

Abstract
A concise and stereoselective total synthesis of the clinically relevant tricyclic prostaglandin D2 metabolite (tricyclic-PGDM) methyl ester in racemic form was accomplished in 8 steps from a readily available known cyclopentene-diol derivative. The synthesis features a nickel-catalyzed Ueno-Stork-type dicarbofunctionalization to generate two consecutive stereocenters, a palladium-catalyzed carbonylative spirolactonization to build the core oxaspirolactone, and a Z-selective cross metathesis to introduce the (Z)-3-butenoate side chain, a group challenging to introduce through traditional Wittig protocols and troublesome for the two previous total syntheses. A general Z-selective cross metathesis protocol to construct (Z)-β,γ-unsaturated esters was also developed that has broad functional group tolerance and high stereoselectivity. Additionally, our synthesis already accumulated 75 mg of valuable material for an 18O tricyclic-PGDM based assay used in clinical settings for inflammation.
Keywords: prostaglandins, carbonylation, olefin metathesis, nickel catalysis, total synthesis
Graphical Abstract
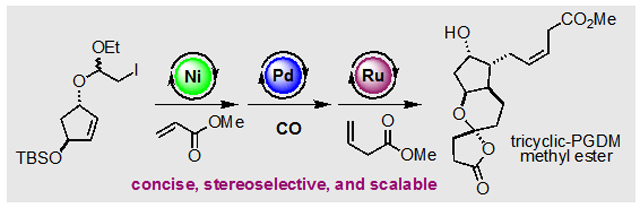
The clinically important tricyclic prostaglandin D2 metabolite (PGDM) methyl ester was synthesized in eight steps from a readily available known compound. The synthesis features Z-selective cross-metathesis to form the challenging Z β,γ-unsaturated ester, palladium-catalyzed carbonylative spirolactonization to build the oxaspirolactone moiety, and nickel-catalyzed Ueno–Stork-type dicarbofunctionalization to form two key C–C bonds and stereocenters.
Prostaglandins are signaling molecules that play a pivotal role in numerous biological pathways. A range of chemical and physical stimuli can activate their biosynthesis, which progresses through an enzymatic cascade involving phospholipase-mediated release of arachidonic acid (AA) from the membrane. Upon release, AA is transformed by cyclooxygenase (COX) enzymes to prostaglandin H2 (PGH2), a common intermediate that gives rise to the four principal prostaglandins by tissue specific isomerases.[1] Prostaglandins serve a crucial regulatory function in the acute inflammatory response. However, upregulation of these pathways spontaneously or extending beyond an initial inflammation event can be detrimental and has been linked to multiple diseases.[2] In addition, other unique non-inflammatory roles of prostaglandins are continuing to emerge further demonstrating that these scaffolds hold a privileged position as biological mediators.[3] Thus, it is to no surprise that the development of sensitive detection methods for prostaglandins has been an active research area. By virtue of their short half-lives, most current methods rely on the detection of metabolites that can be traced to the prostaglandin of interest.[4] Notably, following its recognition as the major urinary metabolite of prostaglandin D2 (PGD2, 1), tricyclic-PGDM (2) was used as a valuable indicator for PGD2 overproduction.[5] Using 18O-labelling, Roberts and colleagues at Vanderbilt University developed an assay for tricyclic-PGDM quantification which has been a valuable tool in clinical settings (Figure 1).[6] However, limited material is a constraint that has restricted this assay from becoming more prevalent since it relies on the fully synthetic tricyclic-PGDM methyl ester (3).
Figure 1.
