

Abstract
Background:
While several interventions can effectively lower lipid levels in people at risk for atherosclerotic cardiovascular disease (ASCVD), cardiovascular event (CVE) risks remain, suggesting an unmet medical need to identify factors contributing to CVE risk. Monocytes and macrophages play central roles in atherosclerosis, but previous work has yet to provide a detailed view of macrophage populations involved in increased ASCVD risk.
Methods:
A novel macrophage foaming analytics tool, AtheroSpectrum, was developed using two quantitative indices depicting lipid metabolism and the inflammatory status of macrophages. Next, a machine-learning algorithm was developed to analyze gene expression patterns in the peripheral monocyte transcriptome of Multi-Ethnic Study of Atherosclerosis participants (MESA-set1, n=911). A list of 30 genes was generated and integrated with traditional risk factors to create an ASCVD risk prediction model (CR-30), which was subsequently validated in the remaining MESA-set2 (n=228); performance of CR-30 was also tested in two independent human atherosclerotic tissue transcriptome datasets (GTEx and GSE43292).
Results:
Using single-cell transcriptomic profiles (GSE97310, GSE116240, GSE97941, FR-FCM-Z23S), AtheroSpectrum detected two distinct programs in plaque macrophages: homeostatic-foaming and inflammatory pathogenic-foaming, the latter was positively associated with severity of atherosclerosis in multiple studies. A pool of 2209 pathogenic foaming genes was extracted and screened to select a subset of 30 genes correlated with CVE in MESA-set1. A CVD risk score model (CR-30) was then developed by incorporating this gene-set with traditional variables sensitive to CVE in MESA-set1 after cross-validation generalizability analysis. The performance of CR-30 was then tested in MESA-set2 (p=2.60×10−4, AUC=0.742), and two independent datasets (GTEx, p=7.32×10−17, AUC=0.664; GSE43292, p=7.04×10−2, AUC=0.633). Model sensitivity tests confirmed the contribution of the 30-gene panel to the prediction model (likelihood ratio test, df=31, p=0.03).
Conclusion:
Our novel computational program (AtheroSpectrum) identified a specific gene expression profile associated with inflammatory macrophage foam cells. A subset of 30 genes expressed in circulating monocytes jointly contributed to prediction of symptomatic atherosclerotic vascular disease. Incorporating a pathogenic foaming gene-set with known risk factors can significantly strengthen the power to predict ASCVD risk. Our programs may facilitate both mechanistic investigations and development of therapeutic and prognostic strategies for ASCVD risk.
Keywords: Atherosclerosis, macrophages, foam cells, MESA, ASCVD risk, single cell transcriptomics
Introduction
Atherosclerosis remains one of the main causes of death worldwide even as several interventions have been shown to reduce risk of atherosclerosis and related atherosclerotic cardiovascular diseases (ASCVD)1, 2. Importantly, residual risk of ASCVD has emerged as a relevant threat in populations with successful lipid and/or hypertension management and interventions to reduce known risk factors (fasting glucose, smoking, etc.)3. Indeed, several recent clinical trials revealed that cardiovascular events (CVE) occur even in populations with no significant elevation of low-density lipoprotein cholesterol (LDL-C), suggesting additional risk factors that could contribute to ASCVD incidence, the leading causes of death worldwide4. Current prediction tools for assessing ASCVD risk rely on traditional risk factors such as LDL-C, fasting glucose, smoking, blood pressure, triglycerides, and high-density lipoprotein cholesterol (HDL-C)5. The existence of residual CVE risk emphasizes the need for novel risk prediction tools.
Given the central role of monocytes/macrophages in atherogenesis, extensive efforts have been invested in understanding their actions during recruitment and after infiltration into atherogenic foci, especially their lipid uptake/efflux relevant to this unique environment6–12. Currently, plaque-resident macrophages have been defined as pro-inflammatory “M1”-like macrophages that release pro-atherogenic cytokines7 and accelerate atherosclerosis progression13, 14, or alternatively activated “M2”-like macrophages that are believed to reduce plaque inflammation and suppress atherosclerosis progression7. As a major component of atherosclerotic plaques, foam cells not only accumulate as fatty streaks, but also release pro-inflammatory cytokines and orchestrate pathological tissue remodeling7. Indeed, specific macrophage/foam cell phenotypes can impact stability of atherosclerotic plaques differently15.
However, many studies fall short in explaining key features that distinguish symptomatic and asymptomatic outcomes in patients, or depicting a comprehensive landscape that captures the intertwined inflammation and lipid handling aspects that are unique to macrophage-derived foam cells. Such crucial information in circulating monocytes is difficult to extract from transcriptomics of bulk RNA-seq data, primarily due to the relatively subtle gene expression differences across cells and the lack of available high-resolution tools to characterize their dynamic and plastic actions7–9. Associations between either monocyte counts or transcriptome profiles and ASCVD incidence are only found to be moderate with limited if any predictive capability16, 17, and have never been investigated in the context of ASCVD risk. Herein, we describe an innovative macrophage-derived foam cell analytics program, AtheroSpectrum, which can infer pathogenic inflammatory foam cell-specific genetic programs contributing to ASCVD risk. By combining this gene-set with known ASCVD risk factors, we are able to enhance ASCVD risk prediction compared with currently used models.
Methods
Data availability
To minimize the possibility of unintentionally sharing information that could be used to re-identify private information, data from this study are available from the following resources: monocyte transcriptome profiles of MESA participants are accessible through GEO Series accession number GSE5604716. The other datasets used in this study were accessed with the GEO Series accession number GSE9731018, GSE9794119, GSE11624020, GSE11623920, GSE4329221, or from FR-FCM-Z23S22 (https://figshare.com/s/c00d88b1b25ef0c5c788, DOI: 10.6084/m9.figshare.9206387). GTEx data were from gtexportal.org (dbGaP Accession phs000424.v8.p2).
MESA Participants
Please see additional information about MESA cohort in Expanded Methods. All measurements and monocyte collection were obtained at MESA Exam 516. Participants were followed for CVE until 2017. Of the 1269 MESA participants, 1207 (male=597, female=610) had valid records of CVE on the MESA Events datasets (2017). CVE was defined as “cvda” in MESA, including Myocardial Infarction, Resuscitated Cardiac Arrest, Definite Angina, Probable Angina (if followed by Revascularization), Stroke, Stroke Death, Coronary Heart Disease Death, Other Atherosclerotic Death, and Other Cardiovascular Disease Death. Of the 173 participants who experienced CVE between Exam 1 and 2017 provided by MESA, 68 participants only had events prior to Exam 5, and were thus excluded from predictive modeling. Participants with CVE records after Exam 5 (total participants=1139, including CVE=0, n=1034; CVE=1, n=105) were used for model training, testing, and the survival analyses with indicated prediction scores. We randomly divided the full dataset (n=1139) into two subsets: MESA-set1 (n=911, 466 females and 445 males) that was used for gene-set selection and model training; and MESA-set2 (n=228, 121 females and 107 males) that was used exclusively for model validation. The present analyses are primarily based on transcriptomic data collected from the previous report16 with the approval from Institutional Review Boards of the four institutions. All participants signed informed consent16 (details in the Expanded Methods).
Creation of MDFI for each cell
MDFI (Macrophage-Derived Foam cell Index) was created using a similar method as MPI (Macrophage Polarization Index)23. Calculation for rchow and rathero values of three atherosclerosis macrophage transcriptome profiles (atherogenesis diet mice) and three normal artery macrophages profiles (chow diet mice)20 were calculated were detailed in Expanded Method. Linear regression was performed on the macrophage profiles on the rchow - rathero plot, producing an adjusted R2 value of 1.000. The regression line was defined as the foaming axis. Let the equation of the regression line be: ax + by + c = 0.
The coordinates of each sample’s projection on the regression line (P) were calculated as follows:
The correlation r values were always within the range −1 to 1; we then set the left-most point on the plot P0 (−1,) as the reference zero-point. Accordingly, the right-most point Pmax (1,) was set as the reference Pmax point. The distance l from the reference 0 point to a given macrophage sample’s P (xp, yp) was calculated as the following:
The distance between Pmax and P0 was scaled to create a 0 (P0) to 100 (Pmax) range and the l value of each macrophage sample was rescaled accordingly, resulting in its MDFI.
Model training and testing with 5-fold cross validation
MESA data with valid CVE record after Exam 5 (n=1139) were randomly split to MESA-set1 (n=911) and MESA-set2 (n=228) and adjusted for stratification of sex. MESA-set1 was used for gene screening (EPIC) and CR-30 modeling; whereas MESA-set2 (n=228) was exclusively used for CR-30 validation.
