Lack of Prognostic Significance of P53 and K-Ras Mutations in Primary Resected Non-Small-Cell Lung Cancer on E4592: A Laboratory Ancillary Study on an Eastern Cooperative Oncology Group Prospective Randomized Trial of Postoperative Adjuvant Therapy. J Clin Oncol (2001) 19:448–57. doi: 10.1200/JCO.2001.19.2.448 [DOI] [PubMed] [Google Scholar]
- 58. Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, et al. Mutations in the Epidermal Growth Factor Receptor and in KRAS Are Predictive and Prognostic Indicators in Patients With Non-Small-Cell Lung Cancer Treated With Chemotherapy Alone and in Combination With Erlotinib. J Clin Oncol (2005) 23:5900–9. doi: 10.1200/JCO.2005.02.857 [DOI] [PubMed] [Google Scholar]
- 59. Tsao MS, Aviel-Ronen S, Ding K, Lau D, Liu N, Sakurada A, et al. Prognostic and Predictive Importance of P53 and RAS for Adjuvant Chemotherapy in Non Small-Cell Lung Cancer. J Clin Oncol (2007) 25:5240–7. doi: 10.1200/JCO.2007.12.6953 [DOI] [PubMed] [Google Scholar]
- 60. Shepherd FA, Domerg C, Hainaut P, Jänne PA, Pignon J-P, Graziano S, et al. Pooled Analysis of the Prognostic and Predictive Effects of KRAS Mutation Status and KRAS Mutation Subtype in Early-Stage Resected Non-Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J Clin Oncol (2013) 31:2173–81. doi: 10.1200/JCO.2012.48.1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Metro G, Chiari R, Bennati C, Cenci M, Ricciuti B, Puma F, et al. Clinical Outcome With Platinum-Based Chemotherapy in Patients With Advanced Nonsquamous EGFR Wild-Type Non-Small-Cell Lung Cancer Segregated According to KRAS Mutation Status. Clin Lung Cancer (2014) 15:86–92. doi: 10.1016/j.cllc.2013.08.002 [DOI] [PubMed] [Google Scholar]
- 62. Kranenburg O, Gebbink MF, Voest EE. Stimulation of Angiogenesis by Ras Proteins. Biochim Biophys Acta (2004) 1654:23–37. doi: 10.1016/j.bbcan.2003.09.004 [DOI] [PubMed] [Google Scholar]
- 63. Tang Y, Kim M, Carrasco D, Kung AL, Chin L, Weissleder R. In Vivo Assessment of RAS-Dependent Maintenance of Tumor Angiogenesis by Real-Time Magnetic Resonance Imaging. Cancer Res (2005) 65:8324–30. doi: 10.1158/0008-5472.CAN-05-0027 [DOI] [PubMed] [Google Scholar]
- 64. Sparmann A, Bar-Sagi D. Ras-Induced Interleukin-8 Expression Plays a Critical Role in Tumor Growth and Angiogenesis. Cancer Cell (2004) 6:447–58. doi: 10.1016/j.ccr.2004.09.028 [DOI] [PubMed] [Google Scholar]
- 65. Matsuo Y, Campbell PM, Brekken RA, Sung B, Ouellette MM, Fleming JB, et al. K-Ras Promotes Angiogenesis Mediated by Immortalized Human Pancreatic Epithelial Cells Through Mitogen-Activated Protein Kinase Signaling Pathways. Mol Cancer Res (2009) 7:799–808. doi: 10.1158/1541-7786.MCR-08-0577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sunaga N, Imai H, Shimizu K, Shames DS, Kakegawa S, Girard L, et al. Oncogenic KRAS-Induced Interleukin-8 Overexpression Promotes Cell Growth and Migration and Contributes to Aggressive Phenotypes of Non-Small Cell Lung Cancer. Int J Cancer (2012) 130:1733–44. doi: 10.1002/ijc.26164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, et al. RAS Interaction With PI3K P110α Is Required for Tumor-Induced Angiogenesis. J Clin Invest (2014) 124:3601–11. doi: 10.1172/JCI74134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Al-Abd AM, Alamoudi AJ, Abdel-Naim AB, Neamatallah TA, Ashour OM. Anti-Angiogenic Agents for the Treatment of Solid Tumors: Potential Pathways, Therapy and Current Strategies - A Review. J Adv Res (2017) 8:591–605. doi: 10.1016/j.jare.2017.