

Abstract
Background/Aims:
Molecular mechanisms underlying the different susceptibility of men and women to NAFLD are poorly understood. The TTC39B locus encodes a scaffolding protein, associates with gynecological disorders and its deletion protects mice from diet-induced steatohepatitis. This study aimed to elucidate the molecular mechanisms linking TTC39B (T39) to the expression of lipogenic genes and to explore sex-specific effects.
Methods:
Co-expression in HEK293A cells validated the novel T39/pRb interaction predicted by a protein-protein interaction algorithm. T39 was knocked down using antisense oligonucleotide (ASO) in dietary NAFLD mice with genetic deficiency of pRb or its downstream effector E2F1 and in primary human hepatocytes.
Results:
T39 interacts with pRb via its C-terminal TPR domain and promotes its proteasomal degradation. In female mice T39 deficiency reduces the mRNA of lipogenic genes, especially Pnpla3, in a pRb- and E2F1-dependent manner. In contrast, in male mice, T39 deficiency results in a much smaller reduction in lipogenic gene expression that is independent of pRb/E2F1. T39 also interacts with VAPB via an N-terminal FFAT motif and stabilizes the interaction of VAPB with SCAP. Ovariectomy abolishes the effect of T39 knockdown on the hepatic pRb/E2F1/Pnpla3 axis. In both sexes T39 knockdown reduces SCAP independent of pRb. In human primary hepatocytes, T39 knockdown reduces expression of PNPLA3 and other lipogenic genes in women but not men.
Conclusions:
We have uncovered a conserved sexual dimorphism in the regulation of hepatic lipogenic genes with effects of T39 mediated through pRb/E2F1 in females and VAPB/SCAP in both sexes. T39 inhibition could be a novel strategy to downregulate PNPLA3 and treat NAFLD in women.
Keywords: TTC39B, lipogenesis, retinoblastoma protein, non-alcoholic fatty liver disease, sterol regulatory element binding protein 1, PNPLA3, SCAP, E2F1, sex characteristics
LAY SUMMARY
In females, TTC39B degrades a tumor suppressor in the liver to promote the synthesis of new fat and expression of a major genetic risk factor for nonalcoholic fatty liver disease. TTC39B is a potential therapeutic target for nonalcoholic fatty liver disease, especially in women.
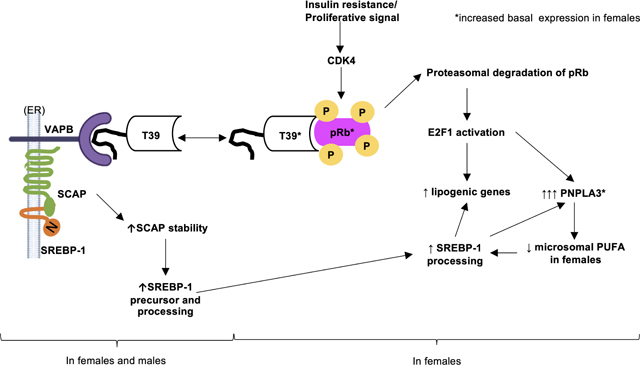
Graphical Abstract
INTRODUCTION
The liver is the most sexually dimorphic metabolic organ [1]. While non-invasive measures indicate a higher NAFLD prevalence in men, females are more likely to have histologically-defined non-alcoholic steatohepatitis (NASH) and women over age 50 have excessive risk of developing advanced hepatic fibrosis [2]. Since the discovery of its association with hepatic fat and inflammation in 2008, the patatin-like phospholipase domain containing 3 (PNPLA3) I148M variant has emerged as the most common genetic determinant of the sequelae of NAFLD [3]. Interestingly, PNPLA3 I148M has a greater impact on NAFLD in women than in men [4, 5]. A deeper understanding of sex-specific differences in molecular pathogenesis and identifying novel pathways that regulate PNPLA3 expression could facilitate the development of therapies targeting NAFLD, especially in women.
Cyclin dependent kinases 4/6 (CDK4/6) catalyze the initial phosphorylation of pRb that inactivates its ability to bind and inhibit E2F signaling. Hepatic CDK4 activity is increased in insulin resistance and in patients with NAFLD [6, 7]. Consistently, there is elevated E2F1 mRNA in obese, insulin-resistant individuals [8]. NAFLD features a three-fold increase in de novo lipogenesis (DNL) and the level of DNL positively correlates with hepatic fat content [9]. Thus pathways that typically regulate cell proliferation may be recruited in the control of lipogenesis in insulin resistant livers.
Tetratricopeptide repeat domain protein 39B (T39) is a scaffolding protein that in genome-wide association studies has been associated with high density lipoprotein (HDL) cholesterol levels [10], gallstone disease [11], endometriosis [12] and ovarian cancer survival [13]. We showed that mice deficient in T39 have a remarkable protection from diet-induced steatohepatitis and death [14]. Both genetic deficiency and pharmacological inhibition of T39 resulted in decreased hepatic lipogenic gene expression and decreased DNL [14, 15]. On a high fat/high cholesterol/bile salt (HF/HC/BS) diet, T39-deficient mice had less hepatocellular ballooning and fewer inflammatory cell infiltrates along with fewer Ki67-positive cells in the liver, suggesting decreased hepatocellular proliferation [14]. However, these studies did not elucidate the link between lipid metabolism and cell proliferation and possible sex-specific effects. Here we describe a novel set of protein-protein interactions for T39 mediating its regulation of hepatic lipogenesis and the early pathogenesis of NAFLD especially in females.