A 5-fold cross validation strategy was used to assess model generalizability. The participants (n=911) in MESA-set1 were randomly shuffled and divided into 5 equal groups (n=182 or 183 in each group); 4 groups were used for training and 1 was used for testing. This random shuffling was conducted 5 times. To evaluate the classification accuracy of models, we considered the average receiver operating characteristic (ROC) under the curve (AUC) of each testing group from 5-fold cross validation. Overall performance of the model was presented as average AUC of the 5 groups. For comparison, ROC curves were also plotted for 10y risk score (JAMA 2001)24, Framingham risk score 200825 (FRS 2008), and the Pooled Cohort Equation for ASCVD risk (PCE 2013)26 in the same 5-fold testing sets. Please see Expanded Methods for additional information for machine learning-powered CVD risk signature gene identification.
Creation of the CR-30 model
A logistic regression model was trained in MESA-set1 using the 30 gene-set and lipid-lowering medication, hypertension medication, diastolic blood pressure, diabetes, sex, age, and MPI. Covariates were rescaled using a standard scaler prior to model training. Each MESA subject was input into the model to generate a regression value between 0 and 1 for their probability of having a CVEs (0=no event; 1=event). The regression values were linearly rescaled to a 0–10 range, and reported as the CR-30 score.
For the analysis of independent validation data (MESA-set2, GTEx, and GSE43292), transcriptome profiles were first normalized using the limma (microarry), or EdgeR (RNA-seq) package. Expression levels of 30 genes in CR-30 were centralized and rescaled using the same scaler (standard scaler) with the model-reference set (MESA-set1). A fixed value of 0 was assigned to the covariate values that were not collected in the independent validation studies. Reshaped data was then uploaded to the model to generate a CR-30 score for each sample.
Methods for signaling enrichment analyses and PCE 2013 risk score calculation are provided in Expanded Methods.
CVE-free survival analyses
Cox proportional hazards regression was conducted on MESA-set2 participants (121 females, 107 males) who had valid record of CVE (yes or no) after monocyte collection date until the date of record in 2017 MESAEvent data. The median CR-30 score of participants in MESA-set2 was used as the cutoff for CVE risk predictions. The analyses were done using R packages “survival” (github.com/therneau/survival) and “survminer” (rpkgs.datanovia.com/survminer/index.html).
Statistics
Unless otherwise stated, p values of gene differential expression were determined by Mann-Whitney U test. ROC test was performed using pROC package (https://search.r-project.org/CRAN/refmans/pROC/html/roc.test.html). p values of enrichment of pathways, upstream regulators, and gene ontology terms were generated by the corresponding bioinformatics tools. All statistics calculations were conducted using R unless otherwise stated.
Results
Residual ASCVD risk not entirely explained by traditional risk factors
The cohort in the present study includes 1207 MESA participants consisting of 610 females and 597 males (Table 1, Fig. 1). Multivariate logistic regression analysis of this cohort confirmed the significant relevance of known factors to CVE, including sex, age, lipid-lowering medication, hypertension medication, diabetes (2003 American Diabetes Association [ADA] fasting criteria, level 1=impaired fasting glucose; level 2=diabetes mellitus, treated or untreated), and diastolic blood pressure (Fig. S1 and Table 2). However, consistent with previous observations4, participants in this cohort who has been treated with lipid-lowering or hypertension medication or additional ASCVD risk factor management may still have significant risk of having cardiovascular event, suggesting an unmet medical need to identify additional factors which may contribute to ASCVD.
Table 1.
Characteristics of participants from the MESA cohort (Exam5, n=1207).
| Female n=610 (50.5%) |
Male n=597 (49.5%) |
|
|---|---|---|
| Age (years), mean±SD | 69.6±9.4 | 69.7±9.3 |
| BMI (kg/m2), mean±SD | 30.1±6.2 | 28.9±4.6 |
| Cholesterol (mg/dL), mean±SD | 191.0±34.4 | 170.1±36.5 |
| LDL-C (mg/dL), mean±SD | 109.8±31.2 | 99.8±32.6 |
| HDL-C (mg/dL), mean±SD | 59.6±16.5 | 48.4±13.0 |
| Triglycerides (mg/dL), mean±SD | 108.1±51.3 | 109.8±50.1 |
| Fasting glucose (mg/dL), mean±SD | 100.6±27.4 | 105.6±28.1 |
| Waist (cm), mean±SD | 99.9±15.9 | 103.1±11.9 |
| SSBP, mean±SD | 125.6±21.6 | 122.0±18.6 |
| SDBP, mean±SD | 66.2±9.4 | 70.6±9.5 |
| Non-smoking, N (%) | 293 (48.0%) | 188 (31.5%) |
| CVE (2017) | 65 | 108 |
SSBP=seated systolic blood pressure; SDBP=seated diastolic blood pressure. BMI=body mass index. Non-smoking=never smoked; CVE is defined by “cvda” in MESA data (details see Methods).
Fig. 1.
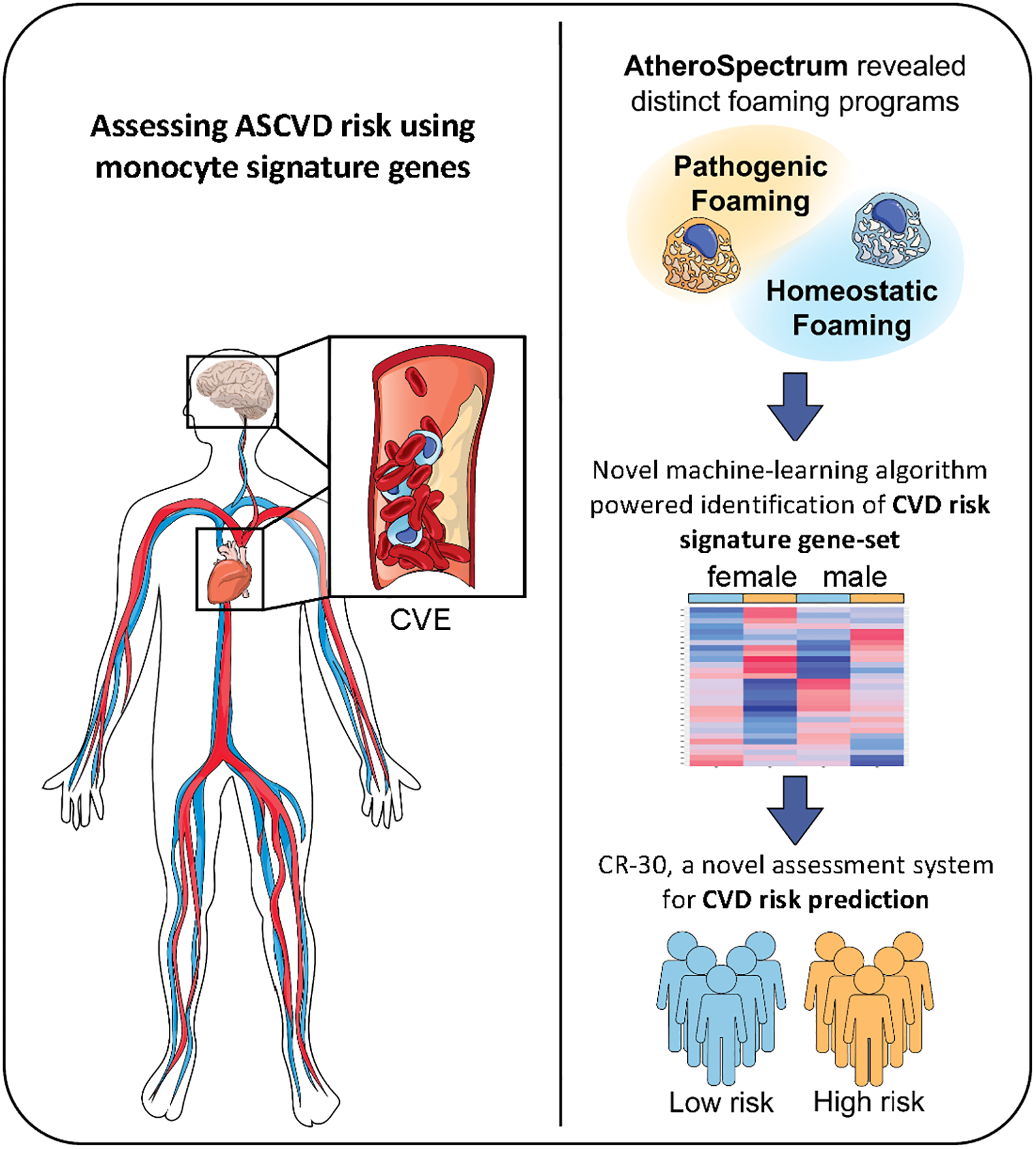
Schematic overview of the study. Substantial ASCVD risk remains in subjects of optimal/near-optimal lipid status. In the present study, our AtheroSpectrum revealed the pathogenic foaming program of atherogenic macrophages. By cross-referencing these genes with genes from peripheral monocytes from MESA participants using our novel algorithm, we identified an ASCVD 30-risk gene-set. By combining this 30 gene-panel with other factors, we created a Cardiovascular Risk score (CR-30) that depicted ASCVD risk.