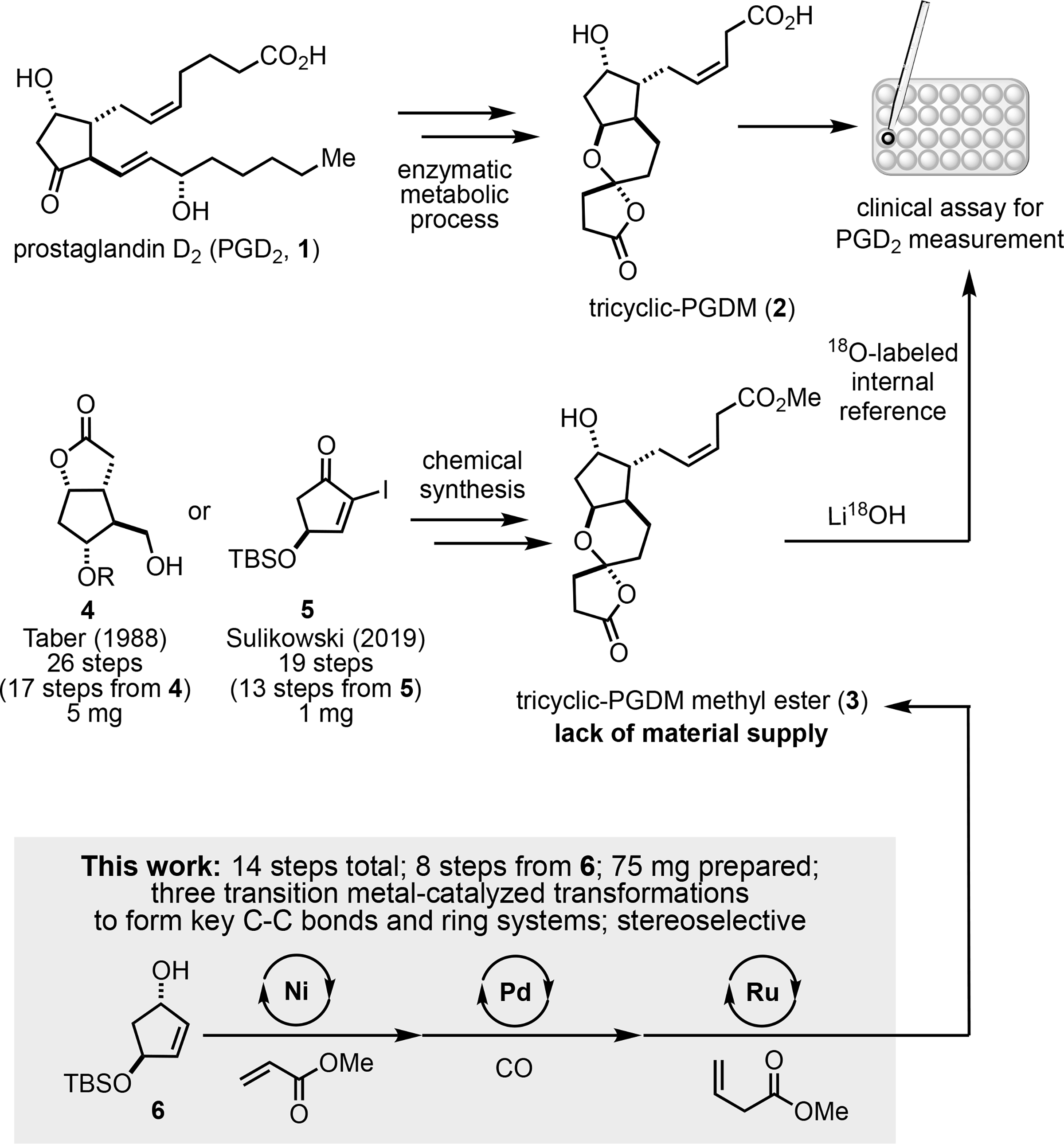
Clinical use of tricyclic-PGDM methyl ester and its synthesis
The prostaglandin molecules and their metabolites have garnered a tremendous amount of interest and efforts from the synthetic community.[7] The complexity of tricyclic-PGDM methyl ester (3) arises from the four contiguous stereocenters on the central cyclopentane ring, the labile oxaspirolactone, and the (Z)-3-butenoate side chain. In 1988, Taber and coworkers surmounted these challenges achieving the first synthesis of (±)-3 in 17 steps from the Corey lactone (4, prepared in 9 steps), with their final step giving 5 mg of 3 for the clinical assay (Figure 1).[8] In their synthesis, the (Z)-3-butenoate side chain proved to be troublesome to install on a hemi-acetal intermediate, with β-phosphoranylidene esters being noncompatible with the Wittig reaction. They overcame this issue by using an orthoester phosphorane.[9] To complete the synthesis, an acid-catalyzed cyclization of a hydroxy ketoacid was used to install the oxaspirolactone. More recently, to help replenish the assay Sulikowski and coworkers developed the second synthesis of (±)-3 from the known α-iodoenone 5 (prepared in 6 steps) in 13 steps, with their final step yielding 1 mg of 3.[10] Their route featured a Lindlar reduction of a β-alkynyl ester to install the (Z)-3-butenoate side chain, albeit in low yield, and an acid-promoted spiroketalization to afford the oxaspirolactone. Herein, we report an efficient synthesis of (±)-3 (75 mg) with three transition metal (Ni, Pd, and Ru)-catalyzed transformations to build strategic C-C bonds and key ring systems.
