

Abstract
Lysosomal acid lipase (LAL), encoded by the gene LIPA, facilitates the intracellular processing of lipids by hydrolyzing cholesteryl esters and triacylglycerols present in newly internalized lipoproteins. Loss-of-function mutations in LIPA result in cholesteryl ester storage disease (CESD) or in Wolman disease when mutations cause complete loss of LAL activity. Although the phenotype of a mouse CESD model has been extensively characterized there has not been a focus on the brain at different stages of disease progression. In the current studies whole-brain mass and the concentrations of cholesterol in both the esterified (EC) and unesterified (UC) fractions were measured in Lal−/− and matching Lal+/+ mice (FVB-N strain) at ages ranging from 14 up to 280 days after birth. Compared to Lal+/+controls at 50, 68-76, 140-142 and 230-280 days of age, Lal−/− mice had brain weights that averaged approximately 6, 7, 18 and 20% less, respectively. Brain EC levels were higher in the Lal−/− mice at every age, being elevated 27-fold at 230-280 days. Brain UC concentrations did not show a genotypic difference at any age. The elevated brain EC levels in the Lal−/− mice did not reflect EC in residual blood. An mRNA expression analysis for an array of genes involved in the synthesis, catabolism, storage and transport of cholesterol in the brains of 141-day old mice did not detect any genotypic differences although the relative mRNA levels for several markers of inflammation were moderately elevated in the Lal−/− mice. The possible sites of EC accretion in the CNS are discussed.
Keywords: Brain mass, Cholesterol-esterifying enzymes, Esterified cholesterol sequestration, Lysosomal acid lipase, Residual blood volume, Unesterified cholesterol
Introduction
In healthy young adult humans the average cholesterol concentration across the entire body is ~2 mg/g wet tissue weight, with ~23% of the body cholesterol pool being present in the brain, spinal cord and peripheral nervous system (Cook, 1958). Over the past decade a series of comprehensive reviews on brain cholesterol metabolism, in both health and disease, have revealed the remarkable advances made in this field while also identifying the major questions that are the subject of ongoing research (Anchisi et al., 2013; Jin et al., 2019; Martin et al., 2014; Orth & Bellosta, 2012; Petrov et al., 2016; Pfrieger & Ungerer, 2011; Vance, 2012; Zhang & Liu, 2015). As detailed in this and earlier literature there are numerous distinguishing features of cholesterol metabolism in the developing and mature brain (Dietschy & Turley, 2004). One of these is the marked variation in cholesterol levels across different areas of the brain, particularly at maturity, with the highest concentrations found in those regions containing the most white matter. Another is that unlike other organs which fulfill their cholesterol needs to varying degrees through de novo synthesis and the uptake of different types of lipoproteins from the circulation, cholesterol in the CNS is essentially all derived from its own biosynthetic pathway mostly throughout fetal and early postnatal development. The brain does have an internal lipoprotein production and transport system operating mostly between astrocytes and neurons as discussed in extensive detail elsewhere (Petrov et al., 2016; Pfrieger & Ungerer, 2011; Orth & Bellosta, 2012; Wang & Eckel, 2014). A third feature is that in the healthy, mature individual more than 99% of the cholesterol in the CNS is unesterified (UC) although during phases of fetal and early postnatal development a transient elevation in the level of esterified cholesterol (EC) has been detected (Alling & Svennerholm, 1969; Kinney et al., 1994; Yusuf et al., 1981).
There are three cholesterol esterifying enzymes in mammals. One of these is sterol O-acyltransferase 1 (SOAT1, previously called ACAT1), which is active in numerous organs where it is expressed in various cell types, including macrophages (Chang et al., 2000; Lee et al., 2000; Lee & Carr, 2004; Lee et al., 2015). Cholesterol in the brain is esterified by SOAT1 and to some extent by lecithin cholesterol acyltransferase (LCAT). In mouse brain SOAT1 is expressed in neurons (Sakashita et al., 2000) at multiple sites including the cerebellum, brain stem, cerebral cortex, thalamus and hippocampus (Bryleva et al., 2010) as well as epithelial cells in the choroid plexus (Zeisel et al.,2018). LCAT, which is bound to circulating lipoproteins, is produced predominantly in the liver (Kunnen & Van Eck, 2012) and to a small extent in astrocytes. Studies in a mouse model showed that LCAT mRNA in the brain was 6% of that in the liver (Hirsch-Reinshagen et al., 2009). A third cholesterol esterifying enzyme, SOAT2 (previously called ACAT2), which is expressed in hepatocytes and enterocytes (Lee and Carr, 2004; Lee et al., 2015; Parini et al., 2004) plays a major role in determining the EC content of lipoproteins produced by the liver and small intestine.
Cellular cholesterol homeostasis in all organs is regulated through the interplay of a constellation of genes that includes LIPA, which encodes lysosomal acid lipase (LAL), as well as Niemann-Pick (NPC) 2 and NPC1. Together these three proteins control cholesterol trafficking through the late endosomal/lysosomal (E/L) compartment of every cell (Goldstein et al., 1975; Kwon et al., 2009). LAL hydrolyzes esterified cholesterol (EC) and triacylglycerol (TAG) contained within various classes of lipoproteins taken up by cells through receptor-mediated and bulk-phase endocytosis (Liu et al., 2007). The unesterified cholesterol (UC) then moves via NPC2 to NPC1 which facilitates its exit from the E/L compartment into a putative pool of metabolically active cholesterol that has input from other sources including from local synthesis. Mutations in LIPA, NPC2, or NPC1 are rare and have profound consequences in multiple organ systems. Disrupted function of NPC2, and especially NPC1, results in UC sequestration to varying degrees in all tissues, with neurodegeneration, liver failure and pulmonary dysfunction being the principal causes of morbidity and mortality (Vance, 2012). In the case of LIPA, mutations that result in almost a complete loss of LAL activity cause Wolman disease (WD) , a rare autosomal recessive storage disorder leading to death in infancy. Other mutations where some residual activity remains lead to cholesteryl ester storage disease (CESD), a milder, later onset disorder. Hence life expectancy is much greater in CESD patients. A recent study reported the pooled prevalence of LAL deficiency to be 1 per 177,452 (Carter et al., 2019). In humans with CESD there is a pronounced increase in hepatic EC levels (Besley et al., 1984; Dinscoy et al., 1984; Hoeg et al., 1984; Sloan & Fredrickson 1972; Todoroki et al., 2000) frequently with accompanying dyslipidemia (Reiner et al., 2014). Dramatic elevations in EC levels in the liver and other organs have been documented in a mouse model of CESD developed by Du et al., 1998, and Du et al., 2001. The same model on different strain backgrounds and given diets of differing composition has been used by other investigators (Aqul et al., 2014; Radović et al., 2016).
One striking difference between CESD and NPC1 disease is that while, in the latter case, neurodegeneration is a major cause of debilitation and death (Patterson et al., 2017), LAL deficiency surprisingly has not been reported to overtly impact the brain (Brown et al, 2014). Although there is a case report of two siblings with CESD that presented with unusual neurological manifestations (Bindu et al., 2007), there is currently no published guidance on the assessment and monitoring of neural defects in children and adults with LAL deficiency (Kohli et al., 2020). There are very few published data pertaining to brain cholesterol metabolism in LAL deficiency. An early study of two Wolman patients, one of whom died at 17 weeks, the other at 60 weeks, noted that their brains were slightly reduced in size (Marshall et al., 1969). In the cerebral cortex of the younger patient ~18% of the total cholesterol was esterified while in the older subject EC accounted for ~11.5% of the total. A later case report documented ultrastructural evidence of lipid inclusions in oligodendrocytes and astrocytes in the CNS of a Wolman patient (Byrd & Powers, 1979). Although a rat model of Wolman disease with a lifespan of just ~120 days has been described, no findings for the CNS were presented (Kuriyama et al., 1990). For CESD patients there appear to be no published data pertaining to brain mass or EC content but in the case of the CESD mouse model developed and characterized by Du et al. (2001) histology of the brain from a 6-month old Lal−/− mouse revealed lipid storage in the epithelial cells of the choroid plexus. Of relevance here is an earlier finding by Du et al., (1996) of a significant level of expression of mRNA and protein for LAL in the choroid plexus epithelium of mouse embryonic brain.
Although our initial studies with a mouse CESD model focused mainly on the liver and small intestine (Aqul et al., 2014; Lopez et al., 2015; Chuang et al., 2017; Lopez et al., 2018) our observation that in the untreated aging Lal−/− mice brain weights were significantly lower than in their matching Lal+/+ controls prompted an investigation of whether there were any discernible changes in an array of molecular markers for cholesterol homeostasis in the CNS of this model. While none were detected, further exploration of why brain mass contracted with advancing disease could potentially raise new questions about brain health in those CESD patients whose diagnosis is not made until late age, or who have mutations in LIPA that make them less responsive to therapy.
Materials and Methods
Animals
Lal+/+ and Lal−/− mice were generated from heterozygous breeding stock on the friend virus B-type susceptibility / NIH background (FVB/N) background, the model that was used for the development of enzyme replacement therapy for lysosomal acid lipase deficiency (Du et al., 2008; Sun et al., 2014).