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kong DH, Kim MR, Jang JH, Na H-J, Lee S. A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy. Int J Mol Sci (2017) 18(8):1786. doi: 10.3390/ijms18081786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Okada F, Rak JW, Croix BS, Lieubeau B, Kaya M, Roncari L, et al. Impact of Oncogenes in Tumor Angiogenesis: Mutant K-Ras Up-Regulation of Vascular Endothelial Growth Factor/Vascular Permeability Factor Is Necessary, But Not Sufficient for Tumorigenicity of Human Colorectal Carcinoma Cells. Proc Natl Acad Sci USA (1998) 95:3609–14. doi: 10.1073/pnas.95.7.3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Grunstein J, Roberts WG, Mathieu-Costello O, Hanahan D, Johnson RS. Tumor-Derived Expression of Vascular Endothelial Growth Factor Is a Critical Factor in Tumor Expansion and Vascular Function. Cancer Res (1999) 59:1592–8. [PubMed] [Google Scholar]
- 72. Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential Role for Oncogenic Ras in Tumour Maintenance. Nature (1999) 400:468–72. doi: 10.1038/22788 [DOI] [PubMed] [Google Scholar]
- 73. Chaft JE, Rusch V, Ginsberg MS, Paik PK, Finley DJ, Kris MG, et al. Phase II Trial of Neoadjuvant Bevacizumab Plus Chemotherapy and Adjuvant Bevacizumab in Patients With Resectable Nonsquamous Non-Small-Cell Lung Cancers. J Thorac Oncol (2013) 8:1084–90. doi: 10.1097/JTO.0b013e31829923ec [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Metro G, Chiari R, Duranti S, Siggillino A, Fischer MJ, Giannarelli D, et al. Impact of Specific Mutant KRAS on Clinical Outcome of EGFR-TKI-Treated Advanced Non-Small Cell Lung Cancer Patients With an EGFR Wild Type Genotype. Lung Cancer (2012) 78:81–6. doi: 10.1016/j.lungcan.2012.06.005 [DOI] [PubMed] [Google Scholar]
- 75. Zer A, Ding K, Lee SM, Goss GD, Seymour L, Ellis PM, et al. Pooled Analysis of the Prognostic and Predictive Value of KRAS Mutation Status and Mutation Subtype in Patients With Non-Small Cell Lung Cancer Treated With Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. J Thorac Oncol (2016) 11:312–23. doi: 10.1016/j.jtho.2015.11.010 [DOI] [PubMed] [Google Scholar]
- 76. Fiala O, Pesek M, Finek J, Benesova L, Belsanova B, Minarik M. The Dominant Role of G12C Over Other KRAS Mutation Types in the Negative Prediction of Efficacy of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Cancer Genet (2013) 206:26–31. doi: 10.1016/j.cancergen.2012.12.003 [DOI] [PubMed] [Google Scholar]
- 77. Mao C, Qiu LX, Liao RY, Du F-B, Ding H, Yang W-C, et al. KRAS Mutations and Resistance to EGFR-TKIs Treatment in Patients With Non-Small Cell Lung Cancer: A Meta-Analysis of 22 Studies. Lung Cancer (2010) 69:272–8. doi: 10.1016/j.lungcan.2009.11.020 [DOI] [PubMed] [Google Scholar]
- 78. Ni D, Li X, He X, Zhang H, Zhang J, Lu S, et al. Drugging K-Ras(G12C) Through Covalent Inhibitors: Mission Possible? Pharmacol Ther (2019) 202:1–17. doi: 10.1016/j.pharmthera.2019.06.007 [DOI] [PubMed] [Google Scholar]
- 79. Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers With a Covalent G12C-Specific Inhibitor. Cell (2018) 172:578–89.e17. doi: 10.1016/j.cell.2018.01.006 [DOI] [PubMed] [Google Scholar]
- 80. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight Toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery (2020) 10:54–71. doi: 10.1158/2159-8290.CD-19-1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nagasaka M, Li Y, Sukari A, Ou S-HI, Al-Hallak MN, Azmi AS. KRAS G12C Game of Thrones, Which Direct KRAS Inhibitor Will Claim the Iron Throne? Cancer Treat Rev (2020) 84:101974. doi: 10.1016/j.ctrv.2020.101974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. D'Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, et al. PD-1 and PD-L1 Expression in Molecularly Selected Non-Small-Cell Lung Cancer Patients. Br J Cancer (2015) 112:95–102. doi: 10.1038/bjc.2014.555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu S-Y, et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res (2017) 23:3012–24. doi: 10.1158/1078-0432.CCR-16-2554 [DOI] [PubMed] [Google Scholar]