The clinical importance of tricyclic-PGDM methyl ester (3) and its intriguing structure, particularly the oxaspirolactone moiety, attracted our attention. We previously developed a palladium-catalyzed carbonylative spirolactonization to synthesize oxaspirolactones from readily available hydroxycyclopropanols (8→9, Figure 2A), and have used this method to efficiently synthesize a multitude of complex natural products.[11] To this end, we were inspired to establish an efficient carbonylative lactonization route to 3 and provide sufficient amount of material to support the clinical assays. Retrosynthetically (Figure 2B), we reasoned that the (Z)-3-butenoate side chain would need to be incorporated late in the synthesis to ensure its integrity. While the Wittig protocol with an orthoester phosphorane employed by Taber et al. gave 50% yield, the Z:E selectivity was only moderate (4:1) and additional orthoester deprotection and esterification steps were required.[8] Compound 12 is a known intermediate in the Sulikowski synthesis, however, six more steps (5% overall yield) including a Lindlar reduction were required to incorporate the (Z)-3-butenoate side chain.[10] Apparently, a direct Z-selective cross metathesis between 12 and methyl 3-butenoate 13 would be ideal to install the side chain, but with foreseeable challenges. While significant advances have been made in Z-selective cross metathesis,[12] β,γ-unsaturated esters including 13 still remain as challenging substrates. Therefore, it is not surprising that no such methods for (Z)-β,γ-unsaturated esters have been disclosed thus far. Olefins with a proximal chelating group, such as esters and amides, are particularly challenging substrates. β,γ-Unsaturated esters are among the worst substrates because the ester can chelate on the metal center after carbenoid formation (cf. A) forming a stable five-membered metallocycle. Furthermore, the cross-metathesis product tends to undergo double bond isomerization to the E-product or migration to the α,β-unsaturated ester. However, the recent Z-selective cross metathesis results between allylic-substituted olefins[13] or acrylamides[14] and common terminal olefins are encouraging. Despite the challenges, a direct Z-selective cross metathesis between 12 and 13 to complete the synthesis of 3 in just one step is attractive and worth developing, which may result in a generalized method for (Z)-β,γ-unsaturated ester