

Abstract
Glucose metabolism comprises numerous amphibolic metabolites that provide precursors for not only the synthesis of cellular building blocks but also for ATP production. In this study, we tested how phosphofructokinase-1 (PFK1) activity controls the fate of glucose-derived carbon in murine hearts in vivo. PFK1 activity was regulated by cardiac-specific overexpression of kinase- or phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase transgenes in mice (termed GlycoLo or GlycoHi mice, respectively). Dietary delivery of 13C6-glucose to these mice, followed by deep network metabolic tracing, revealed that low rates of PFK1 activity promote selective routing of glucose-derived carbon to the purine synthesis pathway to form 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR). Consistent with a mechanism of physical channeling, we found multimeric protein complexes that contained phosphoribosylaminoimidazole carboxylase (PAICS)—an enzyme important for AICAR biosynthesis, as well as chaperone proteins such as Hsp90 and other metabolic enzymes. We also observed that PFK1 influenced glucose-derived carbon deposition in glycogen, but did not affect hexosamine biosynthetic pathway activity. These studies demonstrate the utility of deep network tracing to identify metabolic channeling and changes in biosynthetic pathway activity in the heart in vivo and present new potential mechanisms by which metabolic branchpoint reactions modulate biosynthetic pathways.
Keywords: Glycolysis, Metabolomics, Stable isotope, Anabolism, Metabolons, Channeling
Graphical Abstract
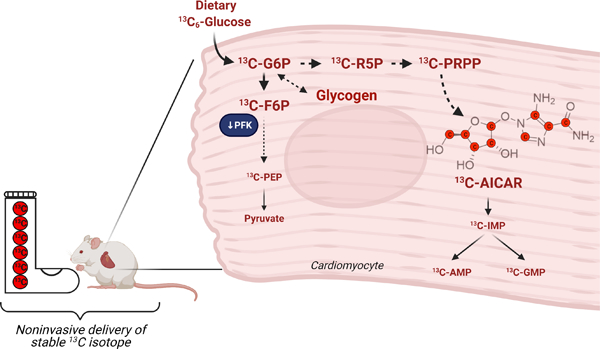
1. Introduction
Several enzymatic steps in glucose metabolism modulate cardiac structure and function. For example, recent studies indicate that lactate dehydrogenase [1,2], pyruvate dehydrogenase [3–5], and the mitochondrial pyruvate carrier [6–8] influence pathological or physiological remodeling of the heart. Phosphofructokinase-1 (PFK1) also influences cardiac remodeling. PFK1 activity is elevated in pressure overloaded hearts [9], and high PFK1 activity appears sufficient to promote mild dilated cardiomyopathy [10]. Conversely, cardiac PFK1 activity is lower during exercise and instigates physiologic cardiac growth [10]. PFK1 activity is modulated both by post-translational modifications and by allosteric effectors, most notably fructose-2,6-bisphosphate (F2,6BP), which is produced by isoforms of phosphofructokinase-2 (PFK2) [10]. Nevertheless, it remains unclear how changes in the activity of critical metabolic enzymes such as PFK1 coordinate energy conversion and anabolic activity.
The networks of metabolism provide conduits for directing nutrients toward multiple fates. Particularly important to nutrient fate are metabolic intermediates that convene at branchpoint sites and direct metabolites for catabolism or building block synthesis. For example, metabolites such as glucose 6-phosphate (G6P), fructose 6-phosphate (F6P), dihydroxyacetone phosphate (DHAP), and 3-phosphoglycerate (3PG) can remain in the glycolytic pathway for transformation to pyruvate, or they can enter into ancillary pathways such the pentose phosphate, hexosamine biosynthetic, glycerolipid, and serine synthesis pathways [11], some of which branch further to direct carbon flow to additional fates (Fig. 1). How these branchpoints are regulated in tissues such as the heart in vivo remains unclear.
Fig. 1.
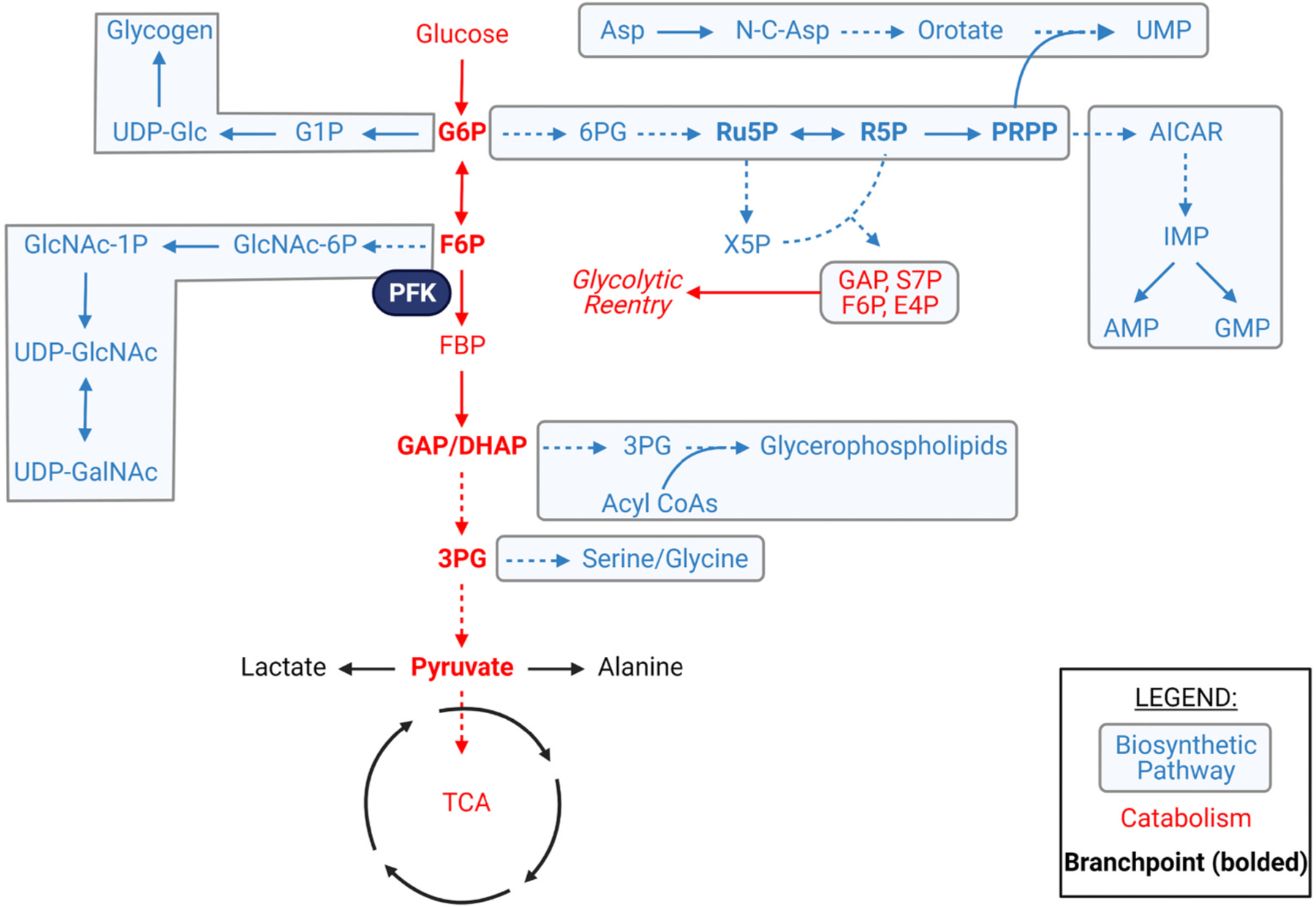
Glucose-derived amphibolic metabolites and biosynthetic pathway enrichment. Metabolic network map highlighting potential modes of glucose utilization and branchpoint intermediates (bolded) which could influence the balance between catabolism and anabolism within a cell. Asp = aspartate, UMP = uridine monophosphate, UDP = uridine diphosphate, Glc = glucose, G1P = glucose-1-phosphate, G6P = glucose-6-phosphate, 6PG = 6-phosphogluconate, Ru5P = ribulose-5-phosphate, R5P = ribose-5-phosphate, PRPP = phosphoribosyl pyrophosphate, AICAR = 5-aminoimidazole-4-carboxamide ribonucleotide, IMP = inosine monophosphate, GMP = guanosine monophosphate, AMP = adenosine monophosphate, F6P = fructose-6-phosphate, FBP = fructose-1,6-bisphosphate, PFK = phosphofructokinase, X5P = xylulose-5-phosphate, GAP = glyceraldehyde-3-phosphate, S7P = seduheptulose-7-phosphate, E4P = erythrose-4-phosphate, DHAP = dihydroxyacetone phosphate, 3PG = 3-phosphoglycerate, TCA = tricarboxylic acid cycle.
Much of our knowledge of the metabolic phenotype of the heart has arisen from isolated organ, cell, or organelle data. In the intact heart, most approaches to measure metabolism fail to provide detailed information on biosynthetic pathways. Furthermore, most in vivo 13C labeling approaches are transient in nature and require bolus injections, gavage, or continuous tracer infusion, which can produce unwanted artifacts or stress responses due to anesthesia or physical restraint [12]. Moreover, these labeling methods are often limited to fast turnover pathways such as glycolysis and the Krebs cycle and may not provide sufficient time for adequate labeling of biosynthetic pathways. Introduction of dietary 13C6-glucose, when coupled with mass spectrometry (MS) and nuclear magnetic resonance (NMR) analysis, can be used to trace the fate of glucose-derived carbon in relatively slower biosynthetic pathways such as the nucleotide and glycerophospholipid synthesis pathways [12,13].
Here, we report dietary administration of 13C-labeled glucose to examine ancillary biosynthetic pathways of glucose metabolism in transgenic mice bearing cardiac-specific isoforms of PFK2. These transgenes code for the bifunctional enzyme defective in either the kinase or phosphatase domains; when expressed, they increase or decrease F2,6BP levels, thereby augmenting or lowering PFK1 activity and glycolytic flux [10,14]. We found that phosphorylated sugars accumulate in mouse hearts with constitutively low PFK1 activity and are associated with elevated glycogen levels; however, hexosamine biosynthetic pathway activity is not affected by high levels of sugar phosphates. Interestingly, we found that low PFK1 activity diverts glucose-derived carbon toward synthesis of the purine biosynthetic pathway intermediate 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), which was associated with the presence of large multimeric complexes containing chaperones and metabolic enzymes, including those required for AICAR biosynthesis. Collectively, these findings illustrate the utility of deep network tracing for gaining new insights into the regulation of cardiac metabolism in vivo and provide evidence of PFK-regulated metabolic channeling in the heart.