The diminished level of mRNA for LIPA in the livers and small intestines of Lal−/− mice of this strain in our facility has been documented previously (Aqul et al., 2014; Chuang et al., 2017). In most but not all of the experiments measurements were made in both males and females, as specified. After the studies with the FVB/N mice had been completed we were able to obtain brain weights in 2 Lal+/+ (1 male / 1 female) and 3 Lal−/− (1 male / 2 female) mice that were all on a pure BALB/c background, and all of which were 83 days of age. For all mice the litters were weaned at 21 days and genotyped before that age using an ear notch. A cereal-based, low-cholesterol rodent diet (No.7001, Envigo:Teklad, Madison, WI) was used in all experiments.. This diet had an inherent cholesterol content of 0.02% (w/w) and a crude fat content of about 4%. All mice were group-housed in plastic colony cages containing wood shavings and had continual access to drinking water. They were kept in a light-cycled room with 12h of light (1200-2400h) and darkness (0-1200h). All mice were studied in the fed state towards the end of the dark phase of the lighting cycle. These studies were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Blood and Brain Collection for Biochemical and Molecular Analyses
The mice were anesthetized and exsanguinated from the vena cava into a heparinized syringe. They were not subsequently perfused. The total amount of blood removed varied with the age of the mouse but was at least half of the circulating volume. The skull was opened and the entire brain was removed, rinsed in saline, blotted and weighed. Those brains intended for mRNA expression analyses were immediately frozen in liquid nitrogen whereas brains used for the measurement of EC and UC concentrations were cut into pieces and added to ~40 ml of chloroform-methanol (2:1 v/v). For the cholesterol synthesis measurements the brains were placed in 5 ml of ethanolic KOH.
Brain Histology
Brains from paired Lal+/+ and Lal−/− mice at 141- and 250-days of age were immersion fixed in 10% v/v buffered formalin as previously described for brains taken from Lal+/+ and Lal−/− mice at 49 days of age (Aqul et al., 2011). Following fixation, hindbrains (brainstem and cerebellum) were amputated from the cerebral hemispheres and diencephalon at the level of the superior colliculus. The cerebral hemispheres were sectioned in the coronal plane at the level of the hypothalamic infundibulum, and the hindbrains were sectioned in the axial plane at the level of the pons. Tissues were dehydrated to xylene, embedded in paraffin, and sectioned on a Leitz ® rotary microtome at 4 μm thicknesses.Paraffin sections were rehydrated, stained with hematoxylin and eosin (H&E), and evaluated by light microscopy. Immunostaining was performed at room temperature on a BenchMarkXT® automated immunostainer using the UltraVIEW ® system with horseradish peroxidase and diaminobenzidine (DAV) chromogen (Ventana Medical Systems, Tucson, AZ). Optimum primary dilutions were predetermined using known positive control sections. Paraffin sections were cut at 4 μm thicknesses on a Leitz ® rotary microtome, mounted on positively-charged glass slides and air-dried overnight. Sections were then placed onto the BenchMarkXT®, deparaffinized and then incubated for 1 h with either primary mouse monoclonal antibody to calbindin (Novocastra®, Newcastle upon Tyne, UC, 1:50 dilution) or rabbit polyclonal antibody to glial fibrillary acidic protein (Biocare®, Concord, CA, 1:400 dilution) diluted in ChemMate® buffer (Ventana Medical Systems, Tucson, AZ) or with buffer alone as a negative reagent control. Following incubation, sections were then washed in buffer, incubated with a freshly-prepared mixture of diaminobenzidine (DAB) and H2O2 in buffer for 8 min, and then washed sequentially in buffer and then water. Sections were then counterstained with hematoxylin, dehydrated in a graded series of ethanols and xylene, and coverslipped. Stained sections were evaluated by light microscopy, with positive reactions identified as a dark brown DAB reaction product.
Brain Esterified and Unesterified Cholesterol Concentrations
These measurements were carried out as described previously for the liver and small intestines in other mouse models using [4-14C]cholesteryl oleate and [1,2-3H(N)]cholesterol to correct for procedural losses associated with the tissue extraction and column separation steps (Turley et al., 2010). Separation of the EC and UC fractions was achieved using an established column chromatography method. The EC and UC fractions were taken to dryness, saponified in alcoholic KOH, and extracted with petroleum ether (PE). At this stage a known quantity of stigmastanol (sitostanol) (usually 1.0 mg) was added to the PE phase. Aliquots of this extract were used to determine recovery of the radiolabeled standards and also to quantitate the mass of cholesterol in each fraction using a Hewlett Packard HP 5890 Series II gas chromatograph (GC) connected to a HP Integrator (Model 3396A). The GC was fitted with an Agilent HP-5 capillary column (No. 19091J-102) lined with (5%-phenyl)-methylpolysiloxane. The column was connected to a flame ionization detector (FID) that used hydrogen as the carrier gas (ie the mobile phase). The material lining the column served as the stationary phase. Hydrogen, together with air, served as the fuel gas in the FID. Nitrogen served as the make-up gas in the detector with the flow rate set at 20 ml/min. The temperature of the oven was set at 310 deg C and for the FID at 350 deg C. Split injection was used because of the generally high levels of cholesterol in the extracts, with the split ratio being 50:1. Cholesterol in the sample extracts was not derivatized before injection onto the column. Losses of sterol during the GC separation and detection steps were corrected for using stigmastanol as the internal standard. With this system the EC and UC concentrations (presented as mg/g wet tissue weight) in whole brain tissue could be accurately determined. However, it could not be applied to making such measurements in different brain regions or cell types given the very small proportion of cholesterol in brain that ordinarily is esterified, and also because structures of particular interest such as the choroid plexus represent only a very small proportion of whole brain mass. This would require much more sophisticated methodology of the kind recently described by Yutuc et al., 2020.
Brain Residual Blood Content
To estimate the contribution of EC in the residual blood present in excised brain tissue to the levels of EC detected in the brain as a whole we used data from studies by Schümann et al., 2007. Those experiments utilized 59Fe to measure the residual blood content of various organs including the brain in normal young adult C57BL/6 mice that had been exsanguinated to different degrees. In mice from which 400 μl of blood had been removed the residual blood content of the brain was 0.8% of wet brain weight. Applying this figure and a blood density of 1.057 g/ml (Riches et al., 1973) we estimated the residual blood content of brains in the present studies to be ~7.5 μl/g wet tissue weight.
Whole Blood and Plasma Cholesterol Concentrations
In one experiment we specifically determined the concentrations of EC and UC in the whole blood of Lal+/+ and Lal−/− mice at 70 to 76 days of age. The blood EC levels from that study, along with the estimated residual blood volume of excised brains were used to calculate the amount of EC detected in the whole brain that was attributable to EC in residual blood. In other studies aliquots of plasma were added directly to 5 ml of alcoholic KOH for determination of the total cholesterol concentration. Whole blood and plasma levels of EC and UC, or just total cholesterol (TC) concentrations, were all determined using gas chromatography (Lopez et al., 2015; Chuang et al., 2017).
Brain Cholesterol Synthesis
The rate of cholesterol synthesis in the whole brain was measured in vivo using tritiated water as described earlier (Lopez et al., 2017a; Lopez et al., 2017b). The mice were given an intraperitoneal injection of [3H]water (approx. 2mCi / g body weight) and then kept in a warm environment under a well ventilated fume hood for 60 min. They were then anesthetized and exsanguinated and the whole brain was excised, weighed and placed in 5 ml of ethanolic KOH. The labeled sterols were extracted, precipitated with digitonin and their activity determined. The rate of cholesterol synthesis was expressed as the nmol of water incorporated into sterol per hour per gram wet weight of tissue (nmol/h/g).
Brain Relative mRNA Expression Analysis
As the biochemical measurements were made using the whole brain, the mRNA analyses were similarly performed using the entire brain rather than just selected regions. At 141-days of age the Lal−/− mice showed overt genotype-related differences in brain mass and EC levels and were therefore considered ideal for exploration of possible changes in levels of expression of a constellation of genes known or believed to regulate brain cholesterol homeostasis. In addition to genes involved in controlling cholesterol synthesis, degradation, esterification and transport, others that serve as markers of inflammation and cell signaling were also included in the analysis. Total RNA was isolated from whole brain extracts and RNA concentrations were determined using RNA STAT-60 (Tel-Test, Inc., Friendswood, TX). Quantitative real-time PCR (qPCR) was performed using an Applied Biosystems 7900HT sequence detection system (Jones et al., 2013). Total RNA was treated with DNase I (RNase-free; Roche Molecular Biochemicals, Indianapolis, IN) and reverse-transcribed with random hexamers using SuperScript II (Invitrogen, Carlsbad, CA) to generate cDNA. The primer sequences for almost all the genes selected are given in several earlier publications (Li et al., 2005; Liu et al., 2009; Taylor et al., 2012). Others were designed using various primer design algorithms as detailed previously (Jones et al., 2013). The mRNA level for each gene in the Lal−/− mice was normalized using the housekeeping gene cyclophilin and then expressed relative to that in the matching Lal+/+ controls, which in each case was arbitrarily set at 1.0.
Analysis of Data
All values are presented as the mean ± SEM of data from the specified number of animals. GraphPad Prism 6.02 software (GraphPad, San Diego, CA) was used to perform all statistical analyses. Differences between means were tested for statistical significance (p < 0.05) using an unpaired Student’s t-test to compare the values for the Lal−/− mice and their age-matched Lal+/+ controls.