preparation. The cross-metathesis precursor 12 could be prepared via olefination of the hemi-acetal derived from hydrolysis of acetal 14. The latter could be prepared from hydroxycyclopropanol 15 using the palladium-catalyzed carbonylative spirolactonization to generate its oxaspirolactone moiety. Hydroxycylopropanol 15 could be generated by the Kulinkovich reaction of lactone 16, which we anticipated to arise from a Ueno-Stork[15] type tandem radical cyclization of iodo-acetal 17. The radical cyclization precursor 17 could be assembled from the readily available known mono-protected diol 6.[16]
Figure 2.
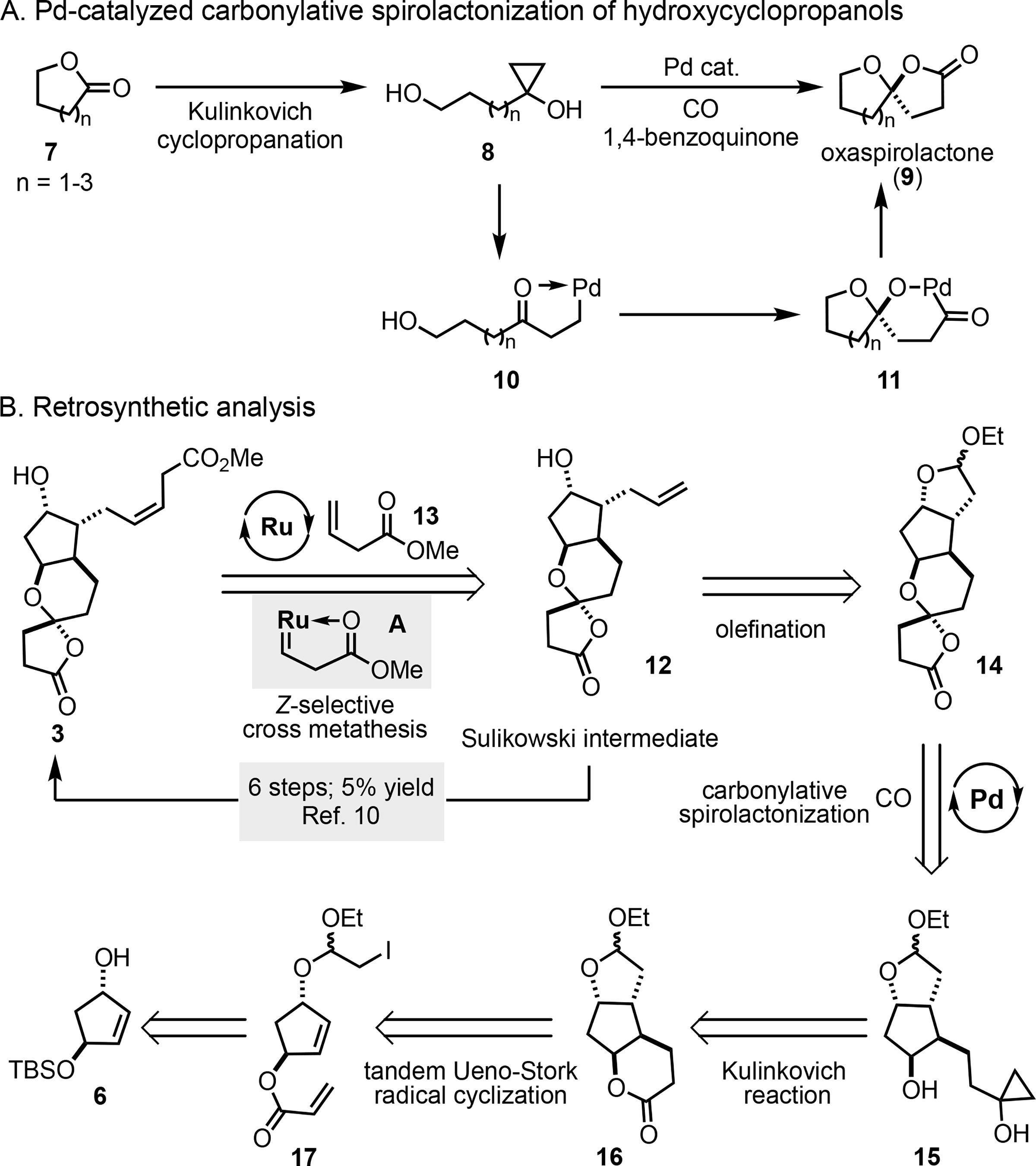
Carbonylative spirolactonization and retrosynthetic analysis.
Our synthesis commenced with the multi-decagram preparation of known compound 6 in six steps in racemic form from cyclopentadiene (Scheme 1). Enantioselective synthesis of 6 with an enzymatic hydrolysis[17] or a Noyori reduction was reported as well.[18] Subsequent conversion to iodo-acetal 18, silyl ether deprotection, and attachment of the acrylate side chain then gave 17, setting the stage for the key Ueno-Stork type cyclization. Unfortunately, tricyclic lactone 16 could not be isolated. Complex mixtures were obtained under various tin-based Ueno-Stork cyclization conditions, and the acrylate tether was cleaved when palladium, nickel and other transition metals were used to promote a dicarbofunctionalization due to its allylic nature.[19] We presumed the terminal 6-endo-trig cyclization was unfeasible for this substrate, and therefore proceeded to an intermolecular variant utilizing intermediate 18 and methyl acrylate. Gratifyingly, with modifications to the original Stork conditions,[19a] product 20 was obtained in 61% yield and excellent stereoselectivity at the newly formed stereogenic centers on up to 10 g scales. While efficient and scalable, the organotin toxicity and complications with t-BuOH during the workup led us to explore more environmentally benign conditions. Accordingly, we developed a novel nickel-catalyzed protocol to obtain dicarbofunctionalization product 20 in 59% yield.[20] While the yield slightly decreased (46%) on larger scale, the procedure was operationally convenient. Next, the Kulinkovich reaction[21] and TBS-ether deprotection proceeded smoothly on multi-gram scale to give cyclopropanol 15, which was unambiguously confirmed by x-ray analysis (CCDC 2089935).[22]
Scheme 1.