2. Results
2.1. Standardization of liquid diet feeding
To examine the utility of in vivo deep network tracing for cardiac metabolic phenotyping, we first standardized dietary delivery of 13C6-glucose in adult C57BL/6J and FVB/NJ mouse strains, which are commonly used in biomedical research [15,16]. Although both strains of mice could consume up to ~20 g of this diet in a 24 h period, consumption of ~14 g of liquid diet was sufficient to maintain body weight in both strains (Supplementary Fig. 1A, 1B). We then placed individual mice and liquid diet in metabolic chambers to assess ambulatory activity and diet-induced changes in the respiratory exchange ratio (RER) (Supplementary Fig. 1C). Although strain-specific differences in ambulatory activity were identified, we found that the liquid diet did not significantly influence ambulatory activity (Supplementary Fig. 1D–F). As expected from the higher carbohydrate levels in liquid diet (Supplementary Table 1), the respiratory exchange ratio was higher in both mouse strains on liquid diet when compared with normal chow (Supplementary Fig. 1G). At the end of the feeding period, we found that the liquid diet did not significantly affect circulating free fatty acid or insulin levels compared with normal chow (Supplementary Fig. 1H, 1I).
2.2. PFK1 activity influences ancillary biosynthetic pathways that branch from G6P and F6P
After standardization of liquid diet delivery, we examined the metabolic fate of dietary 13C6-glucose in FVB mice that constitutively express kinase- or phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in the heart; these mice are termed GlycoLo or GlycoHi mice, respectively. By controlling levels of fructose-2,6-bisphosphate, expression of the GlycoLo or GlycoHi transgenes regulate the activity of cardiac phosphofructokinase [10,14,17,18]. GlycoLo and GlycoHi mice were fed the 13C6-containing liquid diet for 18 h, after which the hearts were freeze-clamped in situ. Metabolite extracts from excised heart tissue were then analyzed by NMR and ion exchange chromatography mass spectrometry (IC-MS) to quantify metabolite abundance and 13C metabolite enrichment (Fig. 2A). The genotypes consumed equivalent amounts of the 13C6-glucose labeled diet during the feeding protocol (Fig. 2B). Moreover, we found no differences in circulating glucose or lactate levels between the groups (Fig. 2C and D).
Fig. 2.
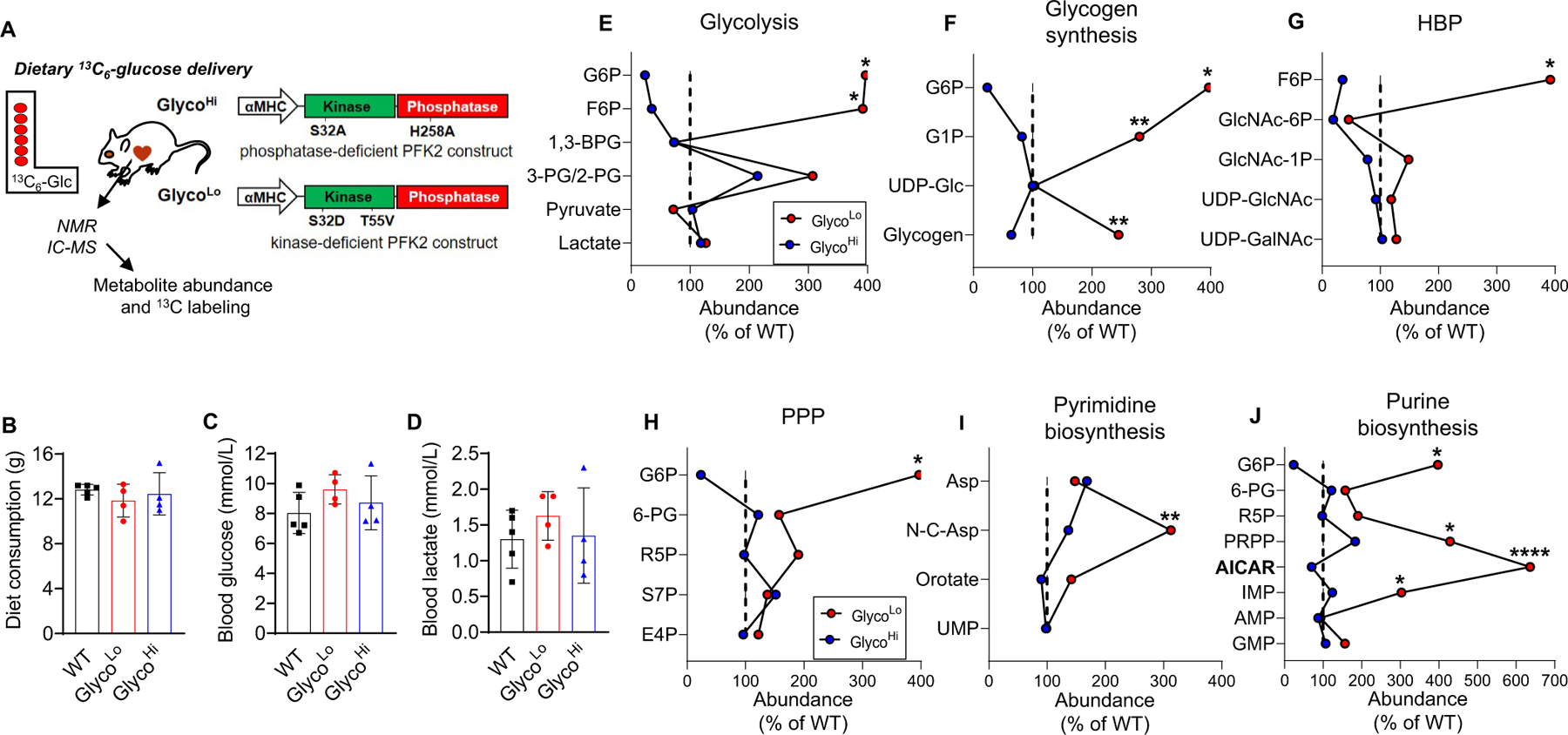
Effect of phosphofructokinase activity on the relative abundances of ancillary biosynthetic pathway metabolites. (A) Schematic of study design and transgenic mouse model. (B) Consumption of 13C6-glucose-containing liquid diet over the course of 18 h. (C) Circulating glucose levels after 18 h of liquid diet feeding. (D) Circulating lactate levels after 18 h of liquid diet feeding. (E–J) Crossover plots: Influence of cardiac phosphofructokinase activity on average relative abundances of metabolites in glycolysis, glycogen synthesis, the hexosamine biosynthetic pathway (HBP), the pentose phosphate pathway (PPP), and the pyrimidine and purine biosynthetic pathways. n = 4–5 hearts/group. *p < 0.05, **p < 0.01, ****p < 0.0001, one-way ANOVA with Tukey post-hoc test. Absolute abundances of each metabolite were used for statistical analysis.
Because PFK is expected to exert strong control over glycolytic metabolite levels [19], we first constructed modified cross-over plots of relative metabolite abundance. These plots have been used to assess the influence of effectors on metabolite levels in enzymatic reaction chains [20]; here, they are used to present metabolite data in graphical form to assess which metabolic pathways are most responsive to differences in PFK1 activity. High PFK activity in GlycoHi hearts reduced levels of G6P and F6P relative to WT littermates, whereas low PFK activity in GlycoLo hearts increased G6P and F6P (Fig. 2E); however, the levels of other branchpoint metabolites, such as 3PG and pyruvate were not different between GlycoLo and GlycoHi hearts.
Because G6P and F6P contribute to glycogen synthesis, the hexosamine biosynthetic pathway (HBP), the pentose phosphate pathway (PPP), and nucleotide biosynthesis pathways, we also constructed cross-over plots for these pathways. As predicted from our previous results [10], low PFK activity is associated with a higher abundance of glycogen synthetic metabolites such as glucose-1-phosphate (G1P) and glycogen (Fig. 2F); however, UDP-glucose levels were not different between the genotypes. Isotopologue analysis revealed higher levels of both unlabeled G1P (m + 0) and fully labeled 13C6-G1P (m + 6) in GlycoLo hearts (Fig. 3A,B). We next performed NMR spectral analysis as represented in Fig. 3C, which indicated higher levels of both unlabeled and 13C-labeled glycogen in GlycoLo hearts (Fig. 3D).
Fig. 3.