- 84. Lee M-H, Yanagawa J, Li R, Walser TC, Krysan K, Wang G, et al. Increased PD-L1 Expression in KRAS Mutated Premalignant Human Bronchial Epithelial Cells Is Enhanced by LKB1 Loss and Mediated by ERK Activation. J ImmunoTher Cancer (2015) 3(Suppl 2):P305. doi: 10.1186/2051-1426-3-S2-P305 [DOI] [Google Scholar]
- 85. Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, et al. KRAS Mutation-Induced Upregulation of PD-L1 Mediates Mmune Escape in Human Lung Adenocarcinoma. Cancer Immunol Immunother (2017) 66:1175–87. doi: 10.1007/s00262-017-2005-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wu ZX, Liu Z, Jiang HL, Pan H-M, Han W-D. Circulating Tumor Cells Predict Survival Benefit From Chemotherapy in Patients With Lung Cancer. Oncotarget (2016) 7:67586–96. doi: 10.18632/oncotarget.11707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Muinelo-Romay L, Vieito M, Abalo A, Nocelo MA, Barón F, Anido U, et al. Evaluation of Circulating Tumor Cells and Related Events as Prognostic Factors and Surrogate Biomarkers in Advanced NSCLC Patients Receiving First-Line Systemic Treatment. Cancers (Basel) (2014) 6:153–65. doi: 10.3390/cancers6010153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Guibert N, Delaunay M, Lusque A, Boubekeur N, Rouquette I, Clermont E, et al. PD-L1 Expression in Circulating Tumor Cells of Advanced Non-Small Cell Lung Cancer Patients Treated With Nivolumab. Lung Cancer (2018) 120:108–12. doi: 10.1016/j.lungcan.2018.04.001 [DOI] [PubMed] [Google Scholar]
- 89. Wang Y, Kim TH, Fouladdel S, Zhang Z, Soni P, Qin A, et al. PD-L1 Expression in Circulating Tumor Cells Increases During Radio(chemo)therapy and Indicates Poor Prognosis in Non-Small Cell Lung Cancer. Sci Rep (2019) 9:566. doi: 10.1038/s41598-018-36096-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nicolazzo C, Raimondi C, Mancini M, Caponnetto S, Gradilone A, Gandini O, et al. Monitoring PD-L1 Positive Circulating Tumor Cells in Non-Small Cell Lung Cancer Patients Treated With the PD-1 Inhibitor Nivolumab. Sci Rep (2016) 6:31726. doi: 10.1038/srep31726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tashima Y, Kuwata T, Yoneda K, Hirai A, Mori M, Kanayama M, et al. Prognostic Impact of PD-L1 Expression in Correlation With Neutrophil-to-Lymphocyte Ratio in Squamous Cell Carcinoma of the Lung. Sci Rep (2020) 10:1243. doi: 10.1038/s41598-019-57321-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zugazagoitia J, Gupta S, Liu Y, Fuhrman K, Gettinger S, Herbst RS, et al. Biomarkers Associated With Beneficial PD-1 Checkpoint Blockade in Non-Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin Cancer Res (2020) 26:4360–8. doi: 10.1158/1078-0432.CCR-20-0175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, et al. Dendritic Cells Dictate Responses to PD-L1 Blockade Cancer Immunotherapy. Sci Transl Med (2020) 12(534):eaav7431. doi: 10.1126/scitranslmed.aav7431 [DOI] [PubMed] [Google Scholar]
- 94. Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, et al. Immune Cell PD-L1 Colocalizes With Macrophages and Is Associated With Outcome in PD-1 Pathway Blockade Therapy. Clin Cancer Res (2020) 26:970–7. doi: 10.1158/1078-0432.CCR-19-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. O'Callaghan DS, Rexhepaj E, Gately K, Coate L, Delaney D, O'Donnell DM, et al. Tumour Islet Foxp3+ T-Cell Infiltration Predicts Poor Outcome in Nonsmall Cell Lung Cancer. Eur Respir J (2015) 46:1762–72. doi: 10.1183/13993003.00176-2014 [DOI] [PubMed] [Google Scholar]
- 96. Swanton C. Take Lessons From Cancer Evolution to the Clinic. Nature (2020) 581:382–3. doi: 10.1038/d41586-020-01347-z [DOI] [PubMed] [Google Scholar]
