

Abstract
Detection of specific proteins using nanopores is currently challenging. To address this challenge, we developed a collection of over twenty nanopore-addressable protein tags engineered as reporters (NanoporeTERs/NTERs). NTERs are constructed with a secretion tag, folded domain, and a nanopore-targeting C-terminal tail in which arbitrary peptide barcodes can be encoded. We demonstrate simultaneous detection of up to nine NanoporeTERs expressed in bacterial or human cells using MinION nanopore sensor arrays.
For nearly four decades, reporter proteins have been used as a means to track biological activities such as genetic regulation1. Although several different reporter strategies have been developed over this period, the typical number of uniquely addressable reporters that can be used together while sharing a common readout is small2–5. This is primarily due to the optical nature of traditional reporters, such as fluorescent protein variants, which have overlapping spectral properties that make simultaneous measurement of unique genetic elements difficult2. An ability to increase the multiplexability of genetically encoded protein reporters would enable more comprehensive and scalable monitoring of biological systems, enabling, for instance, high-dimensional phenotyping6, one-pot parallelized whole-cell biosensing7 and efficient genetic circuit debugging8.
While biomolecular sensing with nanopore sensors has been explored9, only recently have high-throughput nanopore sensor platforms emerged for real-time sequencing of DNA10 and RNA11. The commercial emergence and popularization of these technologies creates an opportunity to build an accessible general nanopore -based platform for direct sensing of engineered reporter proteins. In this context, we present here a new class of genetically-encoded protein reporters, which we call Nanopore-addressable protein Tags Engineered as Reporters (NanoporeTERs, or NTERs), that use commercially-available nanopore sensors (Oxford Nanopore Technologies’ MinION device)10 for multiplexed direct protein reporter detection without the need for any other specialized equipment nor laborious sample preparation prior to analysis (Figures 1a-d).
Figure 1: Nanopore-addressable protein Tags Engineered as Reporters (NanoporeTERs).
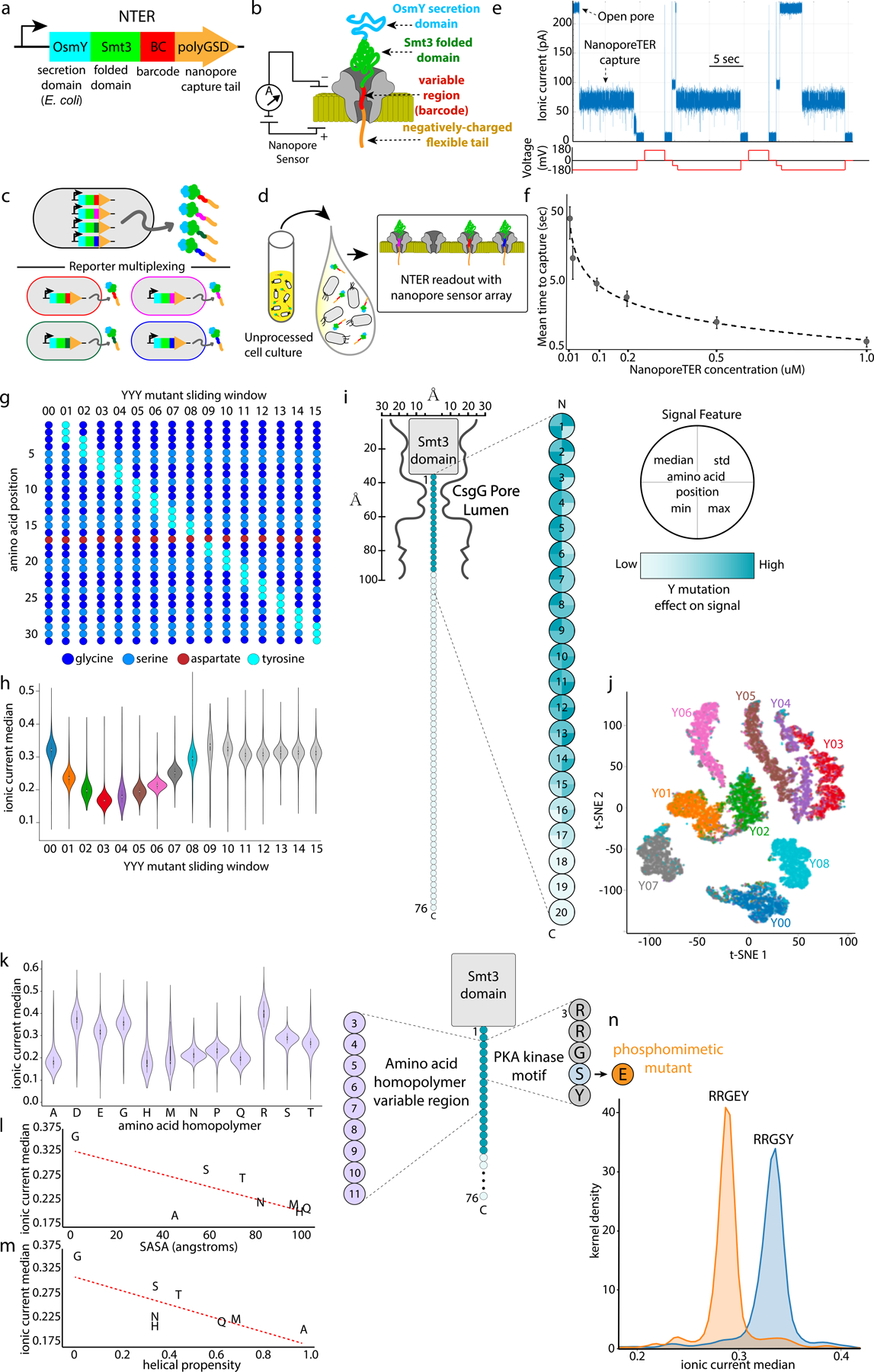
a, Gene schematic of NanoporeTER (NTER) design. b, Schematic of a NanoporeTER captured within a nanopore. c, NanoporeTERs are designed to enable multiplexed readout of protein expression. d, Secretion of the NanoporeTERs into the extracellular medium. e , Example of raw nanopore data generated from a single nanopore showing repeated captures and ejections of NTERY00. f, Concentration curve showing the relationship between NanoporeTER concentration within a flow cell versus the average time between captures or “reads.” Error bars represent SD of n=3 independent nanopore experiments. g, Schematic of the NTERs Y00 −15 sequences. h, Violin plot showing normalized median current level of nanopore capture state for NTERs Y00–15. Center dot: median, black box: 1st/3rd quartiles, whiskers: 1st/3rd quartiles −/+ 1.5 interquartile range. Each NTER distribution is composed of n = ~4000 events per class. i, Model of NTER position within the nanopore during a capture event. Heat map displaying relative change to specific signal features projected onto NTER tail residue positions that were mutated in NTERs Y00–15, showing the relative magnitude of effect tyrosine mutations at each residue have on the NTER’s nanopore signal. j, t-SNE