

Summary
Micronutrients are essential small molecules required by organisms in minute quantity for survival. For instance, vitamins and minerals, the two major categories of micronutrients, are central for biological processes such as metabolism, cell replication, differentiation, and immune response. Studies estimated that around two billion humans worldwide suffer from micronutrient deficiencies, also known as “hidden hunger,” linked to weakened immune responses. While micronutrients affect the immune system at multiple levels, recent studies showed that micronutrients potentially impact the differentiation and function of immune cells as cofactors for epigenetic enzymes, including the 2-oxoglutarate-dependent dioxygenase (2OGDD) family involved in histone and DNA demethylation. Here, we will first provide an overview of the role of DNA methylation in T cells and B cells, followed by the micronutrients ascorbate (vitamin C) and iron, two critical cofactors for 2OGDD. We will discuss the emerging evidence of these micronutrients could regulate adaptive immune response by influencing epigenetic remodeling.
Keywords: T cells, B cells, epigenetics, micronutrients, DNA methylation, vitamin C, iron
Introduction
Micronutrients are low molecular weight molecules essential for the proper physiological functions of an organism. The micronutrients include vitamins and minerals and must be obtained externally via dietary uptake. In contrast to macronutrients that are needed for energy and biosynthesis in relatively larger quantities, micronutrients are required only in small amount. Despite the green revolution has substantially increased the quantity of food and reduced famine, the crops are often low in essential nutrients. The World Health Organization and UNICEF estimated that around two billion people worldwide, mostly in developing countries, suffer from micronutrient deficiencies, the condition also known as “hidden hunger” (1). Micronutrient deficiencies can have devastating consequences to our health, including impaired immune responses, physical growth, and neurological development. If left untreated, severe micronutrient deficiencies can lead to death. In addition to the people in developing countries, certain population in developed countries may develop micronutrient deficiency or insufficiency due to dietary habits or underlying illnesses. In the U.S., data from a national survey (2007–2010) of 16,444 individuals from age four years and older reported that a high percentage of those surveyed had lower micronutrients intakes than recommended: 38.9% for vitamin C (VC), 94.3% for vitamin D, 7.4% for iron, and 9.5% for folate (2). Therefore, micronutrient deficiency and insufficiency are global health issues and might contribute to the spread of infectious diseases due to weakened immune responses.
Adaptive immunity is the defense against infections by the same or related pathogens. Lymphocytes such as B and T cells are responsible for the specific recognition of pathogens via their antigen receptors. During immune responses, these cells are instructed by the signals provided by other cells and the environments to differentiate into specialized lineages intended for optimal protection. For instance, upon receiving signals from antigen receptor and T cells, naïve B cells can differentiate into germinal center (GC) B cells, plasma cells (PCs), and memory B cells (Bmem). Similarly, both CD4 and CD8 T cells can differentiate into various effector and memory T cell subsets. In addition, CD4 T cells can differentiate into regulatory T (Treg), follicular T helper (Tfh), and various polarized helper subsets (Th1, Th2, Th17) that produce specific cytokines. The proper differentiation of cells in response to specific pathogen type is critical for the biological defense of an organism.
The differentiations of these adaptive immune cells are orchestrated by transcription factors, followed by the recruitment of proper epigenetic regulators to remodel the epigenomes in order to maintain the proper lineage identities. Since many epigenetic regulators are enzymes, their activities are influenced by the levels of cofactors, substrates, and products that are often metabolites and micronutrients. Recent studies highlighting the links between metabolisms (mainly metabolites and macronutrients) and the epigenome have been reviewed previously (3–7). Here, we will focus on the role of micronutrients (vitamins and minerals) and how they regulate the epigenome, using the Ten-Eleven Translocation (TET) enzymes and their cofactors ascorbate/VC and iron as examples. We will first discuss the general role of DNA methylation and demethylation in the B and T lymphocytes, followed by the role of VC and iron in adaptive immune responses. Finally, we will discuss how the levels of micronutrients may influence the immune responses via epigenetic remodeling.
1. DNA methylation in Lymphocyte Development and Functions
1-1. DNA methylation and demethylation
DNA methylation is a stable epigenetic modification that enables the cells to annotate the genome and pass down information to daughter cells after cell divisions (8, 9). In mammalian cells, DNA methylation is usually found on cytosine at CpG motifs and has been associated with gene repression and the formation of heterochromatin. Cytosine methylation is catalyzed by DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, and DNMT3B (10). A fourth DNA methyltransferase Dnmt3c was found in mice, but not in humans, that is required to suppress transposon activity in male germ cells (11). DNMTs transfer the methyl group from S-adenosylmethonine (SAM) to the 5th carbon of cytosine residue and generate 5-methyl cytosine (5mC). To establish the methylation pattern, DNMT3A or DNMT3B methylates the unmodified cytosine in a process termed de novo methylation. On the other hand, DNMT1 is responsible for maintenance methylation by replicating the methylation status of CpGs to the newly synthesized unmethylated DNA during cell replication. At the replication fork, UHRF1 recognizes the hemi-methylated CpG motif and recruits DNMT1 to methylate the unmethylated cytosine on the new DNA strand (12).
While DNA methylation is chemically stable, the modification is not static. In fact, recent studies have shown that certain cytosines are continuously being methylated and demethylated by DNMT and TET (13–16) (Fig. 1). TET dioxygenase family consists of three members (TET1, TET2, TET3) and is responsible for DNA demethylation (17–19). While TET1 is mostly expressed in stem cells, TET2 and TET3 are expressed in somatic cells and often have redundant functions. TET enzymes oxidize the methyl group on the 5th position of 5mC in a stepwise manner (20), using α-ketoglutarate (αKG, also known as 2-oxoglutarate or 2-OG), ferrous iron (Fe2+), and molecular oxygen (O2). The first oxidative product is 5-hydroxymethylcytosine (5hmC), a stable epigenetic mark besides being an intermediate for DNA demethylation. The level of 5hmC varies depending on given cell types and is usually less than 10% relative to that of 5mC (17). In general, the amount of 5hmC is lower in proliferative cells as the modification is not replicated to newly synthesized DNA during S phase as DNMT1/UHRF1 does not recognize CpG with oxidized cytosines such as 5hmC. In fact, the inability of DNMT1/UHRF1 to replicate the methyl group is the basis for TET-mediated passive demethylation (Fig. 1). 5hmC can be further oxidized to 5-formylcytosine (5fC) and then to 5-carboxylcytosine (5caC), with each modification being an order of magnitude less than the previous one. Unlike 5hmC, 5fC and 5caC are not stable in the cells as they are recognized and excised by thymine DNA glycosylase (TDG), followed by base-excision repair (BER) and the replacement by an unmodified cytosine. This BER-mediated removal of 5fC and 5caC is also known as active DNA demethylation (Fig. 1).
Figure 1. The DNA methylation-demethylation cycle.