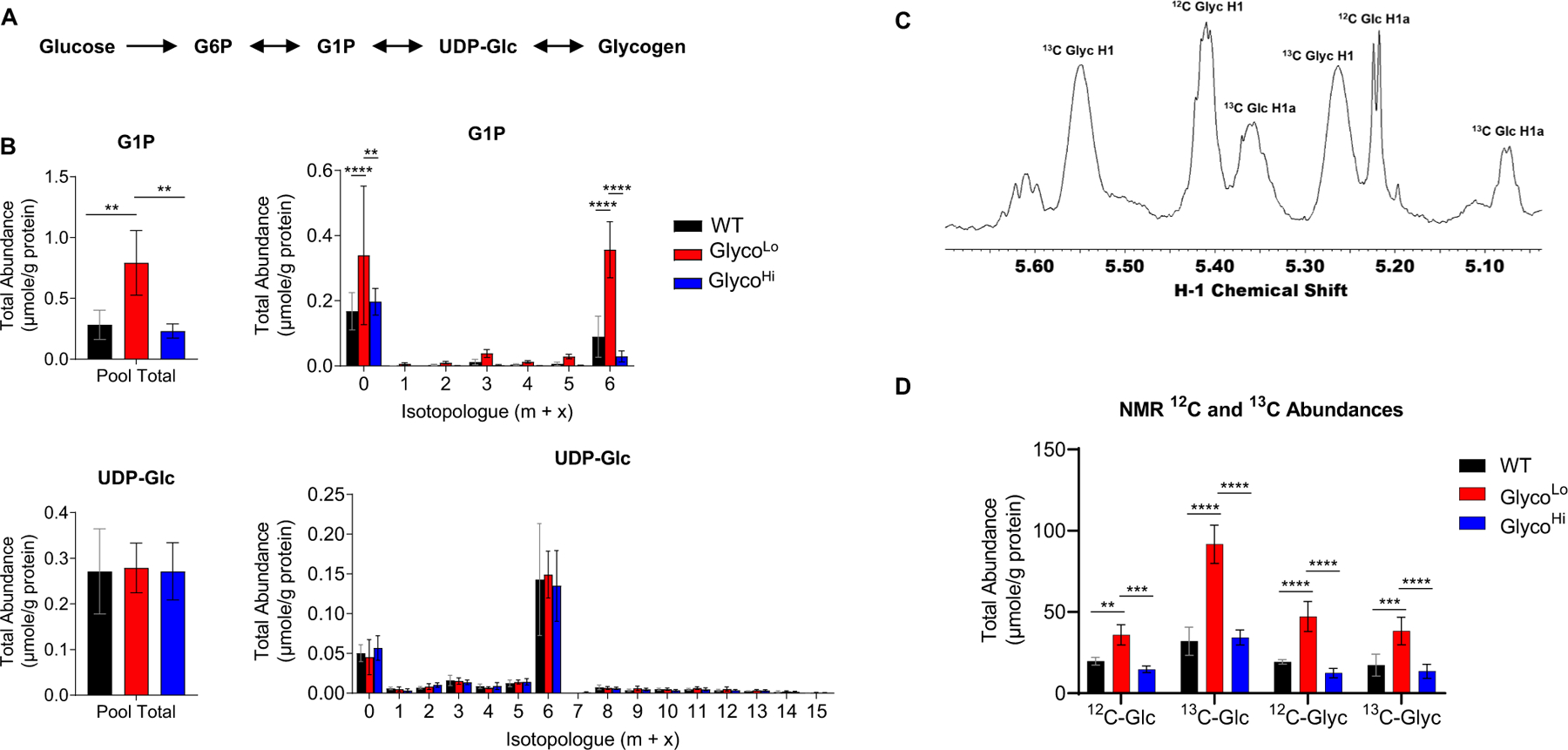
Low phosphofructokinase activity increases glycogen biosynthesis in the heart. (A) Schematic of glycogen biosynthesis pathway. (B) Pool totals and isotopologue abundances for glycogen intermediates. (C) Representative NMR trace for glucose (Glc) and glycogen (Glyc) in WT mouse heart. (D) Abundance of 12C- and 13C-labeled glucose (Glc) and glycogen (Glyc) in the heart. n = 4–5 hearts per group, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way (B) or two-way (D) ANOVA with Tukey’s multiple comparison test.
Although PFK activity influenced glycogen biosynthesis, PFK had little effect on HBP metabolite abundance (Fig. 2G). Despite modest increases in N-acetylglucosamine-1-phosphate (GlcNAc-1P) levels and 13C enrichment, overall abundance and 13C enrichment of UDP-GlcNAc and UDP-GalNAc were largely unaffected by differences in PFK activity (Fig. 4), which is consistent with recent studies suggesting that HBP flux is rather unresponsive to changes in glucose availability in the heart [21]. Similarly, cardiac PFK activity had relatively modest effects on PPP intermediates (Fig. 2H). Nevertheless, we found higher levels of metabolic intermediates in the pyrimidine and purine biosynthetic pathways in GlycoLo hearts (Fig. 2I–J). While the total abundances and 13C labeling of most pyrimidine intermediates and end products were only modestly enhanced by lower PFK activity, total abundance and isotopologue abundance plots showed marked changes in the total levels and 13C labeling of the pyrimidine precursor, carbamoyl aspartate (Fig. 5).
Fig. 4.
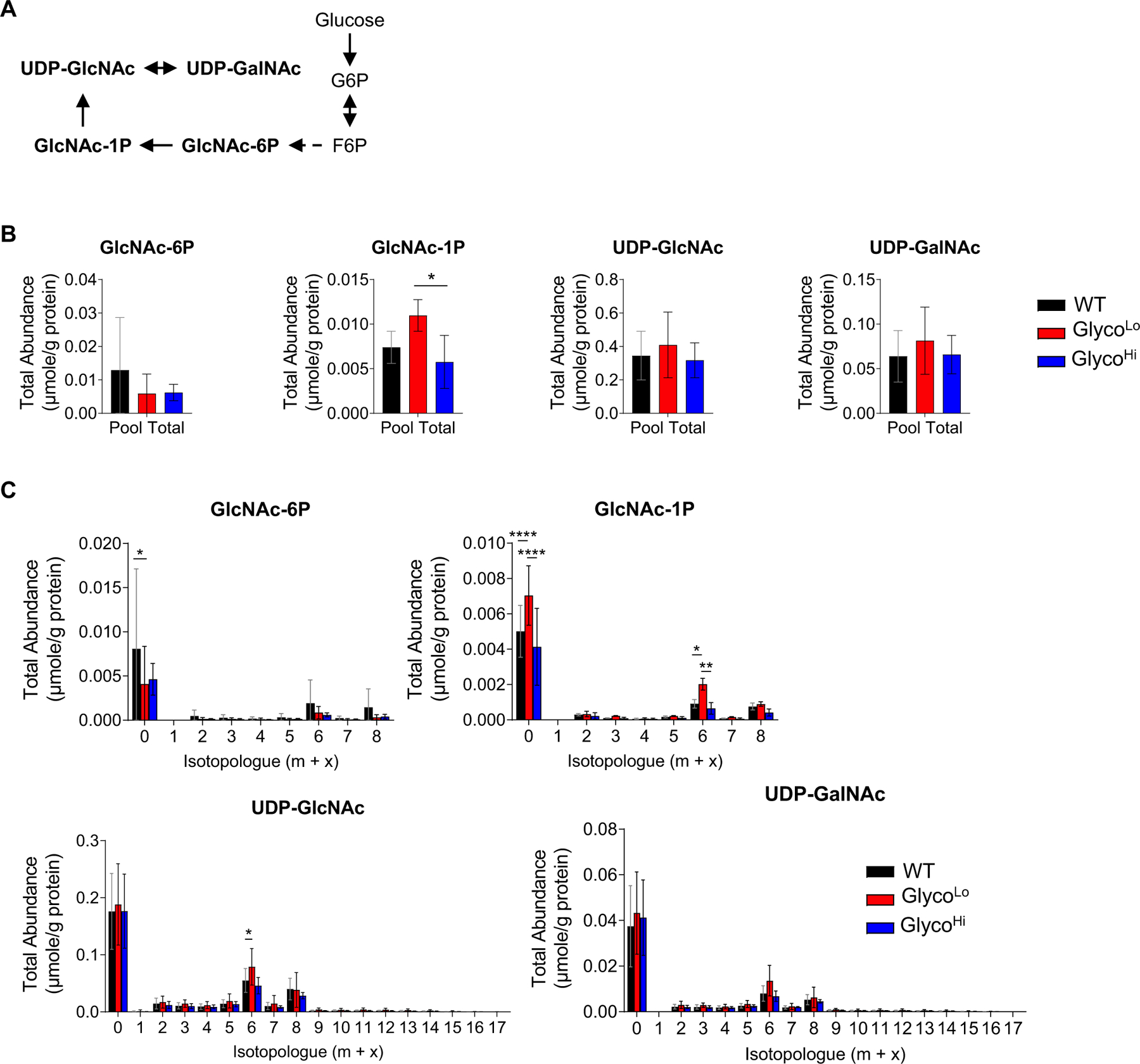
Phosphofructokinase has little effect on hexosamine biosynthetic pathway activity in the heart. (A) Schematic of hexosamine biosynthetic pathway. (B) Pool totals and (C) isotopologue abundances of hexosamine biosynthetic pathway intermediates in hearts of GlycoLo, GlycoHi, and WT hearts. *p < 0.05, **p < 0.01, ****p < 0.0001, (panel B) one-way ANOVA or (panel C) two-way ANOVA with Tukey’s multiple comparison test, n = 4–5 hearts per group.
Fig. 5.
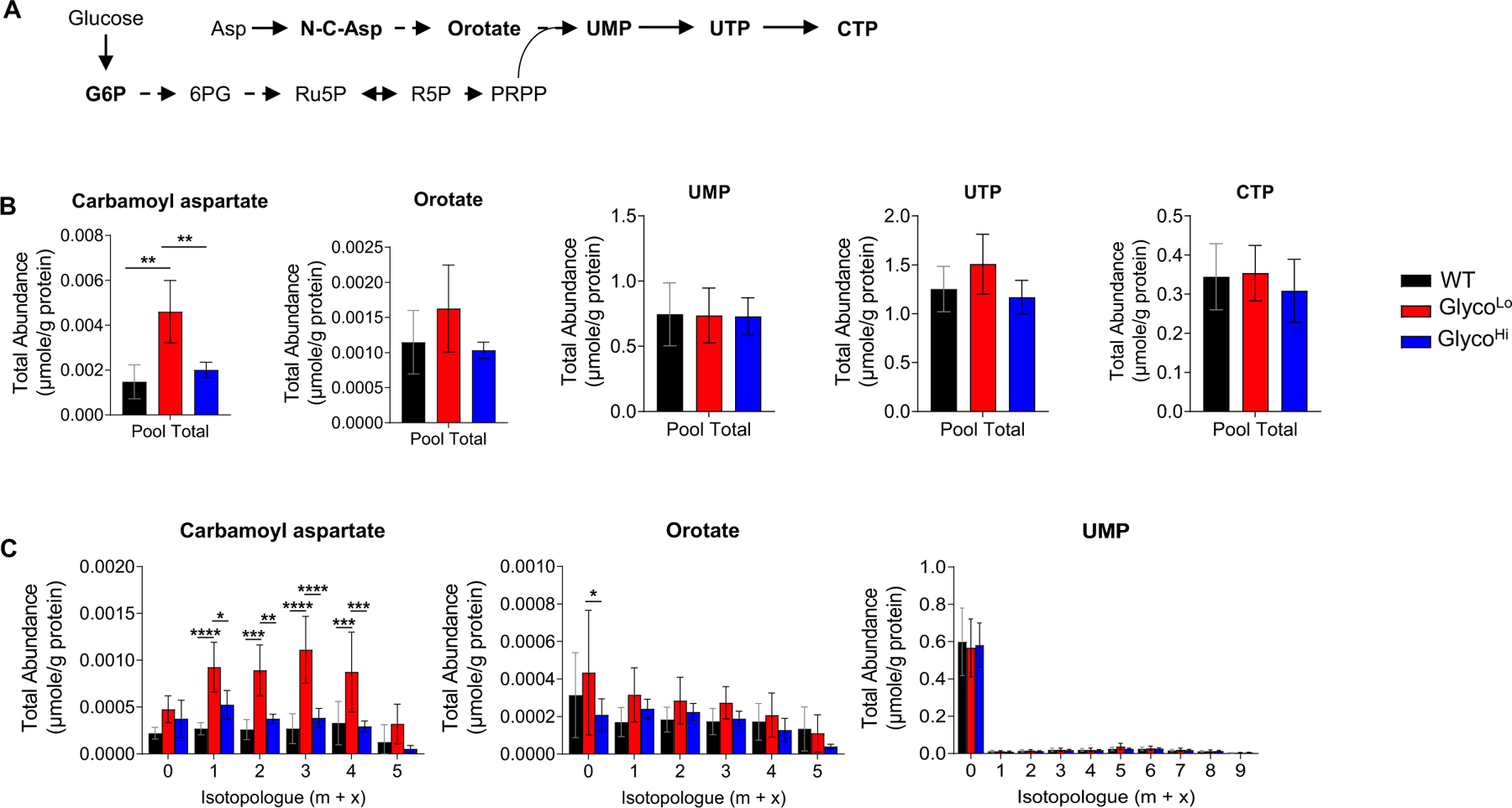
Influence of phosphofructokinase on pyrimidine biosynthesis in the heart. (A) Schematic of pyrimidine biosynthetic pathway. (B) Pool total and (C) isotopologue abundances of pyrimidine biosynthetic pathway metabolites in hearts of GlycoLo, WT, and GlycoHi mice. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, (panel B) one-way ANOVA or (panel C) two-way ANOVA with Tukey’s multiple comparison test, n = 4–5 hearts per group.