plot clustering NTER reads based on ionic current signal features, and colored by the NTER’s barcode identity (Y00–08). n = ~4000 events per class. k, Violin plot showing the normalized median ionic current level of the nanopore capture state for amino acid homopolymer NTERs. Center dot: median, black box: 1st and 3rd quartiles, whiskers: 1st and 3rd quartiles −/+ 1.5 interquartile range. Each NTER distribution is composed of ~1500 single-molecule measurements. l, Scatter plot showing the relationship between amino acid solvent accessible surface area (SASA) versus the respective amino acid homopolymer NTER mutant’s normalized median ionic current level. m, Scatter plot showing the relationship between amino acid helical propensity versus the respective amino acid homopolymer NTER mutant’s normalized median ionic current level. n, Kernel density plot comparing the normalized ionic current median of an NTER containing a PKA phosphorylation motif within its barcode region to those with a phosphomimetic mutation. Each NTER distribution is composed of ~1000 single-molecule measurements.
To develop this method, we first designed a protein (NTERY00) that would be easily detectable by a nanopore sensor (see Figure 1a, Supp. Figures 1 and 2, and Supp. Notes). This protein had three important elements: 1) a long, flexible negatively charged C-terminal domain (polyGSD) to promote capture of the protein in a nanopore sensor12,13, 2) a stable folded domain (Smt3) to sterically inhibit complete translocation of the protein through the pore, and 3) an N-terminal secretion domain (OsmY) to promote transport of the protein to the extracellular media following expression14,15. We expressed and purified NTERY00 from E. coli culture supernatant by immobilized metal affinity chromatography (IMAC) and determined if the NTER could be detected on a MinION. To do this, we used an unmodified R9.4.1 flow cell (which uses a variant of the CsgG pore protein16) and a custom MinION run script (see Methods). The script applies a constant voltage of −180 mV to all the active pores on the flow cell and statically flips the voltage in the reverse direction in 15 second cycles (10 seconds ‘ON’ at −180 mV and 5 seconds ‘OFF’ or in ‘Reverse’, see Figure 1e). The typical R9.4.1 open pore current level at −180 mV and 500 mM KCl is ~220 pA. As expected, when NTERY00 was introduced into the flow cell at a concentration of 0.5 uM in these conditions, the current level during each −180 mV portion of the voltage cycle typically underwent a stepwise drop from the open pore value to a consistent lower ionic current state (Figure 1e and Supp. Figure 3), signaling the putative capture of an NTER within the pore. This current drop was reversible (back to open pore) following reversal of the voltage. We further found that the average time spent in the open pore state before transitioning to the lower ionic current state was dependent on both NTER concentration and the applied voltage (Figure 1f and Supp. Figure 4) . We also explored if shorter C-terminal NTER tail lengths could similarly promote capture in the nanopore and found that a tail length truncated by 20 amino acids captured at a rate similar to the full-length design, while reduction by 40 amino acids substantially reduced capture rates (Supp. Figure 5). Overall, these observations are consistent with a model in which the negatively charged NTER polyGSD tail is electrophoretically captured in the pore under the applied voltage, and can be ejected from the pore by reversal of the electric field.