Results
By 50 Days of Age Lal−/− Mice, Compared to Age-Matched Lal+/+ Controls, Exhibited Lower Whole Brain Weights
In the course of our studies focusing mainly on whole body cholesterol metabolism in Lal−/− mice (Aqul et al., 2014) we noted that, in contrast to the decisive increase in the mass of the liver and other organs like the spleen and small intestine, there was a trend toward lower whole brain weights in the Lal−/− mice as they aged. This was investigated in an experiment with Lal−/− mice and matching Lal+/+ controls, all in the age range of 68-72 days. Measurements were made in both males and females because published data from a study in 25 strains of retired breeder mice (that did not include the FVB/N strain) at approximately 200 days of age showed that in 20 of the strains females had a higher brain weight (Roderick et al., 1973). In the current study there was not a clear genotypic difference in the body weights of the Lal+/+ and Lal−/− mice of either sex (Fig. 1a). However, brain weights, while about the same for males and females within genotype, were significantly lower in both the male and female Lal−/− mice compared to their matching Lal+/+ controls (Fig. 1b). There was also a markedly higher level of EC in the brains of all the Lal−/− mice (Fig. 1c) but no genotypic- or sex–related differences in the UC concentrations (Fig. 1d). These findings prompted similar measurements in the brains of a large number of Lal−/− and matching Lal+/+ mice over a wide age span ranging from pre-weaning to late-stage disease. The body and brain weight data for mice ranging in age from 14 to more than 230 days are presented in Table 1. In many cases these mice were surplus to the needs of other studies and so were used mostly for brain harvesting. Consequently there is significant variation in not only the numbers of mice at each age but also in the proportions of males and females as well as of Lal+/+ and Lal−/− mice. Nevertheless it is clear that from 50 days of age brain weights were lower in the Lal−/− mice, with this feature becoming pronounced in mice at and beyond about 140 days of age. Brain weights were not expressed relative to body weights because in the Lal−/− mice the liver represented an increasing proportion of body mass with advancing disease, reaching about 20% of body weight at 168 days of age compared to only about 4% in matching Lal+/+ controls (Aqul et al., 2014). In evaluating the current data it should be noted also that for our Lal−/− mice on a pure FVB/N background the median age at death was 308 and 377 days, respectively for the males and females (Aqul et al., 2014). In the Lal−/− mice (also pure FVB/N background) used by Sun et al., 2014, a median lifespan of 287 days for Lal−/− mice was found (data for males and females were combined). Shorter lifespans were reported for Lal−/− mice on a mixed strain (129/Sv : CF-1) background (Du et al., 2001). For the five mice on a BALB/c background at 83 days of age the brain weights for the two Lal+/+ mice were 0.453g (female) and 0.475g (male) whereas for their Lal−/− littermates the weights were 0.346 and 0.350g (female), and 0.400g (male). The three Lal−/− mice all exhibited the hallmark hepatomegaly seen in CESD. Clearly these few data make it apparent that the brain mass reduction evident in aging Lal−/− mice of the FVB/N strain was manifest in BALB/c mice with the same mutation.
Fig. 1.
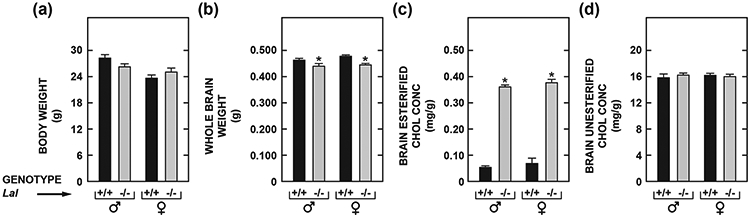
Body (a) and whole-brain (b) weights, and concentrations in brain of esterified (c) and unesterified (d) cholesterol in male and female Lal+/+ and Lal−/− mice at 68 to 72 days of age. The measurements (for c, d) were made in extracts of the entire brain using a combination of column and gas chromatography as described in Materials and Methods. Values represent the mean ± SEM of measurements for 5 Lal+/+ and 6 Lal−/− males, and 4 Lal+/+ and 6 Lal−/− females, all of which were of the FVB/N strain. *Significantly different from the value for Lal+/+ controls of the same gender (p < 0.05).
Table 1.
Whole brain weights in Lal +/+ and Lal −/− mice from before weaning until late-stage disease
| Age (days) |
Genotype | No. of mice ♂ / ♀ |
Body weight (g) |
Whole brain weight (g) |
|---|---|---|---|---|
| 14 | Lal +/+ | 3 / 0 | 7.2 ± 0.4 | 0.381 ± 0.005 |
| Lal −/− | 2 / 3 | 7.0 ± 0.4 | 0.379 ± 0.006 | |
| 21 | Lal +/+ | 3 / 2 | 11.3 ± 0.6 | 0.418 ± 0.005 |
| Lal −/− | 4 / 2 | 10.3 ± 0.6 | 0.402 ± 0.007 | |
| 35 | Lal +/+ | 2 / 4 | 18.6 ± 0.9 | 0.425 ± 0.008 |
| Lal −/− | 1 / 5 | 19.0 ± 0.2 | 0.423 ± 0.006 | |
| 50 | Lal +/+ | 2 / 2 | 23.9 ± 1.2 | 0.457 ± 0.004 |
| Lal −/− | 1 / 3 | 23.1 ± 1.5 | 0.429 ± 0.001* | |
| 68-76 | Lal +/+ | 8 / 5 | 27.3 ± 1.2 | 0.473 ± 0.003 |
| Lal −/− | 7 / 7 | 25.5 ± 0.8 | 0.440 ± 0.005* | |
| 140-142 | Lal +/+ | 0 / 5 | 26.4 ± 0.5 | 0.485 ± 0.008 |
| Lal −/− | 0 / 5 | 23.8 ± 1.0* | 0.400 ± 0.003* | |
| 230-280 | Lal +/+ | 3 / 2 | 34.1 ± 2.2 | 0.499 ± 0.008 |
| Lal −/− | 3 / 3 | 27.9 ± 1.3* | 0.398 ± 0.008* |
After weaning at ~21 days, all mice were fed ad libitum a cereal-based, low-fat rodent diet. All mice were of the FVB/N strain.
Values are mean ± SEM for the number of mice indicated.
p < 0.05 compared to the corresponding value for matching Lal +/+ mice.
Although a Genotypic Difference in Brain Weight Was Not Evident Until Maturity, Elevated Esterified Cholesterol Concentrations in the Brains of Lal−/− Mice Were Seen Before Weaning
Fig. 2 presents the brain EC and UC concentrations and whole brain cholesterol contents for all the groups described in Table 1. Compared to values for matching Lal+/+ mice, the EC level in the Lal−/− mice was elevated 5.7-fold at just 14 days post partum, and 27-fold at 230-280 days (Fig. 2a). At no age was there a discernable genotypic difference in the UC concentration (Fig. 2b). In reviewing these two sets of data it will be evident that even though there were striking increases in the EC levels in the Lal−/− mice, at no age did the amount of EC per gram of tissue exceed more than ~5% of the mass of UC present. Whole brain cholesterol (EC + UC) contents were significantly lower in the Lal−/− mice at and beyond ~140 days of age (Fig. 2c), with this being due to the considerably lower brain weights in the aging mutants.
Fig. 2.
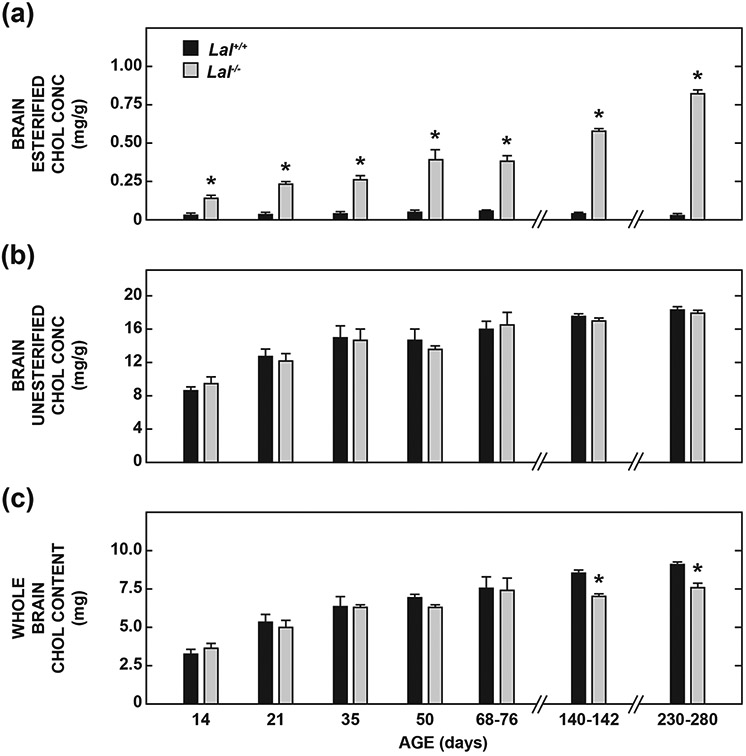
Concentration of esterified (a) and unesterified (b) cholesterol in the brain, and whole-brain cholesterol contents (c) in Lal+/+ and Lal−/− mice from pre-weaning to late-stage disease. All mice were of the FVB/N strain. The brain tissue was derived from the same sets of mice as described in Table 1. Values represent the mean ± SEM of measurements for the number of mice specified. *Significantly different from the value for Lal+/+ controls at that age (p< 0.05).