- 97. Poutahidis T, Erdman SE. Commensal Bacteria Modulate the Tumor Microenvironment. Cancer Lett (2016) 380:356–8. doi: 10.1016/j.canlet.2015.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-Derived Inosine Modulates Response to Checkpoint Inhibitor Immunotherapy. Science (2020) 369:1481–9. doi: 10.1126/science.abc3421 [DOI] [PubMed] [Google Scholar]
- 99. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal Microbiota-Derived Short-Chain Fatty Acids Regulation of Immune Cell IL-22 Production and Gut Immunity. Nat Commun (2020) 11:4457. doi: 10.1038/s41467-020-18262-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ohtani N. Microbiome and Cancer. Semin Immunopathol (2015) 37:65–72. doi: 10.1007/s00281-014-0457-1 [DOI] [PubMed] [Google Scholar]
- 101. Dibra D, Xia X, Mitra A, Cutrera JJ, Lozano G, Li S. Mutant P53 in Concert With an Interleukin-27 Receptor Alpha Deficiency Causes Spontaneous Liver Inflammation, Fibrosis, and Steatosis in Mice. Hepatology (2016) 63:1000–12. doi: 10.1002/hep.28379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS Oncogenes: Weaving a Tumorigenic Web. Nat Rev Cancer (2011) 11:761–74. doi: 10.1038/nrc3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Castellano E, Downward J. RAS Interaction With PI3K: More Than Just Another Effector Pathway. Genes Cancer (2011) 2:261–74. doi: 10.1177/1947601911408079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Smith MJ, Ikura M. Integrated RAS Signaling Defined by Parallel NMR Detection of Effectors and Regulators. Nat Chem Biol (2014) 10:223–30. doi: 10.1038/nchembio.1435 [DOI] [PubMed] [Google Scholar]
- 105. Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, et al. Genomic and Biological Characterization of Exon 4 KRAS Mutations in Human Cancer. Cancer Res (2010) 70:5901–11. doi: 10.1158/0008-5472.CAN-10-0192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Caiola E, Brunelli L, Marabese M, Broggini M, Lupi M, Pastorelli R. Different Metabolic Responses to PI3K Inhibition in NSCLC Cells Harboring Wild-Type and G12C Mutant KRAS. Oncotarget (2016) 7:51462–72. doi: 10.18632/oncotarget.9849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, et al. The Superior Efficacy of Anti-PD-1/PD-L1 Immunotherapy in KRAS-Mutant Non-Small Cell Lung Cancer That Correlates With an Inflammatory Phenotype and Increased Immunogenicity. Cancer Lett (2020) 470:95–105. doi: 10.1016/j.canlet.2019.10.027 [DOI] [PubMed] [Google Scholar]
- 108. Nieto P, Ambrogio C, Esteban-Burgos L, Gómez-López G, Blasco MT, Yao Z, et al. A Braf Kinase-Inactive Mutant Induces Lung Adenocarcinoma. Nature (2017) 548:239–43. doi: 10.1038/nature23297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, et al. Lung Cancer: Intragenic ERBB2 Kinase Mutations in Tumours. Nature (2004) 431:525–6. doi: 10.1038/431525b [DOI] [PubMed] [Google Scholar]
- 110. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov (2018) 8:822–35. doi: 10.1158/2159-8290.CD-18-0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Alessi JV, Ricciuti B, Spurr LF, Gupta H, Li YY, Glass C, et al. SMARCA4 and Other SWItch/Sucrose NonFermentable Family Genomic Alterations in NSCLC: Clinicopathologic Characteristics and Outcomes to Immune Checkpoint Inhibition. J Thorac Oncol (2021) 16:1176–87. doi: 10.1016/j.jtho.2021.03.024 [DOI] [PubMed] [Google Scholar]
- 112. Liu L, Ahmed T, Petty WJ, Grant S, Ruiz J, Lycan TW, et al. SMARCA4 Mutations in KRAS-Mutant Lung Adenocarcinoma: A Multi-Cohort Analysis. Mol Oncol (2021) 15:462–72. doi: 10.1002/1878-0261.12831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-Occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma With Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov (2015) 5:860–77. doi: 10.1158/2159-8290.CD-14-1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Shackelford DB, Shaw RJ. The LKB1-AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nat Rev Cancer (2009) 9:563–75. doi: 