If this model is correct, we postulated that the ionic current characteristics of the NTERY00 capture state would be dependent upon the amino acid sequence of the NTER residues residing within the pore’s sensitive limiting constriction. To test this, we made a series of NTER mutants (NTERY01–15) in which a sliding three residue region of the polyGSD sequence was mutated to tyrosines (Figure 1g). Tyrosines were chosen because their large side chain structure was predicted to decrease the ionic current flow through the pore relative to the glycines and serines of NTERY00 when captured within the pore. Following purification and MinION analysis of NTERs 01–15, we found the capture state to be NTER mutant-dependent up to NTERY08, after which we observed NTER mutants 09–15 to have signal characteristics indistinguishable from NTERY00 (Figures 1h, i, and j, and Supp. Figures 3 and 6). These results support a model in which the first ~17 amino acids of the polyGSD tail reside within the CsgG nanopore’s sensitive region and contribute to its ionic current signature during a capture event, essentially demarcating this segment of the NTER as a nanopore-addressable amino acid ‘barcode’. We then further characterized this barcode signal space by assessing how different amino acid types and phosphomimetic point mutations17 can modulate both the NTER nanopore ionic current signal (Figures 1k,l,m,n, Supp. Figure 7, and Supp. Notes) and NTER capture efficiency into the pore (Supp. Figure 8).
Having explored the potential NTER barcode sequence and signal space, we sought to demonstrate proof-of-principle NTER applications for multiplexed tracking of gene expression. To do this, we first used supervised machine learning to train classifiers that could accurately discriminate amongst combinations of the NTER barcodes explored above (Figures 2a and 2b, and Supp. Notes). Using either a set of engineered signal features as input to a Random Forest (RF) classifier or the raw ionic current signal directly into a Convolutional Neural Network (CNN) (Figure 2a), we used our purified NTER datasets for model training and validation. Both models achieved similar accuracies that ranged from ~80–90% depending on the model hyperparameters and barcode set (Figure 2b, see Methods). Next, we used the CNN classifier to assess if NTERs were being immediately re-captured in the nanopore at a non-negligible frequency following their initial capture and ejection, which could lead to molecules being double counted, affecting relative NTER quantification. To investigate this, we analyzed the frequency with which successive captures of the same or