Table 2.
Multivariate logistic regression analysis showing association between CVE as the outcome and sex, age, HDL-C, LDL-C, total cholesterol, triglycerides, systolic and diastolic blood pressure, diabetes 2003 ADA fasting criteria (level 1=impaired fasting glucose; level 2=diabetes mellitus, treated or untreated), waist circumference, body-mass index (BMI), total body fat, lipid-lowering medication, and hypertension medication as the variables. p values calculated from multivariable logistic regression.
| Variable | Coefficient | p-value |
|---|---|---|
| Age | 0.050 | 1.11E-05 |
| Hypertension medication | 0.586 | 3.98E-03 |
| Sex | 0.370 | 8.90E-02 |
| Diabetes 2003 criteria levels1 | −0.353 | 1.36E-01 |
| Lipid-lowering medication | 0.199 | 3.05E-01 |
| Seated diastolic blood pressure | 0.012 | 3.17E-01 |
| Diabetes 2003 criteria levels2 | −0.196 | 4.91E-01 |
| Triglyceride | 0.053 | 3.85E-01 |
| Waist | −0.008 | 3.93E-01 |
| Total cholesterol | −0.258 | 3.96E-01 |
| LDL-C | 0.251 | 4.08E-01 |
| HDL-C | 0.238 | 4.32E-01 |
| Fasting glucose | 0.001 | 7.50E-01 |
| Smoking | −0.056 | 8.65E-01 |
| BMI | 0.003 | 9.04E-01 |
| Seated systolic blood pressure | 0.000 | 9.87E-01 |
| Intercept | −4.674 | 2.04E-01 |
AtheroSpectrum identifies the dynamic transition from macrophages to foam cells
To comprehensively characterize the diversified status of monocytes and macrophages in the context of cardiovascular disease (CVD) and identify the significant cell features that associate with ASCVD risks, we developed a novel computational program called AtheroSpectrum. AtheroSpectrum was developed by extending our recently published program MacSpectrum23 that was designed to fine-map monocyte and macrophage activation and inflammatory features in complex tissue settings or diseases (Fig. S2). AtheroSpectrum is designed to capture the heterogeneity of macrophage-derived foam cells in atherogenic foci with two indices that address key aspects of these cells: Macrophage Polarization Index (MPI, inherited from MacSpectrum to annotate the degree of inflammation), and the novel Macrophage-Derived Foam cell Index (MDFI) that depicts macrophage foam cells (Fig. 2A). To create the MDFI, we developed a randomized feature selection approach to screen for a gene-set that could effectively separate the previously reported macrophage foam cells and non-foam cells, as indicated by their lipid deposition and cellular granularity in atherosclerotic vs. normal mice (GSE116239)20, while retaining adequate inclusion of the most differentially expressed genes in macrophage foam cell formation (detailed in the Methods section). Higher MPI suggests more inflammatory cells and higher MDFI suggests more differentiation towards foam cells.
Fig. 2.
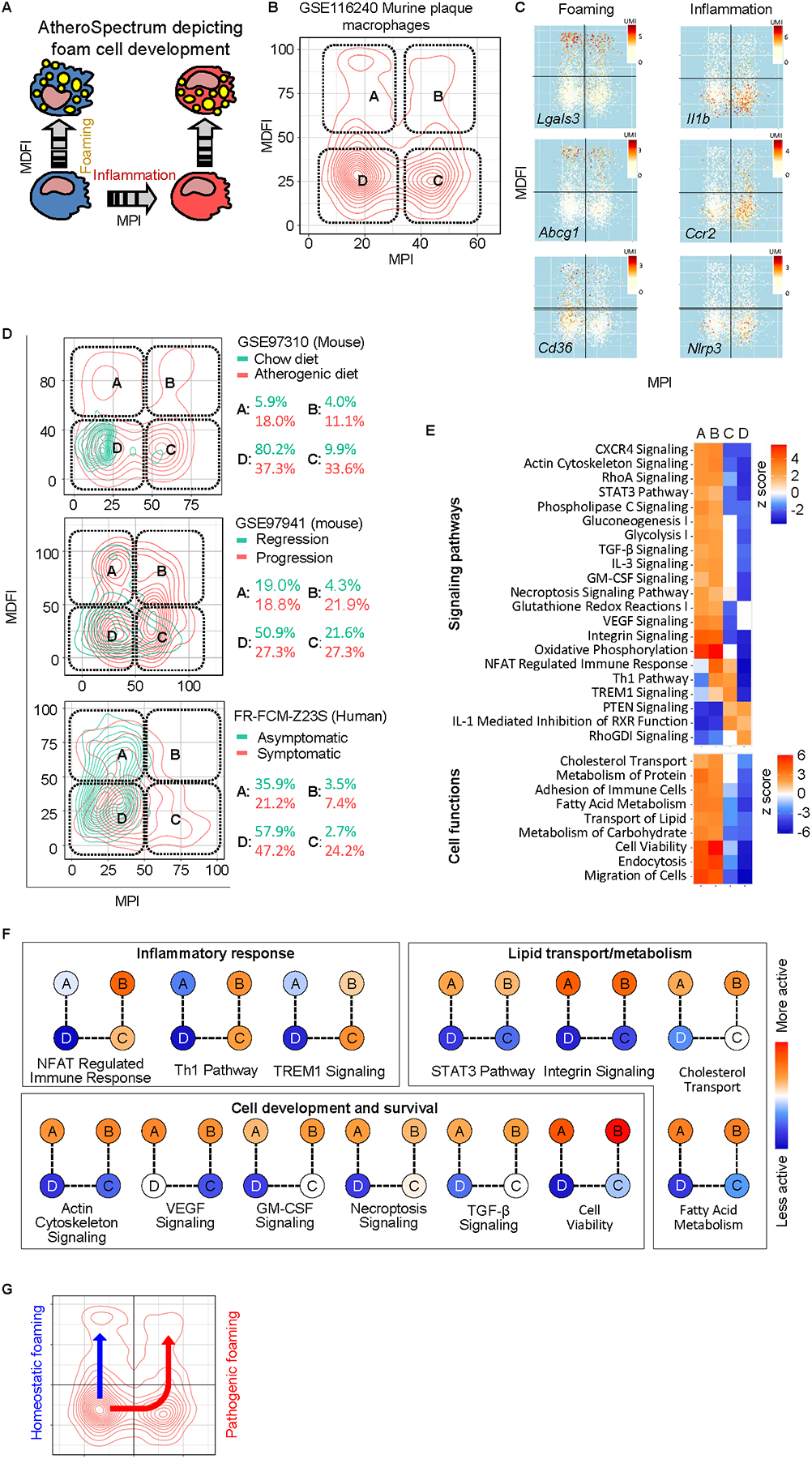
AtheroSpectrum identified inflammatory / macrophage foam cell populations associated with severity of atherosclerosis. A, Scheme showing the transition of different types of atherogenic macrophages by AtheroSpectrum. B,C, AtheroSpectrum depicted 4 major sub-populations of murine plaque macrophages with different foaming and inflammatory properties and marker gene expression(UMI). D, AtheroSpectrum identified different artery macrophage sub-population distributions in atherogenic vs. control mice (GSE97310), progression vs. regression mice (GSE97941), and symptomatic vs. asymptomatic humans with atherosclerosis (FR-FCM-Z23S, 2019). E, Heatmaps showing signaling pathways and cell functions enriched in each of the artery macrophage sub-populations. F, Representative pathways fall into three main categories: inflammatory response, lipid transport/metabolism, and cell development and survival. G, Scheme showing predicted cell differentiation pathways towards homeostatic foaming and pathogenic (inflammatory) foaming phenotypes.
Application of AtheroSpectrum to murine atherogenic plaque macrophages (GSE116240)20 correctly identified foam cells, and further revealed four subpopulations that we have termed non-inflammatory foam cell (A), inflammatory foam cell (B), inflammatory non-foam cell (C), and non-inflammatory/non-foam cell (D) (Fig. 2B). AtheroSpectrum accurately identified a previously reported foam cell population20 and its depiction was supported by expression of inflammatory and foam cell marker genes in the sub-populations depicted by MPI and MDFI, including Lgals3, Abcg1, Cd36, Il1b, Ccr2, Trem2, Lipa, Srebf1(Srebp1), Nlrp3, and others20, 27, 28(Fig. 2C, Fig. S3A).