MATERIALS AND METHODS
Experimental animals
Procedures in mice were approved by Columbia University’s Institutional Animal Care and Use Committee. Studies conformed to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
Additional Methods
Detailed methods, additional assays and statistical analyses are described in Supplementary Information.
RESULTS
T39 interacts with phosphorylated Rb to promote lipogenic gene expression
We have previously shown markedly decreased expression of Pnpla3 mRNA in T39-knockout mice in the postprandial state [14]. In mice treated with a hepatocyte-targeted N-acetylgalactosamine (GalNAc) T39 ASO and fed a diabetogenic HFSC diet [16], the Pnpla3-lowering effect of hepatic T39 deficiency was increased (Figure 1A) as insulin sensitivity became impaired (Supplemental Figure 1A). There was no difference in body weight or adiposity between hepatocyte-targeted T39 ASO-treated mice and control mice (data not shown). In contrast to the HF/HC/BS diet which produces actively proliferating hepatocytes [14], T39 deficiency did not affect hepatic liver X receptor (LXR) target gene expression (Supplemental Figure 1B) nor hepatic LXRα protein levels (Supplemental Figure 1C) in mice fed the HFSC diet. Proliferating cultured primary hepatocytes from HFSC diet fed mice did upregulate LXR targets when treated with the T39 ASO (Supplemental Figure 1D), similar to earlier findings [14]. However, T39 suppression in HFSC diet-fed LXRα knockout mice led to reduced hepatic lipogenic gene expression (Supplemental Figure 1E). Therefore, in insulin resistant, quiescent hepatocytes, T39 affects lipogenic gene expression independently of LXR and likely involves different interacting proteins.
Figure 1. T39 interacts with phosphorylated pRb.

(A) Hepatic Pnpla3 expression in mice treated with T39 GalNac ASO and fed (HFSC) diet for various durations. N= 8 per group, males, **p<0.01 by two-tailed nonparametric t-test corrected for multiple comparisons. (B) Co-immunoprecipitation of wild type T39 and wild type Rb, (C) co-immunoprecipitation of T39 and pRb N-terminus, (D) Co-immunoprecipitation of pRb N-terminus with T39 containing mutations surrounding the hydrophobic pocket and in the N-terminus with the structural model shown on top (colored residues are red = D483, green = F495, blue = S509, and yellow = F515), (E) Co-immunoprecipitation of T39 and phosphorylation-deficient pRb, and (F) co-immunoprecipitation of T39 and pRb with palbociclib in subconfluent HEK293A cells. (G) Hepatic lipogenic gene expression in refed mice fed the HFSC diet for 14 weeks, then administered palbociclib, n=5 per group, **p<0.05, **p<0.001 by two-way ANOVA with Sidak’s multiple comparisons test.
A structure-based algorithm predicted that T39 directly interacts with pRb [17]. This interaction was verified by co-immunoprecipitation in HEK293A cells expressing epitope-tagged T39 and pRb, in both anti-T39 (lane 1) and anti-pRb (lane 2) pulldowns (Figure 1B). In similar experiments T39 did not interact with retinoblastoma-like protein 1 (RBL1/p107) or RBL2 (p130) (data not shown). The N-terminal domain of pRb is distinct from that of RBL1 and RBL2, and indeed it is this domain that associates with T39 (Figure 1C). Structure homology-based modeling [18] of human T39 revealed a putative hydrophobic concave surface in the TPR domain, featuring D483 (magenta) and F495 (green) residue deep within the pocket, along with a S509 (blue) and N654 (orange) at the entrance of the pocket (Figure 1D). To assess their importance in Rb-binding, these residues were conservatively mutated. F495Y failed to co-IP with the N-terminal domain of pRb, suggesting the importance of hydrophobicity of the T39 pocket. In contrast, mutating F515 (cyan), which does not face into the pocket, had no effect. The D483E and D483N mutations abolished the co-IP of T39 with N-terminal Rb, suggesting an important electrostatic interaction. Notably, D483 is a glutamate residue in the mouse orthologue of T39, indicating this negative charge is conserved between species. Diminished co-IP of pRb occurred when S509 and N654 were extended by a single methylene group to become threonine and glutamine residues, suggesting steric hindrance (Figure 1D). These findings support a direct interaction of T39 with pRb in which the pRb N-terminal domain binds to a hydrophobic pocket on the concave face of T39.
While T39 co-immunoprecipitated with pRb in a dose-dependent manner (left three lanes, Figure 1E), T39 did not co-immunoprecipitate ΔCDK-pRb, a mutant that has all the serine/threonine phosphorylation sites mutated to alanine [19] (right three lanes, Figure 1E). The CDK4/6 inhibitor palbociclib, which prevents pRb phosphorylation, impeded T39’s interaction with pRb in a dose-dependent manner (Figure 1F). These cell culture data collectively indicate that T39 interacts specifically with phosphorylated pRb.
To assess the role of CDK4/6-mediated Rb phosphorylation in lipogenic gene expression and the effects of T39, we treated HFSC-diet fed mice with palbociclib. HFSC diet-fed mice treated with the T39 ASO had lower hepatic lipogenic gene expression, including Pnpla3, Acaca, Fasn, Gpam and Acly. Palbociclib administration similarly repressed lipogenic genes in control ASO-treated mice, but had no additive effect in T39 ASO-treated mice (Figure 1G), consistent with the hypothesis that the effects of T39 on lipogenic gene expression are mediated through its interaction with phosphorylated Rb.