different barcode occurred in a mixed barcode experiment with 5 different NTER barcodes mixed at varying concentrations (Y00: 0.05uM, Y02: 0.1uM, Y05: 0.05uM, Y07: 0.2uM, and Y08: 0.1uM). We found that successive NTER captures of the same barcode were not disproportionately represented, suggesting that immediate re-capture of the same NTER molecule was not occurring at a high frequency (Supp. Figure 9).
Figure 2: Classification and multiplexed detection of NanoporeTER expression levels with a MinION.
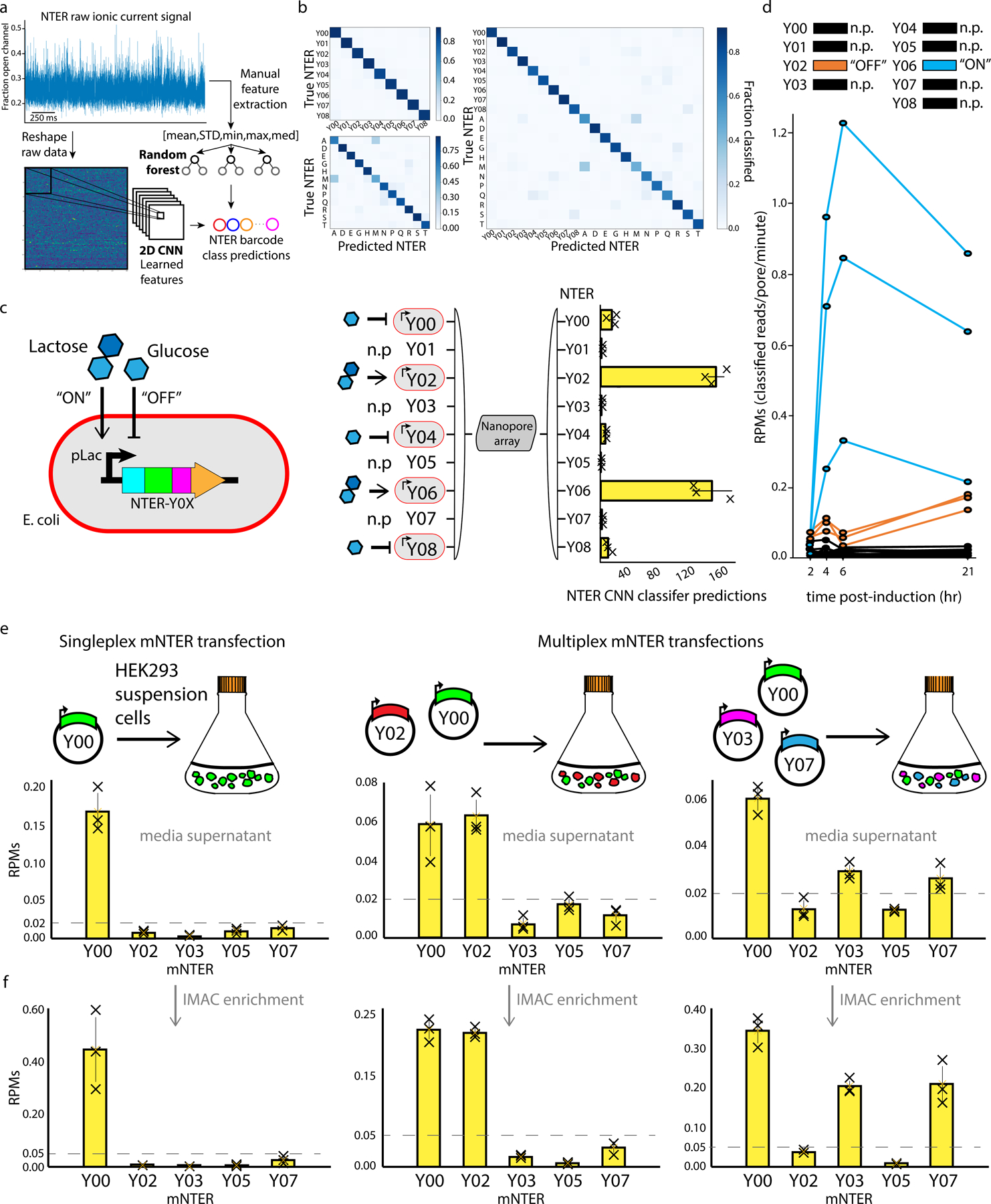
a, Raw ionic current data was classified using either a set of engineered features (mean, std, min, max, and median) or the unprocessed signal directly, and input into either a Random Forest or Convolutional Neural Network classifier, respectively. b, Confusion matrices showing the Random Forest test set classification accuracies on models using different combination of NTER barcodes. Top left: NTERs Y00–08. Bottom left: amino acid homopolymer mutants A, D, E, G, H, M, N, P, Q, R, S, and T. Right: Both the NTERs Y00–08 and amino acid homopolymer mutants. c, Schematic showing the gene construct used for controllable NTER expression. Lactose is used to induce NTER expression (“ON”), while glucose inhibits expression (“OFF”). The diagram and bar plot on the right shows the results of a mixed culture experiments in which NTER expression was induced for NTERs Y02 and Y04, and inhibited for NTERs Y00, Y02, and Y08. NTERs Y01, Y03, Y05, and Y07 were held out of the experiment as negative controls. Bars show the average total number of reads classified as each NTER barcode during MinION analysis of three technical replicates (error bars show SD). d, Line plot showing a time course of NTER expression levels as determined by the rate of classified reads (RPMs: reads/pore/min) for each NTER barcode. NTER Y06 was induced, while NTER Y02 was inhibited. The other NTERs were held out as negative controls and show false-positive classification rates. Three technical replicates for each condition are plotted. e, Bar plots show the results of singleplex and multiplexed HEK293 transfection experiments. For each experiment, a culture of HEK293 suspension cells was transfected with a different barcode combination of vectors containing mNTER proteins (Y0, Y0+Y02, or Y0+Y03+Y07) under the control of a constitutive CMV promoter. Bars show the average rate of classified reads (RPMs) for each barcode during MinION analysis. Three technical replicates for each experiment are plotted (error bars show SD). f, Same as in e, but with the addition of an IMAC purification step prior to MinION analysis.