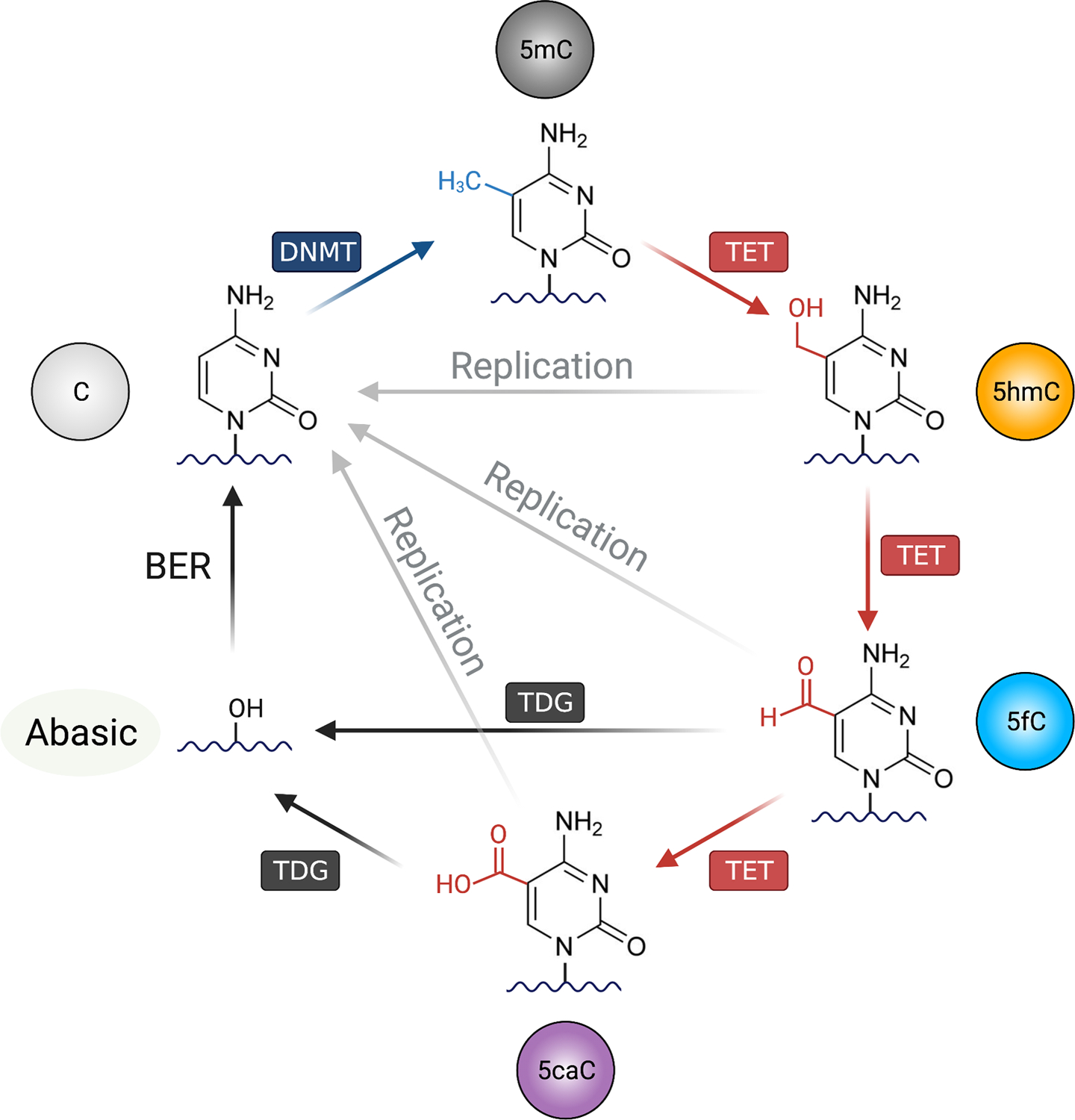
Unmodified cytosine (C) on the DNA can be methylated by the DNA methyltransferases (DNMTs), forming 5-methylcytosine (5mC). For DNA demethylation, TET (Ten-Eleven Translocation) oxidizes 5mC into oxidized cytosines including 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). There are mainly two mechanisms for TET-mediated DNA demethylation: replication-independent (active) and replication-dependent (passive) pathways. In active DNA demethylation, 5fC and 5caC are recognized and excised by thymine DNA glycosylase (TDG), creating an abasic site that is replaced with unmethylated cytosine by base excision repair (BER) pathway. In passive demethylation, while the DNMT1/UHRF1 complex recognize hemi-methylated DNA CpG on replicating DNA, the complex is not able to recognize and methylate the hemi-methylated CpG containing oxidized cytosines (5fC, 5fC and 5caC), thus inhibiting the methylation of the CpG motif on the newly replicated DNA. Figure was created with BioRender.
1-2. DNA Methylation in B cells
Following B cell differentiation, the DNA is progressively demethylated across the genome, with terminally differentiated PCs having the lowest level of DNA methylation (21, 22). Previous reports have indicated that the enhancers of key genes for PCs, including Prdm1, Irf4, Xbp1, are demethylated in both mouse (22) and human (23) B cells during PC differentiation. DNA methylation similarly decreased when naïve B cells differentiate into GCB cells and Bmem (24, 25), suggesting DNA demethylation may have important functions in B cells. Indeed, the deletions of Tet2 or Tet2/Tet3 resulted in impaired bone marrow B cell differentiation, class switch recombination (CSR), and PC differentiation (26–31). Consistent with the notion that DNA methylation at some enhancers is reciprocally regulated by DNMT and TET, the mutation of the de novo DNMTs in B cells (Cd19-Cre Dnmt3a/b-flox) resulted in increased PC differentiation by potentially tipped the balance to favor TET-mediated demethylation (32). Multiple targets of TET-facilitated PC differentiation have been proposed, including elements at Irf4 and Prdm1 loci (33–35). Whether and how TETs and DNMTs co-regulate these elements remains to be determined. In humans, mutations in DNMT3B caused the immunodeficiency-centromeric instability-facial anomalies (ICF) syndrome, an autosomal recessive disorder associated with facial anomalies, cognitive disability, and recurrent infections (36). More importantly, analysis of the peripheral blood showed that patients have drastically lower Bmem and PC (37, 38). Note that the PC phenotypes differ between mice with B-cell-specific Dnmt3a/b-deficiency and human patients with germline DNMT3B mutation. Whether the difference is due to DNMT3A or other B cell-extrinsic factors is unclear.
A recent study has revealed one of the target genes of TET in PC differentiation. By immunizing mice adoptively transferred with B cells from Tet2/3-deficient B1–8hi BCR (specific for hapten 4-hydroxy-3-nitrophenylacetyl, NP), Fujii et al. showed that these B cells are defective in differentiating into PCs in response to T-dependent (NP-CGG) or T-independent antigens (NP-Ficoll) (34). These cells were unable to express high levels of IRF4, which is required to induce Prdm1 and PC differentiation. Interestingly, overexpression of IRF4 was able to bypass the requirement of TET and induced Prdm1 expression, suggesting that IRF4 is one of the main targets of TET2/3 during PC differentiation. The link between TET and IRF4 was also observed in developing B cells in the bone marrow, where TET2/3 were required for IRF4 expression (31). It will be interesting to determine if IRF4 is similarly regulated by TET in other immune cell types.