As shown in Fig. 6, analysis of purine biosynthetic pathway metabolites revealed marked changes in GlycoLo hearts. We found higher levels of unlabeled ribose 5-phosphate (m + 0), as well as higher levels of unlabeled and 13C5-phosphoribosyl pyrophosphate (PRPP) in GlycoLo hearts (Fig. 6A). Furthermore, isotopologue analysis suggests routing of glucose-derived carbon to the purine synthesis pathway, with marked increases in unlabeled and 13C-labeled 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR). However, the end products of the purine biosynthesis pathway (IMP, AMP, GMP) were much less affected by differences in PFK activity, and there were no differences in overall cardiac energy charge (WT, 0.73 ± 0.03; GlycoHi; 0.75 ± 0.04, GlycoLo, 0.77 ± 0.03; p = 0.22). In GlycoHi hearts, the abundances of unlabeled and 13C labeled AICAR were lower than that in WT and GlycoLo hearts.
Fig. 6.
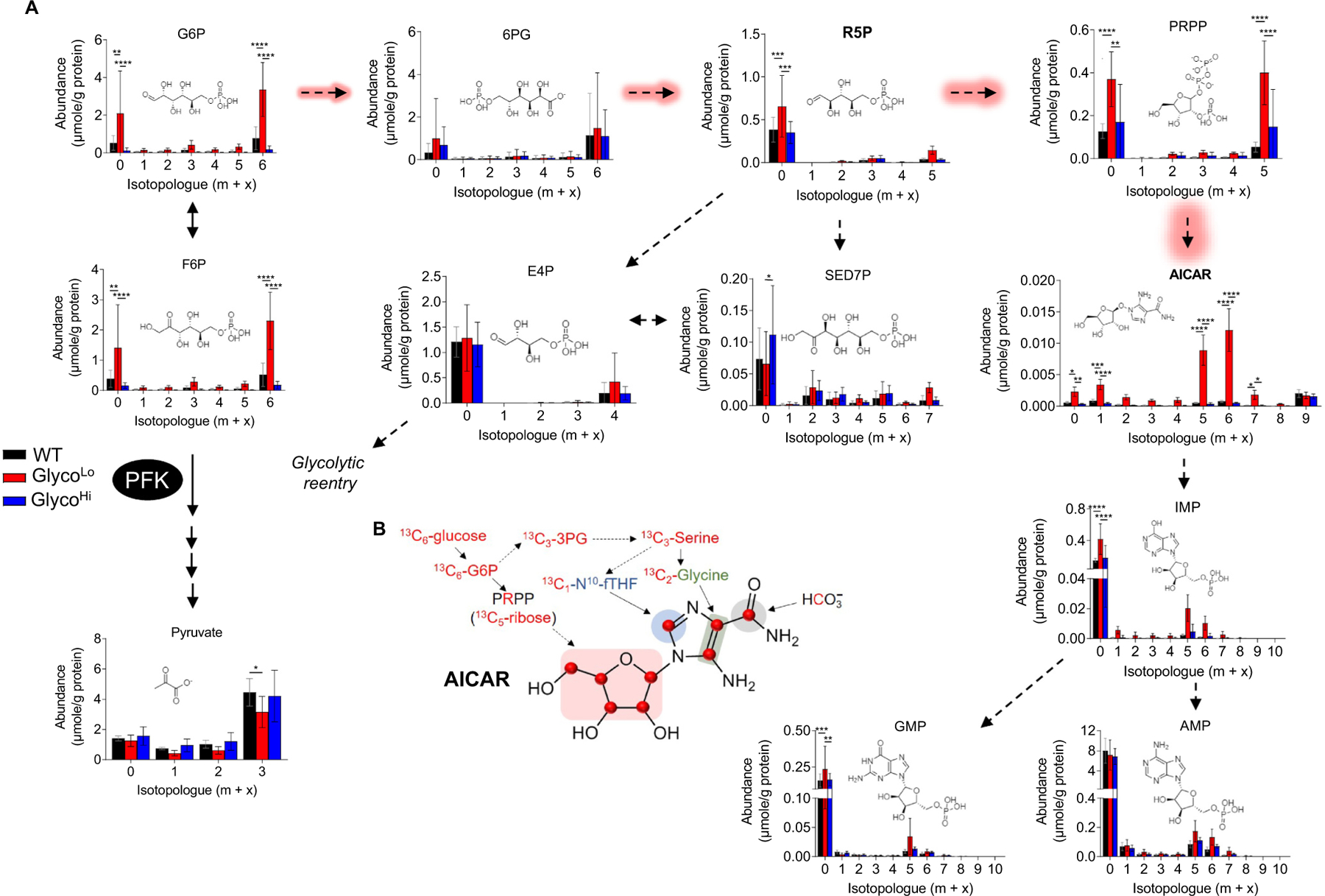
Low cardiac phosphofructokinase activity promotes channeling of glucose-derived carbon to form AICAR. (A) Metabolite isotopologue abundances for intermediates in the pentose phosphate and purine biosynthetic pathways in hearts of GlycoLo, WT, and GlycoHi mice. (B) Schematic of 13C6-glucose-derived carbon incorporation into AICAR biosynthesis. n = 4–5 hearts per group,*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA with Tukey’s multiple comparison test.
2.3. PFK activity influences cytosolic enzyme complexes in the heart
The accumulation of unlabeled and labeled R5P, PRPP, and AICAR caused by low PFK activity could be due to a decrease in AICAR utilization or an increase in its formation. Although many possibilities exist, findings from studies in cultured cells suggest the presence of cytosolic, multi-enzyme complexes (e.g., purinosomes) that could facilitate carbon channeling in the purine biosynthetic pathway [22,23]. To determine whether such complexes might exist in vivo and whether they could explain the preferential routing of glucose carbon to AICAR, we separated cytosolic cardiac proteins in their native state via BN-PAGE and identified protein complexes migrating to molecular masses between ~480–1000 kDa. As shown in Supplementary Fig. 2A, we identified at least seven bands, designated by Greek letters. Proteomics analysis of each band revealed that each complex contains structural, metabolic, and chaperone proteins, with metabolic proteins comprising a large proportion of the highly abundant proteins. To determine whether PFK activity influences these complexes, we isolated cytosolic fractions from WT, GlycoLo and GlycoHi hearts and examined gross changes in protein complex migration. Interestingly, samples from GlycoHi hearts showed molecular mass shifts in the β1 and β2 complexes (Fig. 7A). To see how the composition of these bands differed between the genotypes, we excised each band and identified proteins by mass spectrometry followed by iBAQ normalization and univariate or multivariate analyses. ANOVA results suggested the purine biosynthetic enzyme phosphoribosylaminoimidazole carboxylase (PAICS) to be the most significantly different constituent between the groups (Fig. 7B). To further ensure that PAICS is present in complexes at or near this size, we separated cardiac cytosolic complexes by size exclusion chromatography (SEC) and assessed PAICS in the fractions by immunoblotting. As shown in Supplementary Fig. 2B, PAICS was present in SEC fraction 2, which contained complexes between ~500–1000 kDa. Also markedly different by ANOVA were transgene-derived PFKFB1, as well as several metabolic enzymes involved in fatty acid metabolism (fatty acid synthase, FASN; ATP citrate lyase, ACLY; carnitine palmitoyl transferase 1b, CPT1B), in caveolae formation (caveolae associated protein 5, MURC; caveolae associated protein 1, PTRF), and in components of the chaperonin-containing T-complex (TCP1, CCT3, CCT6A, CCT7, CCT5) (Fig. 7B).
Fig. 7.