To validate if similar cell sub-populations are consistently observed in atherosclerosis from different studies, we applied AtheroSpectrum to several atherosclerosis datasets generated under different scenarios. In mice with on-going atherosclerosis (GSE97310)18, AtheroSpectrum identified similar distribution patterns of athero-plaque macrophages as the four sub-populations mentioned above (Fig. 2D, top panel). In contrast, artery macrophages from healthy, chow diet mice primarily concentrated in the “D” region, suggesting a non-inflammatory/non-foamy, unstimulated phenotype in healthy tissues.
Interestingly, compared to mice exhibiting improving/regressing atherosclerosis, mice with progressing atherosclerosis (GSE97941)19 had markedly increased macrophage populations in regions “B” (21.9% vs. 4.3%) and “C” (27.3% vs. 21.6%), with a similar portion in region “A” (18.8% vs 19.0%) (Fig. 2D, middle panel). Similar patterns were also observed in human atherogenic plaque-resident macrophages22 (Fig. 2D, bottom panel): plaque macrophages of asymptomatic patients predominately concentrated in regions “A” (35.9%) and “D” (57.9%), while those from symptomatic patients showed a marked increase in populations “B” (7.4% vs. 3.5%) and “C” (24.2% vs. 2.7%). On average there was 3.9 fold increase (p=0.02) in populations “B” and “C” in more severe vs. healthy/less-severe conditions across the three datasets. These analyses revealed that the two novel inflammatory-foam cell/ inflammatory subpopulations (“B” and “C”) that were identified by AtheroSpectrum were associated with CVD severity.
We further conducted gene function analyses on the four cell sub-populations (Fig. 2E). Many of the enriched signaling pathways and cell functions could be categorized into one of three functional groups: inflammatory response, lipid transport/metabolism, and cell development and survival (Fig. 2F). Pro-inflammatory signaling, such as the Th1 pathway, was enriched in populations “B” and “C” (Fig. 2E, Fig. 2F). Pathways of lipid metabolism (e.g., STAT3, RXR signaling pathways), lipid transport (e.g., integrin signaling, cholesterol transport, endocytosis), and cell development (e.g., GM-CSF signaling, TGF-β signaling) presented higher activation z-scores in populations “A” and “B” (Fig. 2E, Fig. 2F) – consistent with their predicted status as macrophage foam cells. Of note, the foam cells “A” and “B” showed both activated cell viability and necroptosis signaling, suggesting those foam cells were proliferating and differentiating in the tissue, but died at the end of their foam cell fate.
The pathogenic foam cell program signature genes display distinct expression patterns in a population with ASCVD
Based on our analyses, we proposed two cell programs of atherogenic macrophages: the “homeostatic foaming” (“D” to “A”) that was present in both benign and severe disease conditions, and the “pathogenic foaming” (“D” to “C” to “B”) that was associated with severity of disease (Fig. 2G).
Genes that had significantly different expression (p< 0.05) in population “B” vs. “C” but not in “A” vs. “D”, and in population “B” vs. “A” but not in “C” vs. “D” resulted in a list of 2209 enriched genes that were associated with the inflammatory foam cell development of macrophages. This gene list included known signaling pathways involved in inflammation (e.g., tumor necrosis alpha [TNF-α] signaling, NF-kappa B signaling pathway), lipid transport (lysosome pathway), and lipid metabolism (MAPK signaling). A major portion of these genes (1566, 70.9%) presented significant differential expression in atherosclerotic vs. non-atherosclerotic artery tissues of human subjects deposited at the GTEx Portal (gtexportal.org), supporting the pathological relevance of these genes with ASCVD (Fig. S3).
We next designed a self-optimizing signature gene-set feature selection algorithm (Exploration system of Process-Incorporated features in Cells [EPIC]) to screen the 2209 enriched genes using peripheral blood monocyte transcriptomes from MESA participants (Illumina bead array transcriptomics16, Table 1). To assess generalization performance, we randomly divided the MESA dataset (n=1139) into two subsets: MESA-set1 (n=911, 466 females and 445 males with 84 CVE cases) that was used for gene-set selection and model training; and MESA-set2 (n=228, 121 females and 107 males with 21 CVE cases) that was used exclusively for model validation. Our algorithm incorporated multiple steps of self-optimizing cycles that gradually selected a panel of 30 CVE associated genes (see Expanded Methods).
These genes showed differential expression patterns between the two sexes based on standardized gene expression levels (Fig. 3A). Further, we fit a logistic regression model using the standardized expression levels of the 30 risk signature genes to predict CVE and observed significant associations between gene expression and CVE (Fig. 3B). Of note, ATP8B4 and SLC1A3 were positively associated with occurrence of CVE (Fig. 3B), and were expressed at higher levels in atherosclerotic vs. non-atherosclerotic human artery tissues of the same subjects in the GTEx portal (Fig. 3C) and another human carotid atheroma dataset (GSE43292)21 (Fig. 3D), while R3HCC1 and MRPL1, which were negatively associated with occurrence of CVE (Fig. 3B), were expressed at lower levels in atherosclerotic vs. non-atherosclerotic human artery tissues of the same subjects in these artery tissue datasets (Fig. 3C,D). For these genes, similar patterns were observed in both female and male subjects (Fig. S4).
Fig. 3.
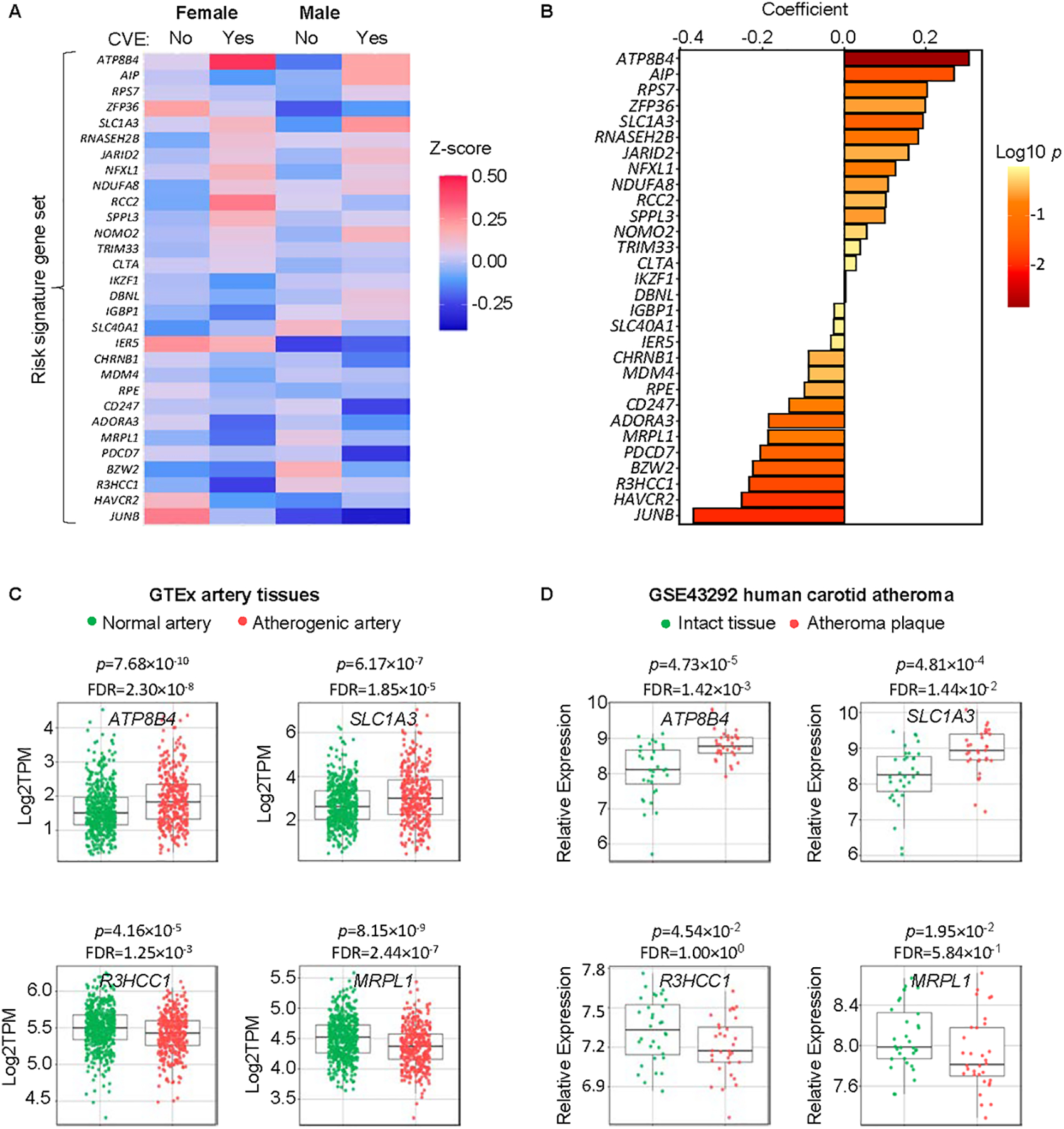
Our novel algorithm identified an ASCVD signature gene-panel associated with CVEs in the MESA (n=1207). A, Relative expression of the 30 risk signature genes (Z-score, standardized expression) in circulating monocytes of female or male MESA participants who had a CVE (Yes) or not (No). B, Coefficients of the 30 signature genes in the regression analysis. C, ATP8B4, SLC1A3, R3HCC1, and MRPL1 are associated with CVEs and had significantly different expression levels in atherosclerotic vs. non-atherosclerotic artery tissues of human subjects deposited at GTEx portal.org (537 non-atherosclerotic samples, 362 atherosclerotic samples). D, Relative expression (Log 2 RMA signal values) of ATP8B4, SLC1A3, R3HCC1, and MRPL1 in atheroma plaque vs. intact tissue from 32 patients (microarray, GSE43292). p values in C and D were calculated by Mann-Whitney U test with false discovery rate (FDR) adjustment.