Besides terminal differentiations, the early bone marrow development of B cells are also regulated by TET enzymes. Deletion of Tet2 and Tet3 at early B cell stage using Mb1-Cre (expressed around pre-pro B stage) resulted in the developmental blockade from pro-B to pre-B stage that was due to decreased light chain Igκ rearrangement (27, 31). Concordantly, deletion of Dnmt3a/b with Mb1-Cre resulted in precocious rearrangement of Igκ. Several studies using Cd19-Cre to delete either Dnmt3a or Dnmt3a/b have demonstrated that de novo methylation is largely dispensable for the major checkpoints during B cell development (39, 40). Other alternative B cell lineages are also regulated by TET enzymes. For instance, the numbers of peritoneal B1a and splenic marginal zone B cells dramatically decreased in the Cd19-Cre Tet2/Tet3-deficient mice (27, 31). Conversely, B-cell-specific Dnmt3a-deficient (Cd19-Cre) mice showed selective expansion of B1a cells (41), suggesting the methylation status of certain cis-elements is critical for B1a cell differentiation. However, another study using Cd19-Cre to delete both Dnmt3a/b showed no difference in marginal zone B cells (32). In summary, depending on the timing of Cre expression, conditional deletion TET or DNMT3 may cause developmental blockade in multiple B cell subsets.
While TET and DNMT3 appear to have opposite functions, their deficiencies could occasionally result in similar phenotypes. For instance, both B-cell-specific Tet2- and Dnmt3a/b-deficient mice displayed GC hyperplasia after immunization (28, 32), suggesting both methylation and demethylation are required for the proper expression of genes related to cell cycle and/or apoptosis. A similar overlapping phenotype has been observed in hematopoietic stem cells, where the mutations of Dnmt3a (42), Dnmt3a/b (43), or Tet2 (44) resulted in increased self-renewal. In humans, mutations in DNMT3A and TET2 similarly resulted in a selective advantage of hematopoietic stem cells and are the two major mutations in clonal hematopoiesis (45).
In contrary to the de novo methyltransferase DNMT3, the maintenance methyltransferase DNMT1 has a fundamentally different mode of action. In most cases, de novo methyltransferases and TETs are akin to pencils and erasers that are important to annotate the genome for accurate interpretation. The maintenance methyltransferase DNMT1 is similar to a copier where the edit is propagated. Therefore, mutation in DNMT1 often causes catastrophic consequences in replicating cells due to global DNA hypomethylation, reactivation of repressed genes, and genomic instability (46–48). Mice with hypomorphic mutation of DNMT1, or deletion of UHRF1 in activated B cells (Aicda-Cre), resulted in decreased GC B cells frequency, affinity maturation, and antibody responses (49, 50). Mutation of UHRF1 in activated B cells impaired the control of chronic lymphocytic choriomeningitis virus (LCMV) infection. Interestingly, mutation of UHRF1 resulted in cell cycle arrest that is in part due to the DNA demethylation and expression of the cyclin-dependent kinase (CDK) inhibitor Cdkn1a (p21) (50). Altogether, these observations are consistent with the notion that DNA methylation is a major epigenetic modification that requires constant remodeling during cell development.
1-3. DNA methylation in T cell
T cells developed in the thymus where they undergo substantial genomic reorganization to rearrange the T cell receptor genes. The developmental stages can be generally categorized by the surface expression of CD4 and CD8: from CD4−CD8− double-negative (DN), to CD4+CD8+ double-positive (DP), followed by either CD4+CD8− or CD8+CD4− single-positive (SP) stages. T cells constantly remodel their epigenome to adjust to their current development stage and lineage identity. Studies have shown that DNA methylation is essential for the thymic development, peripheral differentiation, and functions of T cells (7, 51, 52). Consistent with the importance of DNA methylation in the overall genomic stability, the deletion of Dnmt1 in early DN T cells with Lck-Cre resulted in thymic hypoplasia that may cause by the decreased survival of αβ T cells. In addition, an alternative lineage CD8+ γδ T cells was increased in the knockout (KO) mice. Interestingly, deletion of Dnmt1 with Cd4-Cre at the DP stage had no significant effect on the peripheral T cell numbers. Instead, the T cells from the Cd4-Cre Dnmt1-KO mice showed impaired activation-induced proliferation but paradoxically had enhanced cytokine production in both “naïve” CD4 and CD8 T cells (53). The stronger phenotype in the Lck-Cre Dnmt1-KO is potentially due to the lack of maintenance DNA methylation in the proliferating T cells at DN and DP stages, whereas the SP thymocytes and naïve T cells are relatively quiescence without TCR stimulation.
De novo DNA methylation is also required for proper T cell development and function. It was observed that TCR signal induced Dnmt3a expression in CD4 T cells(54). While the germline deletion of Dnmt3b resulted in embryonic lethality, the Dnmt3a-deficient mice survive until around 4 weeks of age (55). The germline Dnmt3a-KO mice displayed an impaired DN-to-DP stage transition and decreased thymocytes number. Interestingly, the germline and conditional deletion of Dnmt3a (Cd4-Cre) resulted in the appearance of “bipolar” CD4 T cells with both the characteristics of Th1 and Th2 that produced both IL-4 and IFNγ (54). In addition, in vitro differentiated Th2, Th17, and the induced Treg (iTreg) from Dnmt3a-conditional KO mice failed to methylate and repress IFNγ expression, demonstrating the importance of de novo DNA methylation in defining lineage identity (54, 56). Similar to CD4 T cells, the deletion of Dnmt3a in cytotoxic CD8 T cells resulted in the hypomethylation at Tcf7 that encodes TCF1, the transcription factor important for memory cell. These CD8 T cells had resulted in an increased memory cell differentiation and reduced effector cell differentiation (57).
Localized DNA modification and demethylation often occur at critical lineage genes during T cell differentiation (58–60). Similar to de novo DNA methylation, TET-mediated DNA demethylation is essential for T cell development and function. For instance, disruption of TET2 and TET3 using CD4-Cre caused the thymic T cells to develop into the invariant natural killer T cell lineage (61). In peripheral CD4 T cells, TET2 is required for the transcription of the lineage-specific cytokines in vitro (e.g. IFNγ in Th1 cells and IL-17 in Th17 cells) (62). Interestingly, Tet2 deficient mice developed a more severe disease in the autoimmune experimental encephalomyelitis model due to a lower level of IL-10, suggesting the regulation of these cytokines in T cells is more complex in vivo (62). Besides CD4 T-helper cells, TET2 and TET3 are important for Treg cell lineage stability. TET2 and TET3 are required for the demethylation of the Foxp3 intronic enhancer CNS2. The mutations of Tet2/3 resulted in decreased Foxp3 expression, lineage instability, and autoimmunity (63, 64). In cytotoxic CD8 T cells, Tet2 deficiency enhanced the IFNγ response and increased number of memory cells in an LCMV infection model (65). Strikingly, in a human clinical immunotherapy trial, the mutation of TET2 in the T cells with chimeric antigen receptor (CAR-T cells) resulted in a central memory phenotype and improved tumor clearance (66). Altogether, these observations are consistent with the function of TET enzymes in promoting cell differentiation and lineage stability.