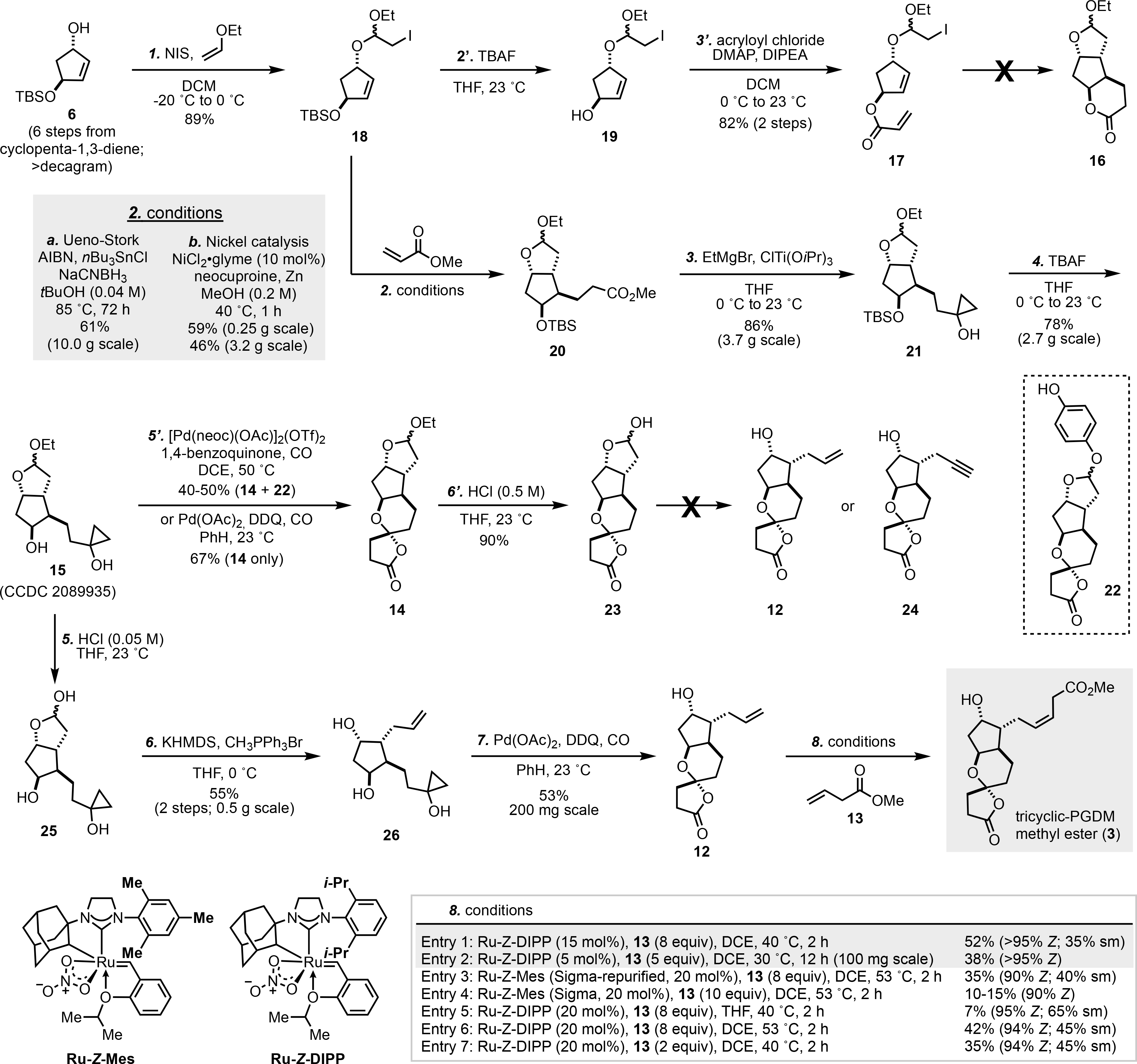
Total synthesis of tricyclic-PGDM methyl ester (3). TBS = tert-butyldimethylsilyl, NIS = N-iodosuccinimide, DCM = dichloromethane, TBAF = tetrabutylammonium fluoride, DMAP = 4-(dimethylamino)pyridine, DIPEA = N,N-diisopropylethylamine, AIBN = 2,2’-azobis(2-methylpropionitrile), THF = tetrahydrofuran, DCE = 1,2-dichloroethane, DDQ = 2,3-dichloro-5,6-dicyano-p-benzoquinone, KHMDS = potassium bis(trimethylsilyl)amide.
With 15 in hand, we next pursued the key carbonylative cyclization. Under our previously reported conditions for oxaspirolactone synthesis,[11a] tetracyclic product 14 was formed with an acceptable yield (40–50%). However, traces of acetic acid generated from the catalyst resulted in partial trans-acetalization of the ethyl acetal with hydroquinone to give 22. The resulting mixture was nontrivial to separate. Hydrolysis of the acetal mixture did not simplify the purification, with the resulting hydroquinone being equally difficult to separate. We then tested our reported conditions (Pd(OAc)2 and DDQ) for synthesizing bicyclic lactones[23] and were delight to find these conditions were also efficient for synthesizing oxaspirolactones. After replacing 1,4-benzoquinone with DDQ, 14 was cleanly produced in even higher yield (67%) without any evidence of trans-acetalization. Also, the use of commercially available Pd(OAc)2 to replace [Pd(neoc)(OAc)]2(OTf)2 simplified the procedure. Under both reaction conditions, only the desired stereochemistry was obtained at the spirocenter with the lactone oxygen in the axial position due to the anomeric effect. Subsequent HCl-promoted hydrolysis to hemi-acetal 23 proceeded smoothly, setting the stage for the seemingly simple one-carbon homologation. To our surprise, under various Wittig conditions, olefination of hemi-acetal 23 was unsuccessful. Using standard Wittig conditions, no reaction occurred at low temperatures and complex mixtures were obtained at higher temperatures; under Lebel’s salt free Wittig conditions,[24] no reaction occurred after days of stirring. Alkynylation to 24 using the Seyferth or Bestmann reagent was also pursued, however, only meager yields were obtained (<5%).