10.1038/nrc2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-Cell Activity in the Lung Tumor Microenvironment. Cancer Res (2016) 76:999–1008. doi: 10.1158/0008-5472.CAN-15-1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, et al. Whole-Exome Sequencing and Immune Profiling of Early-Stage Lung Adenocarcinoma With Fully Annotated Clinical Follow-Up. Ann Oncol (2017) 28:75–82. doi: 10.1093/annonc/mdw436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. La Fleur L, Falk-Sorqvist E, Smeds P, Berglund A, Sundström M, Mattsson JS, et al. Mutation Patterns in a Population-Based Non-Small Cell Lung Cancer Cohort and Prognostic Impact of Concomitant Mutations in KRAS and TP53 or STK11. Lung Cancer (2019) 130:50–8. doi: 10.1016/j.lungcan.2019.01.003 [DOI] [PubMed] [Google Scholar]
- 118. Schabath MB, Welsh EA, Fulp WJ, Chen L, Teer JK, Thompson ZJ, et al. Differential Association of STK11 and TP53 With KRAS Mutation-Associated Gene Expression, Proliferation and Immune Surveillance in Lung Adenocarcinoma. Oncogene (2016) 35:3209–16. doi: 10.1038/onc.2015.375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Skoulidis F, Heymach JV. Co-Occurring Genomic Alterations in Non-Small-Cell Lung Cancer Biology and Therapy. Nat Rev Cancer (2019) 19:495–509. doi: 10.1038/s41568-019-0179-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chen X, Su C, Ren S, Zhou C, Jiang T. Pan-Cancer Analysis of KEAP1 Mutations as Biomarkers for Immunotherapy Outcomes. Ann Transl Med (2020) 8:141. doi: 10.21037/atm.2019.11.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, et al. High-Throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell (2016) 30:214–28. doi: 10.1016/j.ccell.2016.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Cancer Genome Atlas Research Network . Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature (2014) 511:543–50. doi: 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 Interaction in Non-Small-Cell Lung Cancer. PloS Med (2006) 3:e420. doi: 10.1371/journal.pmed.0030420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. Keap1 Loss Promotes Kras-Driven Lung Cancer and Results in Dependence on Glutaminolysis. Nat Med (2017) 23:1362–8. doi: 10.1038/nm.4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bauer AK, Cho HY, Miller-Degraff L, Walker C, Helms K, Fostel J, et al. Targeted Deletion of Nrf2 Reduces Urethane-Induced Lung Tumor Development in Mice. PloS One (2011) 6:e26590. doi: 10.1371/journal.pone.0026590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature (2011) 475:106–9. doi: 10.1038/nature10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Satoh H, Moriguchi T, Takai J, Nilsson JA, Lindahl P, Bergo MO. Nrf2 Prevents Initiation But Accelerates Progression Through the Kras Signaling Pathway During Lung Carcinogenesis. Cancer Res (2013) 73:4158–68. doi: 10.1158/0008-5472.CAN-12-4499 [DOI] [PubMed] [Google Scholar]
- 128. Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci Transl Med (2014) 6:221ra15. doi: 10.1126/scitranslmed.3007653 [DOI] [PubMed] [Google Scholar]
- 129. Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell (2016) 166:963–76. doi: 10.1016/j.cell.2016.06.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Satoh H, Moriguchi T, Saigusa D, Baird L, Yu L, Rokutan H, et al. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, But After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res (2016) 76:3088–96. doi: 10.1158/0008-5472.CAN-15-1584 [DOI] [PubMed] [Google Scholar]