2. Micronutrients in Immunity
2-1. Vitamin C
VC is a critical micronutrient functioning as an antioxidant to limit the damage from free radicals. In animals, primates (including humans) and guinea pigs are unable to synthesize VC due to a deficiency of the enzyme L-gulonolactone oxidase (GULO) that is responsible for the last enzymatic step for VC biosynthesis. Therefore, humans require a regular dietary intake of VC. Long-term VC deficiency results in scurvy, a condition associated with immunodeficiency and pneumonia among other symptoms (67). Although scurvy is rare nowadays, a survey estimated that the rate of VC insufficiency is around 7.1% in the U.S. (68) potentially due to diets, habits, and underlying diseases or conditions. For instance, advanced age, smoking, and obesity are often associated with lower VC levels (69–72), which may contribute to immunodeficiency and variation in immune responses toward infections and vaccinations.
The dietary uptake of VC by the organism depends on the sodium dependent VC transporter 1 (SVCT1) expressed on the intestinal epithelial cells (73). VC is then translocated from the intestinal lumen to circulation. The concentration of VC in plasma is around 50–80 μM in healthy individual. Strikingly, depending on the cell type, VC concentration can be as high as 0.5 to 10 mM (10–200×), with blood mononuclear cells having around 80× more VC that that in the plasma (74). A recent study has shown that the average amount of VC in mouse hematopoietic stem cells is around 2.5 fmole per cell, which is about 5 times higher than regular bone marrow cells (75). In the body, cells mainly rely on two pathways to uptake VC from the circulation (Fig. 2). The reduced VC (ascorbate, or ASC) is transported to cells via SVCT1 or SVCT2, which is expressed in most cells. Although the level of the oxidized VC (dehydroascorbate, or DHA) is low in the circulation (76), DHA can be taken up via glucose transporters (GLUT1, GLUT3, GLUT4)(77). The intracellular DHA can be reduced back to ASC by oxidizing glutathione via the glutathione-ascorbate cycle (Fig. 2).
Figure 2. Ascorbate and iron in epigenetic regulation.
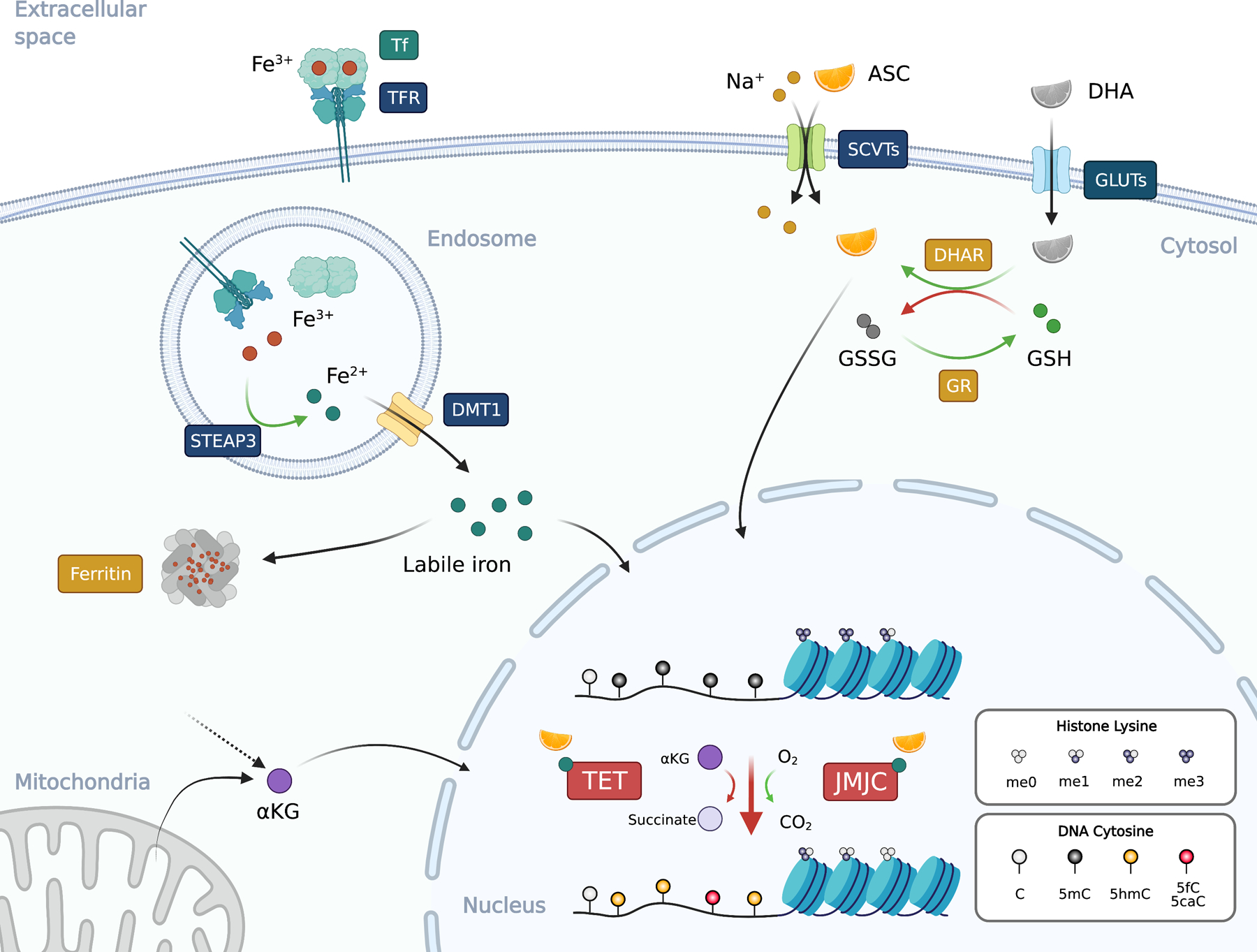
Ascorbate (ASC; vitamin C or VC) is co-transported with sodium ion (Na+) from extracellular space to the cytosol via SVCTs (sodium-ascorbate co-transporters) at the ratio of 1-to-2 (ASC to sodium). Dehydroascorbate (DHA), the oxidized form of ASC, enters the cells via the glucose transporters (GLUTs). DHA in the cells is reduced to ASC by dehydroascorbate reductase (DHAR) by oxidizing two molecules of reduced (GSH) into oxidized glutathione (GSSG). GSSG can be reduced back to GSH by glutathione reductase (GR). Iron usually exists in the circulation as the oxidized ferric iron (Fe3+) bound to transferrin (Tf) at a ratio of 2:1. Transferrin receptor (TFR) binds to and internalize the Tf:iron complex to endosome. The ferric iron is released from Tf in the acidic environment and reduced by the metalloreductase STEAP3 (Six-Transmembrane Epithelial Antigen of Prostate 3) into ferrous iron (Fe2+), which then translocates from the endosome to cytosol via DMT1 (Divalent Metal Transporter 1) to form the labile iron pool. Since ferrous iron can catalyze the formation of ROS, its concentration is tightly regulated and is stored as ferric iron in the ferritin complex that consists of 24 subunits. The ferritin complex can hold around 4,500 molecules of Fe3+. Both ASC and ferrous iron are cofactors for 2OGDDs including TET and JMJC proteins that used αKG (2-OG) and molecular oxygen to oxidize the target. TET oxidizes 5mC into 5hmC, 5fC, and 5caC, while various JMJC proteins demethylate histone lysine (and arginine, not depicted). αKG can be derived from mitochondria or cytosol as an intermediate metabolite. Black arrows, movement of molecules; green arrows, reduction; red arrows, oxidation. Blue text boxes represent transmembrane proteins and boxes with other colors indicate the location of the proteins. Figure was created with BioRender.