The unsuccessful one-carbon homologation of 23 prompted us to rethink our route, and we surmised the olefination would likely be more feasible without the sensitive oxaspirolactone present. Accordingly, cyclopropanol 15 was chosen as an appropriate candidate, since it would not require us to extensively modify our route. We suspected that the cyclopropanol would likely survive mild acetal hydrolysis conditions, however, we anticipated the methylenation to be a formidable challenge. To this end, hydrolysis of 15 proceeded smoothly in dilute HCl (0.05 M) to give hemi-acetal 25, but as expected the olefination was not as straightforward. Finally, with careful monitoring of reaction temperature and time, the methylenation product 26 was obtained in acceptable yield on fairly large scales. Moving forward, the carbonylative spirolactonization was once again reliable. Oxaspriolactone 12 was delivered as a single stereoisomer due to the anomeric effect and in good yield despite the potential competition of a 5-exo-trig oxypalladation and other unproductive pathways.
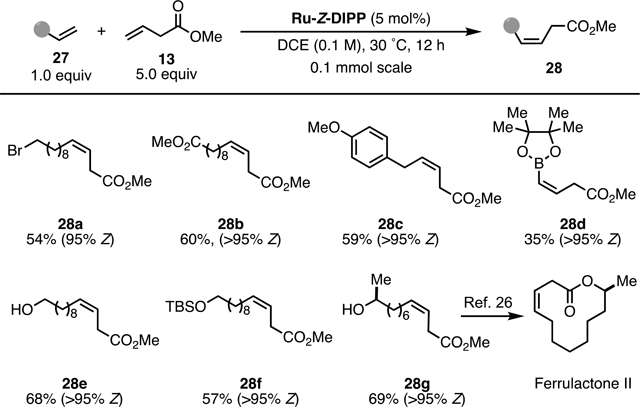
Our next task was to find a suitable Z-selective cross metathesis condition to complete the total synthesis in just one step.[25] We reasoned a cross metathesis using the commercially available Ru-Z-Mes as catalyst and methyl 3-butenoate (13) as the partner would be a suitable starting point. After extensive investigations, we found that about 10–15% (90% Z) of the cross-metathesis product could be obtained with the commercially available Ru-Z-Mes, but the results were not consistent. We further learned that re-purification of the Ru-Z-Mes was beneficial and increased the yield to 35%. To further enhance the yield and Z selectivity, the Ru-Z-DIPP catalyst was evaluated and found to be superior to the Ru-Z-Mes catalyst. Final product 3 was produced in better yield (52%) and higher selectivity (>95% Z). Interestingly, while both Ru-Z-Mes and Ru-Z-DIPP usually perform best in THF, only negligible yields (<10%) were obtained for this case. The reaction could also be conducted at 100-mg scale to give 38% (>95% Z) yield of 3 at a lower catalyst loading (5 mol%) and less amount of 13 (5 equiv.).
With the total synthesis of 3 secured, we next sought to generalize the Z-selective cross metathesis with the challenging β,γ-unsaturated ester substrate 13 (Table 1). Various terminal olefins with a wide range of functional groups were investigated. Under the aforementioned conditions (15 mol% Ru-Z-DIPP, 8 equiv of 13, 40 °C), although cross metathesis products were obtained in good yield, the Z/E selectivity dropped slightly (90%−92% Z). Gratifyingly, lower the catalyst loadings (5 mol% Ru-Z-DIPP) and temperature (30 °C) gave satisfactory yields and excellent Z selectivity (>95%) for a range of olefin substrates. Notably, free alcohol, TBS ether, alkyl bromide, ester, and boronate are all tolerated and cross metathesis product 28g is a known precursor for synthesis of the beetle pheromone Ferrulactone II.[26]
Table 1.