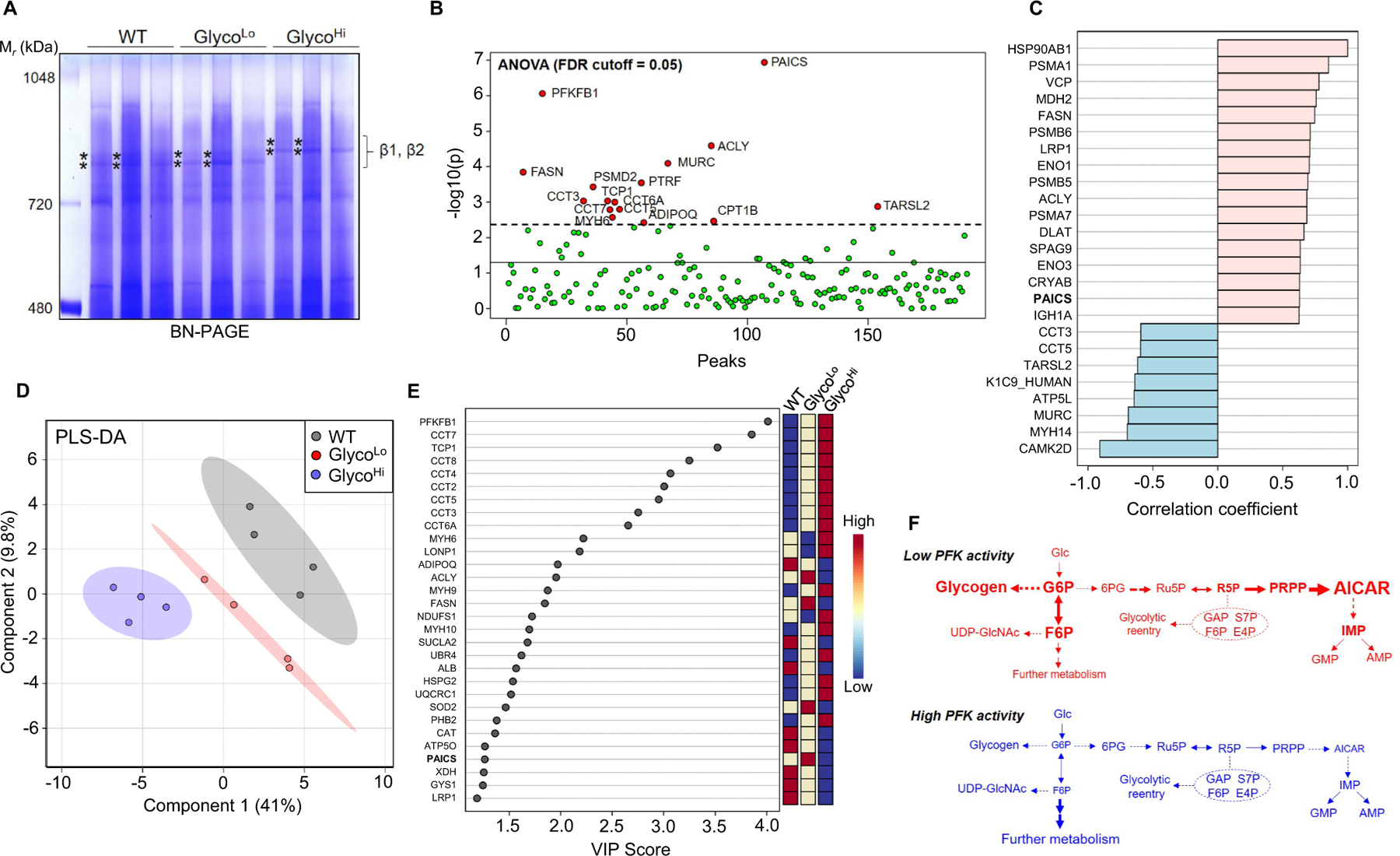
Phosphofructokinase coordinates metabolic complex assembly in the heart. (A) Blue Native (BN)-PAGE of cytosolic fractions from WT, GlycoLo, and GlycoHi hearts. Asterisks indicate bands of the β1 and β2 complexes that were excised for proteomic analyses. n = 3 hearts per group. (B) ANOVA analyses using iBAQ normalized protein abundances for β complex bands. n = 4 bands per genotype (2 hearts per group). (C) Correlation of protein abundances in the β complex with HSP90. (D) Partial least squares discriminant analysis (PLS-DA) of protein abundances in the β complex. (E) Variable importance in projection (VIP) scores showing the contribution of proteins in the β complexes to group separation in panel D. (F) Working model suggesting the role of phosphofructokinase in ancillary biosynthetic pathway activity in the heart.
Because chaperones such as HSP90 are required for purinosome formation [24,25], we next determined which proteins in the complexes were most strongly associated with HSP90. As shown in Fig. 7C, correlation analysis demonstrated high correlation coefficients for PAICS as well as several other cytosolic metabolic enzymes (FASN, ENO1, ACLY, ENO3), some mitochondrial and endoplasmic reticulum enzymes (MDH2, DLAT, VCP), chaperones (CRYAB), and components of the proteasome (PSMA1, PSMB6, PSMB5, PSMA7), among others (LRP1, SPAG9). Negatively correlating with HSP90 were components of the chaperonin-containing T-complex (CCT3, CCT5), TARSL2, MURC, MYH14, CAMK2D, and ATP5L, which suggests that these proteins were not associated with HSP90 in the protein complexes.
Given that PAICS in the β protein complexes was most significantly different between the genotypes and was associated strongly with HSP90, we next performed multivariate analyses to identify which protein constituents contributed most to phosphofructokinase-mediated differences in β complex composition. As shown in Fig. 7D and E, partial least square discriminant analysis followed by variable importance in projection (VIP) score analysis suggested PFKFB1 contributed most to group separation, which is not surprising given that PFKFB1 mutants comprise the overexpressed, mutant transgenes. Mirroring contribution by PFKFB1, several members of the T-complex were elevated in GlycoHi and GlycoLo hearts. Interestingly, PAICS also contributed largely to group separation, with higher levels of PAICS in GlycoLo hearts and lower levels in GlycoHi hearts; other metabolic enzymes such as FASN and ACLY paralleled the PAICS pattern. Collectively, these findings insinuate phosphofructokinase-driven compositional changes in multimeric protein complexes, which support the idea that metabolic channeling identified by deep network tracing in vivo could be due to the formation of protein assemblies that influence glucose-derived carbon fate.
3. Discussion
In this study, we delivered 13C-labeled glucose via diet to mice with low or high cardiac glycolytic activity and then performed in vivo deep network tracing to examine ancillary biosynthetic pathway activity. We found that the amphibolic metabolites G6P and F6P accumulated in hearts of mice with constitutively low PFK activity and were associated with elevated glycogen levels; however, HBP activity was relatively insensitive to differences in PFK activity. Interestingly, we found that PFK activity influences routing of glucose-derived carbon to the purine biosynthetic pathway intermediate, AICAR (Fig. 7F). These changes were associated with the presence of large multimeric complexes containing chaperones and metabolic enzymes, including PAICS, which is required for AICAR biosynthesis. Collectively, these studies illustrate the utility of deep network tracing for cardiac metabolic phenotyping and for developing new insights into the differential regulation of biosynthetic pathways in vivo.
Of practical importance, we found that both the C57 and FVB genetic mouse backgrounds consumed similar amounts of diet and that the diet did not significantly affect circulating levels of free fatty acids or insulin. These standardization studies were important because a preponderance of genetic mouse models is on these backgrounds [15,16]. Moreover, our studies suggest that deep network tracing using dietary 13C-glucose introduction can be used to measure biosynthetic pathway activity in the heart. Typically, fast turnover pathways such as glycolysis and the TCA cycle require relatively short durations of labeling to achieve isotopic steady state, whereas some biosynthetic pathways may take numerous hours or even days to see significant labeling and reach isotopic steady state [13,26]. For the purposes of assessing biosynthetic pathway activity in the heart in vivo, we strove to incorporate enough glucose-derived 13C into the biosynthetic pathway metabolite pools for confident isotopologue detection, while also staying within the dynamic phase of labeling, which allows assessment of relative biosynthetic pathway activity [13,26]. Indeed, in pilot studies comparing 18 h versus 36 h 13C-glucose feeding, we found that 36 h of feeding led to generally higher 13C labeling enrichment in intermediates and end products of ancillary biosynthetic pathways compared with the 18 h feeding group (Supplementary Fig. 3). Collectively, these findings indicate that 18 h of dietary 13C-glucose provision is sufficient to label biosynthetic pathway pools for adequate detection and to stay within the dynamic phase of labeling. Furthermore, the findings suggest that this duration of feeding should be adequate for analysis of biosynthetic pathway activity in other genetic models or in mouse models of heart failure or physiologic cardiac growth.
We chose to examine how PFK regulates biosynthetic pathways because of its relevance to glucose metabolism and because changes in its activity influence cardiac remodeling. The reaction catalyzed by PFK1 is a strongly exergonic, committed step in glycolysis [27,28] and appears to exert strong control on glycolytic flux [19]. Our findings suggest that PFK activity strongly influences the abundance of G6P and F6P, thereby controlling levels of precursors for several biosynthetic pathways, including the PPP, nucleotide biosynthetic pathways, glycogen synthesis, and the HBP. Consistent with the idea that low PFK activity reroutes glucose to glycogen synthesis [29–32], high levels of sugar phosphates in GlycoLo hearts were associated with accumulation of unlabeled and 13C-glycogen. While glycogen levels were influenced by levels of sugar phosphates, HBP activity was mostly unaffected by increased levels of G6P and F6P, which is consistent with recent studies showing that HBP flux is independent of changes in glucose availability [21]. This may be due to the multiple metabolic inputs required for UDP-GlcNAc synthesis, which effectively diminish PFK control over the pathway.
By tracking the fate of 13C through the PPP and nucleotide biosynthetic pathways, we found that low PFK activity was associated with increased glucose carbon partitioning into PRPP and AICAR. We interpret this metabolic phenotype as a form of metabolic channeling for several reasons. A general flooding paradigm of pathways by elevated G6P levels would predict more uniform 13C deposition in all possible pathways, including pentose phosphate products and the final nucleotide products; however, enhanced 13C labeling only became apparent at the amphibolic metabolite R5P and increased successively through PRPP and AICAR, while 13C labeling of the final nucleotide products was not altered. Thus, the occurrence of such selective partitioning of glucose-derived carbon downstream of metabolic branchpoints provides evidence of metabolic channeling.
It remains unclear if the form of channeling observed in this study fits the simple substrate channeling paradigm. The general paradigm of substrate channeling implicates stoichiometric assemblies of enzymes that catalyze sequential transfer of metabolic intermediates from one enzyme to the next without dilution of products in the bulk medium. Such substrate channeling has been proposed to promote pathway efficiency, regulate entry of intermediates into competing metabolic pathways, and provide a means to segregate and microcompartmentalize metabolites in the cell [33–36]. Consistent with the idea of spatial organization of metabolism, we found evidence of metabolic enzymes in high molecular weight complexes in cytosolic heart extracts. In particular, higher molecular weight α, β1, β2, and γ complexes contained numerous glycolytic enzymes, as well as fatty acid metabolism enzymes, and the lower molecular weight δ, ε, and ζ complexes showed generally higher levels of glycogen metabolism enzymes (e.g., GYS1, AGL, UGP2, PYGM) as well as enzymes important for nucleotide biosynthesis (e.g., PAICS, GOT1). We also found by proteomics that PAICS in β complexes was influenced by PFK activity and correlated strongly with the presence of the purinosome chaperone HSP90 [24,25]. In addition, some of the complexes also contained relatively high levels of myofibrillar components, the sarcoplasmic reticulum calcium ATPase, and mitochondrial proteins, indicating spatial coordination of energy consuming and providing processes in the heart. These data are consistent with the presence of higher order, quinary protein structures consisting of functionally related proteins, some of which respond to changes in PFK activity.