Besides being an antioxidant, VC is a cofactor for a variety of enzymes including the 2-oxoglutarate-dependent dioxygenase (2OGDD) family that includes the prolyl-4-hydroxylases (PHD1/2/3), collagen prolyl 4-hydroxylases (C-P4Hs), TET methylcytosine oxidases, and the Jumonji C (JmjC)-domain containing histone demethylases. For instance, during normoxia (normal oxygen level), PHD1/2/3 hydroxylate the proline residue of hypoxia-inducible factor 1-α (HIF1-α), facilitating its degradation and suppress the hypoxic response (78). VC is also essential for collagen synthesis by promoting C-P4H-mediated proline hydroxylation required for the stabilization of collagen triple helix (79). Importantly, VC is critical for the activity of various epigenetic enzymes, including TET and JmjC histone demethylases (80, 81). Mechanistically, one of the functions of VC on the 2OGDD is to maintain the iron at the active site at a reduced state (Fe2+). Therefore, VC has a broad effect to a number of biological processes.
2-1a. Animal models of VC deficiency and VC supplementation
To model VC deficiency, animals such as Gulo deficient mice and guinea pigs are often used due to their inability to synthesize VC. Several studies using these models have suggested a role of VC in immune responses. Two studies have investigated the anti-viral response in the VC deficient Gulo-KO mice but the results were inconsistent. In one study, influenza infection resulted in decreased pro-inflammatory cytokine (TNFα, IL-1β) but no effect on viral titer or the survival of the Gulo-KO mice (82). Another study showed increases in the same pro-inflammatory cytokines but decreased type I interferon (83). Surprisingly, half of the VC-depleted mice died one day after infection. The cause of death of these mice remained to be determined as the kinetics was significantly faster even when compared to lethal influenza infection (84). In another model of T cell-dependent liver injury, VC-deficient Gulo-KO mice displayed increased TNFα and IFNγ and severe liver damage (85). How VC-deficiency affects lung immune response and inflammation remained unresolved.
VC supplement has been used for treatment of various diseases such as cancers and sepsis in humans (86–88). Numerous animal studies have investigated VC supplement in vivo and in vitro, we will focus our discussion on several studies that have characterized the adaptive immune cells. Note that the upper physiological plasma VC concentration is tightly controlled by adsorption and excretion. Therefore, only delivering the VC by injections, but not by ingestion, can result in a temporary supraphysiologic VC concentration (“megadose”). Counterintuitively, VC at extremely high level in the plasma will instead facilitate the generation of reactive oxygen species (ROS) rather than acting as an anti-oxidant. This is potentially caused by reducing the blood ferric (Fe3+) to ferrous iron (Fe2+) and promotes the Fenton reaction where ferrous iron reacts with hydrogen peroxide and produces hydroxide or hydroxyl radicals. For instance, studies have used VC as a potential combined-treatment in cancer immunotherapy through the enhancement of anti-tumor CD8 T cell responses (86, 89). VC supplement is found to promote Th1 response and CD8+ memory T cell production through promoting IL-12 secretion by bone marrow-derived dendritic cells (90–93). In addition to promoting immune response, VC has been used to enhance the generation of the in vitro iTreg for immunosuppression (discussed below) (64, 94). In a skin graft mouse models, the iTreg generated in vivo in the presence of VC showed increased lineage stability and promoted the survival of the skin allograft (95, 96). In an allogeneic heart transplant model, however, the iTreg differentiated in the presence or absence of VC functioned similarly in promoting the survival of heart allograft (97). Therefore, the effect of VC on epigenome remodeling warrants further investigation to improve adoptive immune cell therapy, especially for specific immunosuppression.
2-1b. VC-mediated and Epigenome Regulation in T and B cells
VC is a cofactor for many epigenetic enzymes including TETs and histone demethylases, therefore exerting a strong influence on the epigenome (80, 98–100). For instance, VC has been shown to strongly promote the reprogramming of induced pluripotent stem cells and other cells (101, 102) by enhancing the activities of TET (103, 104) and other histone demethylases (e.g., KDM2A/2B for H3K36me2/3(105); KDM3A/3B/4 for H3K9me2/3 (106, 107)). In the in vitro T cell differentiation system using OP9-DL1 stromal cells, the addition of VC increased the transition from CD4−CD8− DN to CD4+CD8+ DP stage and facilitated the rearrangement of TCRβ (108). Further in vivo evidence supporting the role of VC on thymic development is from the SVCT2 (Slc23a2)-KO mice. As the Slc23a2-KO mice are perinatal lethal, their fetal liver cells were used as donors to generate chimeras. These mice displayed decreased number of T cells without affecting the numbers of myeloid and B cells (75, 108). The inhibitions of histone methyltransferases and DNMTs facilitated the DN-to-DP transition, which is consistent with the hypothesis that part of the functions of VC is to facilitate the histone and DNA demethylation (108). Besides thymic T cell development, VC also promote Th cell differentiation such as the in vitro Th17 polarization. In this system, VC has no effect on DNA demethylation of IL-17A promoter and the phenotype was not affected by the knockdowns of Tet2 and Tet3. Instead, the effect of VC is dependent on KDM4 (JMJD2A/B/C), the enzymes responsible for the demethylation of the repressive mark H3K9me3 (109).
Among the studies of VC on immune cells, perhaps the most notable example is the VC-mediated enhancement of Treg cells stability (63, 64, 94, 110, 111). Naïve CD4+ T cells can be induced to differentiate into iTreg cells by the presence of TGFβ during cell activation. However, these iTreg cells are relatively unstable. Notably, the Treg-specific demethylating region (at the Foxp3 intronic enhancer CNS2) remained methylated (112, 113). Studies have found that the addition of VC can enhance the stability of iTreg cells by facilitating TET2/3-dependent DNA demethylation at CNS2 (64, 94). Recently, a comprehensive study of the role of VC on iTreg cell has revealed that the TET2 and TET3 are essential for the Treg-specific signatures of gene expression, demethylation, and chromatin accessibility (110). The addition of VC facilitates the accessibility of chromatin regions, many of which contain STAT5 motifs. This is in accordant with the increased expression of IL-2 receptor α, STAT5 phosphorylation, and STAT5 binding to Foxp3 CNS2 (110). It will be of interest to determine if VC deficiency has detrimental effect on self-tolerance due to the destabilized Treg cells.