Brain Histology in Lal−/− Mice at 141- and 250-Days of Age Was Unremarkable
We previously conducted a comparative study of brain histology in 49-day old Lal−/− mice (FVB strain) and Npc1−/−nih mice (BALB/c strain) (Aqul et al., 2011). Unlike the profound changes seen in the Npc1−/−nih mice, the brains of the matching Lal−/− mice showed normal neuronal morphology, Purkinje cell calbindin reactivity, and GFAP (glial fibrillary acidic protein) immunoreactivity. Nevertheless the markedly lower brain weights and dramatic rise in EC levels seen in the aging Lal−/− mice prompted an examination of the general histological features of brains from matching Lal−/− and Lal+/+ mice at 141 and 250 days of age. Nothing remarkable was evident at either age except in the 250-day old mutant which had one xanthogranuloma and a few isolated lipid-laden macrophages in the choroid plexus but no parenchymal abnormalities.
Elevation of Brain Esterified Cholesterol Levels in Lal−/− Mice Was Not Attributable to Residual Blood in Resected Tissue
To better interpret the brain EC data presented in Fig. 2a we needed to obtain an estimate of how much of this EC was present in residual blood. While the bulk of UC in resected brain tissue is contained within myelin sheaths (Snipes & Suter, 1997), a comparatively minor amount is present in erythrocytes in retained blood which also has a variable quantity of EC present in lipoproteins. The two main factors determining the amount of EC present in residual blood are the completeness of exsanguination and the plasma EC concentration which is elevated in all the non-HDL lipoprotein fractions in CESD patients (Gomaraschi et al., 2016). In the studies of Schümann et al., 2007 it was determined that, after removing 400 μl of blood from young adult mice, the residual blood content of the brain was 0.8% of wet brain weight. Using a density for blood of 1.057 g/ml (Riches et al., 1973) it can be calculated that the residual blood content of the brain in healthy adult mice after this degree of exsanguination was 7.6 μl/g. We measured the total cholesterol (TC) concentrations (as the sum of the UC and EC levels) in the whole blood of Lal+/+ and Lal−/− mice at 70 to 76 days of age (Fig. 3a). While the TC levels did not vary with genotype, the proportion of the blood cholesterol in the Lal−/− mice that was esterified (17%) was only about half of that in their Lal+/+ controls (37%). Using the blood EC concentrations in Fig. 3a, along with a residual blood content of 7.6 μl/g, it can be estimated that in the brains from Lal−/− and Lal+/+ mice at 70 to 76 days the amount of EC present in residual blood was 1.5 and 3.1 μg/g, respectively. Thus, from the brain EC data for similarly aged mice (Fig. 2a) it can be concluded that in the Lal+/+ mice (which typically had a brain EC level of 0.056 mg/g) about 6% of the detected EC was in residual blood. In the matching Lal−/− mice (with an average brain EC level of 0.383 mg/g) this proportion was only 0.4%. This low value partly reflects the fact that Lal−/− mice on a pure FVB background have lower plasma TC levels compared to their Lal+/+ controls (Fig. 3b). This contrasts with Lal−/− mice on a mixed strain (129/Sv : CF-1) background where plasma TC levels do not differ from those in their Lal+/+ controls (Du et al., 2001), and more particularly with Lal−/− mice on either a pure C57BL/6 (Radović et al., 2016) or BALB/c (Posey, Lopez, & Turley, unpublished data) background which have elevated plasma TC levels.
Fig. 3.
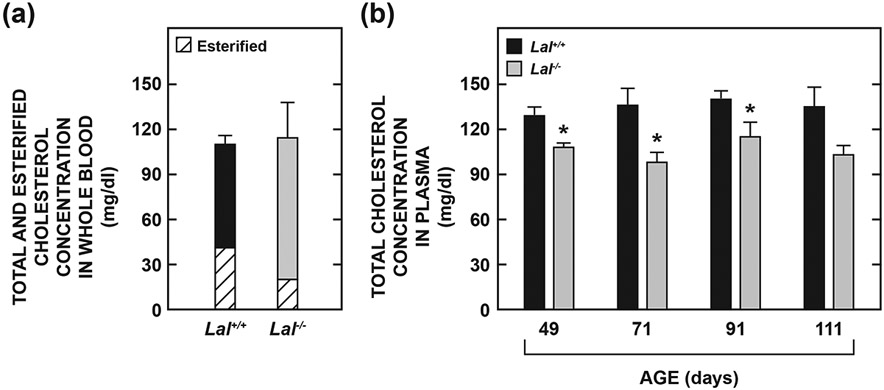
Concentration of total cholesterol in whole blood (a) and in plasma (b) of Lal+/+ and Lal−/− mice in different studies. In one experiment the concentrations of EC and UC in whole blood were measured in mice aged from 70 to 76 days and summed to obtain the total cholesterol (TC) level. In another study plasma TC data were obtained directly without separation of the EC and UC fractions from mice at four different ages ranging from 49 to 111 days. In both studies all mice were of the FVB/N strain. Values for whole blood represent the mean ± SEM of data from 4 Lal+/+ (3 males /1 female) and 4 Lal−/− (3 males /1 female) mice, while those for the plasma TC levels at four age points were from males only. The numbers of mice at each age were respectively: 49 d: 12 Lal+/+ and 15 Lal−/−, 71 d: 5 Lal+/+ and 6 Lal−/−, 91 d: 5 Lal+/+ and 6 Lal−/−, 111 d: 4 Lal+/+ and 3 Lal−/−.*Significantly different from the value for the matching Lal+/+ controls (p < 0.05).
Brain Cholesterol Synthesis Rates Showed a Prototypical Decline With Age But Did Not Differ Between Lal−/− Mice and Their Lal+/+ Controls
One well documented feature of the developing brain is its exceptionally high rate of cholesterol synthesis which provides the UC needed for myelin formation (Jurevics & Morell, 1995; Snipes & Suter, 1997; Quan et al., 2003). Two other features of the brain cholesterol synthesis rate, based on studies in a rat model, are that it does not vary diurnally and is not influenced by the level of dietary cholesterol intake (Jurevics et al., 2000). In our next study we measured the rate of brain cholesterol synthesis in vivo in newly weaned Lal+/+ and Lal−/− mice (23 days), and in others that were approaching early adulthood (50 days). As shown in Fig.4, at neither age was a genotypic difference in the synthesis rate discernible. However, as expected, there was a significant decline in the rate as a function of the level of maturity.
Fig. 4.
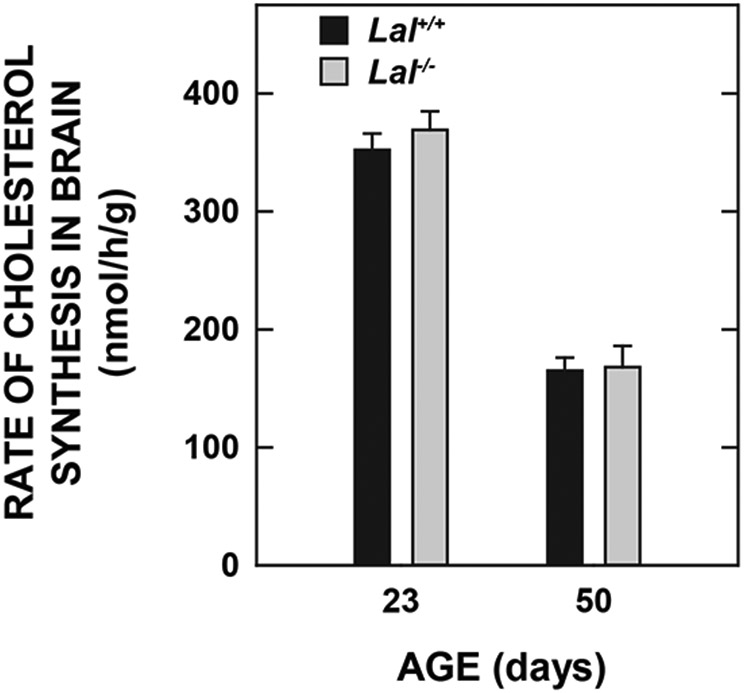
Rate of cholesterol synthesis in the brains of Lal+/+ and Lal−/− mice at two different ages. These rates were measured in vivo using [3H]water as described in Materials and Methods. They reflect the rate of incorporation of water into new sterols throughout the entire brain over the course of 1 h. Values represent the mean ± SEM of measurements in 3 Lal+/+ (1 male /2 females) and 5 Lal−/− (3 males /2 females) mice at 23 days, and in 6 Lal+/+ (all females) and 6 Lal−/− (all females) mice at 50 days. All mice were of the FVB/N strain. At neither age was the value for the Lal−/− mice significantly different from that for the matching Lal+/+ controls (p >0.05).