We then used the best performing classifier that was trained on NTERs Y00–08 (in addition to a background/noise class, see Methods) to determine the relative NTER expression levels within bacterial cultures composed of mixed populations of strains engineered with different NTER-tagged plasmid-based circuits. Specifically, we grew independent mono-barcoded cultures overnight with NTER expression either induced or inhibited (by autoinduction media containing lactose or LB supplemented with glucose, respectively). In the morning, just prior to nanopore readout, the cultures were mixed into a single solution and diluted into MinION running buffer and loaded directly into a flow cell for analysis. These cell cultures underwent no processing or purification prior to analysis, in contrast to our previous experiments. Results from these experiments showed higher classification counts for the NTER barcodes for which expression was induced (NTERs 02 and 06), and lower counts for strains that were inhibited (glucose: NTERs Y00, Y04 and Y08) or not present at all in the mixed population (NTERs Y01, Y03, Y05, and Y07) for all replicates (Figure 2c) over a ten minute MinION runtime. We then conducted a time course experiment in which we tracked expression of two different NTERs over multiple hours, one of which was induced with IPTG (NTERY06), and the other of which NTER expression was inhibited with glucose (NTERY02). Again, cultures were grown independently, but then mixed just prior to nanopore readout. Figure 2d shows the results of this time course (and replicates) during ten minutes of MinION analysis at 2, 4, 6, and 21-hour timepoints following induction (NTERY06) or inhibition (NTERY02) of the NanoporeTER circuit. Again, the rate of NTER classification (reads/pore-minute) was substantially higher for the induced NTERY06 circuits compared to the uninduced NTERY02 circuits. Leaky expression of NTERY02 was still detectable over the background false-positive classification rates for the NTER barcodes that were not present at all in the experiment (Y00, Y01, Y03, Y04, Y05, Y07 and Y08). These results demonstrate that NanoporeTERs can be used as reliable reporters of relative protein expression levels in bacterial cell culture and that cell-based assays can be performed directly on the flow cell itself.
To show that NanoporeTERs can also be used for detecting gene expression in alternative cell types, such as mammalian cell culture, we modified the E. coli NTER design to function in HEK293 cells (Supp. Figure 1). We did this by making two key changes: 1) we replaced the bacterial secretion domain (OsmY) with a human secretion tag (IFNα2)18, and 2) made two mutations to the Smt3 domain that increase its resistance to intracellular degradation in mammalian cells (SUMOstar)19. We then cloned several different barcoded versions of this mammalian-optimized NanoporeTER design (mNTER) into a vector under the control of a constitutive CMV promoter, and performed experiments in which varying combinations of these vectors were transfected into HEK293 suspension cultures. Results from these experiments are shown in Figure 2e and 2f. Specifically, we detected mNTERs directly from media supernatant collected 3 days after the cultures were transfected with one (Y00), two (Y00,Y02), or three (Y00,Y03,Y07) different mNTER barcodes. The number of mNTER counts for each barcode class was reflective of the barcode combinations that were introduced into each of the cultures, as shown by the substantially higher classification counts for the transfected barcode classes relative to classes included in the classifier but absent from the experiment. We observed that while mNTERs could be detected over background classification levels directly from the raw media supernatant with no further processing (Figure 2e), superior classification results were obtained from media samples post-IMAC enrichment (Figure 2f), indicating media contaminants in cell culture led to higher levels of mNTER misclassification events. These results, together with the previous results obtained from E. coli, suggest that NanoporeTERs will be applicable to a wide diversity of cell types and model systems.
In conclusion, we have laid the foundations for a class of multiplexable protein reporters that can be analyzed using a commercially available nanopore sensor array, the ONT MinION. To support future applications, we have also performed preliminary assessments of the experimental throughput of NTER measurements on the MinION and discuss the scalability of NTER barcode space for future work (see Supp. Figures 10–13 and Supp. Notes). We foresee many potential NanoporeTER applications, including simultaneously reading the protein-level outputs of many genetically engineered circuit components in one-pot, enabling more efficient debugging and tuning than current analysis methods. For instance, in comparison to traditional sets of fluorescent protein reporters, NanoporeTERs have a (potentially much) larger sequence and signal space that allows for the simultaneous analysis of a greater number of unique genetic elements in a single experiment (multiplexing). While RNA-seq can be used to measure the transcriptional output of many circuits in parallel20, our method has the advantages of 1) little to no sample preparation, which makes it more amenable to automation21–23 and reduces both time to analysis (latency) and cost, 2) a non-destructive readout, enabling the profiling of expression dynamics in living cells, and 3) direct detection of outputs at the protein level. The last advantage opens new opportunities to engineer reporters with NTER barcodes that can report on both protein expression and specific post translational modifications simultaneously. We anticipate this capability will be especially useful for synthetic protein-level circuit engineering24.
Methods:
Code availability:
Codes are available upon request and on github.com/uwmisl/NanoporeTERs. Custom MinION MinKNOW runscripts can also be obtained from Oxford Nanopore Technologies upon request.
Data availability:
Data are available upon request and on github.com/uwmisl/NanoporeTERs.
Materials availability:
All plasmids and cells lines used in this study are available upon request.