T39 promotes pRb degradation
Co-expression of pRb with increasing amounts of T39 led to decreased recovery of pRb while the proteasome inhibitor bortezomib markedly increased recovery of both pRb and coimmunoprecipitated T39 (Figure 2A). T39 levels were not affected by bortezomib when coexpressed with ΔCDK-pRb (Supplemental Figure 2A), suggesting that phosphorylated Rb and T39 are co-degraded as a complex by the proteasome. In a cycloheximide turnover study pRb had a half-life of approximately 8–12 h (lanes 1–4, Figure 2B) consistent with a previous report [20]. T39 overexpression accelerated the decay of pRb (t1/2 < 4 h, lanes 9–12), and this effect was abolished by bortezomib pretreatment (lanes 13–16) (Figure 2B). The lower levels of Rb at t=0 h likely reflects the effects of T39 on pRb prior to the addition of bortezomib. Together these findings suggest that T39 increases pRb turnover by the proteasome. A role for destabilizing pRb has been described for MDM2 [21] and gankyrin [20], but neither had an impact on T39-mediated pRb degradation (Supplemental Figure 2B and 2C). As reported [21], we were unable to detect pRb ubiquitination with or without T39 expression (data not shown). In HFSC diet-fed mice, treatment with the T39 ASO increased hepatic pRb protein levels by 33% (Figure 2C), while in mice fed the HF/HC/BS diet, genetic deletion of hepatic T39 was associated with a 41% increase in pRb protein levels (Figure 2D). The increase in pRb protein was accompanied by a decrease in its mRNA, consistent with the negative feedback loop mediated by E2F1 binding sites in the Rb promoter [22]; moreover, the decrease in Rb mRNA was abolished by the proteasome inhibitor ixazomib (Supplemental Figure 2D). These in vivo findings are consistent with the cell culture data showing that T39 facilitates the proteasomal degradation of Rb protein and suggest that this leads to increased E2F1 activity tending to restore pRb levels.
Figure 2. T39 promotes the proteasomal degradation of Rb and destabilizes Rb in vivo.

(A) Anti-pRb pulldown of T39-pRb complexes in HEK293A cells treated with bortezomib. (B) Cycloheximide turnover study in subconfluent HEK293A cells. (C) Hepatic pRb protein in refed mice fed the HFSC diet for 10 weeks. N=10 per group, **p<0.01 by two-tailed t-test. (D) Hepatic pRb protein in refed mice fed the HF/HC/BS diet. N=8–13 per group, females, **p<0.01 by two-tailed t-test.
T39 and pRb regulate hepatic lipogenic gene expression through SREBP-1
To determine if pRb has an essential role in mediating these effects of T39 deficiency in early NAFLD, we administered AAV8-TBG-Cre to Rb1flox/flox mice. In GFP-treated Rb1foxl/fox female mice fed the HFSC diet for 10 weeks, the T39 ASO caused significant reductions in the hepatic mRNA levels of Pnpla3, Acacb, Scd1, Thrsp, and Pcsk9, as expected. In contrast, in mice with hepatic pRb knockdown, T39 ASO treatment did not significantly decrease any of these lipogenic genes (Figure 3A). In female mice, the protein levels of the precursor form of SREBP-1 were reduced by almost 40% while the transcriptionally active form of SREBP-1 was decreased by 60% by the T39 ASO and the differences were abrogated by hepatic pRb deficiency (Figure 3B–D), without any alteration in Srebf1 mRNA levels (Figure 3A).
Figure 3. T39 knockdown suppresses hepatic lipogenic gene expression and mature SREBP-1 in a pRb-dependent fashion.

(A) Hepatic lipogenic gene expression, *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA per gene, (B) Representative immunoblots of hepatic SREBP-1 protein, (C) Quantification of SREBP-1 precursor form from the immunoblot shown in Figure 3B and (D) Quantification of mature SREBP-1 from the immunoblot shown in Figure 3B, ***p<0.001 by two-way ANOVA with Sidak’s multiple comparisons test where the column factor is T39 in refed female Rb1fl/fl mice administered AAV8-GFP or AAV8-TBG-Cre then fed the HFSC diet for 10 weeks, n=9–10 per group. (E) Hepatic lipogenic gene expression, *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA per gene, (F) Representative blots of hepatic SREBP-1 protein, (G) Quantification of SREBP-1 precursor form from the immunoblot shown in Figure 3F and (H) Quantification of mature SREBP-1 from the immunoblot shown in Figure 3F, *p<0.05, **p<0.01 by two-way ANOVA with Sidak’s multiple comparisons test where the column factor is T39 ASO in refed male Rb1fl/fl mice, n=10–11 per group. (I) Hepatic Cd9 mRNA expression in refed mice of both sexes. N=9–11 per group, **p<0.01, ****p<0.0001 by two-way ANOVA with Sidak’s multiple comparisons test.
In male mice, administration of the T39 ASO had only moderate effects on lipogenic gene expression, and these effects appeared to be independent of pRb expression (Figure 3E). Likewise, the column factor of the two-ANOVA analysis indicated that T39 ASO had a significant effect to reduce the precursor and mature forms of SREBP-1 regardless of pRb (Figure 3F–H). These data indicate a major effect of T39 on SREBP-1 precursor and mature forms and lipogenic gene expression in females that is primarily mediated through pRb, while suggesting a smaller effect in males that seems to be independent of pRb.