Although the channeling to AICAR, a known signaling molecule [37], observed by deep network tracing could occur via purinosome-like metabolons, several issues and limitations must be addressed for further elucidation. Metabolons found in mammalian systems, such as glucosomes [38–40] and purinosomes [22,23], are thought to be loosely associated and thus could dissociate during extraction or native separation. Although loose associations may help provide sensitivity for altering carbon fate in response to stimuli in vivo, it limits our ability to isolate an intact, active metabolon, which could otherwise be tested in vitro for confirmation of direct substrate channeling. Also, it is possible that direct channeling represents only one form of spatial regulation used to direct carbon flow in tissues. It is possible that metabolic enzymes could become sequestered or made unavailable to some metabolic conduits, resulting in accumulation of upstream metabolic intermediates. Nevertheless, our in vivo tracer and native separation experiments insinuate control over cardiac glucose-derived carbon allocation, dependent on phase-separated condensates of metabolic complexes. We propose that this is the first evidence of cytosolic metabolons in the heart, which have been previously inferred to exist from experimental results of in vitro and cultured cell studies [35,41–46].
Findings of this study are also important in light of the evidence that PFK activity influences cardiac remodeling. For example, low PFK activity during exercise appears to activate the cardiac growth transcriptional program [10], and high levels of PFK activity, such as that occurs in the pressure-overloaded heart [9], are sufficient to promote a deleterious form of remodeling [10]. Such metabolic regulation of cardiac remodeling may be due in part to the ability of PFK to influence biosynthetic pathways, which not only provide building blocks for myocyte structure, but also regulate redox state and cell signaling [11,47]. Understanding how PFK-mediated changes in biosynthetic pathways activity coordinate anabolic pathways and transcriptional programs to elicit cardiac remodeling remains an exciting challenge for future studies.
3.1. Study limitations
Several limitations of this study deserve mention. First, these studies used mice with constitutive overexpression of kinase- or phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase transgenes in the heart to regulate PFK1 activity. This creates a metabolic phenotype from birth and promotes metabolic inflexibility at the level of PFK1, which may not mimic the dynamic changes in PFK activity that occur with physiological or pathological stress [10]. Nevertheless, these studies reveal how different levels of PFK activity influence cardiac biosynthetic pathway activity, which may be important for understanding how even transient changes in PFK activity may influence biosynthetic pathways in other contexts. Also, because the heart is not composed exclusively of cardiomyocytes, it is possible that some of the results could be influenced by non-cardiomyocyte contributions to metabolism; however, the mutant transgenes used to modulate PFK1 activity are expressed only in cardiac myocytes (under the αMHC promoter), which suggests that the findings represent changes in predominantly cardiomyocytes. Second, although label scrambling can be a significant impediment in in vivo 13C labeling, our principal focus on anabolic pathways of glucose metabolism largely negates this issue because a primary endpoint was the net incorporation of glucose-derived 13C into biosynthetic pathway metabolites. Furthermore, because cardiac myocytes lack significant phosphoenolpyruvate carboxykinase activity [48], there is little concern of incorporation of 13C from circulating lactate or pyruvate, especially in those isotopologues in biosynthetic pathway metabolites that branch from G6P or F6P. Finally, although our deep network tracing studies demonstrate apparent metabolic channeling, identifying the exact composition of the complexes that contributed to the elevations in AICAR remains a difficult, yet exciting challenge.
4. Materials and methods
4.1. Resource table
| Reagent | Source | Catalog |
|---|---|---|
| Rabbit polyclonal Anti-PAICS antibody | Millipore Sigma | AV46049 (1:1000 dilution) |
| HRP-linked Anti-Rabbit antibody | Cell Signaling Technologies | #7074 (1:1000 dilution) |
| Basal diet base | Envigo | TD.150344.PWD |
| 12C6-glucose | Sigma Aldrich | #G7528 |
| 13C6-glucose | Sigma Aldrich | #389374 |
| Acetonitrile | ThermoFisher | A956-1 |
| Chloroform | Sigma Aldrich | 650498 |
| MOPS | Fisher | BP308 |
| Mannitol | VWR | BDH9248 |
| Sucrose | VWR | BDH9308 |
| EGTA | Sigma Aldrich | E4378 |
| Digitonin | Sigma Aldrich | 300410 |
| Coomassie Blue G250 | Fisher | BP100 |
| Polyacrylamide (acrylamide:bis-acrylamide 37.5:1) | BioRad | #1610158 |
| Tricine | Sigma Aldrich | T0377 |
| Bis-Tris | VWR | #0715 |
| Trypsin | Promega | V511A |
| DTT | Sigma Aldrich | D0632 |
| TEA-BC | Sigma Aldrich | T7408 |
| Iodoacetamide | Sigma Aldrich | I1149 |
| Formic Acid | Fisher | A117-50 |
| Acetonitrile | Fisher | A955-4 |
| LC-MS grade Water | Fisher | W6-4 |
| Free Fatty-Acid Quantitation Kit | Sigma Aldrich | MAK044-1KT |
| Rat/Mouse Insulin ELISA | Millipore Sigma | EZRMMI-13 K |
| 0.45 μm Regenerated Cellulose Syringe Filter | Fisher | F2504-7 |
| Equipment | Source | Catalog |
|
| ||
| Orbitrap Fusion Tribrid mass Tribrid mass spectrometer | ThermoFisher | https://www.thermofisher.com/order/catalog/product/IQLAAEGAAPFADBMBCX#/IQLAAEGAAPFADBMBCX |
| Dionex ICS 5000 | Dionex | https://www.thermofisher.com/us/en/home/industrial/chromatography/ion-chromatography-ic/ion-chromatography-systems.html |
| Orbitrap Elite - ETD mass spectrometer | ThermoFisher | https://assets.thermofisher.com/TFS-Assets/CMD/brochures/BR-30232-LC-MS-Orbitrap-Elite-BR30232-EN.pdf |
| Q Exactive HF Hybrid Quadrupole-Orbitrap Mass Spectrometer | ThermoFisher | https://assets.thermofisher.com/TFS-Assets/CMD/Specification-Sheets/PS-64048-LC-MS-Q-Exactive-HF-Orbitrap-PS64048-EN.pdf |
| MestReNova v.12.0.0 | Mestrelab Research S.L. | https://mestrelab.com/download/mnova/ |
| Agilent Varian DD2 spectrometer | Agilent | https://www.agilent.com/cs/library/slidepresentation/public/2.DD2console.pdf |
| Software | Source | Link |
|
| ||
| MetaboAnalyst 5.0 | MetaboAnalyst | https://www.metaboanalyst.ca/ |
| Prism 9.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Proteome Discoverer v1.4.1.114 | ThermoFisher | https://www.thermofisher.com |
| Xcalibur v2.2 | ThermoFisher | https://www.thermofisher.com |
| TraceFinder 3.3 | ThermoFisher | https://assets.thermofisher.com/TFS-Assets/CMD/manuals/Man-XCALI-97728-TraceFinder-33-User-Quan-ManXCALI97728-A-EN.pdf |
4.1.1. Mice
All procedures were approved by the Institutional Animal Care and Use Committee at the University of Louisville. For liquid diet standardization, adult, male FVB/6J and C57BL/6J mice (16–18 weeks of age) were used. For studies addressing the influence of PFK on metabolism, adult, male mice on the FVB background (22–24 weeks of age) overexpressing kinase- or phosphatase-deficient 6-phosphofructo-2-ki-nase/fructose-2,6-bisphosphatase transgenes in the heart (termed GlycoLo or GlycoHi mice, respectively) and wild-type (WT) littermates were used. All mice were kept on a 12 h:12 h light: dark cycle. Normal chow and water were provided ad libitum unless provided liquid diet for deep network tracing experiments. Upon completion of each experiment, mice were anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and mice were euthanized via excision of the heart. Following, tissues were harvested. These procedures are consistent with the AVMA Guidelines on Euthanasia.
4.2. Detailed methods
4.2.1. Stable isotope-resolved metabolomics (SIRM)
Powdered, basal diet base (TD.150344.PWD) was purchased from Envigo Teklad Diets (Madison, WI) and dissolved in water, with addition of the glucose source (12C6-glucose and 13C6-glucose purchased from Sigma Aldrich, #G7528 and #389374, respectively) to create liquid diet. For isotope tracing, adult, male GlycoLo and GlycoHi mice, alongside WT littermates, were provided ~20 g of 12C6-glucose-containing liquid diet (0.167 g glucose/g diet) per day over the course of 54 h to acclimate to feeding on liquid diet. Then, diet was replaced with 13C6-glucose-containing liquid diet (~20 g; 0.173 g glucose/g diet) for the final 18 h of feeding. Each mouse was singly housed during the liquid diet feeding protocol. Following the feeding protocol, mice were anesthetized with pentobarbital and euthanized. Hearts were freeze-clamped in situ using liquid N2-cooled Wollenberger tongs for analyses while additional tissues were snap-frozen in liquid N2. Frozen heart tissue was pulverized in liquid N2 into small particles, and 20 mg of each sample was extracted in acetonitrile/water/chloroform (V/V 2:1.5:1) to separate into polar, lipid, and protein fractions. Polar fractions were lyophilized and reconstituted in 30 μl nanopure water for analysis via Dionex ICS-5000 + ion chromatograph interfaced to Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). Peak areas were integrated and exported to Excel via TraceFinder 3.3 (Thermo) software package, and natural abundance was corrected for each isotopologue as described previously [49,50]. Fractional enrichment and total abundance (μmole/g protein) were calculated to quantify 13C enrichment.
4.2.2. Nuclear magnetic resonance
The polar extracts were additionally analyzed via NMR spectroscopy. NMR spectra were recorded at 15 °C on 14.1 T on a Varian DD2 spectrometer equipped with a 3 mm inverse triple resonance (HCN) cold probe as previously described [51]. 1H PRESAT spectra were recorded with an acquisition time of 2 s with a relaxation delay of 4 s, during which a weak transmitter pulse was applied to suppress the residual HDO resonance. 1H[13C] HSQC spectra were recorded with an acquisition time of 0.25 s and a relaxation delay of 1.75 s, with GARP decoupling during the detection period. Free induction decays were transformed using MNOVA software with zero-filling to 128 k points (PRESAT) or 8 k points (HSQC), anodizing with a cosine-squared function and a 1 Hz (PRESAT) or 4 Hz (HSQC) line broadening exponential function. Spectra were phased, baseline corrected using third order Bernstein polynomials, and referenced to internal DSS-d6 (27.5 nmol) at 0 ppm. Resonances of individual metabolites were integrated using mixed Lorentzian-Gaussian line fitting, and the areas were normalized to the DSS resonance. This normalized value was then corrected for the amount of material in the extract and further normalized to the tissue protein level. Isotopomer levels, F, were calculated as: F = A(13C)/[A (13C) + A(12C)] where A(13C), A(12C) are the areas of the protons attached to 13C or 12C, respectively.