Relative Level of Expression of mRNA For Multiple Genes Involved in Regulating Brain Cholesterol Homeostasis Was Unchanged in Lal−/− Mice With Advanced Disease
In the next study we used the whole brains from Lal−/− mice at 141 days of age to measure the relative levels of mRNA for an array of genes that play key roles in regulating cholesterol homeostasis in the CNS. At this age the brain mass in the Lal−/− mice was ~18% less than in their matching Lal+/+ controls (Table 1), and the brain EC levels in the Lal−/− mice were elevated ~15-fold (Fig. 2a). This age approximated that at which Du et al., 2001 carried out oil red O staining of the choroid plexus in this model (~182 days). The expression level for each gene in the Lal+/+ mice was arbitrarily set at 1.0 (dashed line) so bars reflect the fold change in the Lal−/− mice. The data in Fig. 5a are for genes involved in various pathways in cholesterol metabolism. A comment is needed here regarding the finding that the Lipa mRNA results do not show a complete absence of this transcript in the Lal−/− mice. This simply reflects the fact that our qPCR primers detect and amplify mRNA sequence upstream of the genetic disruption (downstream of exon 4) in this targeted allele, and thus likely detect a small amount of short-lived, truncated Lipa mRNA which does not encode for the functional protein in the knockout mice. In no case was a significant genotypic difference observed in the mRNA for any of the cholesterol regulatory proteins. For Hmgcs and Hmgcr this result is fully consistent with the finding that the rate of brain cholesterol synthesis did not show a genotypic difference at a much earlier stage of disease progression (Fig. 4). In regard to cholesterol esterifying enzymes, the data presented are for Soat1 and Lcat but not Soat2. The quantitative PCR Ct value for Soat2 was many fold greater than that for the other two esterifying enzymes, in keeping with its negligible level of detection in mouse brain (Zeisel et al., 2018), and its reported absence in human brain (Lee et al., 2015). The data in Fig. 5b are for four genes that encode proteins that are markers of inflammation. In all cases these were elevated in the Lal−/− mice but to only a moderate degree compared to what has been previously reported for the liver in this model (Aqul et al., 2014).
Fig. 5.
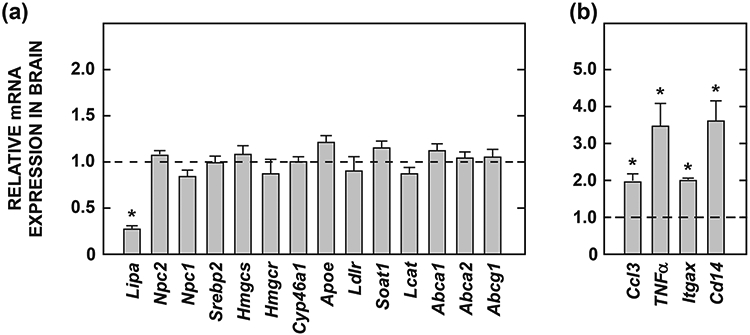
Relative mRNA levels for multiple genes involved in the regulation of cholesterol homeostasis (a), or that serve as markers of inflammation (b) in the brains of 141-day old Lal+/+ and Lal−/− mice. The details of these analyses are given in Materials and Methods. For each genotype brains were obtained from 2 male and 2 female mice, all of the FVB/N strain. The expression level for each gene in the Lal+/+ mice was arbitrarily set at 1.0 (dashed line) so bars reflect fold-change observed in the Lal−/− mice. Values represent the mean ± SEM. *Significantly different from the value for the Lal+/+ controls (p<0.05). The full name of each gene in the order as it appears in the figure is: Lipa, gene that encodes LAL; Npc2, Niemann-Pick type C2; Npc1, Niemann-Pick type C1; Srebp2, sterol regulatory element-binding protein 2; Hmgcs, 3-hydroxy-3-methylglutaryl coenzyme A synthase; Hmgcr, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; Cyp46a1, cholesterol 24-hydroxylase; Apoe, apolipoprotein E; Ldlr, low-density lipoprotein receptor; Soat1, sterol O-acyltransferase 1 (previously called Acat1); Lcat, lecithin:cholesterol acyltransferase; Abca1, ATP-binding cassette transporter A1; Abca2, ATP-binding cassette transporter A2; Abcg1, ATP-binding cassette transporter G1; Ccl3, chemokine (C-C motif) ligand 3 (also known as Mip1α); TNFα, tumor necrosis factor alpha; ltgax, integrin subunit alpha X (also known as CD11c); Cd14 (Cluster of differentiation 14 antigen).
Discussion
The brain does not clear lipoproteins from the general circulation but has its own lipoprotein production and transport system involving mostly astrocytes and neurons (Pfrieger & Ungerer, 2011;Orth & Bellosta, 2012; Vitali et al., 2014; Wang & Eckel, 2014). Astrocytes synthesize cholesterol and also produce apolipoprotein E (apo E). These components, along with phospholipids, are used in the assembly of high-density lipoprotein (HDL)-like particles that are subsequently taken up by neurons through various receptors belonging to the low-density lipoprotein family. Even though EC ordinarily represents just a fraction of cellular cholesterol content throughout the CNS, its formation, mostly by SOAT1, is a key part of this internal lipoprotein transport system. Presumably, when Lipa is absent or mutated the EC contained in these internally produced lipoproteins is not hydrolyzed and therefore accumulates in the late endosomal/lysosomal compartment of neurons and possibly of oligodendrocytes and astrocytes as well. Compared to the substantial literature on the impact of disrupted Npc1 function on brain mass and cholesterol homeostasis in the CNS, relatively little is known about how mutations in Lipa affect these two parameters in humans and animal models of CESD or WD. Certainly, the findings of lipid inclusions in oligodendrocytes and astrocytes in the CNS of a Wolman patient (Byrd & Powers,1979), and later of lipid-laden epithelial cells in the choroid plexus of a CESD mouse model (Du et al., 2001), together suggested that further exploration of how the absence of Lipa impacted brain mass, histology and cholesterol homeostasis was needed. The data presented here represent an initial step in meeting this need.
In evaluating these data a number of points relating to the use of the whole brain and not separate regions warrant comment. One reason for this approach is the very low level of EC in normal brain tissue. While these levels could be determined precisely in the brain as a whole using the conventional gas chromatographic technique that we employed, it was not sensitive enough to measure the EC content of individual regions, especially those that constitute a minor proportion of brain mass. There are very few published data for EC levels in different regions of the brain in either humans or animal models. However, one study using autopsied brain tissue from neonates at 1 to 26 months of age found similar EC concentrations in the forebrain, cerebellum, and brain stem (Yusuf et al., 1981). These concentrations were mostly in the range of 40 to 70 ug/g wet tissue weight (ie 0.04 to 0.07 mg/g) which are comparable to the EC levels found in the Lal+/+ mice across all ages in the current study (Fig. 2a). In the Lal−/− mice at 140-142 and 230-280 days of age the brain EC levels had risen 15- and 27-fold, respectively. A second reason we used the whole brain was that it facilitated a precise way of determining how much of the EC detected in the tissue was present in residual blood. This level of contamination was found to be low because of the considerable degree of exsanguination and the fact that Lal−/− mice of the FVB/N strain are not hypercholesterolemic (Fig. 3b), unlike their counterparts on C57BL/6 background (Radović et al., 2016) and CESD patients (Reiner et al., 2014). Yet another advantage of using the whole brain in this type of study is that it circumvents regional differences throughout the CNS in the expression levels of genes involved in the maintenance of multiple aspects of brain cholesterol homeostasis (Lund et al. 1999; Prasad et al., 2002; Sullivan et al., 2004; Segatto et al., 2012).
One of the main questions arising from the EC concentration data presented here is whether the dramatic increment found in the aging Lal−/− mice is distributed across the bulk of the brain or localized to a specific region(s). In 2001 Du et al. published a detailed study of the histopathological and biochemical features of their Lal−/− mouse model on a mixed 129Sv : CF-1 background that had been maintained on a basal rodent diet. The plasma total cholesterol levels in the Lal−/− mice were not elevated although the proportion of cholesterol in LDL increased while that in HDL decreased compared to levels in matching Lal+/+ mice. A particularly notable finding was lipid storage, as detected by oil red O staining, in the epithelial cells of the choroid plexus in a 6-month old Lal−/− mouse. In earlier studies by the Farese laboratory (Accad et al., 2000) it was discovered that the selective deficiency of Soat1 (called Acat1 at that time) in two mouse models of atherosclerosis resulted in an extensive deposition of unesterified cholesterol (UC) in the skin and brain. These models, which were deficient in either apolipoprotein E (Apoe) or the Ldl receptor, as well as in Acat1, were fed lipid-enriched dietary formulations for extended periods resulting in extreme dyslipidemia. In the Apoe−/− : Acat1−/− mice UC crystal deposits were found in the brain, specifically near the choroid plexus. Similarly, in the Ldlr−/− : Acat1−/− mice there was crystalline UC in the cerebellum and near the choroid plexus. Together these findings raise the question of what proportion of the marked increase in EC levels in the brains of the 140-142 -day old Lal−/− mice in the current studies might have been localized in or near the choroid plexus. This calculation can be made using precise morphometric data for the choroid plexus (embracing its four regions) in normal rat brain (Quay, 1972). In that study data are presented for male rats, the postnatal age of which ranged from 3 to 25 weeks. Specifically, it was found that in the 25-week old rats the whole brain wet weight was 1.707 g, with the wet weight of the four choroid plexuses combined being 4.921 mg (0.29% of wet brain weight). The water content of the choroid plexuses averaged 80.4%. Extrapolation of these data to the brains of the Lal−/− mice at 140-142 days of age allows for an estimation of the mass of their choroid plexus (wet and dry weights), and then a comparison of this mass to that of the EC contained within their brains as a whole. These calculations are as follows. From the rat data it can be estimated that the mass of the choroid plexus in the Lal−/− mice at 140-142 days of age was around 1.16 mg, wet weight. The brains of these mice had an EC concentration of 0.576 mg / g wet weight (Fig.2A) which equates to 0.230 mg / brain, wet weight. On this basis the entire mass of EC in the brain is about 20% of the estimated wet weight of the choroid plexus. However, when taking into account a water content of about 80%, the dry weight of the choroid plexus is ~ 0.227 g. Although some of the sequestered EC may have been contained in epithelial cells within the choroid plexus this appears to be at most a minor site of its sequestration in this model. However, it is possible that the accumulated EC was present in cells in close proximity to the choroid plexus. Be that as it may, the challenge of localizing the region(s) of the CNS where the EC is concentrated resides partly in the fact that the absolute mass of EC is minute compared to that of the entire brain in the Lal−/− mice. However, recent studies by Yutuc et al (2020) described new technology using an advanced mass spectrometry imaging platform to conduct quantitative measurements of sterols in minute amounts of brain tissue. Application of such methodology to the choroid plexus and adjacent brain regions from aging Lal−/− mice with and without Soat1 or Lcat may not only localize the sites of EC sequestration but also shed more light on the respective roles of these two enzymes in generating the EC in the CNS.