These studies involved a short term HFSC diet to avoid inflammatory responses that may influence lipogenic gene expression [23]. However, T39 knockdown significantly reduced expression of Cd9, a hematopoetic cell marker that increases in early NASH [24] in female but not male mice (Figure 3I), suggesting a possible enhanced hepatoprotective effect of T39 deficiency in early NASH.
T39 regulates hepatic lipogenic gene expression via E2F1
In the canonical pathway of pRb action, E2F1 is preferentially bound and inhibited by pRb [25]. The T39 ASO-induced decreases in Pnpla3, Acaca, Fasn, Scd1, Thrsp and Pcsk9 were abolished in E2F1 knockout mice (Figure 4A). Notably, E2F1 knockout alone reduced Pnpla3 expression and T39 ASO treatment had no additional effect (Figure 4A) consistent with a primary effect of E2F1 on Pnpla3 gene expression. Similar to the effects of pRb knockdown, female T39 ASO-treated wild type mice had lower hepatic levels of both precursor (Figure 4B and 4C) and mature (Figure 4B and 4D) forms of SREBP-1. These differences were abolished in E2f1−/− mice without changes in Srebf1 mRNA levels (Figure 4A). Therefore, in female mice, T39 increases hepatic lipogenic gene expression via the canonical CDK4/Rb/E2F1 pathway by increasing the protein levels of the precursor and mature forms of SREBP-1c.
Figure 4. T39 knockdown suppresses hepatic lipogenic gene expression and mature SREBP-1 in an E2F1-dependent fashion.

(A) Hepatic lipogenic gene expression, n=11–17 per group, *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA per gene, (B) Representative immunoblots of SREBP-1 protein, (C) Quantification of SREBP-1 precursor form from the immunoblot shown in Figure 4B and (D) Quantification of mature SREBP-1 from the immunoblot shown in Figure 4B, n=11–13 per group, *p<0.05 by two-way ANOVA with Sidak’s multiple comparisons test in refed female E2F1 knockout mice and wild type littermates fed the HFSC diet for 10 weeks. (E) Hepatic lipogenic gene expression, n=12–23 per group, *p<0.05, ****p<0.0001 by one-way ANOVA per gene, (F) Hepatic expression of SREBP-1 precursor form and (G) mature SREBP-1 based on immunoblotting, n=12 per group in refed male E2F1 knockout mice and wild type littermates fed the HFSC diet for 10 weeks.
In male mice, hepatic lipogenic gene expression and SREBP-1 was not strongly affected by T39 ASO or E2F1 deficiency (Figure 4E–G). It should be noted however that there was greater variability in lipogenic gene expression in this experiment than in the pRb knockdown experiment (Figure 3), probably because the E2F1 knockout mice were on a mixed genetic background.
T39 knockdown increases PUFA in phosphatidylethanolamine (PE) in an Rb-dependent fashion
Previous studies have suggested that E2F1 may increase expression of various lipogenic genes including Srebf1 by binding and increasing enhancer activity [8]. Increased levels of long chain PUFA decreased SREBP-1 processing and we found an increase in long chain PUFA in ER phospholipids, especially in phosphatidylethanolamine (PE), in T39 KO mice [14]. We confirmed a moderate PUFA enrichment in PE with T39 ASO treatment and showed that this effect was pRb-dependent (Supplemental Figure 3A). Since PNPLA3 has been shown to affect phospholipid fatty acid saturation [26, 27], we analyzed microsomal lipids in female Pnpla3−/− mice. Similar to T39 ASO-treated mice, PNPLA3 knockout mice had moderate PUFA enrichment specifically in PE (Supplemental Figure 3B). Pnpla3 deficiency also had a moderate allele dose-dependent effect to reduce expression of hepatic SREBP-1 targets, including Fasn, Acly and Me1 (Supplemental Figure 3C). Cholesterol has been shown in cell culture models to affect SREBP-1 processing, but hepatic cholesterol levels were not affected by the T39 ASO on the HFSC diet (Supplemental Figure 3D). Therefore, when T39 is knocked down decreases in the mature form of SREBP-1 may be partly attributed to the increase in PUFA-containing PE secondary to the marked reduction in Pnpla3 expression.
Conserved sex-specific differences in T39, pRb and PNPLA3 expression
Seeking to understand the sex specific effects of T39 knockdown, we noted that female mice had higher levels of hepatic T39 mRNA (2.20 ± 0.17 AU vs. 1.06 ± 0.06 AU) (Figure 5A) and Rb1 mRNA (0.44 ± 0.03 AU vs. 0.38 ± 0.02 AU) (Figure 5B) compared to males. The difference was even greater for hepatic pRb protein levels, in which females had 3-fold the pRb protein (Figure 5C). Interrogation of the GTex liver gene expression data revealed that in humans, TTC39B and RB1 are similarly higher in females (Supplemental Figure 4) [28], which may explain the greater impact of the pRb/E2F1 pathway in females. We discovered that HFSC diet-fed female mice had almost six-fold higher hepatic Pnpla3 expression than male mice in both the fasting and postprandial state (Figure 5D). In addition, Pnpla3 is dramatically induced during gestation independent of nutritional status (Supplemental Figure 4B). This finding is consistent with a previous report in rats [29]. PNPLA3 mRNA was not significantly higher in women compared to men (Supplemental Figure 4A), but this may be because 75% of the women donors contained in the GTex sample set were aged 60–69 and therefore postmenopausal. Together these findings indicate an important role of female hormones in the induction of PNPLA3 expression, independent of nutritional status.