- 131. Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras Copy Number Defines Metabolic Reprogramming and Therapeutic Susceptibilities. Nature (2016) 531:110–3. doi: 10.1038/nature16967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cancer Genome Atlas Research Network .Comprehensive Genomic Characterization of Squamous Cell Lung Cancers. Nature (2012) 489:519–25. doi: 10.1038/nature11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Jaramillo MC, Zhang DD. The Emerging Role of the Nrf2-Keap1 Signaling Pathway in Cancer. Genes Dev (2013) 27:2179–91. doi: 10.1101/gad.225680.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Konstantinopoulos PA, Spentzos D, Fountzilas E, Francoeur N, Sanisetty S, Grammatikos AP, et al. Keap1 Mutations and Nrf2 Pathway Activation in Epithelial Ovarian Cancer. Cancer Res (2011) 71:5081–9. doi: 10.1158/0008-5472.CAN-10-4668 [DOI] [PubMed] [Google Scholar]
- 135. Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, et al. Genetic Alteration of Keap1 Confers Constitutive Nrf2 Activation and Resistance to Chemotherapy in Gallbladder Cancer. Gastroenterology (2008) 135:1358–1368, 1368.e1-4. doi: 10.1053/j.gastro.2008.06.082 [DOI] [PubMed] [Google Scholar]
- 136. Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, et al. Oncogenic NRF2 Mutations in Squamous Cell Carcinomas of Oesophagus and Skin. J Pathol (2010) 220:446–51. doi: 10.1002/path.2653 [DOI] [PubMed] [Google Scholar]
- 137. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated Molecular Analysis of Clear-Cell Renal Cell Carcinoma. Nat Genet (2013) 45:860–7. doi: 10.1038/ng.2699 [DOI] [PubMed] [Google Scholar]
- 138. Fabrizio FP, Costantini M, Copetti M, la Torre A, Sparaneo A, Fontana A, et al. Keap1/Nrf2 Pathway in Kidney Cancer: Frequent Methylation of KEAP1 Gene Promoter in Clear Renal Cell Carcinoma. Oncotarget (2017) 8:11187–98. doi: 10.18632/oncotarget.14492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Muscarella LA, Barbano R, D'Angelo V, Copetti M, Coco M, Balsamo T, et al. Regulation of KEAP1 Expression by Promoter Methylation in Malignant Gliomas and Association With Patient's Outcome. Epigenetics (2011) 6:317–25. doi: 10.4161/epi.6.3.14408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hanada N, Takahata T, Zhou Q, Ye X, Sun R, Itoh J, et al. Methylation of the KEAP1 Gene Promoter Region in Human Colorectal Cancer. BMC Cancer (2012) 12:66. doi: 10.1186/1471-2407-12-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Goldstein LD, Lee J, Gnad F, Klijn C, Schaub A, Reeder J, et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep (2016) 16:2605–17. doi: 10.1016/j.celrep.2016.08.010 [DOI] [PubMed] [Google Scholar]
- 142. Krall EB, Wang B, Munoz DM, Ilic N, Raghavan S, Niederst MJ, et al. KEAP1 Loss Modulates Sensitivity to Kinase Targeted Therapy in Lung Cancer. Elife (2017) 6:e33173. doi: 10.7554/eLife.33173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming With Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res (2019) 79:3251–67. doi: 10.1158/0008-5472.CAN-18-3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Singh A, Daemen A, Nickles D, Jeon S-M, Foreman O, Sudini K, et al. NRF2 Activation Promotes Aggressive Lung Cancer and Associates With Poor Clinical Outcomes. Clin Cancer Res (2021) 27:877–88. doi: 10.1158/1078-0432.CCR-20-1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of Co-Occurring Genomic Alterations on Outcomes in Patients With KRAS-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res (2018) 24:334–40. doi: 10.1158/1078-0432.CCR-17-1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Negrao MV, Lam VK, Reuben A, Rubin ML, Landry LL, Roarty EB, et al. PD-L1 Expression, Tumor Mutational Burden, and Cancer Gene Mutations Are Stronger Predictors of Benefit From Immune Checkpoint Blockade Than HLA Class I Genotype in Non-Small Cell Lung Cancer. J Thorac Oncol (2019) 14:1021–31. doi: 10.1016/j.jtho.2019.02.008 [DOI] [PubMed] [Google Scholar]