The finding that Lipa deficiency did not lead to changes in the expression levels of mRNA for multiple key regulatory genes in cholesterol metabolism within the CNS (Fig 5a) contrasts with what we previously reported for several genes in the liver of the same CESD mouse model at 50 days of age (Aqul et al., 2014). In that study there were, for example, marked elevations in the hepatic mRNA levels for Npc2 and Abcg1 in the Lal−/− mice. Such changes were not unexpected given the profound effect that disrupted Lipa expression is known to have in the liver. Although the mRNA levels for Hmgcs and Hmgcr (two major regulatory enzymes in the cholesterol synthetic pathway) showed little change in the livers of the Lal−/− mice, their rates of hepatic cholesterol synthesis, measured in vivo using the same technique employed for the brain in the current studies, were elevated about 5-fold. This clearly reflected a response to a major perceived cellular deficit of cholesterol in hepatocytes owing to the sequestration of EC in the E/L compartment. At 50 days of age hepatic EC levels in the Lal−/− mice reached ~50 mg/g which represented ~100-fold higher level than in the livers of matching Lal+/+ controls (Aqul et al., 2014). In the brains of 50-day old Lal−/− mice the EC level (0.297 ± 0.011 mg/g) was nearly 7-fold higher than in matching Lal+/+ controls (0.043 ± 0.001 mg/g) (Fig. 2a). The studies of Quan et al. (2003) demonstrated that in the normal mouse at 50 days after birth the formation of myelin, which is the main driver of the rate of cholesterol synthesis in the CNS (Snipes & Suter, 1997), is still progressing to a small degree. In the mature animal there is a very low basal rate of synthesis that is needed to supply the cholesterol used in generating HDL-like particles for subsequent transfer to neurons and possibly other cell types. When Lipa function is absent in the developing CNS there is no discernable change in the rate of cholesterol synthesis across the entire brain (Fig.4) probably because any compensatory increase in the synthesis rate elicited in response to the entrapment of EC at the sites of lipoprotein uptake are so subtle as to be undetectable in the face of the far higher rates of cholesterol synthesis happening elsewhere as part of myelin production. It should be mentioned here that similar findings have been described for the brain vs the liver in Npc1−/− nih mice ( Liu et al., 2010) although their compensatory increase in hepatic cholesterol synthesis is not nearly as pronounced as that in Lal−/−mice (Aqul et al., 2014).
The next point of discussion centers on the strikingly lower brain weights in the Lal−/− mice in the latter stages of disease progression, a feature not confined to only mice of the FVB/N strain. Here an interesting comparison of brain weight reduction in Lal−/− mice and Npc1−/− nih mice can be made. The latter model, on a pure BALB/c background and maintained on a low cholesterol rodent diet, had a median lifespan of 84 days (Liu et al., 2009), far shorter than that of Lal−/− mice on a mixed strain (129/Sv : CF-1) (Du et al., 2001) or a pure strain (FVB) (Aqul et al., 2014) background. In Npc1−/− nih mice at 49 days of age, both males and females had a 16% lower brain mass compared to their matching Npc1+/+ nih controls (Xie et al., 2000). This was principally the result of Purkinje cell death, a hallmark feature of NPC disease (Vance, 2012). For similarly aged Lal−/− mice in the current studies brain mass was ~6% less than in age-matched Lal+/+ controls (Table 1). This genotypic difference increased to ~18% in Lal−/− mice at 140-142 days of age. However, we found no evidence of altered histology in the brains of Lal−/− mice at different stages of disease progression. This conclusion was reached in an earlier detailed comparison of the cerebellar cortex in 49-day old Lal−/− mice and Npc1−/− nih mice (Aqul et al., 2011). Specifically, in contrast to what was found in the Npc1−/− nih mice, in those lacking LAL there was normal neuronal morphology, Purkinje cell calbindin reactivity, and glial fibrillary acidic protein immunoreactivity. Even in Lal−/− mice as old as ~250 days where brain weight was ~20% lower than in similarly aged Lal+/+ controls there were no clear signs of altered histopathology. Thus, while this reduction in brain mass could have been linked to severe liver disease in aging Lal−/− mice, the lack of overt histological changes in their brains indicates these mice did not have hepatic encephalopathy. The modest increases in the mRNA levels of four markers of inflammation in the brains of Lal−/− mice at 141 days (Fig. 5b) possibly reflected the minor presence of lipid-laden macrophages as observed in the brains of Lal−/− mice at ~250 days. Here it is noteworthy that the mRNA level for Soat1, often taken as an indicator of macrophage presence, was not significantly higher in the brains of the 141-day old Lal−/− mice (Fig. 5a). This was in sharp contrast to the > 20-fold increase in hepatic mRNA levels for Soat1 in 50-day old Lal−/− mice (Aqul et al., 2014). Earlier, Du et al. (2001) demonstrated a massive presence of lipid-laden macrophages in multiple organs, particularly the livers, of aging Lal−/− mice. This difference in findings for the brain and liver is not unexpected given that the amount of EC present in lipoproteins cleared daily from the circulation by the liver is many orders of magnitude greater than the amounts of EC moving through the internal lipoprotein transport system in the brain. Nevertheless it is conceivable that in the course of their lifetime CESD patients not receiving enzyme replacement therapy could sequester appreciable levels of EC in their brains.
Finally, the collection of brain weight data together with histopathological analyses of tissue from different regions of the brain in CESD patients who were not diagnosed until late adulthood and / or whose disease proved too advanced for effective therapeutic intervention may provide useful leads in better defining EC sequestration in the CNS (Pisciotta et al., 2009; Canbay et al., 2018; Rashu et al., 2020). However, the rarity of such cases, together with the challenges of interpreting brain autopsy data (Gonzalez-Riano et al., 2017), mean that further research using animal models of CESD or Wolman disease will be essential. One such project might be a detailed biochemical and histochemical exploration of various regions of the brains of Lal−/− mice given intravenous enzyme replacement therapy, with variations in the dose administered and the stage of disease when treatment is started. Using such a strategy Sun et al., 2014 demonstrated reversal of advanced disease in the liver, spleen and small intestine of Lal−/− mice. The impetus for carrying out more animal model studies derives partly from the likelihood that many more cases of CESD could be detected given the expanding use of reduced LAL activity in blood as a marker for non-alcoholic fatty liver disease in pediatric and adult populations (Baratta et al., 2019).
Acknowledgments
This research was funded by National Institutes of Health Grants R01HL009610 (S.D. Turley) and R01DK078592 (J.J. Repa). The guidance and support of the now late John M. Dietschy, M.D. in the conduct of this research is warmly acknowledged. Drs. G. Grabowski and H. Du at the University of Cincinnati generously supplied the Lal+/− breeding stock used in these studies. Mario Saucedo, Carolyn Crumpton, Jennifer Burg, Monti Schneiderman, and Stephen Ostermann provided excellent technical assistance.
Abbreviations:
- bw
body weight
- CNS
central nervous system
- CESD
cholesteryl ester storage disease
- E/L
endosomal/lysosomal
- EC
esterified cholesterol
- LAL
lysosomal acid lipase
- SOAT1
sterol O-acyltransferase 1
- SOAT2
sterol O-acyltransferase 2
- UC
unesterified cholesterol
- WD
Wolman disease
Footnotes
Conflict of Interest
We declare no conflict of interest.