Figure 5. Hormonal dependence of T39’s effect on hepatic lipogenic genes in females.

(A) Hepatic T39 and (B) Rb1 expression in male and female mice fed HFSC diet in the refed state, n=26–34 per group, ****p<0.001 by Mann-Whitney test. (C) Hepatic pRb protein expression in male and female mie fed the HFSC diet for 10 weeks, n=9–11 per group, *p<0.05 by Mann Whitney U-test. (D) Hepatic Pnpla3 expression in male and female mice fed HFSC diet in the fasting and refed state, n=5–9 per group, **p<0.01, ***p<0.001, ****p<0.001 by two-way ANOVA with Sidak’s multiple comparisons test. (E) Hepatic pRb protein level, (F) hepatic gene expression and (G) hepatic triglyceride content of refed ovariectomized or sham-operated female mice fed HFSC diet for 10 weeks, n=10 per group, *p<0.05, **p<0.01 and ***p<0.001 by two-way ANOVA with Sidak’s multiple comparison’s test where the column factor is T39 ASO.
Specific effects of T39 require female hormonal signaling
We next asked whether ovarian hormones affect T39’s impact on lipogenic genes in females by treating HFSC diet-fed ovariectomized and sham operated mice with T39 ASO. Ovariectomy attenuated the hepatic pRb protein stabilization effect of the T39 ASO (Figure 5E). Likewise, ovariectomy abrogated the suppressive effect of T39 ASO on hepatic Pnpla3, Scd1 and Pcsk9 expression (Figure 5F), which were also pRb-dependent genes (Figure 3A). Notably, ovariectomy halved hepatic Pnpla3 mRNA levels (Figure 5F), suggesting that ovarian hormones are important for high PNPLA3 expression in females. The decrease in lipogenic gene expression created an anti-steatotic phenotype in which T39 ASO decrease hepatic TG accretion in females (Figure 5G) but not males (Supplemental Data 4C).
T39 regulates SREBP-1 via SCAP
Changes in E2F1-dependent Pnpla3 expression and subsequent effects on phospholipid fatty acids and SREBP-1c processing do not fully explain the marked reduction in the immature form of SREBP-1c in response to T39 ASO, nor its moderate effects on lipogenic gene expression in males which appeared to be independent of pRb/E2F1 (Figure 3E and 3G). Since reductions in both the precursor and mature forms of SREBP-1 occur in SCAP knockout mice [30], we interrogated whether T39 affected hepatic SCAP protein levels. In males (Figure 6A), and to a lesser degree in females (Figure 6B), SCAP protein was significantly decreased with the T39 ASO. T39’s effect on hepatic SCAP was enhanced in the postprandial state (Supplemental Figure 5A). There was no associated change in Scap mRNA (Figure 3A). To facilitate further mechanistic studies, we employed the Huh-7 human HCC cell line. Like the murine liver, Huh-7 cells downregulate lipogenic genes in response to T39 ASO and E2F1 knockdown (Supplemental Figure 5B). For FASN and SCD1, E2F1 knockdown abrogated the T39 ASO-mediated decrease. However, the T39 ASO still had a significant lowering effect on PNPLA3 and PCSK9 in the presence of E2F1 siRNA (Supplemental Figure 5B), once again suggesting that T39 promotes lipogenic gene expression via an additional, pRb/E2F1-independent mechanism. SREBP-1c is the primary regulator of PNPLA3 expression in human hepatocyte cell lines [31]. The T39 ASO significantly lowered mature SREBP-1 levels (Figure 6C and 6D). T39 knockdown decreased SCAP protein (Figure 6C and Figure 6E), also without a corresponding change in SCAP mRNA (Figure 6F). Since SCAP also affects SREBP-2, we measured cholesterogenic enzyme transcripts and found them to be lower in T39 ASO-treated cells in the presence of bortezomib (Supplemental Figure 5C). The failure to observe changes in cholesterogenic genes in T39 deficient mice [14] could be due to the use of high cholesterol diets. Interestingly, SCAP protein levels were unaffected by proteasomal inhibition and the effect of T39 knockdown on SCAP was still observable in the presence of bortezomib (Figure 6C and 6E), suggesting that T39 protects SCAP from a non-proteasomal degradative pathway.
Figure 6. T39 knockdown posttranslationally decreases SCAP in both males and females.

(A) Hepatic SCAP protein levels of refed male (n=10–11 per group) and (B) female (n=8 per group) Rb1fl/fl mice administered AAV8-GFP or AAV8-TBG-Cre then fed the HFSC diet for 10 weeks. (Top) representative immunoblots and (bottom) quantification, *p<0.05 by two-way ANOVA with Sidak’s multiple comparisons test where column factor is T39 ASO. (C) Immunoblot of SREBP-1 and SCAP protein expression with (D) Quantification of mature SREBP-1 protein, (E) Quantification of SCAP protein and (F) SCAP mRNA levels in Huh-7 cells treated with bortezomib for 3 h in complete media (10% FBS), n=4 per group, *p<0.05, **p<0.01, ***p<0.001 by two-way ANOVA with Tukey’s multiple comparisons test. (G) Co-immunoprecipitation of T39 and VAPB in HEK293A cells expressing WT T39 or T39 containing mutations of the acidic residues of the N-terminus (Δacid). (H) Co-immunoprecipitation of VAPB and SCAP in HeLa cells with/without 1 μM nocodazole.