A Novel ASCVD Risk score by 30 gene-panel (CR-30) can predict ASCVD risk
To determine if the 30 gene-panel would enhance ASCVD risk prediction, we developed a novel risk score model, CR-30 (CVD Risk score by a 30 gene-set). CR-30 is a logistic regression model that integrates the 30-gene panel revealed by AtheroSpectrum and EPIC, with clinical variables from MESA (Exam 5) including sex, age, lipid-lowering medication status, hypertension medication status, diabetes, and diastolic blood pressure based on their association to CVEs from multivariate regression analyses in this cohort (Table 2), and our defined inflammatory index, MPI. A 5-fold cross validation analysis demonstrated that CR-30 coefficients were stable across folds with an average AUC=0.670 (p=1.53×10−2) (Fig. 4A). In parallel, this analysis also demonstrated reasonable generalizability of other commonly used risk scores in MESA-set1: 10y risk score (JAMA 2001)24 (average AUC=0.628, p=8.62×10−2), Framingham risk score 200825 (FRS 2008) (average AUC=0.667, p=1.62×10−2), and the Pooled Cohort Equation for ASCVD risk (PCE 2013)26 (average AUC=0.645, p=4.16×10−2).
Fig. 4.
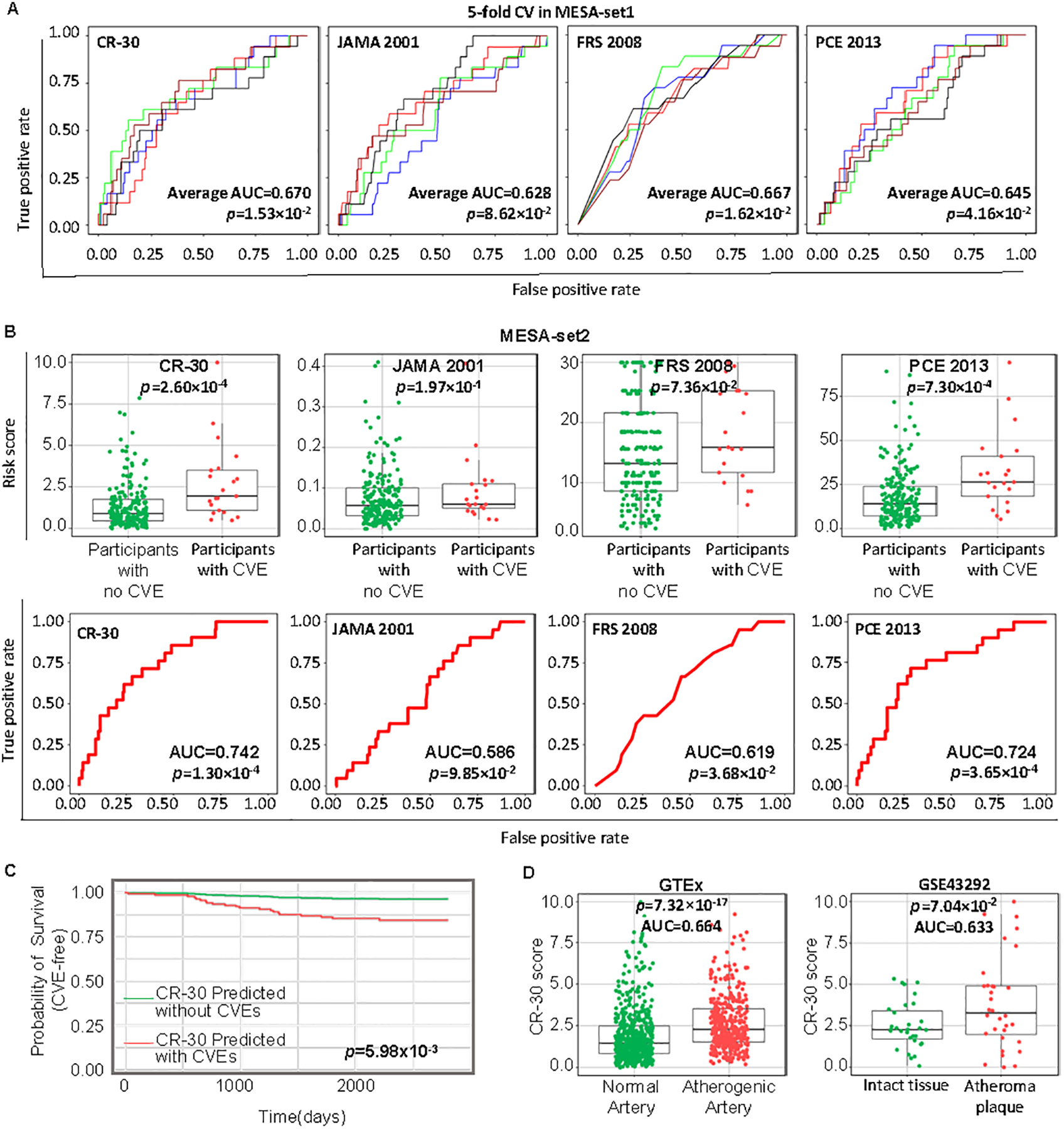
CR-30 score effectively depicts CVD risks. A, ROC plots and AUC values of 5-fold cross for CR-30 score, 10y risk scores (JAMA 2001), Framingham 2008 risk score (FRS 2008), and the 2013 Pooled Cohort Equation for ASCVD risk score (PCE 2013) for the MESA-set1 participants (n=911). Differently colored curves indicate individual folds of the 5-fold cross validation (n=182–183), and the average AUC and p value of the 5 folds were shown. B, CR-30, JAMA 2001, FRS 2008, and PCE 2013 risk scores and ROC plots for MESA-set2 participants (n=228). p values were calculated by Mann-Whitney U test. C, Probability of survival (CVE-free) of MESA-set2 participants (n=228) who were predicted to have CVE or not by CR-30 score since monocyte collection was calculated by Cox regression with Wald test for p values. D, CR-30 scores and AUC values for two independent tissue gene expression datasets, GTEx tissue transcriptomics (n=899), and GSE43292 (n=64). p values were calculated by Mann-Whitney U test.
Next, we trained the CR-30 model in the MESA-set1 data (Table 3). The significance of MPI in the multivariate regression further suggested the importance of inflammation in CVD, consistent with increased inflammatory populations (“B” and “C”) in symptomatic patients relative to asymptomatic patients (Fig. 2D). The performance of CR-30 was then tested in three independent datasets, including the MESA-set2 (n=228) that was not used in model development (Fig. 4B), and two independent datasets (GTEx and GSE4329221) (Fig. 4D). The CR-30 score range is set to 0–10, with 0 and 10 indicating lowest and highest risk for a CVE, respectively. Although trained in a moderate-sized data (MESA-set1, n=911), CR-30 predicted CVE in the MESA-set2 with an accuracy of AUC=0.742 (p=1.30×10−4) and a significant discrimination of subjects with CVE from those without CVE after sample collection (p=2.60×10−4). Other models in this cohort (MESA-set2, n=228) were also analyzed for their performance, with JAMA 2001 (p=1.97×10−1, AUC=0.586), FRS 2008 (p=7.36×10−2, AUC=0.619), PCE 2013 (p=7.30×10−4, AUC=0.724). Furthermore, paired ROC performance analyses demonstrated better prediction accuracy of CR-30 in MESA-set2 than JAMA 2001 (p=2.61×10−2) and FRS 2008 (p =2.62×10−2), and comparable to PCE 2013 (p=3.67×10−1). In addition, in MESA-set2, those predicted to have CVE based on their CR-30 scores indeed were more likely to have an event compared to those with prediction of no-event (Wald test, p=5.98×10−3) after monocyte sample collection (Fig. 4C). Similarly, CR-30 displayed a prediction of CVEs in females (Wald test, p=4.10×10−2) and males (Wald test, p=8.90×10−2) by Cox regression analyses (Fig. S5B). CR-30 was also tested in two tissue gene expression datasets (Fig 4D). CR-30 scores significantly discriminated atherosclerotic lesions from normal tissue among patients with an AUC of 0.664 in GTEx (n=899, p=7.32×10−17) and AUC of 0.633 in GSE4329221 (n=64, p=7.04×10−2).