NanoporeTER construction, cloning, expression, and purification
The initial NanoporeTER protein was constructed with a gBlock (Integrated DNA Technologies) composed of the Smt3 and tail sequence and cloned into plasmid pCDB180 downstream of the OsmY domain. The Q5 site-directed mutagenesis method (New England Biolabs) was used to generate the different NTER barcode mutants. All cloning was performed using the 5-alpha competent E. coli strain following NEB’s cloning protocol (New England Biolabs). Sequence verification was obtained through Genewiz Inc. Expression of the NanoporeTER protein was done in BL21 (DE3) E. coli strain using Overnight Express instant TB medium (Novagen).
Proteins were purified via immobilized metal affinity chromatography (IMAC) using TALON metal affinity cobalt resin (Takara). The purification used the associated buffer set from Takara, following their specified protocol. Proteins were concentrated using Amicon Ultra 0.5 mL centrifugal filters with Ultracel 30K (Amicon). The final concentration of proteins averaged ~7 mg/ml from 5 mL overnight cultures. The purified proteins were stored for long-term storage at −80C in 10 uL aliquots, as well as for short-term storage at 4C.
E.coli raw culture mixing experiments
Cultures were picked from single colonies on plates and used to inoculate 3mL LB supplemented with 0.5 mM IPTG and kanamycin (induced), or 3mL LB supplemented with 0.2% glucose and kanamycin (inhibited). After overnight incubation at 37C with shaking, cultures were equally mixed together in a total volume of 45uL, 50uL 4X C17 buffer (2 M KCl, 100 mM HEPES, pH 8), and 105 uL water (total volume 200uL). This solution was then immediately loaded into a MinION flow cell for analysis.
E. coli expression time course
Time course experiments were performed by diluting 30uL of overnight cultures (LB) into 3mL fresh LB supplemented with 0.5 mM IPTG and kanamycin (induced), or 3mL fresh LB supplemented with 0.2% glucose and kanamycin (inhibited). The cultures were placed in a shaker/incubator at 37C to allow for culture growth. Samples were then collected at 2, 4, 6, and 21-hour time-points. At each time-point, cultures were equally mixed together in a total volume of 10 uL, 50uL 4X C17 buffer, and 140 uL water (total volume 200uL). This solution was then immediately loaded into a MinION flow cell for analysis.
HEK293 transfection experiments
To clone the mammalian NanoporeTERs, we used a mammalian expression vector consisting of a CMV enhancer and CMV promoter driving expression of mCherry and a N-terminal nuclear localization signal (NLS). First, we replaced the NLS by inverse PCR of the vector at the edges of the NLS and assembled via Gibson assembly (NEB) with a gBlock (IDT) comprising of the sequence for an IFNalpha2 secretion tag and 10x His tag. To add the mNTER to the C-terminus, we used inverse PCR at the mCherry stop codon and assembled via Gibson assembly (NEB) with a gBlock (IDT) synthesized with the NTERY00 sequence. To generate mNTER variants, we inverse PCRed at the variable site using primers with overlapping extensions containing the new NTER barcode and compatible overhangs. All mNTERs were transformed and cultured in DH5a electrocompetent cells (NEB) and verified with Sanger sequencing through Genewiz Inc.
Cells used for transfection experiments were FreeStyle 293-F cells (Gibco, ThermoFisher R79007) and were grown in FreeStyle 293 expression medium (Gibco) with no added antibiotic. The day before transfection, cells were seeded at a density of 500k cells/mL in order to reach a density of 1 million cells/mL the next day. On the day of transfection, cells were transfected with 1ug of DNA per 1 million cells using a lipid-based method of transfection. Cells were then left to express for 3 days on a shaker platform shaking at 135RPM at 37C supplemented with 8% CO2 before collecting media supernatant for subsequent nanopore analysis or IMAC purification.
Nanopore analysis of HEK293 mNTER expression was conducted by mixing 5–10 uL of raw supernatant, 50uL 4X C17 buffer, and 140–145 uL water (total volume 200uL). This solution was then immediately loaded into a MinION flow cell for analysis. For IMAC-purified samples, protein was diluted to a final concentration of 0.02 uM total protein in 1X C17 before loading into a MinION flow cell for analysis.
MinION experiments
All experiments were performed with unmodified R9.4.1 MinION flow cells (Oxford Nanopore Technologies) by diluting analyte solution into C17 buffer for a final concentration of 0.5M KCl and 25mM HEPES (pH 8), into the flow cell priming port. Flow cells were run on the MinION at a temperature of 30°C and a run voltage of −180mV with a 10khz sampling frequency and 15 second static flip frequency. Use of a modifiable MinKNOW script (available from ONT) enabled voltage flipping cycle parameters to be set as well as collection of raw current data across the entire run. Individual flow cells could be reused for different analytes after flushing them with 1mL C17 buffer three times between experiments. Flow cells were stored at 4°C in C18 buffer (150mM potassium ferrocyanide, 150mM potassium ferricyanide, 25mM potassium phosphate, pH8) when not in use.