4.2.3. Circulating substrate measurements
Blood samples for circulating glucose and lactate were obtained via tail clip and measured in about 0.7 μl blood using the Accu-Check Aviva meter (Roche) and the Lactate Plus meter (Nova Biomedical), respectively.
4.2.4. Indirect calorimetry
Respiratory exchange ratio (VCO2/VO2) and locomotion, ambulatory and fine movements were measured using a metabolic cage system (TSE PhenoMaster System, Bad Homberg, Germany). Mice were acclimated to metabolic cage system and then monitored for 24 h while consuming either normal chow or liquid diet.
4.2.5. Blue-native PAGE separation of protein complexes
Multimeric enzyme complexes were separated in their native state using Blue Native PAGE (BN-PAGE). Briefly, hearts were homogenized in 5 mM MOPS buffer containing 220 mM mannitol, 70 mM sucrose, and 1 mM EGTA (pH 7.2) using a Teflon-coated Glass-Col homogenizer. The homogenate was then centrifuged at 10,000g for 10 min. To the supernatant, we added digitonin to give a detergent/protein ratio of 8:1 (gram:gram) and incubated the samples on ice for 20 min. Following centrifugation at 14,000g for 20 min, we added Coomassie dye for protein separation by BN-PAGE. The BN-PAGE gels were prepared using polyacrylamide (acrylamide:bis-acrylamide, 37.5:1) at either a 5–15% or a 3–12% gradient. The cathode buffers contained 50 mM Tricine, 15 mM Bis-Tris, pH 7.0, and 0.02% Coomassie Blue G250. The anode buffer consisted of 50 mM Bis-Tris pH 7.0. Electrophoresis was performed at 4 °C using high blue buffer at 100 V for 1 h, followed by low blue buffer at 250 V for 1.5 h.
4.2.6. Size exclusion chromatography (SEC)
SEC was carried out at room temperature on a Sepharose 6 Increase 10/300 column (GE Healthcare) equilibrated with PBS at a flow rate of 0.5 ml/min. The column was calibrated using both low and high molecular weight calibration kits (GE Healthcare) by plotting molecular weights of standards versus their relative elution volume. Elution position was determined by measuring the absorbance of the eluted fractions at 280 nm. Eluted peaks were concentrated approximately 10-fold on Amicon Ultra-4 spin columns. Following SEC separation, the fractions were analyzed either by Blue Native PAGE or by immunblotting following SDS-PAGE. For the latter, the fractions were loaded onto a denaturing SDS-PAGE gel and the proteins were transferred to PVDF membranes. An anti-PAICS polyclonal antibody (Millipore Sigma, AV46049) was used as the 1° antibody (1:1000 dilution). A streptavidin-linked anti-rabbit 2°antibodies (1:1000 dilution) was used to detect PAICS immunoreactivity.
4.2.7. Mass spectrometric protein identification
Protein excised from BN-PAGE gels were identified by liquid chromatograph (LC) ESI MS/MS, after the in-gel trypsin digestion as previously described with modifications [52]. For in-gel digestion, Coomassie-stained BN-PAGE gel bands were cut into 1-mm3 plugs and incubated in 100 mM triethylammonium bicarbonate (TEA-BC; Sigma) at room temperature for 15 min. Acetonitrile was then added to the TEA-BC solution, and the gel plugs were incubated at room temperature for 15 min with gentle vortexing. The solvent was removed, and the washing process was repeated until the Coomassie Blue stain was no longer visible. Solvent was removed, and the gel plugs were dried in a SpeedVac for 5 min. The dried plugs were incubated in DTT (20 mM DTT, 100 mM TEA-BC) at 56 °C for 45 min, followed by iodoacetamide (55 mM iodoacetamide, 100 mM TEA-BC) at room temperature for 30 min. Iodoacetamide was removed, and gels were washed in 50 mM TEA-BC at room temperature for 15 min, followed by gentle vortexing in acetonitrile for 15 min at room temperature. The gel plugs were again dried for 5 min in a SpeedVac and incubated in digestion buffer [20 ng/μl modified Trypsin (Promega) in 50 mM TEA-BC] for 10 min. Then, 50 mM TEA-BC was added to the plugs, followed by 37 °C overnight incubation in a shaker. Peptides were extracted from the gel pieces [53] and the digest was diluted with one volume of LC-MS grade water, four volumes of 1:2 5% v/v formic acid in water:acetonitrile, and incubated for 15 min in a shaker at 100 RPM. The extract was dried in a SpeedVac, dissolved in 20 μl 2% v/v acetonitrile/0.1% v/v formic acid, and filtered through a 0.45 μm regenerated cellulose syringe filter (ThermoFisher); 4 μl were used for analysis.
For protein identification, peptides were analyzed using an Easy-nLC 1000 system and an Orbitrap Elite - ETD mass spectrometer (ThermoFisher Scientific, Waltham, MA) with 1.6 kV of spray voltage and 225 °C at the heated desolvation capillary. A 100 μm ID fused silica needle tip packed in-house with 12 cm of Aeris Peptide 3.6 μm XB-C18 material (Phenomenex, Torrance, CA) was used with the EASY-nLC 1000. Samples were separated with a 45 min linear gradient from 0% B (100% A) to 50% B at 250 nl/min, followed by a 5 min linear gradient from 50% B to 95% B and linear flow ramp from 250 to 300 nl/min, and a 10 min wash with 95% B at 300 nl/min. Solvent A = A 2% v/v acetonitrile / 0.1% v/v formic and B = 80% v/v acetonitrile / 0.1% v/v formic acid. A Nanospray Flex source (ThermoFisher) was used to introduce sample to the Orbitrap Elite. An Nth Order Double Play method was created in Xcalibur v2.2 (ThermoFisher) to acquire data with the Elite. Scan event one obtained an FTMS MS1 scan (normal mass range; 240,000 resolution, full scan type, positive polarity, profile data type) for the range 300–2000 m/z. Scan event two obtained ITMS MS2 scans (normal mass range, rapid scan rate, centroid data type) on up to twenty peaks with a minimum signal threshold of 5000 counts from scan event one. The lock mass option was enabled (0% lock mass abundance) using the 371.101236 m/z polysiloxane peak. The data was analyzed in Proteome Discoverer v1.4.1.114 (ThermoFisher) using Mascot v2.5.1 and SequestHT and the 2/6/2017 version of the UniprotKB Mus musculus reference proteome canonical and isoform sequences (Proteome ID UP000000589). The enzyme specified was trypsin (maximum two missed cleavages; inhibition by P) with Carbamidomethyl(C) as static and Oxidation(M) as dynamic modifications. Tolerances were 1.0 Da (monoisotopic) for fragments and 50 ppm (monoisotopic) for parent masses. The result files from Proteome Discoverer were loaded into Scaffold Q + S v4.4.5. The false discovery rate was calculated using the Scaffold Local FDR and Protein Prophet algorithms. Peptides were accepted if the identification had probability greater than 99.9% and parent mass error within 2 ppm. Proteins were accepted if they had a probability greater than 99.9% and at least one peptide. Proteins were grouped into clusters to satisfy the parsimony principle. Following iBAQ normalization, proteins in selected bands were subjected to univariate and multivariate tests.
4.3. Statistical analyses
No data were excluded for statistical purposes. Differences in pool total abundances and isotopologue total abundances were calculated using one-way or two-way ANOVA, respectively, with Tukey’s multiple comparison. For examining protein complexes, ANOVA was used with an FDR cutoff of 0.05. Correlation coefficients, PLS-DA analysis, and VIP score analyses were assessed using Metaboanalyst software. Analyses were performed using GraphPad Prism 9 (La Jolla, CA, USA) and significance was assigned when p < 0.05.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the NIH (HL154663, HL147844, HL130174, HL078825, GM127607, and S10OD020106). The SIRM analysis was additionally supported by NIH 1U24DK097215-01A1 and Shared Resource(s) of the University of Kentucky Markey Cancer Center P30CA177558. We thank Andrew N. Lane, Rick Higashi, Xiang Zhang, and Steven P. Jones for their insightful contributions. The Graphical Abstract was created with BioRender.com.
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2021.08.013.