T39 stabilizes SCAP via an interaction with VAPB
A high throughput affinity-purification mass spectrometry study identified the VAMP-associated protein B (VAPB) as a protein interaction partner of T39, involving VAPB’s major sperm protein (MSP) domain [32]. We confirmed that T39 interacts with both overexpressed and endogenous VAPB in HEK293A cells (Figure 6G). VAPB overexpression adversely affected T39 cellular solubility (data not shown), which may explain why T39 was nearly undetectable in the whole cell lysate input (Figure 6G). The MSP domain binds proteins via electrostatic interactions with the acidic residues of the FFAT motif. Human and mouse T39 contain a stretch of acidic residues in the N-terminus. Conservatively eliminating the negative charge by mutating these residues to asparagine and glutamines (Δacid) abolished T39’s interaction with VAPB (Figure 6G), but not pRb (Figure 1D), indicating this is a functional FFAT motif. The T39/VAPB interaction persisted in the presence of bortezomib (Figure 6G).
SCAP has been reported to interact with VAPB outside of the MSP domain [33]. Indeed, SCAP co-immunoprecipitated with endogenous VAPB in HeLa cells (Figure 6H), and this interaction was enhanced with a brief nocodazole treatment that prevents ER-to-Golgi transport. T39 depletion with the ASO decreased the amount of SCAP that co-immunoprecipitated with VAPB, particularly in nocodazole-treated cells (Figure 6H). This suggests that T39 strengthens the SCAP/VAPB interaction in the ER, which may enhance SCAP stability.
T39 knockdown decreases hepatic lipogenic gene expression in human primary hepatocytes
Primary hepatocytes from approximately age- and BMI-matched human donors of both sexes (n=7 each) were treated with T39 ASO, depleting T39 by over 80% (Figure 7A). In female primary hepatocytes, the T39 ASO significantly reduced PNPLA3 mRNA (Figure 7B), along with mRNA levels of PCSK9, SCD1 THRSP (Figure 7C), but not SREBF1 (Figure 7C). In male hepatocytes T39 suppression did not significantly impact lipogenic gene expression. The female primary human hepatocytes had over 2-fold higher expression of PNPLA3 mRNA (Figure 7B) and a trend to higher TTC39B expression. Therefore, in both humans and mice, antagonizing T39 lowers hepatic lipogenic gene expression more prominently in females than males.
Figure 7. Enhanced suppressive effect of T39 knockdown on lipogenic gene expression in female human hepatocytes.

(A) TTC39B mRNA, (B) PNPLA3 mRNA and (C) lipogenic gene expression in primary hepatocytes from approximately age- and BMI-matched donors treated with T39 ASO. N=7 per sex, *p<0.05, **p<0.01, ***p<0.001 by paired t-test.
CONCLUSIONS
We discovered that T39 binds the paradigmatic tumor suppressor pRb via the hydrophobic pocket in its C-terminal TPR domain to promote the proteasomal degradation of pRb in proliferating cells, in the liver during the insulin resistance of mild NAFLD, and in the hepatic proliferative response of advanced NASH. In the postprandial state, hyperinsulinemia activates CDK4 [34], leading to pRb phosphorylation which renders it vulnerable to T39-mediated proteasomal degradation and liberates E2F1 to increase expression of hepatic lipogenic genes especially Pnpla3. The induction of Pnpla3 by E2F1 amplifies the pro-lipogenic effect of T39 by increasing the saturation of ER phospholipids to stimulate SREBP-1 processing. The effects of T39 on lipogenic gene expression via pRb/E2F1 are much more prominent in female mice and in human female primary hepatocytes. In a parallel pathway that occurs in both sexes, T39 stabilizes SCAP likely through its interaction with VAPB at its N-terminal FFAT motif. The increased SCAP abundance preserves SREBP-1 precursor levels and facilitates SREBP-1 transport to the Golgi for proteolytic activation. In females, the integration of both these pathways augments the impact of T39 on hepatic lipogenic gene expression. The molecular pathway we have identified using cell culture and mouse models appears to be relevant to human pathophysiology: NAFLD patients have higher TTC39B mRNA compared to healthy subjects [35], while patients whose NAFLD activity score improved in response to lifestyle intervention had lower hepatic TTC39B expression [36].
Our studies unveil a central role of T39-Rb interactions in mediating the link between CDK4, E2F1 and hepatic lipogenic gene expression. Studies by Denechaud et al showed that E2F1 directly promoted enhancer activity of lipogenic genes, including Srebf1 [8]. While not mutually exclusive, our suggested mechanism places E2F1-mediated induction of Pnpla3 upstream of increased processing of SREBP-1c contributing to increased expression of other lipogenic gene expression. This model is consistent with increased fatty acid synthesis in PNPLA3-overexpressing mice [37] and decreased lipogenic gene expression in PNPLA3 ASO-treated mice [38].
With cell culture and in vivo evidence, we showed that T39 knockdown also caused a reduction in SCAP in a posttranscriptional mechanism. In addition to promoting SREBP processing, SCAP is essential for maintaining SREBP stability through interactions with the C-terminal domain [39]. A lysosomal degradation pathway has been described for SCAP [40]. SCAP interaction with VAPB, an abundant ER resident protein, was enhanced when microtubule-based transport out of the ER was disrupted (Figure 5H). Following SREBP cleavage, SCAP needs to be recycled by retrograde transport from the Golgi to the ER to enable it to escort additional SREBP molecules for processing [41]. The decreased SCAP/VAPB interaction with T39 knockdown could indicate inefficient SCAP recycling leading to increased SCAP degradation.