Unlike those for T cells, studies of the VC in B cells have been limited. Early studies have shown that VC supplementation could enhance antibody response in guinea pigs (114, 115). However, the effect of VC in the steady state antibody levels in human was inconclusive (116–119). In vitro, VC is able to increase the numbers of IgM secreting cells from mice (120) and in humans (121). However, the molecular mechanism remained unclear. Several recent studies, including ours, have shown that VC promotes PC differentiation in vitro and in vivo (33, 35). In vitro, the addition of VC can strongly enhance the differentiation of PC from naïve B cells using an IL-21-dependent co-culture system (122). Similarly, the VC-deficient Gulo-deficient mice showed impaired antibody response and PC differentiation after immunization (33). The effect of VC on PC differentiation is conserved between mice and humans and was not recapitulated by other antioxidants (35). Interestingly, VC is required during early B cell activation prior to the induction of PC differentiation and has limited effect on the transcriptome and proximal IL-21 signaling (35). Instead, the effect of VC on PC is likely on the epigenome and dependent on TET2 or TET3 (33, 35). By analyzing the genome-wide distribution of 5hmC, we identified many ascorbate-responsive elements (EAR) at the locus of Prdm1 (encoding BLIMP1, important transcription factor for PC), including previously undescribed distal elements, that displayed increased 5hmC modification after VC treatment (35). VC treatment resulted in TET2/3-dependent demethylation of intronic Prdm1 elements (33). Together, these observations strongly suggest that VC is important for humoral immune response by TET-mediated epigenome remodeling.
2-2. Iron
Iron is one of the most abundant elements on Earth’s crust and is an essential micronutrient. However, most of the iron exists in the oxidized form that is low in solubility and not readily available to organisms. Worldwide, it has been estimated that more than 1.2 billion people suffered from iron deficiency (123), the major cause of anemia and has been linked to weakened immunity (124, 125). For instance, in randomized controlled vaccine trials of African infants, iron deficiency at the time of vaccination associated with inferior antibody response while iron supplement enhances the response (126), strongly suggesting a role of iron in adaptive immune response. In addition, a missense mutation in the transferrin receptor (TFR/CD71, encoded by TFRC) was identified to cause combined immunodeficiency (discussed below)(127).
Iron is essential for biological processes due to its chemical property (128). While a number of oxidative states are possible, the ‘reduced’ ferrous (Fe2+) and ‘oxidized’ ferric (Fe3+) iron are the most common states in living organisms and are interchangeable via oxidation/reduction. Both forms of iron are highly reactive and can generate cytotoxic ROS. Therefore, besides the intracellular level of iron is tightly controlled, the metal is usually chelated or bound by proteins such as ferritin in the cells. Iron usually associates with proteins in the forms of ion, heme, or iron-sulfur (Fe-S) cluster to catalyze the biochemical reactions.
After dietary absorption or recycling from heme, the ferric irons in the circulation are bound by transferrin at a 2:1 ratio (Fe3+:Tf, Fig. 2). Expressed by most cells, TFR binds to and internalize the Tf-Fe3+ complex, from which the Fe3+ dissociate in the acidic environment at early endosome. The ferric iron is then reduced to ferrous iron Fe2+ by STEAP3 (Six-Transmembrane Epithelial Antigen of Prostate 3) and transported into the cytosol via DMT1 (Divalent Metal Transporter 1; encoded by SLC11A2) (129). Due to its ability to generate ROS, the ferrous iron Fe2+ is usually sequester and oxidized into ferric iron Fe3+ by ferritin in the cytosol. Ferritin forms a 24-subunit complex that is able to contain around 4,500 ferric iron ions (130). Alternatively, increased cytosolic iron may be stored as hemosiderin, an insoluble complex composed of ferritin and other proteins. The free cytosolic ferrous iron is collectively known as the “labile iron pool”, which may be used to supply heme and Fe-S biosynthesis in mitochondria or associated with other iron binding enzymes as a cofactor.
2-2a. Iron and Adaptive Immune Response
Iron is essential for both innate and adaptive immune response and its systemic level is critically controlled during infections (129). For instance, iron-deficiency has been linked to immunodeficiencies in humans (124–126) and decreased inflammatory responses in mouse models of atherosclerosis and experimental autoimmune encephalomyelitis (131–133). In B cell response, iron-deficiency in mice resulted in decreased mature B cells and impaired GC response after immunization (134). In vitro, iron chelation impaired B cell proliferation and PC differentiation. Human patients with iron deficiency also show significantly weakened antibody responses after challenged with the measles vaccine (134). These observations argue the importance iron in adaptive immunity.
Genetic mutations affecting the iron homeostasis are often associated with impaired immune response. For example, TFR is important for cells to uptake iron via transferrin in the circulation and the expression is induced after T and B cells activation (135, 136). Early in vitro studies using TFR-blocking antibody or iron chelator showed that TFR is required for human T cell DNA synthesis in response to mitogenic signal (137, 138). Consistent with the essential role of iron in major biological processes including mitochondria metabolism, germline mutation of TFR (encoded by Tfrc) is embryonic lethal due to defective erythropoiesis and neurological development (139). Using blastocysts complementation, it was shown that TFR is required for lymphocyte development. In the absence of TFR, T cells are arrested at CD4−CD8−CD3− DN stage in the thymus, while B cells were less affected and able to progress at least to IgM+ stage (140).
TFR is essential for iron uptake and therefore the germline loss-of-function mutation TFR is lethal. However, a genetic study in patients with combined immunodeficiencies has identified a missense mutation in TFRC (127), where the mutant TFR is defective in internalization and the cells from patients have high surface TFR. These patients showed mild anemia and hypogammaglobulinemia (low serum IgG) but otherwise normal lymphocyte numbers. Despite presented at relatively normal numbers, the T and B cells are defective in mitogen induced proliferation. Consistent with the hypogammaglobulinemia, B cells from patient were defective in CSR after stimulation despite expressing similar level of AICDA, which encodes the essential enzyme activation-induced cytidine deaminase (AID). Mice with the same Tfrc point mutation (TfrcY20H/Y20H) were viable and displayed similar phenotypes. Therefore, this observation suggests that while iron is essential for every cell in the body, iron deficiency may preferentially affect lymphocytes and thus contribution to immunodeficiency.
After iron uptake, the ferric iron is reduced to ferrous iron in the endosome by the metalloreductase STEAP3 before exporting to the cytosol through DMR1 (Fig. 2). Similar to the mice deficient in iron, Steap3-deficient mice had reduced mature B cell population but otherwise normal B cell development (134). Steap3-deficient mice were defective in mounting humoral immune response and the B cells were defective in antigen-triggered proliferation, demonstrating the iron trafficking pathway is important for B cell response. After reduced by Steap3, ferrous iron translocated from endosome into cytosol and form the labile iron pool (Fig. 2). The excessive ions are usually oxidized and sequestered by the ferritin complex (130). Therefore, the loss of ferritin can result in an increased in the labile iron pool (Fe2+), which are reactive and can generate ROS. In mice, the deletion of ferritin heavy chain (FTH) in hematopoietic cells with Mx1-Cre resulted in a moderate decrease in the number of peripheral lymphocytes but no significant difference in myeloid cells. This phenotype is cell-intrinsic as confirmed using Cd4-Cre (T cells) and Cd19-Cre (B cells)(141). Consistent with its role in intracellular iron storage, the deletion of Fth resulted in increased the pool of labile ferrous iron, ROS, and mitochondria polarization. Fth-deficiency preferentially affect the developing lymphocytes, which showed increased percentage of cells in cell cycle, potentially either caused by increased intracellular ferrous iron or compensatory expansion due to ROS-induced cell loss. Although a large increase in intracellular free ferrous iron is cytotoxic, a moderate increase in iron has been associated with pro-inflammatory reaction in CD4 T cells. A recent study demonstrated that a higher intracellular iron concentration promoted the production of cytokines (GM-CSF and IL-2) in CD4 T cells, while iron depletion resulted an opposite phenotype. Iron inhibited the proteolysis of an RNA binding protein PCBP1 that can stabilize the CSF2 (encoding GM-CSF) mRNA (142), providing a mechanistic explanation of how iron levels may affect inflammation. Whether the change in labile iron pool affect the epigenome and its relative contribution to the observed phenotypes remained to be determined.