- 147. Jeong Y, Hellyer JA, Stehr H, Hoang NT, Niu X, Das M, et al. Role of KEAP1/NFE2L2 Mutations in the Chemotherapeutic Response of Patients With Non-Small Cell Lung Cancer. Clin Cancer Res (2020) 26:274–81. doi: 10.1158/1078-0432.CCR-19-1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wilson BG, Roberts CW. SWI/SNF Nucleosome Remodellers and Cancer. Nat Rev Cancer (2011) 11:481–92. doi: 10.1038/nrc3068 [DOI] [PubMed] [Google Scholar]
- 149. Becker PB, Workman JL. Nucleosome Remodeling and Epigenetics. Cold Spring Harb Perspect Biol (2013) 5(9):a017905. doi: 10.1101/cshperspect.a017905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and Bioinformatic Analysis of Mammalian SWI/SNF Complexes Identifies Extensive Roles in Human Malignancy. Nat Genet (2013) 45:592–601. doi: 10.1038/ng.2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Helming KC, Wang X, Roberts CWM. Vulnerabilities of Mutant SWI/SNF Complexes in Cancer. Cancer Cell (2014) 26:309–17. doi: 10.1016/j.ccr.2014.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the Hallmarks of Lung Adenocarcinoma With Massively Parallel Sequencing. Cell (2012) 150:1107–20. doi: 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hodges HC, Stanton BZ, Cermakova K, Chang C-Y, Miller EL, Kirkland JG, et al. Dominant-Negative SMARCA4 Mutants Alter the Accessibility Landscape of Tissue-Unrestricted Enhancers. Nat Struct Mol Biol (2018) 25:61–72. doi: 10.1038/s41594-017-0007-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Shain AH, Pollack JR. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PloS One (2013) 8:e55119. doi: 10.1371/journal.pone.0055119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Rodriguez-Nieto S, Cañada A, Pros E, Pinto AI, Torres-Lanzas J, Lopez-Rios F, et al. Massive Parallel DNA Pyrosequencing Analysis of the Tumor Suppressor BRG1/SMARCA4 in Lung Primary Tumors. Hum Mutat (2011) 32:E1999–2017. doi: 10.1002/humu.21415 [DOI] [PubMed] [Google Scholar]
- 156. Orvis T, Hepperla A, Walter V, Song S, Simon J, Parker J, et al. BRG1/SMARCA4 Inactivation Promotes Non-Small Cell Lung Cancer Aggressiveness by Altering Chromatin Organization. Cancer Res (2014) 74:6486–98. doi: 10.1158/0008-5472.CAN-14-0061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Bell EH, Chakraborty AR, Mo X, Liu Z, Shilo K, Kirste S, et al. SMARCA4/BRG1 Is a Novel Prognostic Biomarker Predictive of Cisplatin-Based Chemotherapy Outcomes in Resected Non-Small Cell Lung Cancer. Clin Cancer Res (2016) 22:2396–404. doi: 10.1158/1078-0432.CCR-15-1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Dagogo-Jack I, Schrock AB, Kem M, Jessop N, Lee J, Ali SM, et al. Clinicopathologic Characteristics of BRG1-Deficient NSCLC. J Thorac Oncol (2020) 15:766–76. doi: 10.1016/j.jtho.2020.01.002 [DOI] [PubMed] [Google Scholar]
- 159. Gupta M, Concepcion CP, Fahey CG, Keshishian H, Bhutkar A, Brainson CF, et al. BRG1 Loss Predisposes Lung Cancers to Replicative Stress and ATR Dependency. Cancer Res (2020) 80:3841–54. doi: 10.1158/0008-5472.CAN-20-1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Lissanu Deribe Y, Sun Y, Terranova C, Khan F, Martinez-Ledesma J, Gay J, et al. Mutations in the SWI/SNF Complex Induce a Targetable Dependence on Oxidative Phosphorylation in Lung Cancer. Nat Med (2018) 24:1047–57. doi: 10.1038/s41591-018-0019-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Schoenfeld AJ, Bandlamudi C, Lavery JA, Montecalvo J, Namakydoust A, Rizvi H, et al. The Genomic Landscape of SMARCA4 Alterations and Associations With Outcomes in Patients With Lung Cancer. Clin Cancer Res (2020) 26:5701–8. doi: 10.1158/1078-0432.CCR-20-1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Xue Y, Meehan B, Fu Z, Wang XQD, Fiset PO, Rieker R, et al. SMARCA4 Loss Is Synthetic Lethal With CDK4/6 Inhibition in Non-Small Cell Lung Cancer. Nat Commun (2019) 10:557. doi: 10.1038/s41467-019-08380-1 [DOI] [PMC free article] [PubMed] [Google Scholar]