The use of inbred mouse strains to study sexual dimorphism should be approached with caution, as profiling of hepatic lipids in a panel of inbred mouse strains revealed that the magnitude of sex-specific differences varied widely across strains [42]. However, it should be noted the directionalities of the differences were conserved across mouse strains, and we observed a similarly enhanced response to the T39 ASO in female versus male human primary hepatocytes. Consideration of sexually dimorphic effects could be important in the development of effective treatments for NASH. For example, the CCR2/5 antagonist cenicriviroc, which failed to meet the primary outcome overall, reduced fibrosis in men but not women [43]. The conserved increase in PNPLA3 expression in females of multiple species and the strong induction of PNPLA3 in pregnancy suggests that PNPLA3 may have an important role in female reproduction. Our studies suggest that a strategy of T39 inhibition to lower PNPLA3 I148M expression might be more effective in women. Alterations in G1/S transition genes occur specifically in female NASH [44], in agreement with our finding that the T39/Rb/E2F1 pathway plays a more important role in females. T39-mediated tumor suppressor degradation provides a new paradigm for investigating the sex-specific pathogenesis of liver diseases.
Supplementary Material
HIGHLIGHTS.
TTC39B is a scaffolding protein that interacts with and promotes the proteasomal degradation of pRb.
TTC39B deficiency increases hepatocyte pRb which inhibits E2F1 activity and lipogenic gene expression in females.
In both sexes, TTC39B deficiency decreases hepatic SCAP protein levels to posttranslationally inhibit SREBP-1.
TTC39B inhibition could be a novel strategy to target PNPLA3 and treat NAFLD, especially in women.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Mark Graham and Rosanne Crooke from Ionis Pharmaceuticals for providing the T39 ASOs and reviewing the manuscript, respectively. Lipidomics analyses were supported by the Biomarkers Core Laboratory at the Irving Institute for Clinical and Translational Research.
FINANCIAL SUPPORT STATEMENT
NIH R01HL119830 (A.R.T.)
NIH K01DK114380 (J.H.)
NIH P30DK636608 (J.H.)
Gilead Sciences Liver Scholar program (J.H.)
Naomi Berrie Diabetes Center Russell Berrie Foundation Fellow Award (K.M.M.)
List of abbreviations:
- ACACA
acetyl-CoA carboxylase alpha
- ASO
antisense oligonucleotide
- BMI
body mass index
- CDK4/6
cyclin-dependent kinase 4/6
- Co-IP
coimmunoprecipitation
- DNL
de novo lipogenesis
- E2F1
E2F transcription factor 1
- ER
endoplasmic reticulum
- FASN
fatty acid synthase
- GalNAc
N-acetylgalactosamine
- HCC
hepatocellular carcinoma
- HDL
high density lipoprotein
- HF/HC/BS
high fat high cholesterol bile salt
- HFSC
high fat sucrose cholesterol
- LXR
liver X receptor
- MFA
monounsaturated fatty acids
- MSP
major sperm protein
- NAFLD
nonalcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- PC
phosphatidylcholine
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PE
phosphatidylethanolamine
- PNPLA3
patatin-like phospholipase domain containing 3
- pRB
retinoblastoma protein
- SCAP
SREBP chaperone protein
- SCD1
stearoyl-CoA desaturase 1
- SFA
saturated fatty acids
- SREBP-1
sterol regulatory element binding protein-1
- T39
Tetratricopeptide repeat domain containing protein 39B
- THRSP
thyroid hormone responsive/SPOT14
- VAPB
VAMP-associated protein B
Footnotes
CONFLICT OF INTEREST STATEMENT
P.D-J., A.R.T. and J.H. are cofounders and shareholders of Fortico Biotech.