Substrate scope for the Z-selective cross metathesis with 13.
|
In summary, a concise synthesis of tricyclic-PGDM methyl ester (3) was achieved in 8 steps from readily available known compound 6. The synthesis features a Z-selective cross metathesis to form the challenging (Z)-β,γ-unsaturated ester, a palladium-catalyzed carbonylative spirolactonization to build the oxaspirolactone moiety, and a nickel-catalyzed Ueno-Stork-type dicarbofunctionilzation to form two key C-C bonds and stereocenters. A general cross-metathesis protocol for (Z)-β,γ-unsaturated esters was established. Through this route, we have accumulated over 75 mg of pure 3. We anticipate this material will be useful for the tricyclic-PGDM assay and further its availability as a clinical tool.
Supplementary Material
Acknowledgements
This work was supported by NSF 2102022. The NIH CA023168 is acknowledged for supporting shared NMR resources to Purdue Center for Cancer Research. H.S. gratefully acknowledges support from the Purdue Drug Discovery Training Program (NIH T32 GM125620). The XRD data was collected on a new single crystal X-ray diffractometer supported by the NSF through the Major Research Instrumentation Program under Grant No. CHE 1625543.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- [1].FitzGerald GA, Nat. Rev. Drug Discov, 2003, 2, 879–890. [DOI] [PubMed] [Google Scholar]
- [2].Ricciotti E, Fitzgerald GA, Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].For selected examples: Urade Y, Hayaishi O, Sleep Med. Rev, 2011, 15, 411–418;22024172Lee ML, Matsunaga H, Sugiura Y, Hayasaka T, Yamamoto I, Ishimoto T, Imoto D, Suematsu M, Iijima N, Kimura K, Diano S, Toda C, Nat. Commun, 2021, 12, 2330;33879780Palla AR, Ravichandran M, Wang YX, Alexandrova L, Yang AV, Kraft P, Holbrook CA, Schürch CM, Ho ATV, Blau HM, Science, 2021, 371, eabc8059.33303683
- [4].Tsikas D, Chromatogr J B Biomed. Sci. Appl, 1998, 717, 201–245. [DOI] [PubMed] [Google Scholar]
- [5].Liston TE, Roberts LJ, J. Biol. Chem, 1985, 260, 13172–13180. [PubMed] [Google Scholar]
- [6].Morrow JD, Prakash C, Awad JA, Duckworth TA, Zackert WE, Blair IA, Oates JA, Roberts LJ, Anal. Biochem, 1991, 193, 142–148. [DOI] [PubMed] [Google Scholar]
- [7].a) Das S, Chandrasekhar S, Yadav JS, Grée R, Chem. Rev. 2007, 107, 3286–3337; [DOI] [PubMed] [Google Scholar]; b) Peng HH, Chen FE, Org. Biomol. Chem. 2017, 15, 6281–6301. [DOI] [PubMed] [Google Scholar]
- [8].Prakash C, Saleh S, Roberts LJ, Blair IA, Taber DF, J. Chem. Soc. Perkin Trans 1, 1988, 2821–2826. [Google Scholar]
- [9].Corey EJ, Shimoji K, J. Am. Chem. Soc, 1983, 105, 1662–1664. [Google Scholar]
- [10].Kimbrough JR, Austin Z, Milne GL, Sulikowski GA, Org. Lett, 2019, 21, 10048–10051. [DOI] [PubMed] [Google Scholar]
- [11].a) Davis DC, Walker KL, Hu C, Zare RN, Waymouth RM, Dai M, J. Am. Chem. Soc, 2016, 138, 10693–10699; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ma K, Yin X, Dai M, Angew. Chem. Int. Ed, 2018, 57, 15209–15212; Angew. Chem., 2018, 130, 15429–15432; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yin X, Ma K, Dong Y, Dai M, Org. Lett, 2020, 22, 5001–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Endo K, Grubbs RH, J. Am. Chem. Soc, 2011, 133, 8525–8527; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Keitz BK, Endo K, Patel PR, Herbert MB, Grubbs RH, J. Am. Chem. Soc, 2012, 134, 693–699; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rosebrugh LE, Herbert MB, Marx VM, Keitz BK, Grubbs RH, J. Am. Chem. Soc, 2013, 135, 1276–1279; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Meek SJ, O’Brian RV, Llaveria J, Schrock RR, Hoveyda AH, Nature, 2011, 471, 461–466; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mann TJ, Speed AWH, Schrock RR, Hoveyda AH, Angew. Chem. Int. Ed, 2013, 52, 8395–8400; Angew. Chem., 2013, 125, 8553–8558; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Koh MJ, Khan RKM, Torker S, Yu M, Mikus MS, Hoveyda AH, Nature, 2015, 517, 181–186; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Occhipinti G, Hansen FR, Törnroos KW, Jensen VR, J. Am. Chem. Soc, 2013, 135, 3331–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Quigley BL, Grubbs RH, Chem. Sci, 2014, 5, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xu Y, Wong JJ, Samkian AE, Ko JH, Chen S, Houk KN, Grubbs RH, J. Am. Chem. Soc, 2020, 142, 20987–20993. [DOI] [PubMed] [Google Scholar]
- [15].For a comprehensive review on the Ueno-Stork reaction see: Salom-Roig XJ, Dénès F, Renaud P, Synthesis, 2004, 12, 1903–1928.