2-2b. Iron and Epigenome
The 2OGDD family are non-heme iron proteins that associate with iron ion and includes many epigenetic enzymes such as TET and histone demethylases containing Jumonji C (JmjC) domains (143). 2OGDDs use the conserved histidine residues at the HxE/D…H triad to bind to an Fe2+ ion that is essential for the enzymatic activity (143, 144). Therefore, the activities of these enzymes are likely influenced by the intracellular ferrous iron concentration.
A recent study showed the link between iron deficiency and epigenetic remodeling in B cells. As described above, iron deficiency leads to lower antibody response both in human and mouse. The lack of iron resulted in the inability of demethylating the H3K9me2 at the promoter of cyclin E1 important for cell proliferation (134). The effect of iron was largely mediated by the H3K9me2 lysine demethylases KDM2B, KDM3B, and KDM4C that regulate cyclin E1 expression (134). Another recent study of mouse and human MBC revealed that the cells may have a more active heme-related metabolic program compared to naïve B cells (145). The addition of hemin (an iron-containing complex) increased oxidative phosphorylation and facilitated PC differentiation in human and mouse B cells (145). Therefore, iron is critical for humoral immune response through mediating activity of epigenetic enzymes.
2-3. Other micronutrients and epigenetic regulation
Besides VC and iron, other micronutrients likely influence the epigenome and immune responses via different mechanisms. For example, the roles of vitamins A and D in immune regulation have been well established and have been extensively reviewed (146–148). Each vitamin has specific nuclear receptors that bind to specific sequences. Therefore, the effect of these vitamins on the epigenome may be caused by the recruitment of epigenetic factors by the nuclear receptor or secondary effect by the differentially expressed genes.
Members of the vitamin B family, including folate (B9) and cobalamin (B12), are involved in folate-methionine cycle and one-carbon metabolism that are essential for the production of SAM (149, 150), the essential substrate for DNA and histone methylation. One study has shown that the in vitro generated bone-marrow-derived dendritic cells are less mature when differentiated in a folate-deficient environment (151). These dendritic cells produced less IL-12 in response to lipopolyscharride and are less competent in activating T cells. Since dendritic cells differentiation is independent of massive proliferation and thus may require folate-dependent nucleoside synthesis, it will be of interest to investigate the contribution of DNA and histone methylation in this setting.
3. Concluding Remarks
In this review, we have discussed how micronutrients may regulate the adaptive immune responses by facilitating the enzymatic remodeling of the epigenome. First, we have described the role of DNA methylation in adaptive immune responses. Followed by the discussion of two micronutrients (VC and iron) that have long been implicated in immunoregulation. Both VC and iron are critical cofactors for a number of enzymes, including the 2OGDD family that includes DNA methylcytosine oxidases (TETs) and histone demethylases (JmjC-containing proteins), strongly suggesting the levels of these micronutrients will have a major impact on the adaptive immune system.
As the effects of micronutrients are extremely broad and may affect numerous biological processes, enzymes, and cell types, the outcome of micronutrient deficiencies (or overloads) on immune responses will likely be the sum of the total effect on the system. In order to specifically determine the effect of micronutrients on the overall response, future work will require temporal- and cell-type-specific methods to deplete or increase the level of the micronutrient of interest. Furthermore, it will be important to discern whether the effect of micronutrients on the cell is temporary or inheritable after cell division. Heritability, whether it is between parents-offspring or mother-daughter cells, is the fundamental definition of epigenetic process. One example is the effect of VC on iTreg cells (64, 81, 94, 110, 111), where the effects (stable Treg lineage and demethylated Foxp3 enhancer) are inherited by daughter cells after division. Studies have convincingly demonstrated that the Treg-stabilizing effect of VC is dependent on TET-mediated DNA demethylation, strongly supporting the notion that this is an epigenetic event. Since the effect of micronutrients (deficiency) might be ingrained in the epigenome, it is possible that a previous episode of micronutrient disturbance may cause a long lasting effect to subsequent immune response, as in the case of trans-generational inheritance of metabolic diseases (152).
The activity of epigenetic enzymes critically depends on the concentrations of substrates, cofactors, and products. It is possible that organisms may evolve to utilize some of these enzymes as sensors to gauge the availability of micronutrients and metabolites. The change in activity will in turn alter the way epigenetic information is edited and may lead to a different transcriptional program. For instance, in the case of PC differentiation, the level of VC correlates with the probability of whether the activated B cells will become PC (33, 35). Therefore, TET and other enzymes are capable of directing the lineage decision when the cells are in a permissive microenvironment and cellular state: sufficient oxygen, low oxidative stress (VC), and active metabolism (αKG). In addition, each epigenetic enzyme may vary in its sensitivity to the concentrations of cofactors, substrates, and products. Thus, the combination of these factors may have heterogeneous effects on the epigenome depending on the activities of a given set of epigenetic enzymes.
While the major function of micronutrients is to sustain many basic biological functions of an organism, increasing evidence have shown that these small molecules also have essential missions at the command center of the cells. It may be a truism to say that a balanced diet is necessary for good health, but recent findings highlighted the potential long-lasting impacts of micronutrient on the epigenome. Perhaps something micro in quantity may be mighty in capability.
Acknowledgements
C.W.-J.L. is supported by NIH National Cancer Institute K22 Award (K22CA241290), startup funds from the Department of Microbial Infection and Immunity and the Pelotonia Institute of Immuno-Oncology (PIIO) at the Ohio State University. The authors declare no related conflict of interest.