DATA AVAILABILITY
All data that support the findings of this study are included in the main figures and the supplementary material.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
Author names in bold designate shared co-first authorship
- [1].Della Torre S, Maggi A. Sex Differences: A Resultant of an Evolutionary Pressure? Cell metabolism 2017;25:499–505. [DOI] [PubMed] [Google Scholar]
- [2].Balakrishnan M, Patel P, Dunn-Valadez S, Dao C, Khan V, Ali H, et al. Women Have a Lower Risk of Nonalcoholic Fatty Liver Disease but a Higher Risk of Progression vs Men: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 2021;19:61–71 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Romeo S, Sanyal A, Valenti L. Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease. Cell Metab 2020;31:35–45. [DOI] [PubMed] [Google Scholar]
- [4].Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011;53:1883–1894. [DOI] [PubMed] [Google Scholar]
- [5].Speliotes EK, Butler JL, Palmer CD, Voight BF, Consortium G, Consortium MI, et al. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010;52:904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jin J, Valanejad L, Nguyen TP, Lewis K, Wright M, Cast A, et al. Activation of CDK4 Triggers Development of Non-alcoholic Fatty Liver Disease. Cell Rep 2016;16:744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 2014;510:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Denechaud PD, Lopez-Mejia IC, Giralt A, Lai Q, Blanchet E, Delacuisine B, et al. E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. The Journal of clinical investigation 2016;126:137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ferkingstad E, Oddsson A, Gretarsdottir S, Benonisdottir S, Thorleifsson G, Deaton AM, et al. Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat Commun 2018;9:5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Steinthorsdottir V, Thorleifsson G, Aradottir K, Feenstra B, Sigurdsson A, Stefansdottir L, et al. Common variants upstream of KDR encoding VEGFR2 and in TTC39B associate with endometriosis. Nat Commun 2016;7:12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].French JD, Johnatty SE, Lu Y, Beesley J, Gao B, Kalimutho M, et al. Germline polymorphisms in an enhancer of PSIP1 are associated with progression-free survival in epithelial ovarian cancer. Oncotarget 2016;7:6353–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hsieh J, Koseki M, Molusky MM, Yakushiji E, Ichi I, Westerterp M, et al. TTC39B deficiency stabilizes LXR reducing both atherosclerosis and steatohepatitis. Nature 2016;535:303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McCabe KM, Hsieh J, Thomas DG, Molusky MM, Tascau L, Feranil JB, et al. Antisense oligonucleotide treatment produces a type I interferon response that protects against diet-induced obesity. Mol Metab 2020;34:146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Subramanian S, Han CY, Chiba T, McMillen TS, Wang SA, Haw A 3rd, et al. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol 2008;28:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012;490:556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zimmermann L, Stephens A, Nam SZ, Rau D, Kubler J, Lozajic M, et al. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol 2018;430:2237–2243. [DOI] [PubMed] [Google Scholar]
- [19].Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Higashitsuji H, Itoh K, Nagao T, Dawson S, Nonoguchi K, Kido T, et al. Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat Med 2000;6:96–99. [DOI] [PubMed] [Google Scholar]
- [21].Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, et al. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell 2005;20:699–708. [DOI] [PubMed] [Google Scholar]
- [22].Shan B, Chang CY, Jones D, Lee WH. The transcription factor E2F-1 mediates the autoregulation of RB gene expression. Mol Cell Biol 1994;14:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Miller AM, Wang H, Bertola A, Park O, Horiguchi N, Ki SH, et al. Inflammation-associated interleukin-6/signal transducer and activator of transcription 3 activation ameliorates alcoholic and nonalcoholic fatty liver diseases in interleukin-10-deficient mice. Hepatology 2011;54:846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell 2019;75:644–660 e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Julian LM, Palander O, Seifried LA, Foster JE, Dick FA. Characterization of an E2F1-specific binding domain in pRB and its implications for apoptotic regulation. Oncogene 2008;27:1572–1579. [DOI] [PubMed] [Google Scholar]
- [26].Luukkonen PK, Nick A, Holtta-Vuori M, Thiele C, Isokuortti E, Lallukka-Bruck S, et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mitsche MA, Hobbs HH, Cohen JC. Patatin-like phospholipase domain-containing protein 3 promotes transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J Biol Chem 2018;293:9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bustamante JJ, Copple BL, Soares MJ, Dai G. Gene profiling of maternal hepatic adaptations to pregnancy. Liver Int 2010;30:406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Moon YA, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab 2012;15:240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dubuquoy C, Robichon C, Lasnier F, Langlois C, Dugail I, Foufelle F, et al. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J Hepatol 2011;55:145–153. [DOI] [PubMed] [Google Scholar]
- [32].Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015;162:425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002;110:489–500. [DOI] [PubMed] [Google Scholar]
- [34].Lagarrigue S, Lopez-Mejia IC, Denechaud PD, Escote X, Castillo-Armengol J, Jimenez V, et al. CDK4 is an essential insulin effector in adipocytes. J Clin Invest 2016;126:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [35].Liu MJ, Jin H, Chen YB, Yu JJ, Guo ZY, He SQ, et al. Screening of non-alcoholic steatohepatitis (NASH)-related datasets and identification of NASH-related genes. Int J Clin Exp Pathol 2021;14:567–581. [PMC free article] [PubMed] [Google Scholar]
- [36].Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Molendi-Coste O, Fleury S, et al. Transcriptional Network Analysis Implicates Altered Hepatic Immune Function in NASH development and resolution. Nat Metab 2019;1:604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest 2012;122:4130–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Linden D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andreasson AC, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab 2019;22:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kober DL, Xu S, Li S, Bajaj B, Liang G, Rosenbaum DM, et al. Identification of a degradation signal at the carboxy terminus of SREBP2: A new role for this domain in cholesterol homeostasis. Proc Natl Acad Sci U S A 2020;117:28080–28091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shao W, Espenshade PJ. Sterol regulatory element-binding protein (SREBP) cleavage regulates Golgi-to-endoplasmic reticulum recycling of SREBP cleavage-activating protein (SCAP). J Biol Chem 2014;289:7547–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Takashima K, Saitoh A, Funabashi T, Hirose S, Yagi C, Nozaki S, et al. COPI-mediated retrieval of SCAP is crucial for regulating lipogenesis under basal and sterol-deficient conditions. J Cell Sci 2015;128:2805–2815. [DOI] [PubMed] [Google Scholar]
- [42].Norheim F, Bjellaas T, Hui ST, Chella Krishnan K, Lee J, Gupta S, et al. Genetic, dietary, and sex-specific regulation of hepatic ceramides and the relationship between hepatic ceramides and IR. J Lipid Res 2018;59:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018;67:1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vandel J, Dubois-Chevalier J, Gheeraert C, Derudas B, Raverdy V, Thuillier D, et al. Hepatic molecular signatures highlight the sexual dimorphism of Non-Alcoholic SteatoHepatitis (NASH). Hepatology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.