Nanopore Signal Analysis, Quantification, and Classification
The analysis pipeline for a NanoporeTER sequencing run begins with extracting the segments of the raw nanopore signal that contain capture events. A capture is defined as a region where the signal current falls below 70% of the open pore current for a duration of at least one millisecond. The fractional current values (as compared to open pore current) computed from the segmentation process, as well as the start and end times of each capture, are saved in separate data files. This information is then passed through a general filter that separates putative NanoporeTER captures from noise captures based on features of the normalized raw current (mean, standard deviation, minimum, maximum, median) as well as the duration of the capture. Captures that pass this initial filter are then fed into a classifier and classified as a specific NTER barcode or a background/noise blockade. The metadata for the captures within each NTER class are subsequently fed to a quantifier which calculates the average time elapsed between those captures and (optionally) converts this time to the predicted NTER concentration using a standard curve. An alternative method of quantification is to calculate the number of reads per class per active pore per minute (reads/pore-minute or RPMs). In addition to the NTER data sets, a background/noise class data set was also used in training the models to recognize data generated from non-NTER-specific pore blockages that made it through the filtering step. This data was collected from experiments in which only running buffer, LB media, and/or NTER-free E. coli or HEK293 cultures were loaded into the flow cell.
We explored two different classifiers for NTER barcode discrimination. The first, a Random Forest model, was implemented in scikit-learn (sklearn.ensemble.RandomForestClassifier). The second classifier was a CNN implemented in PyTorch. An 80/20 train/test split was used to generate the classification accuracy estimates and confusion matrix results. For both models, only the first two seconds of each capture were considered for analysis. The Random Forest was trained on an array composed of the mean, standard deviation, minimum, maximum, and median of that two second window. Default Random Forest hyperparameters were modified to: n_estimators=300 and max _depth=100. The CNN used the two seconds of raw signal directly as input following reshaping of the 1D signal into a 2D structure. The neural network was composed of four 2D convolutional layers each with ReLU activation and max pooling. These were followed by a fully connected layer which had a log-sigmoid activation function, and then a final output layer of the same size as the number of NTER classes (plus noise class) considered in the experiment. This output layer can be interpreted as the confidence scores associated with each class, which can also be applied as a confidence threshold filter (e.g. only assigning labels for events with >95% confidence in a single class). Confidence thresholds of 95% and 90% were used for the E. coli and HEK293 cell culture experiments, respectively. Full model details and code can be found at https://github.com/uwmisl/NanoporeTERs.
While both classifiers (Random Forest and CNN) achieved similar accuracies on the train/test data, the CNN is more likely to be extensible as we increase the number of barcodes. With the Random Forest, the information content available for classification is reduced to just a few manually extracted summary statistics (ionic current blockade mean, median, maximum, minimum, and standard deviation), leaving other distinguishing signal characteristics behind. In contrast, the CNN classifies barcodes directly from the ionic current traces. As the number of barcodes increases, a CNN will likely be able to take better advantage of features embedded in the signal itself, like variation in the noise pattern for a specific barcode.
Supplementary Material
Acknowledgments:
We thank additional members of the Molecular Information Systems Lab for helpful discussion and feedback on this work. The OsmY expression plasmid was generously provided by Cassie Bryan and Lauren Carter (Institute for Protein Design, University of Washington). We also thank Andy Heron and Rich Gutierrez (Oxford Nanopore Technologies) for providing the configurable MinION run script and discussions on its use, and Miten Jain (UCSC) for a custom Matlab script that facilitated visualization of the raw MinION data.
Funding:
This work was supported in part by NSF EAGER Award 1841188 and NSF CCF Award 2006864 to LC and JN, an NIH/NCI Cancer Center Support Grant P30 CA015704 Pilot Award and NSF Award 2021552 to JN, and a sponsored research agreement from Oxford Nanopore Technologies.
Footnotes
Competing interests: A provisional patent has been filed by the University of Washington covering aspects of this work (Patent Application 17/283,007). KS is an employee of Microsoft. JN is a consultant to Oxford Nanopore Technologies. The remaining authors declare no competing interests.