References
- Accad M, Smith SJ, Newland DL, Sanan DA, King LE Jr., Linton MF, Fazio S, Farese RV Jr. (2000) Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. The Journal of Clinical Investigation, 105:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alling C, & Svennerholm L (1969) Concentration and fatty acid composition of cholesteryl esters of normal human brain. Journal of Neurochemistry, 16:751–759. [DOI] [PubMed] [Google Scholar]
- Anchisi L, Dessi S, Pani A, & Mandas A (2013) Cholesterol homeostasis: a key to prevent or slow down neurodegeneration. Fronteirs in Physiology, 3:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqul A, Liu B, Ramirez CM, Pieper AA, Estill SJ, Burns DK, Liu B, Repa JJ, Turley SD, & Dietschy JM (2011) Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. The Journal of Neuroscience, 31:9404–9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqul A, Lopez AM, Posey KS, Taylor AM, Repa JJ, Burns DK, & Turley SD (2014) Hepatic entrapment of esterified cholesterol drives continual expansion of whole body sterol pool in lysosomal acid lipase-deficient mice. American Journal of Physiology-Gastrointestinal and Liver Physiology, 307:G836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratta F, Pastori D, Ferro D, Carluccio G, Tozzi G, Angelico F, Violi F,& Del Ben M (2019) Reduced lysosomal acid lipase activity: A new marker of liver disease severity across the clinical continuum of non-alcoholic fatty liver disease. World Journal of Gastroenterology, 25:4172–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besley GTN, Broadhead DM, Lawlor E, McCann SR, Dempsey JD, Drury MI, & Crowe J (1984) Cholesterol ester storage disease in an adult presenting with sea-blue histiocytosis. Clinical Genetics, 26:195–203. [DOI] [PubMed] [Google Scholar]
- Bindu PS, Taly AB, Christopher R, BharatKumar PV, Panda S, Netravathi M, Ravishankar S, Mahadevan A & Yasha TC (2007) Cholesterol ester storage disease with unusual neurological manifestation in two siblings: A report from South India. Journal of Child Neurology, 22:1401–1404. [DOI] [PubMed] [Google Scholar]
- Brown WV, Desnick RJ, & Grabowski GA (2014) JCL Roundtable: Enzyme replacement therapy for lipid storage disorders. Journal of Clinical Lipidology, 8:463–472. [DOI] [PubMed] [Google Scholar]
- Bryleva EY, Rogers MA, Chang CCY, Buen F, Harris BT, Rousselet E, Seidah NG, Oddo S, LaFerla FM, Spencer TA, Hickey WF, and Chang TY,(2010) ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proceedings of the National Academy of Sciences of the United States of America, 107:3081–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd JC III, & Powers JM (1979) Wolman’s disease: Ultrastructural evidence of lipid accumulation in central and peripheral nervous systems. Acta Neuropathologica, 45:37–42. [DOI] [PubMed] [Google Scholar]
- Canbay A, Müller MN, Philippou S, Gerken G, & Tromm A (2018) Cholesteryl ester storage disease:fatal outcome without causal therapy in a female patient with the preventable sequelae of progressive liver disease after many years of mild symptoms. American Journal of Case Reports, 19:577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter A, Brackley SM, Gao J, & Mann JP (2019) The global prevalence and genetic spectrum of lysosomal acid lipase deficiency: A rare condition that mimics NAFLD. Journal of Hepatology, 70:142–150. [DOI] [PubMed] [Google Scholar]
- Chang CCY, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, Lin S, Lee C-YG, Strom SC, Kashyap R, Fung JJ, Farese RJ Jr., Patoiseau J-F, Delhon A, & Chang TY (2000) Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. The Journal of Biological Chemistry, 275:28083–28092. [DOI] [PubMed] [Google Scholar]
- Chuang J-C, Lopez AM, & Turley SD (2017) Quantitation of the rates of hepatic and intestinal cholesterol synthesis in lysosomal acid lipase-deficient mice before and during treatment with ezetimibe. Biochemical. Pharmacology,135:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook RP (1958) Distribution of sterols in organisms and in tissues. In: Cook RP (Ed.), Cholesterol: Chemistry, biochemistry, and pathology (pp. 145–180). New York, NY: Academic. [Google Scholar]
- Dietschy JM, & Turley SD (2004) Cholesterol metabolism in the central nervous system during early development and in the mature animal. Journal of Lipid Research, 45:1375–1397. [DOI] [PubMed] [Google Scholar]
- Dincsoy HP, Rolfes DB, McGraw CA, & Schubert WK (1984) Cholesterol ester storage disease and mesenteric lipodystrophy. American Journal of Clinical.Pathology, 81:263–269. [DOI] [PubMed] [Google Scholar]
- Du H, Witte DP, & Grabowski GA (1996) Tissue and cellular specific expression of murine lysosomal acid lipase mRNA and protein. Journal of Lipid Research, 37:937–949. [PubMed] [Google Scholar]
- Du H, Duanmu M, Witte D, & Grabowski GA (1998) Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Human Molecular Genetics, 7:1347–1354. [DOI] [PubMed] [Google Scholar]
- Du H, Heur M, Duanmu M, Grabowski GA, Hui DY,Witte DP, & Mishra J (2001) Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. Journal of Lipid Research, 42:489–500. [PubMed] [Google Scholar]
- Du H, Cameron TL, Garger SJ, Pogue GP, Hamm LA, White E, Hanley KM, & Grabowski GA (2008) Wolman disease /cholesteryl ester storage disease: efficacy of plant-produced human lysosomal acid lipase in mice. Journal of Lipid Research, 49:1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Dana SE, Faust JR, Beaudet AL, & Brown MS (1975) Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease.The Journal of Biological Chemistry, 250:8487–8495. [PubMed] [Google Scholar]
- Gomaraschi M, Arnaboldi L, Abello F, Pavanello C, Zimetti F, Favari E, Calabresi L, & Guardamagna O (2016) Plasma lipoproteins of patients with genetic LAL deficiency are enriched in cholesteryl esters: Relevance of cholesterol esterification by Lcat. Atherosclerosis, 252, Abstract EAS16-0497, e74–e75. [Google Scholar]
- Gonzalez-Riano C, Tapia-González S, Garcia A, Muñoz A, DeFelipe J, & Barbas C (2017) Metabolomics and neuroanatomical evaluation of post-mortem changes in the hippocampus. Brain Structure and Function, 222:2831–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Donkin J, Stukas S, Chan J, Wilkinson A, Fan J, Parks JS, Kuivenhoven JA, Lütjohann D, Pritchard H, & Wellington CL (2009) LCAT synthesized by primary astrocytes esterifies cholesterol on glia-derived lipoproteins. Journal of Lipid Research, 50:885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeg JM, Demosky SJ Jr., Pescovitz OH, & Brewer HB Jr. (1984) Cholesteryl ester storage disease and Wolman disease: phenotypic variants of lysosoaml acid cholesteryl ester hydrolase deficiency. American Journal of Human Genetics, 36:1190–1203. [PMC free article] [PubMed] [Google Scholar]
- Jin U, Park SJ,& Park SM (2019) Cholesterol metabolism in the brain and its association with Parkinson’s disease. Experimental Neurobiology, 28:554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RD, Taylor AM, Tong EY, & Repa JJ (2013) Carboxylesterases are uniquely expressed among tissues and regulated by nuclear hormone receptors in the mouse. Drug Metabolism and Disposition, 41:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevics H, & Morell P (1995) Cholesterol for synthesis of myelin is made locally, not imported into brain. Journal of Neurochemistry, 64:895–901. [DOI] [PubMed] [Google Scholar]
- Jurevics H, Hostettler J, Barrett C, Morell P, & Toews AD (2000) Diurnal and dietary-induced changes in cholesterol synthesis corelate with levels of mRNA for HMG-CoA reductase. Journal of Lipid Research, 41:1048–1053. [PubMed] [Google Scholar]
- Kinney HC, Karthigasan J, Borenshteyn NI, Flax JD, & Kirschner DA (1994) Myelination in the developing human brain: biochemical correlates. Neurochemical Research, 19:983–996. [DOI] [PubMed] [Google Scholar]
- Kohli R, Ratziu V, Fiel MI, Waldmann E, Wilson DP, & Balwani M (2020). Initial assessment and ongoing monitoring of lysosomal acid lipase deficiency in children and adults: Consensus recommendations from an international collaborative working group. Molecular Genetics and Metabolism, 129:59–66. [DOI] [PubMed] [Google Scholar]
- Kunnen S,& Van Eck M (2012) Lecithin:cholesterol acyltransferase: old friend or foe in atherosclerosis? Journal of Lipid Research, 53:1783–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama M, Yoshida H, Suzuki M, Fujiyama J, & Igata A (1990) Lysosomal acid lipase deficiency in rats: lipid analyses and lipase activities in liver and spleen. Journal of Lipid Research, 31:1605–1612. [PubMed] [Google Scholar]
- Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, & Infante RE (2009) Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell, 137:1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RG, Willingham MC, Davis MA, Skinner KA, & Rudel LL (2000) Differential expression of ACAT1 and ACAT2 among cells within the liver, intestine, kidney, and adrenal of nonhuman primates. Journal of. Lipid Research, 41:1991–2001. [PubMed] [Google Scholar]
- Lee J-Y, & Carr TP (2004) Dietary fatty acids regulate acyl-CoA:cholesterol acyltransferase and cytosolic cholesteryl ester hydrolase in hamsters. The Journal of Nutrition, 134:3239–3244. [DOI] [PubMed] [Google Scholar]
- Lee JW, Huang J-D, & Rodriguez IR (2015) Extra-hepatic metabolism of 7-ketocholesterol occurs by esterification to fatty acids via cPLA2α and SOAT1 followed by selective efflux to HDL. Biochimica et Biophysica. Acta, 1851:605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Repa JJ, Valasek MA, Beltroy EP, Turley SD, German DC, & Dietschy JM (2005) Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick Type C disease. Journal of Neuropathology and Experimental Neurology, 64:323–333. [DOI] [PubMed] [Google Scholar]
- Liu B, Xie C, Richardson JA, Turley SD, & Dietschy JM (2007) Receptor-mediated and bulk-phase endocytosis cause macrophage and cholesterol accumulation in Niemann-Pick C disease. Journal of Lipid Research, 48:1710–1723. [DOI] [PubMed] [Google Scholar]
- Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, & Dietschy JM (2009) Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proceedings of the National Academy of Sciences of the United States of America, 106:2377–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Ramirez CM, Miller AM, Repa JJ, Turley SD, & Dietschy JM (2010) Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. Journal of Lipid Research, 51:933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez AM, Chuang J-C, Posey KS, Ohshiro T, Tomoda H, Rudel LL, & Turley SD (2015) PRD125, a potent and selective inhibitor of sterol O-acyltransferase 2 markedly reduces hepatic cholestesteryl ester accumulation and improves liver function in lysosomal acid lipase-deficient mice. The Journal of Pharmacology and Experimental Therapeutics, 355:159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez AM, Chuang J-C, Posey KS, & Turley SD (2017a) Suppression of brain cholesterol synthesis in male Mecp2-deficient mice is age dependent and not accompanied by a concurrent change in the rate of fatty acid synthesis. Brain Research, 1654:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez AM, Chuang J-C, Turley SD (2017b) Measurement of rates of cholesterol and fatty acid synthesis in vivo using tritiated water. In Gelissen IC. & Brown AJ (Eds.), Cholesterol homeostasis. Methods and protocols (pp. 241–256). New York, NY: Humana. [DOI] [PubMed] [Google Scholar]
- Lopez AM, Chuang J-C, & Turley SD(2018) Impact of loss of SOAT2 function on disease progression in the lysosomal acid lipase-deficient mouse. Steroids, 130:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM, & Russell DW (1999) cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proceedings of the National Academy of Sciences of the United States of America, 96:7238–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WC, Ockenden BG, Fosbrooke AS, & Cumings JN (1969) Wolman’s Disease. A rare lipidosis with adrenal calcification. Archives of Disease in Childhood, 44:331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MG, Pfrieger F, & Dotti CG (2014) Cholesterol in brain disease: Sometimes determinant and frequently implicated. EMBO Reports, 15:1036–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth M, & Bellosta S (2012) Cholesterol: Its regulation and role in central nervous system disorders. Cholesterol, 2012:292598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parini P, Davis M, Lada AT, Erickson SK, Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin B, Tomoda H, Omura S, Willingham MC, & Rudel LL (2004) ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation 110:2017–2023. [DOI] [PubMed] [Google Scholar]
- Patterson M, Clayton P, Gissen P, Anheim M, Bauer P, Bonnot O, Dardis A, Dionisi-Vici C, Klünemann H-H, Latour P, Lourenco CM, Ory DS, Parker A, Pocoví M, Strupp M, Vanier MT, Walterfang M, & Marquardt T (2017) Recommendations for the detection and diagnosis of Niemann-Pick disease type C. An update: Neurology Clinical Practice, 7:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AM, Kasimov MR, & Zefirov AL (2016) Brain cholesterol metabolism and its defects: Linkage to neurodegerative diseases and synaptic dysfunction. Acta Naturae, 8:58–73. [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, & Ungerer N (2011) Cholesterol metabolism in neurons and astrocytes. Progress in Lipid Research, 50: 357–371. [DOI] [PubMed] [Google Scholar]
- Pisciotta L, Fresa R, Bellocchio A, Pino E, Guido V, Cantafora A, Di Rocco M, Calandra S, & Bertolini S (2009) Cholesteryl ester storage disease (CESD) due to novel mutations in the LIPA gene. Molecular Genetics and Metabolism, 97:143–148. [DOI] [PubMed] [Google Scholar]
- Prasad A, Fischer WA, Maue RA, & Henderson LP (2002) Regional and developmental expression of the Npc1 mRNA in the mouse brain. Journal of Neurochemistry, 75:1250–1257. [DOI] [PubMed] [Google Scholar]
- Quan G, Xie C, Dietschy JM, & Turley SD (2003) Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Developmental Brain Research, 146:87–98. [DOI] [PubMed] [Google Scholar]
- Quay WB (1972) Regional and quantitative differences in the postweaning development of choroid plexuses in the rat brain. Brain Research, 36:37–45. [DOI] [PubMed] [Google Scholar]
- Radović B, Vujić N, Leopold C, Schlager S, Goeritzer M, Patankar JV, Korbelius M, Kolb D, Reindl J, Wegscheider M, Tomin T, Birner-Gruenberger R, Schittmayer M, Groschner L, Magnes C, Diwoky C, Frank S, Steyrer E, Du H, Graier WF, Madl T, & Kratky D (2016). Lysosomal acid lipase regulates VLDL synthesis and insulin sensitivity in mice. Diabetologia, 59:1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashu EB, Junker AE, Danielsen KV, Dahl E, Hamberg O, Borgwardt L, Christensen VB, Wewer Albrechtsen NJ, & Gluud LL (2020) Cholesteryl ester storage disease of clinical and genetic characterization: A case report and review of literature. World Journal of Clinical Cases, 8:1642–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner Z, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, Jones S, Ćorić M, Calandra S, Hamilton J, Eagleton T, & Ros E (2014) Lysosomal acid lipase deficiency -- an under recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis, 235:21–30. [DOI] [PubMed] [Google Scholar]
- Riches AC, Sharp JG, Thomas DB, & Smith SV (1973) Blood volume determination in the mouse. Journal of Physiology, 228:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick TH, Wimer RE, Wimer CC, & Schwartzkroin PA (1973) Genetic and phenotypic variation in weight of brain and spinal cord between inbred strains of mice. Brain Research, 64:345–353. [DOI] [PubMed] [Google Scholar]
- Sakashita N, Miyazaki A, Takeya M, Horiuchi S, Chang CCY, Chang TY, & Takahashi K (2000) Localization of human acyl-coenzyme C: cholesterol acyltransferase-1 (ACAT-1) in macrophages and in various tissues. American Journal of Pathology, 156:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schümann K, Szegner B, Kohler B, Pfaffl MW, & Ettle T (2007) A method to assess 59Fe in residual blood content in mice and its use to correct 59Fe-distribution kinetics accordingly. Toxicology, 241:19–32. [DOI] [PubMed] [Google Scholar]
- Segatto M, Trapani L, Lecis C, & Pallottini V (2012) Regulation of cholesterol biosynthetic pathway in different regions of the rat central nervous system. Acta Physiologica, 206:62–71. [DOI] [PubMed] [Google Scholar]
- Sloan HR, & Fredrickson DS (1972) Enzyme deficiency in cholesteryl ester storage disease. The Journal of Clinical Investigation, 51:1923–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snipes GJ, & Suter U (1997) Cholesterol and myelin. In: Bittman R (Ed.), Cholesterol: Its functions and metabolism in biology and medicine (subcellular biochemistry) (Vol. 28, pp. 173–204). New York, NY: Plenum. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Mace BE, Maeda N, & Schmechel DE (2004) Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience, 124:725–733. [DOI] [PubMed] [Google Scholar]
- Sun Y, Xu Y-H, Du H, Quinn B, Liou B, Stanton L, Inskeep V, Ran H, Jakubowitz P, Grilliot N, & Grabowski GA (2014) Reversal of advanced disease in lysosomal acid lipase deficient mice: a model for lysosomal acid lipase deficiency disease. Molecular Genetics and Metabolism, 112:229–241. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Liu B, Mari Y, Liu B, & Repa JJ (2012) Cyclodextrin mediates rapid changes in lipid balance in Npc1−/− mice without carrying cholesterol through the bloodstream. Journal of Lipid Research, 53:2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todoroki T, Matsumoto K, Watanabe K, Tashiro Y, Shimizu M, Okuyama T, & Imai K (2000) Accumulated lipids, aberrant fatty acid composition and defective cholesterol ester hydrolase activity in cholesterol ester storage disease. Annals of Clinical Biochemistry, 37:187–193. [DOI] [PubMed] [Google Scholar]
- Turley SD, Valasek MA, Repa JJ, & Dietschy JM (2010) Multiple mechanisms limit the accumulation of unesterified cholesterol in the small intestine of mice deficient in both ACAT2 and ABCA1. American Journal of Physiology-Gastrointestinal and Liver Physiology, 299:G1012–G1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance JE (2012) Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Disease Models & Mechanisms, 5:746–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali C, Wellington CL, & Calabresi L (2014) HDL and cholesterol handling in the brain. Cardiovascular Research 103:405–413. [DOI] [PubMed] [Google Scholar]
- Wang H, & Eckel RH (2014) What are lipoproteins doing in the brain? Trends in Endocrinology and Metabolism, 25:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Turley SD, & Dietschy JM (2000) Centripetal cholesterol flow from the extrahepatic organs through the liver is normal in mice with mutated Niemann-Pick type C protein (NPC1). Journal of Lipid Research 41:1278–1289. [PubMed] [Google Scholar]
- Yusuf HKM, Dickerson JWT, Hey EN, & Waterlow JC (1981) Cholesterol esters of the human brain during fetal and early postnatal development: Content and fatty acid composition. Journal of Neurochemistry. 36:707–714. [DOI] [PubMed] [Google Scholar]
- Yutuk E, Angelini R, Baumert M, Mast N, Pikuleva I, Newton J, Clench MR, Skibinski DOF, Howell OW, Wang Y & Griffiths WJ (2020) Localization of sterols and oxysterols in mouse brain reveals distinct spatial cholesterol metabolism. Proceedings of the National Academy of Sciences of the United States of America, 117:5749–5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G, Codeluppi S, Furlan A, Lee K, Skene N, Harris KD, Hjerling-Leffer J, Arenas E, Ernfors P, Marklund U, & Linnarsson S (2018) Molecular architecture of the mouse nervous system. Cell, 174:999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, & Liu Q (2015) Cholesterol metabolism and homeostasis in the brain. Protein & Cell, 6:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]