References
- [1].Cluntun AA, Badolia R, Lettlova S, Parnell KM, Shankar TS, Diakos NA, Olson KA, Taleb I, Tatum SM, Berg JA, Cunningham CN, Van Ry T, Bott AJ, Krokidi AT, Fogarty S, Skedros S, Swiatek WI, Yu X, Luo B, Merx S, Navankasattusas S, Cox JE, Ducker GS, Holland WL, McKellar SH, Rutter J, Drakos SG, The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure, Cell Metab 33 (3) (2020) 629–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dai C, Li Q, May HI, Li C, Zhang G, Sharma G, Sherry AD, Malloy CR, Khemtong C, Zhang Y, Deng Y, Gillette TG, Xu J, Scadden DT, Wang ZV, Lactate dehydrogenase a governs cardiac hypertrophic growth in response to hemodynamic stress, Cell Rep 32 (9) (2020) 108087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liu LX, Rowe GC, Yang S, Li J, Damilano F, Chan MC, Lu W, Jang C, Wada S, Morley M, Hesse M, Fleischmann BK, Rabinowitz JD, Das S, Rosenzweig A, Arany Z, PDK4 inhibits cardiac pyruvate oxidation in late pregnancy, Circ. Res 121 (12) (2017) 1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Atherton HJ, Dodd MS, Heather LC, Schroeder MA, Griffin JL, Radda GK, Clarke K, Tyler DJ, Role of pyruvate dehydrogenase inhibition in the development of hypertrophy in the hyperthyroid rat heart: a combined magnetic resonance imaging and hyperpolarized magnetic resonance spectroscopy study, Circulation 123 (22) (2011) 2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bogh N, Hansen ESS, Omann C, Lindhardt J, Nielsen PM, Stephenson RS, Laustsen C, Hjortdal VE, Agger P, Increasing carbohydrate oxidation improves contractile reserves and prevents hypertrophy in porcine right heart failure, Sci. Rep 10 (1) (2020) 8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang Y, Taufalele PV, Cochran JD, Robillard-Frayne I, Marx JM, Soto J, Rauckhorst AJ, Tayyari F, Pewa AD, Gray LR, Teesch LM, Puchalska P, Funari TR, McGlauflin R, Zimmerman K, Kutschke WJ, Cassier T, Hitchcock S, Lin K, Kato KM, Stueve JL, Haff L, Weiss RM, Cox JE, Rutter J, Taylor EB, Crawford PA, Lewandowski ED, Des Rosiers C, Abel ED, Mitochondrial pyruvate carriers are required for myocardial stress adaptation, Nat. Metab 2 (11) (2020) 1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McCommis KS, Kovacs A, Weinheimer CJ, Shew TM, Koves TR, Ilkayeva OR, Kamm DR, Pyles KD, King MT, Veech RL, DeBosch BJ, Muoio DM, Gross RW, Finck BN, Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice, Nat. Metab 2 (11) (2020) 1232–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fernandez-Caggiano M, Kamynina A, Francois AA, Prysyazhna O, Eykyn TR, Krasemann S, Crespo-Leiro MG, Vieites MG, Bianchi K, Morales V, Domenech N, Eaton P, Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy, Nat. Metab 2 (11) (2020) 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, Tian R, Mechanisms for increased glycolysis in the hypertrophied rat heart, Hypertension 44 (5) (2004) 662–667. [DOI] [PubMed] [Google Scholar]
- [10].Gibb AA, Epstein PN, Uchida S, Zheng Y, McNally LA, Obal D, Katragadda K, Trainor P, Conklin DJ, Brittian KR, Tseng MT, Wang J, Jones SP, Bhatnagar A, Hill BG, Exercise-induced changes in glucose metabolism promote physiological cardiac growth, Circulation 136 (22) (2017) 2144–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gibb AA, Hill BG, Metabolic coordination of physiological and pathological cardiac remodeling, Circ. Res 123 (1) (2018) 107–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun RC, Fan TW, Deng P, Higashi RM, Lane AN, Le AT, Scott TL, Sun Q, Warmoes MO, Yang Y, Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing, Nat. Commun 8 (1) (2017) 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lorkiewicz PK, Gibb AA, Rood BR, He L, Zheng Y, Clem BF, Zhang X, Hill BG, Integration of flux measurements and pharmacological controls to optimize stable isotope-resolved metabolomics workflows and interpretation, Sci. Rep 9 (1) (2019) 13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X, Bhatnagar A, Jones SP, Hill BG, Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes, Biochem. J 474 (16) (2017) 2785–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Taketo M, Schroeder AC, Mobraaten LE, Gunning KB, Hanten G, Fox RR, Roderick TH, Stewart CL, Lilly F, Hansen CT, et al. , FVB/N: an inbred mouse strain preferable for transgenic analyses, Proc. Natl. Acad. Sci. U. S. A 88 (6) (1991) 2065–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Battey J, Jordan E, Cox D, Dove W, An action plan for mouse genomics, Nat. Genet 21 (1) (1999) 73–75. [DOI] [PubMed] [Google Scholar]
- [17].Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN, Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity, J. Biol. Chem 279 (46) (2004) 48085–48090. [DOI] [PubMed] [Google Scholar]
- [18].Wang Q, Donthi RV, Wang J, Lange AJ, Watson LJ, Jones SP, Epstein PN, Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia, Am. J. Physiol. Heart Circ. Physiol 294 (6) (2008) H2889–H2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cortassa S, Caceres V, Bell LN, O’Rourke B, Paolocci N, Aon MA, From metabolomics to fluxomics: a computational procedure to translate metabolite profiles into metabolic fluxes, Biophys. J 108 (1) (2015) 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ichihara K, Abiko Y, Crossover plot study of glycolytic intermediates in the ischemic canine heart, Jpn. Heart J 23 (5) (1982) 817–828. [DOI] [PubMed] [Google Scholar]
- [21].Olson AK, Bouchard B, Zhu WZ, Chatham JC, Des Rosiers C, First characterization of glucose flux through the hexosamine biosynthesis pathway (HBP) in ex vivo mouse heart, J. Biol. Chem 295 (7) (2020) 2018–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].An S, Kumar R, Sheets ED, Benkovic SJ, Reversible compartmentalization of de novo purine biosynthetic complexes in living cells, Science 320 (5872) (2008) 103–106. [DOI] [PubMed] [Google Scholar]
- [23].Pareek V, Tian H, Winograd N, Benkovic SJ, Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells, Science 368 (6488) (2020) 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].French JB, Zhao H, An S, Niessen S, Deng Y, Cravatt BF, Benkovic SJ, Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome, Proc. Natl. Acad. Sci. U. S. A 110 (7) (2013) 2528–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pedley AM, Karras GI, Zhang X, Lindquist S, Benkovic SJ, Role of HSP90 in the regulation of de novo purine biosynthesis, Biochemistry 57 (23) (2018) 3217–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jang C, Chen L, Rabinowitz JD, Metabolomics and isotope tracing, Cell 173 (4) (2018) 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mor I, Cheung EC, Vousden KH, Control of glycolysis through regulation of PFK1: old friends and recent additions, Cold Spring Harb. Symp. Quant. Biol 76 (2011) 211–216. [DOI] [PubMed] [Google Scholar]
- [28].Yalcin A, Telang S, Clem B, Chesney J, Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer, Exp. Mol. Pathol 86 (3) (2009) 174–179. [DOI] [PubMed] [Google Scholar]
- [29].Hue L, Taegtmeyer H, The Randle cycle revisited: a new head for an old hat, Am. J. Physiol. Endocrinol. Metab 297 (3) (2009) E578–E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kemppainen J, Fujimoto T, Kalliokoski KK, Viljanen T, Nuutila P, Knuuti J, Myocardial and skeletal muscle glucose uptake during exercise in humans, J. Physiol 542 (Pt 2) (2002) 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Depre C, Ponchaut S, Deprez J, Maisin L, Hue L, Cyclic AMP suppresses the inhibition of glycolysis by alternative oxidizable substrates in the heart, J. Clin. Invest 101 (2) (1998) 390–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Depre C, Rider MH, Veitch K, Hue L, Role of fructose 2,6-bisphosphate in the control of heart glycolysis, J. Biol. Chem 268 (18) (1993) 13274–13279. [PubMed] [Google Scholar]
- [33].Ovadi J, Physiological significance of metabolic channelling, J. Theor. Biol 152 (1) (1991) 1–22. [PubMed] [Google Scholar]
- [34].Miles EW, Rhee S, Davies DR, The molecular basis of substrate channeling, J. Biol. Chem 274 (18) (1999) 12193–12196. [DOI] [PubMed] [Google Scholar]
- [35].Pedley AM, Benkovic SJ, A new view into the regulation of purine metabolism: the Purinosome, Trends Biochem. Sci 42 (2) (2017) 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fernie AR, Zhang Y, Sweetlove LJ, Passing the baton: substrate channelling in respiratory metabolism, Research (Wash D C) 2018 (2018) 1539325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Visnjic D, Lalic H, Dembitz V, Tomic B, Smoljo T, AICAr, a widely used AMPK activator with important AMPK-independent effects: a systematic review, Cells 10 (5) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Puchulu-Campanella E, Chu H, Anstee DJ, Galan JA, Tao WA, Low PS, Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane, J. Biol. Chem 288 (2) (2013) 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Araiza-Olivera D, Chiquete-Felix N, Rosas-Lemus M, Sampedro JG, Pena A, Mujica A, Uribe-Carvajal S, A glycolytic metabolon in Saccharomyces cerevisiae is stabilized by F-actin, FEBS J 280 (16) (2013) 3887–3905. [DOI] [PubMed] [Google Scholar]
- [40].Jang S, Nelson JC, Bend EG, Rodriguez-Laureano L, Tueros FG, Cartagenova L, Underwood K, Jorgensen EM, Colon-Ramos DA, Glycolytic enzymes localize to synapses under energy stress to support synaptic function, Neuron 90 (2) (2016) 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhao X, Palacci H, Yadav V, Spiering MM, Gilson MK, Butler PJ, Hess H, Benkovic SJ, Sen A, Substrate-driven chemotactic assembly in an enzyme cascade, Nat. Chem 10 (3) (2018) 311–317. [DOI] [PubMed] [Google Scholar]
- [42].Sweetlove LJ, Fernie AR, The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation, Nat. Commun 9 (1) (2018) 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wu F, Minteer SD, Tricarboxylic acid metabolon, Methods Enzymol 617 (2019) 29–43. [DOI] [PubMed] [Google Scholar]
- [44].Jung T, Mack M, Interaction of enzymes of the tricarboxylic acid cycle in Bacillus subtilis and Escherichia coli: a comparative study, FEMS Microbiol. Lett 365 (8) (2018). [DOI] [PubMed] [Google Scholar]
- [45].Bulutoglu B, Garcia KE, Wu F, Minteer SD, Banta S, Direct evidence for metabolon formation and substrate channeling in recombinant TCA cycle enzymes, ACS Chem. Biol 11 (10) (2016) 2847–2853. [DOI] [PubMed] [Google Scholar]
- [46].Almacellas E, Pelletier J, Manzano A, Gentilella A, Ambrosio S, Mauvezin C, Tauler A, Phosphofructokinases axis controls glucose-dependent mTORC1 activation driven by E2F1, iScience 20 (2019) 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hill BG, Shiva S, Ballinger S, Zhang J, Darley-Usmar V, Bioenergetics and translational metabolism: implications for genetics, physiology, and precision medicine, Biol. Chem 401 (1) (2019) 3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Opie LH, Newsholme EA, The activities of fructose 1,6-diphosphatase, phosphofructokinase and phosphoenolpyruvate carboxykinase in white muscle and red muscle, Biochem. J 103 (2) (1967) 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fan TWM, Zhang X, Wang C, Yang Y, Kang WY, Arnold S, Higashi RM, Liu J, Lane AN, Exosomal lipids for classifying early and late stage non-small cell lung cancer, Anal. Chim. Acta 1037 (2018) 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Carreer WJ, Flight RM, Moseley HN, A computational framework for high-throughput isotopic natural abundance correction of omics-level ultra-high resolution FT-MS datasets, Metabolites 3 (4) (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lin P, Lane AN, Fan TW, Stable isotope-resolved metabolomics by NMR, Methods Mol. Biol 2037 (2019) 151–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Larsen CP, Trivin-Avillach C, Coles P, Collins AB, Merchant M, Ma H, Wilkey DW, Ambruzs JM, Messias NC, Cossey LN, Rosales IA, Wooldridge T, Walker PD, Colvin RB, Klein J, Salant DJ, Beck LH Jr., LDL receptor-related protein 2 (Megalin) as a target antigen in human kidney anti-brush border antibody disease, J. Am. Soc. Nephrol 29 (2) (2018) 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M, In-gel digestion for mass spectrometric characterization of proteins and proteomes, Nat. Protoc 1 (6) (2006) 2856–2860. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.