Table 3.
CR-30 model output from the training data (MESA-set1, n=911, standard scaler). Variables in this model include a list of 30 pathogenic foaming genes, as well as MPI (Macrophage Polarization Index), sex, age, seated diastolic blood pressure, diabetes 2003 ADA fasting criteria (level 1=impaired fasting glucose; level 2=diabetes mellitus, treated or untreated), hypertension medication, and lipid-lowering medication. See Methods for model training details.
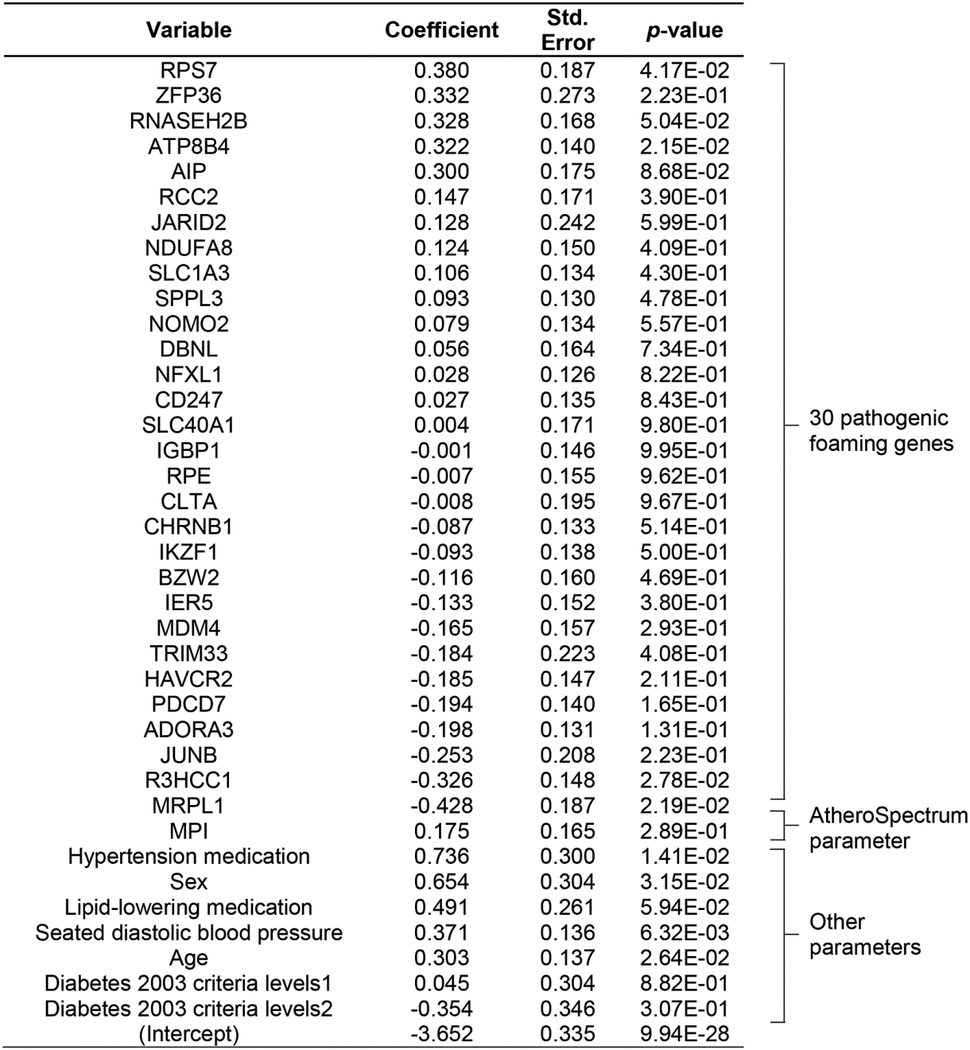
|
To evaluate the contribution of the CR-30 gene panel and MPI in CVE prediction, we performed a model sensitivity test (Fig. S6). We used all variables from the PCE 2013 model to develop 2 models: PCE-Mute (PCE 2013 variables only) and PCE-CR30-Mute (PCE 2013 variables + 30-gene panel + MPI). Generalizability of both models was first evaluated through a 5-fold cross validation strategy (PCE-Mute, average AUC=0.639, p=5.29×10−2; PCE-CR30-Mute, average AUC=0.655, p=2.95×10−2) (Fig. S6B). The two models were trained in MESA-set1 (Fig. S6A) and the likelihood ratio analysis with 31 degrees of freedom found significantly better support for the PCE-CR30-Mute (complex) model over PCE-Mute (null) (p=3.30×10−2). The performance of PCE-CR30-Mute (p=7.89×10−5, AUC=0.762) in MESA-set2 displayed better prediction power than PCE-Mute (p=7.66×10−3, AUC=0.677, Fig. S6C,D). In summary, our CR-30 model, powered by AtheroSpectrum and a novel machine learning algorithm for feature selection, effectively predicted CVD risk and presence of atherosclerotic lesions, exhibiting strong potential for future translational development.
Discussion
CVE risk assessment is critical for determining appropriate interventions and minimizing occurrence of future CVE. For decades, traditional cardiovascular risk factors, such as HDL-C, LDL-C, and triglyceride levels have been targeted in ASCVD patients and/or high-risk groups for preventative interventions. Our analyses confirmed the association of known factors associated with CVE risk in the MESA cohort, including age, sex, diabetes, blood pressure, as well as medications for lipid-lowering and hypertension. However, a substantial number of people may still experience new CVE4, suggesting additional factors contributing to CVE are yet to be revealed.
The monocyte-macrophage lineage plays a critical role in the development of ASCVD. Monocytes sense cues from the microenvironment as they travel through the circulation and adopt specific gene expression profiles in response. Upon recruitment to the artery intima in atherogenic conditions, monocytes differentiate into macrophages and orchestrate pathological progression depending on their activation and foam cell development process. Therefore, this cell lineage carries comprehensive information on subjects with atherogenic conditions, making them ideal targets for analyzing ASCVD risk. However, due to a lack of high-resolution transcriptomic approaches, previous studies of this lineage only found limited if any association with CVE, and monocyte/macrophage-derived predictive factors for ASCVD risk were never investigated until now16, 17.
For decades, lipid metabolism had been extensively investigated for its role in foam cell formation. In contrast, little research has been done on the impact of inflammation on the foaming process. This deficit is largely due to a strategy of defining a cell subpopulation by few known genes followed by their feature characterization. Mouse models provide robust information on atherogenesis but little insight into the role of foaming cells and their inflammatory status in atherosclerosis progression, and even less to their potential roles in plaque instability and rupture. Recent studies also reported the involvement of key regulators for foaming, with or without inflammatory response. For example, ANGPTL4 deletion can enhance foam cell formation primarily by regulating lipid uptake through increased CD36 expression and reducing ABCA1 membrane localization29; whereas Nur77 ablation enhances macrophage inflammation and atherogenesis13. Recent meta-study of nine single cell mouse datasets from the Ley group suggested that four macrophage subsets could be identified, and transcriptomic similarity of the foamy subset (Trem2+ macrophages) are transitioned by inflammatory and interferon inducible macrophage subsets, summarized using UMAP algorithms30. This study supported the low-inflammatory feature of Trem2+ macrophage subset (GSE119240), induction of LXR- and SREBP- depended fatty acid metabolism in foam cells using bulk RNA-seq analyses20, 31. Indeed, distribution of these genes on AtheroSpectrum also supported these observations in regarding to their roles in foaming or inflammation (Fig. 2) and provided further expression pattern along either homeostatic or pathogenic foaming programs.