- [16].Ghosh AK, Xu CX, Osswald HL, Tetrahedron Lett, 2015, 56, 3314–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Deardorff DR, Windham CQ, Craney CL, Org. Synth. 1996, 73, 25. [Google Scholar]
- [18].Singh G, Meyer A, Aubé J, J. Org. Chem. 2014, 79, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Stork G, Sher PM, Chen HL, J. Am. Chem. Soc, 1986, 108, 6384–6385; [Google Scholar]; b) Firmansjah L, Fu GC, J. Am. Chem. Soc, 2007, 129, 11340–11341;17718496 [Google Scholar]; c) Wang GZ, Shang R, Cheng WM, Fu Y, J. Am. Chem. Soc, 2017, 139, 18307–18312;29116777 [Google Scholar]; d) Nguyen JD, D’Amato EM, Narayanam JMR, Stephenson CRJ, Nat. Chem, 2012, 4, 854–859;23001000 [Google Scholar]; e) Kim H, Lee C, Org. Lett, 2011, 13, 2050–2053.21417402 [Google Scholar]
- [20].For a review: Qi X, Diao T, ACS Catal. 2020, 10, 8542–8556.33732540
- [21].The reaction was unsuccessful under Kulinkovich’s originally reported catalytic conditions: Kulinkovich OG, Sviridov SV, Vasilevski DA, Synthesis, 1991, 234;when using stoichiometric amounts of ClTi(OiPr)3, the reaction proceeded smoothly:Kingsbury JS, Corey EJ, J. Am. Chem. Soc, 2005, 127, 13813–13815;16201801 also see reference 11b.
- [22].CCDC 2089935 contains the supplementary crystallographic data for compound 15.
- [23].Cai X, Liang W, Liu M, Li X, Dai M, J. Am. Chem. Soc, 2020, 142, 13677–13682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Lebel H, Paquet V, Proulx C, Angew. Chem. Int. Ed, 2001, 40, 2887–2890; Angew. Chem., 2001, 113, 2971–2974; [PubMed] [Google Scholar]; b) Lebel H, Paquet V, J. Am. Chem. Soc, 2004, 126, 320–328. [PubMed] [Google Scholar]
- [25].Herbert MB, Marx VM, Pederson RL, Grubbs RH, Angew. Chem. Int. Ed, 2013, 52, 310–314; Angew. Chem., 2013, 125, 328–332;Cannon JS, Grubbs RH, Angew. Chem. Int. Ed, 2013, 52, 9001–9004; Angew. Chem., 2013, 125, 9171–9174;Bronner SM, Herbert MB, Patel PR, Marx VM, Grubbs RH, Chem. Sci, 2014, 5, 4091–4098;25346842Mangold SL, O’Leary DJ, Grubbs RH, J. Am. Chem. Soc, 2014, 136, 12469–12478;25102124 For reviews see: Herbert MB, Grubbs RH, Angew. Chem. Int. Ed, 2015, 54, 5018–5024; Angew. Chem., 2015, 127, 5104–5110;Montgomery TP, Johns AM, Grubbs RH, Catalysts, 2017, 7, 87. For examples in total synthesis see: Chung WJ, Carlson JS, Bedke DK, Vanderwal CD, Angew. Chem. Int. Ed, 2013, 52, 10052–10055; Angew. Chem., 2013, 125, 10236–10239;Li J, Ahmed TS, Xu C, Stoltz BM, Grubbs RH, J. Am. Chem. Soc, 2019, 141, 154–158;30537831Li J, Stoltz BM, Grubbs RH, Org. Lett, 2019, 21, 10139–10142;31808699De Léséleuc M, Godin É, Parisien-Collette S, Lévesque A, Collins SK, Org J Chem, 2016, 81, 6750–6756.
- [26].a) Mori K, Tomioka H, Liebigs Ann. Chem, 1992, 1011–1017; [Google Scholar]; b) Pawar AS, Sankaranarayanan S, Chattopadhyay S, Tetrahedron Asymmetry, 1995, 6, 2219–2226. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.