References:
- 1.Ghim CM, Lee SK, Takayama S & Mitchell RJ The art of reporter proteins in science: Past, present and future applications. BMB Reports (2010). doi: 10.3858/BMBRep.2010.43.7.451 [DOI] [PubMed]
- 2.Rodriguez EA et al. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends in Biochemical Sciences (2017). doi: 10.1016/j.tibs.2016.09.010 [DOI] [PMC free article] [PubMed]
- 3.Martin L, Che A & Endy D Gemini, a bifunctional enzymatic and fluorescent reporter of gene expression. PLoS One (2009). doi: 10.1371/journal.pone.0007569 [DOI] [PMC free article] [PubMed]
- 4.Parrello D, Mustin C, Brie D, Miron S & Billard P Multicolor whole-cell bacterial sensing using a synchronous fluorescence spectroscopy-based approach. PLoS One (2015). doi: 10.1371/journal.pone.0122848 [DOI] [PMC free article] [PubMed]
- 5.Shimo T, Tachibana K & Obika S Construction of a tri-chromatic reporter cell line for the rapid and simple screening of splice-switching oligonucleotides targeting DMD exon 51 using high content screening. PLoS One (2018). doi: 10.1371/journal.pone.0197373 [DOI] [PMC free article] [PubMed]
- 6.Wroblewska A et al. Protein Barcodes Enable High-Dimensional Single-Cell CRISPR Screens. Cell 175, (2018). doi: 10.1016/j.cell.2018.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He W, Yuan S, Zhong WH, Siddikee MA & Dai CC Application of genetically engineered microbial whole-cell biosensors for combined chemosensing. Applied Microbiology and Biotechnology (2016). doi: 10.1007/s00253-015-7160-6 [DOI] [PubMed]
- 8.Nielsen AAK et al. Genetic circuit design automation. Science 352, aac7341–aac7341 (2016). doi: 10.1126/science.aac7341 [DOI] [PubMed] [Google Scholar]
- 9.Shi W, Friedman AK & Baker LA Nanopore Sensing. Anal. Chem 89, 157–188 (2017). doi: 10.1021/acs.analchem.6b04260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain M, Olsen HE, Paten B & Akeson M The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol (2016). doi: 10.1186/s13059-016-1103-0l [DOI] [PMC free article] [PubMed]
- 11.Garalde DR et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods (2018). doi: 10.1038/nmeth.4577 [DOI] [PubMed]
- 12.Nivala J, Marks DB & Akeson M Unfoldase-mediated protein translocation through an α-hemolysin nanopore. Nat. Biotechnol (2013). doi: 10.1038/nbt.2503 [DOI] [PMC free article] [PubMed]
- 13.Nivala J, Mulroney L, Li G, Schreiber J & Akeson M Discrimination among protein variants using an unfoldase-coupled nanopore. ACS Nano 8, 12365–12375 (2014). doi: 10.1021/nn5049987 [DOI] [PubMed] [Google Scholar]
- 14.Yim HH & Villarejo M osmY, a new hyperosmotically inducible gene, encodes a periplasmic protein in Escherichia coli. J. Bacteriol (1992). doi: 10.1128/jb.174.11.3637-3644.1992 [DOI] [PMC free article] [PubMed]
- 15.Kotzsch A et al. A secretory system for bacterial production of high-profile protein targets. Protein Sci (2011). doi: 10.1002/pro.593 [DOI] [PMC free article] [PubMed]
- 16.Goyal P et al. Structural and mechanistic insights into the bacterial amyloid secretion channel CsgG. Nature (2014). doi: 10.1038/nature13768 [DOI] [PMC free article] [PubMed]
- 17.Taylor SS et al. PKA: A portrait of protein kinase dynamics. in Biochimica et Biophysica Acta - Proteins and Proteomics (2004). doi: 10.1016/j.bbapap.2003.11.029 [DOI] [PubMed]
- 18.Román R et al. Enhancing heterologous protein expression and secretion in HEK293 cells by means of combination of CMV promoter and IFNα2 signal peptide. J. Biotechnol 239, 57–60 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Peroutka RJ, Elshourbagy N, Piech T & Butt TR Enhanced protein expression in mammalian cells using engineered SUMO fusions: Secreted phospholipase A 2. Protein Sci (2008). doi: 10.1110/ps.035576.108 [DOI] [PMC free article] [PubMed]
- 20.Gorochowski TE et al. Genetic circuit characterization and debugging using RNA‐seq. Mol. Syst. Biol (2017). doi: 10.15252/msb.20167461 [DOI] [PMC free article] [PubMed]
- 21.Gach PC et al. A Droplet Microfluidic Platform for Automating Genetic Engineering. ACS Synth. Biol 5, 426–433 (2016). doi: 10.1021/acssynbio.6b00011 [DOI] [PubMed] [Google Scholar]
- 22.Chao R, Mishra S, Si T & Zhao H Engineering biological systems using automated biofoundries. Metab. Eng 42, 98–108 (2017). doi: 10.1016/j.ymben.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madison AC et al. Scalable Device for Automated Microbial Electroporation in a Digital Micro fluidic Platform. ACS Synth. Biol 1701–1709. (2017). doi: 10.1021/acssynbio.7b00007 [DOI] [PubMed]
- 24.Chen Z & Elowitz EB Programmable protein circuit design. Cell S0092–8674(21)00292–0. (2021). doi: 10.1016/j.cell.2021.03.007. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request and on github.com/uwmisl/NanoporeTERs.