Of note, methods using conventional computational algorithms for single-cell RNAseq analyses (such as tSNE, UMAP, PCA, and others) often produce a connective spreading population for one given cell type (macrophages in these studies) and rely on subsequent “marker” gene expression detection to define subsets. We and others have pointed out that current unbiased clustering methods for single-cell sequencing analyses may poorly depict subsets within one cell lineage23 32. Thus, in the present study, we took a different strategy where we annotated macrophage actions without aiming to define macrophage subsets during the foaming process. We leveraged our experience computationally depicting this lineage with MacSpectrum23 to develop the novel AtheroSpectrum system to profile foam-cell-developing macrophages during atherogenesis. This system features two indices, MPI and MDFI, that depict the inflammation and foam cell formation of atherogenic macrophages, respectively. We validated performance of AtheroSpectrum with multiple datasets from separate studies (Fig. 2D). In murine plaque macrophages (GSE116240)20, AtheroSpectrum accurately depicted the foam cell population, which was indicated by expression of foam cell signature genes Lgals3 and Abcg1 reported in the original publication (Fig. 2C). In another study (GSE97310)18, AtheroSpectrum revealed expansion of foamy and inflammatory macrophage populations in atherogenic diet-induced atherosclerotic mice compared to chow diet mice (Fig. 2D), which was consistent with previous knowledge of atherogenic macrophages33–35. These results support the efficacy of AtheroSpectrum, which is tailored for this disease.
AtheroSpectrum not only correctly identified known features of atherogenic macrophages but also identified a novel program consisting of both inflammatory and foam cell populations that were consistently higher in more severe ASCVD conditions: increased inflammatory/foamy populations in mice with progressing vs. regressing atherosclerosis, and in human subjects of symptomatic vs. asymptomatic atherosclerosis (Fig. 2D). For the first time, our discoveries suggest that not all foam cells contribute to atherogenesis equally, and the inflammatory foamy process – which we termed “pathogenic foaming” – is associated with detrimental outcomes.
We next extracted the 2209 genes that were enriched in the pathological foaming program of atherogenic macrophages followed by gene-set screening using our original computational program EPIC (Expanded Methods) with a selection guidance for gene-sets association with CVE. However, even within the 2209 enriched gene pool, there could be enormous combinations of genes (e.g., taking 30 from 2209 genes can produce 1.73×10100 different gene-sets), and it is impossible to screen each of them in a feasible amount of time. To solve this challenge, we created a novel algorithm, EPIC, that features recursive random sampling and self-optimization; through 7 stages of screening, our algorithm gradually narrowed down a candidate gene list and eventually produced a final 30-gene panel correlated with occurrence of CVE. In addition to the 30 signature genes, the MPI of monocytes also showed significant association with CVE (Table 3), suggesting an important role of inflammation for the increased pathological foaming program in more severe CVD conditions.
Based on the 30 signature genes as well as MPI, sex, age, hypertension medication, lipid-lowering medication, diabetes, and diastolic blood pressure, we developed a novel score system, named CR-30 (CVD Risk score by 30 genes), for assessing CVD risk by monocyte gene expression profiles (Table 3). Cross validation analysis of MESA-set1 for modeling suggested reasonable generalization of CR-30 model training with stable AUCs, as well as JAMA 2001, FRS 2008, and the PCE 2013 risk scores in MESA-set1 (Fig. 4A). The efficacy of CR-30 was then validated in remaining MESA-set2 (p=2.60×10−4) and two independent data (GTEx, p=7.32×10−17 and GSE43292, p=7.04×10−2) (Fig. 4). A likelihood ratio test of model fit of PCE-Mute model (null) to the alternative PCE-CR30-Mute on the MESA-set1 data was performed with 31 degrees of freedom which further supports the PCE-CR30-Mute model (p=3.30×10−2). Our work established an approach for assessing CVD risk with monocytes from peripheral blood, and therefore could be used for longitudinal study and the follow-up evaluation of CVD medications.
To date, few monocyte profiles are available with comprehensive clinical measures from large cohorts, which significantly limited the robust validation and optimization of the CR30 prediction tool. There are also several limitations of the current CR-30 score that could be further developed when more monocyte transcriptomics data are available. For example, in this study, feature selection and CR-30 model training was using MESA-set1 and tested in MESA-set2 and two independent datasets; whereas FRS 2008, PCE 2013 and JAMA 2001 models were trained using independent datasets during their development. It would be ideal to conduct feature selection, modeling training in different dataset when they are available in the future. In addition, features (including the gene panel and physiological variables from MESA cohort) could be selected using various machine learning algorithms and/or stepwise strategy for clinical variables and gene features. Although cross validation analysis suggested a reasonable generalizability of MESA-set1 (n=911) for CR-30 model training, the moderate sample size with imbalanced case/control ratio (CVE0:CVE1 ratio is about 9:1) could also be adjusted using imbalanced sampling strategies to improve model sensitivity or larger monocyte profiles if available in the future. The CR-30 score is not ethnic-specific, given the limited size of our dataset and the number within each ethnic group with CVE (Supplementary Table S1). In addition, CR-30 was not applied to smaller subsets (e.g., ethnic group) due to the modest sample size in the current cohort. Lack of monocyte profiles from human subjects from different age groups also restricted age-related risk factor identification. When more monocyte datasets with CVE and detailed time courses become available, the CR-30 score can be evaluated and improved to provide more precise and robust prediction power of CVD risks within specified periods of time. CR-30 was developed using circulating monocyte gene profiles which can be challenging to translate seamlessly to clinical practice at this time, primarily due to the cost and technical difficulties associated with monocyte isolation from participant blood samples as well as RNA-sequencing. It is also a technical challenge to incorporate and cross-compare datasets generated using different platforms, which requires standardizing data normalization. Given the rapid advances of technologies in transcriptomic and genomic profiling, as well as fast developing bioinformatics tools, we are optimistic that application of CR-30 and other gene signature-based CVD risk assessment tools will ultimately be feasible, and will benefit patients for precision therapy and prevention care in the near future.
Supplementary Material
Clinic Perspective.
What is new?
A novel macrophage foaming computational program, AtheroSpectrum, revealed two distinct programs during atherosclerosis: homeostatic foaming and pathogenic foaming programs.
CR-30, an ASCVD risk assessment tool developed using monocyte transcriptomes and known risk factors, provided enhanced prediction power.
What are the clinical implications?
Atherosclerosis remains one of the main causes of death in US and worldwide, demanding precision ASCVD risk assessment.
Pathogenic foaming program was positively associated with of the incidence of atherosclerosis in multiple studies.
Acknowledgement
We are grateful for the scientific editing support from Dr. Christopher Bonin and Dr. Geneva Hargis UConn School of Medicine. We are also thankful for the support and valuable suggestions from members of the Immuno-Cardio Group at UConn Health.
BZ conceived the project; CL, LQ and AJM performed computational analyses; CL, LQ, AJM, DA, YL, DH, JR, WP, and BZ developed analytic strategies; YL performed monocyte isolation and microarray profiling; PM helped signature gene validation in GTEx database; CL, LQ, AJM, BZ, DA, YL, DH, JR, WP, AR, AV, and PM contributed to data interpretation. DA, YL, DH, WP, AR, and AM helped with statistical analyses of MESA data. WP, DH, JR, SR, DV, and CJ contributed to MESA data collection. CL, LQ, AJM and BZ wrote the manuscript. The manuscript has been revised by all authors.
Source of Funding
The study is supported by AHA 19TPA34910079, NIH/NIDDK R01DK121805 to Dr. Beiyan Zhou. Dr. Rodriguez was supported by NIH HL1381862 grant and an endowment by the Linda and David Roth Chair for Cardiovascular Research.
The MESA study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420. Also supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The MESA Epigenomics & Transcriptomics Studies were funded by NIH grants 1R01HL101250, 1RF1AG054474, R01HL126477, R01DK101921, and R01HL135009.
Nonstandard Abbreviations and Acronyms
- ASCVD
Atherosclerotic cardiovascular disease
- CVD
Cardiovascular disease
- CVE
Cardiovascular event
- MESA
Multi-Ethnic Study of Atherosclerosis
- GTEx
Genotype-Tissue Expression
- MDFI
Macrophage-Derived Foam cell Index
- MPI
Macrophage Polarization Index
- FRS
Framingham risk score
- PCE
Pooled Cohort Equation
- ADA
American Diabetes Association
- EPIC
Exploration system of Process-Incorporated features in Cells
- LDL-C
Low-density lipoprotein cholesterol
- HDL-C
High-density lipoprotein cholesterol
- CR-30
CVD Risk score by a 30 gene-set
Footnotes
Disclosures
Dr. Rodriguez is the founder of Lipid Genomics.
Supplemental Materials
Reference
- 1.Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgözoğlu L and Lewis EF. Atherosclerosis. Nature Reviews Disease Primers. 2019;5:56. [DOI] [PubMed] [Google Scholar]
- 2.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawes CM, Parag V, Bennett DA, Suh I, Lam TH, Whitlock G, Barzi F, Woodward M and Asia Pacific Cohort Studies C. Blood glucose and risk of cardiovascular disease in the Asia Pacific region. Diabetes Care. 2004;27:2836–42. [DOI] [PubMed] [Google Scholar]
- 4.Matsuura Y, Kanter JE and Bornfeldt KE. Highlighting Residual Atherosclerotic Cardiovascular Disease Risk. Arterioscler Thromb Vasc Biol. 2019;39:e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sampson UK, Fazio S and Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep. 2012;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yancey PG, Bortnick AE, Kellner-Weibel G, de la Llera-Moya M, Phillips MC and Rothblat GH. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–9. [DOI] [PubMed] [Google Scholar]
- 7.Moore KJ, Sheedy FJ and Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrett TJ. Macrophages in Atherosclerosis Regression. Arterioscler Thromb Vasc Biol. 2020;40:20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore KJ and Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pourcet B and Staels B. Alternative macrophages in atherosclerosis: not always protective! J Clin Invest. 2018;128:910–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rader DJ and Pure E. Lipoproteins, macrophage function, and atherosclerosis: beyond the foam cell? Cell Metab. 2005;1:223–30. [DOI] [PubMed] [Google Scholar]
- 12.Batista-Gonzalez A, Vidal R, Criollo A and Carreno LJ. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front Immunol. 2019;10:2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K and Hedrick CC. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110:416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamers AA, Vos M, Rassam F, Marinkovic G, Kurakula K, van Gorp PJ, de Winther MP, Gijbels MJ, de Waard V and de Vries CJ. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ Res. 2012;110:428–38. [DOI] [PubMed] [Google Scholar]
- 15.Newby AC, George SJ, Ismail Y, Johnson JL, Sala-Newby GB and Thomas AC. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb Haemost. 2009;101:1006–11. [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Reynolds LM, Ding J, Hou L, Lohman K, Young T, Cui W, Huang Z, Grenier C, Wan M, Stunnenberg HG, Siscovick D, Hou L, Psaty BM, Rich SS, Rotter JI, Kaufman JD, Burke GL, Murphy S, Jacobs DR Jr., Post W, Hoeschele I, Bell DA, Herrington D, Parks JS, Tracy RP, McCall CE and Stein JH. Blood monocyte transcriptome and epigenome analyses reveal loci associated with human atherosclerosis. Nat Commun. 2017;8:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berg KE, Ljungcrantz I, Andersson L, Bryngelsson C, Hedblad B, Fredrikson GN, Nilsson J and Bjorkbacka H. Elevated CD14++CD16- monocytes predict cardiovascular events. Circ Cardiovasc Genet. 2012;5:122–31. [DOI] [PubMed] [Google Scholar]
- 18.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE and Zernecke A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res. 2018;122:1661–1674. [DOI] [PubMed] [Google Scholar]
- 19.Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, Loke P and Fisher EA. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, Jang MY, Seok Jang H, Yun TJ, Lee SH, Yoon WK, Prat A, Seidah NG, Choi J, Lee SP, Yoon SH, Nam JW, Seong JK, Oh GT, Randolph GJ, Artyomov MN, Cheong C and Choi JH. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ Res. 2018;123:1127–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayari H and Bricca G. Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J Biosci. 2013;38:311–5. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, Khan NS, Wong CK, Shamailova R, Hill CA, Wang Z, Remark R, Li JR, Pina C, Faries C, Awad AJ, Moss N, Bjorkegren JLM, Kim-Schulze S, Gnjatic S, Ma’ayan A, Mocco J, Faries P, Merad M and Giannarelli C. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C, Menoret A, Farragher C, Ouyang Z, Bonin C, Holvoet P, Vella AT and Zhou B. Single cell transcriptomics based-MacSpectrum reveals novel macrophage activation signatures in diseases. JCI Insight. 2019;5(10):e126453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Agostino RB Sr., Grundy S, Sullivan LM, Wilson P and Group CHDRP. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA. 2001;286:180–7. [DOI] [PubMed] [Google Scholar]
- 25.D’Agostino RB Sr., Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM and Kannel WB. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743–53. [DOI] [PubMed] [Google Scholar]
- 26.Goff DC Jr., Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Gibbons R, Greenland P, Lackland DT, Levy D, O’Donnell CJ, Robinson JG, Schwartz JS, Shero ST, Smith SC Jr., Sorlie P, Stone NJ, Wilson PW, Jordan HS, Nevo L, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC Jr., Tomaselli GF and American College of Cardiology/American Heart Association Task Force on Practice G. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S49–73. [DOI] [PubMed] [Google Scholar]
- 27.Chen L, Yao Q, Xu S, Wang H and Qu P. Inhibition of the NLRP3 inflammasome attenuates foam cell formation of THP-1 macrophages by suppressing ox-LDL uptake and promoting cholesterol efflux. Biochem Biophys Res Commun. 2018;495:382–387. [DOI] [PubMed] [Google Scholar]
- 28.Wang R, Wu W, Li W, Huang S, Li Z, Liu R, Shan Z, Zhang C, Li W and Wang S. Activation of NLRP3 Inflammasome Promotes Foam Cell Formation in Vascular Smooth Muscle Cells and Atherogenesis Via HMGB1. J Am Heart Assoc. 2018;7:e008596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aryal B, Rotllan N, Araldi E, Ramirez CM, He S, Chousterman BG, Fenn AM, Wanschel A, Madrigal-Matute J, Warrier N, Martin-Ventura JL, Swirski FK, Suarez Y and Fernandez-Hernando C. ANGPTL4 deficiency in haematopoietic cells promotes monocyte expansion and atherosclerosis progression. Nat Commun. 2016;7:12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, Robbins CS, Monaco C, Park I, McNamara CA, Binder CJ, Cybulsky MI, Scipione CA, Hedrick CC, Galkina EV, Kyaw T, Ghosheh Y, Dinh HQ and Ley K. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ Res. 2020;127:402–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, Raetz CR, Wang EW, Kelly SL, Sullards MC, Murphy RC, Merrill AH Jr., Brown HA, Dennis EA, Li AC, Ley K, Tsimikas S, Fahy E, Subramaniam S, Quehenberger O, Russell DW and Glass CK. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151:138–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiselev VY, Andrews TS and Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet. 2019;20:273–282. [DOI] [PubMed] [Google Scholar]
- 33.Stoger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E and de Winther MP. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–8. [DOI] [PubMed] [Google Scholar]
- 34.Seneviratne AN, Cole JE, Goddard ME, Park I, Mohri Z, Sansom S, Udalova I, Krams R and Monaco C. Low shear stress induces M1 macrophage polarization in murine thin-cap atherosclerotic plaques. J Mol Cell Cardiol. 2015;89:168–72. [DOI] [PubMed] [Google Scholar]
- 35.Fang S, Xu Y, Zhang Y, Tian J, Li J, Li Z, He Z, Chai R, Liu F, Zhang T, Yang S, Pei C, Liu X, Lin P, Xu H, Yu B, Li H and Sun B. Irgm1 promotes M1 but not M2 macrophage polarization in atherosclerosis pathogenesis and development. Atherosclerosis. 2016;251:282–290. [DOI] [PubMed] [Google Scholar]
- 36.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, Greenland P, Jacob DR Jr., Kronmal R, Liu K, Nelson JC, O’Leary D, Saad MF, Shea S, Szklo M and Tracy RP. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–81. [DOI] [PubMed] [Google Scholar]
- 37.Ong KL, Morris MJ, McClelland RL, Maniam J, Allison MA and Rye KA. Lipids, lipoprotein distribution and depressive symptoms: the Multi-Ethnic Study of Atherosclerosis. Transl Psychiatry. 2016;6:e962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeboah J, Folsom AR, Burke GL, Johnson C, Polak JF, Post W, Lima JA, Crouse JR and Herrington DM. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation. 2009;120:502–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To minimize the possibility of unintentionally sharing information that could be used to re-identify private information, data from this study are available from the following resources: monocyte transcriptome profiles of MESA participants are accessible through GEO Series accession number GSE5604716. The other datasets used in this study were accessed with the GEO Series accession number GSE9731018, GSE9794119, GSE11624020, GSE11623920, GSE4329221, or from FR-FCM-Z23S22 (https://figshare.com/s/c00d88b1b25ef0c5c788, DOI: 10.6084/m9.figshare.9206387). GTEx data were from gtexportal.org (dbGaP Accession phs000424.v8.p2).