References
- 1.In: Howson CP, Kennedy ET, Horwitz A, eds. Prevention of Micronutrient Deficiencies: Tools for Policymakers and Public Health Workers. Washington (DC), 1998. [PubMed] [Google Scholar]
- 2.Carr AC, Rowe S. Factors Affecting Vitamin C Status and Prevalence of Deficiency: A Global Health Perspective. Nutrients.2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lio CJ, Huang SC. Circles of Life: linking metabolic and epigenetic cycles to immunity. Immunology.2020;161:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dai Z, Ramesh V, Locasale JW. The evolving metabolic landscape of chromatin biology and epigenetics. Nature reviews Genetics.2020;21:737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Izzo LT, Affronti HC, Wellen KE. The Bidirectional Relationship Between Cancer Epigenetics and Metabolism. Annu Rev Cancer Biol.2021;5:235–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chisolm DA, Weinmann AS. Connections Between Metabolism and Epigenetics in Programming Cellular Differentiation. Annual review of immunology.2018;36:221–246. [DOI] [PubMed] [Google Scholar]
- 7.Van Acker HH, Ma S, Scolaro T, Kaech SM, Mazzone M. How metabolism bridles cytotoxic CD8(+) T cells through epigenetic modifications. Trends in immunology.2021;42:401–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature reviews Genetics.2010;11:204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nature reviews Molecular cell biology.2019;20:590–607. [DOI] [PubMed] [Google Scholar]
- 10.Lyko F The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nature reviews Genetics.2018;19:81–92. [DOI] [PubMed] [Google Scholar]
- 11.Barau J, et al. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science (New York, NY).2016;354:909–912. [DOI] [PubMed] [Google Scholar]
- 12.Liu X, et al. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nature communications.2013;4:1563. [DOI] [PubMed] [Google Scholar]
- 13.Wu W, et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature.2021;593:440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ginno PA, et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity. Nature communications.2020;11:2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charlton J, et al. TETs compete with DNMT3 activity in pluripotent cells at thousands of methylated somatic enhancers. Nature genetics.2020;52:819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parry A, Rulands S, Reik W. Active turnover of DNA methylation during cell fate decisions. Nature reviews Genetics.2021;22:59–66. [DOI] [PubMed] [Google Scholar]
- 17.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (New York, NY).2009;324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (New York, NY).2011;333:1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nature reviews Genetics.2017;18:517–534. [DOI] [PubMed] [Google Scholar]
- 20.Crawford DJ, Liu MY, Nabel CS, Cao X-J, Garcia BA, Kohli RM. Tet2 Catalyzes Stepwise 5-Methylcytosine Oxidation by an Iterative and de novo Mechanism. Journal of the American Chemical Society.2016;138:730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kulis M, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nature genetics.2015;47:746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barwick BG, Scharer CD, Bally APR, Boss JM. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nature immunology.2016;17:1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caron G, et al. Cell-Cycle-Dependent Reconfiguration of the DNA Methylome during Terminal Differentiation of Human B Cells into Plasma Cells. Cell reports.2015;13:1059–1071. [DOI] [PubMed] [Google Scholar]
- 24.Kulis M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nature genetics.2012;44:1236–1242. [DOI] [PubMed] [Google Scholar]
- 25.Oakes CC, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nature genetics.2016;48:253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature.2008;454:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orlanski S, et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proceedings of the National Academy of Sciences of the United States of America.2016;113:5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dominguez PM, et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer discovery.2018;8:1632–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoeler K, et al. TET enzymes control antibody production and shape the mutational landscape in germinal centre B cells. FEBS J.2019;286:3566–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lio CJ, et al. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Science immunology.2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lio CW, Zhang J, Gonzalez-Avalos E, Hogan PG, Chang X, Rao A. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. eLife.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barwick BG, et al. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nature communications.2018;9:1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi T, Sun M, Zhang C, Chen P, Xiao C, Chang X. Ascorbic Acid Promotes Plasma Cell Differentiation through Enhancing TET2/3-Mediated DNA Demethylation. Cell reports.2020;33:108452. [DOI] [PubMed] [Google Scholar]
- 34.Fujii K, et al. Tet DNA demethylase is required for plasma cell differentiation by controlling expression levels of IRF4. International immunology.2020;32:683–690. [DOI] [PubMed] [Google Scholar]
- 35.Chen H-Y, et al. Epigenetic Remodeling by Vitamin C Potentiates the Differentiation of Mouse and Human Plasma Cells. bioRxiv.2021. [Google Scholar]
- 36.Gagliardi M, Strazzullo M, Matarazzo MR. DNMT3B Functions: Novel Insights From Human Disease. Front Cell Dev Biol.2018;6:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blanco-Betancourt CE, et al. Defective B-cell-negative selection and terminal differentiation in the ICF syndrome. Blood.2004;103:2683–2690. [DOI] [PubMed] [Google Scholar]
- 38.Xu GL, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature.1999;402:187–191. [DOI] [PubMed] [Google Scholar]
- 39.Manoharan A, Du Roure C, Rolink AG, Matthias P. De novo DNA Methyltransferases Dnmt3a and Dnmt3b regulate the onset of Igkappa light chain rearrangement during early B-cell development. European journal of immunology.2015;45:2343–2355. [DOI] [PubMed] [Google Scholar]
- 40.Duncan CG, et al. Base-Resolution Analysis of DNA Methylation Patterns Downstream of Dnmt3a in Mouse Naive B Cells. G3 (Bethesda).2018;8:805–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahajan VS, et al. B1a and B2 cells are characterized by distinct CpG modification states at DNMT3A-maintained enhancers. Nature communications.2021;12:2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Challen GA, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nature genetics.2011;44:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Challen GA, et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell stem cell.2014;15:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moran-Crusio K, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell.2011;20:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bowman RL, Busque L, Levine RL. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell stem cell.2018;22:157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen T, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nature genetics.2007;39:391–396. [DOI] [PubMed] [Google Scholar]
- 47.Sharif J, et al. Activation of Endogenous Retroviruses in Dnmt1(−/−) ESCs Involves Disruption of SETDB1-Mediated Repression by NP95 Binding to Hemimethylated DNA. Cell stem cell.2016;19:81–94. [DOI] [PubMed] [Google Scholar]
- 48.Kim M, Trinh BN, Long TI, Oghamian S, Laird PW. Dnmt1 deficiency leads to enhanced microsatellite instability in mouse embryonic stem cells. Nucleic acids research.2004;32:5742–5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaknovich R, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood.2011;118:3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen C, et al. Uhrf1 regulates germinal center B cell expansion and affinity maturation to control viral infection. The Journal of experimental medicine.2018;215:1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frias AB, Boi SK, Lan X, Youngblood B. Epigenetic regulation of T cell adaptive immunity. Immunological reviews.2021;300:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Correa LO, Jordan MS, Carty SA. DNA Methylation in T-Cell Development and Differentiation. Crit Rev Immunol.2020;40:135–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee PP, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity.2001;15:763–774. [DOI] [PubMed] [Google Scholar]
- 54.Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA methyltransferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differentiation. Journal of immunology.2009;183:2267–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell.1999;99:247–257. [DOI] [PubMed] [Google Scholar]
- 56.Thomas RM, Gamper CJ, Ladle BH, Powell JD, Wells AD. De novo DNA methylation is required to restrict T helper lineage plasticity. The Journal of biological chemistry.2012;287:22900–22909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ladle BH, et al. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proceedings of the National Academy of Sciences of the United States of America.2016;113:10631–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nestor CE, et al. 5-Hydroxymethylcytosine Remodeling Precedes Lineage Specification during Differentiation of Human CD4(+) T Cells. Cell reports.2016;16:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsagaratou A, et al. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proceedings of the National Academy of Sciences of the United States of America.2014;111:E3306–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsagaratou A Deciphering the multifaceted roles of TET proteins in T-cell lineage specification and malignant transformation. Immunological reviews.2021;300:22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsagaratou A, et al. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nature immunology.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ichiyama K, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity.2015;42:613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yue X, Lio CJ, Samaniego-Castruita D, Li X, Rao A. Loss of TET2 and TET3 in regulatory T cells unleashes effector function. Nature communications.2019;10:2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yue X, et al. Control of Foxp3 stability through modulation of TET activity. The Journal of experimental medicine.2016;213:377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carty SA, et al. The Loss of TET2 Promotes CD8(+) T Cell Memory Differentiation. Journal of immunology.2018;200:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fraietta JA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature.2018;558:307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hemila H, Louhiala P. Vitamin C may affect lung infections. J R Soc Med.2007;100:495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schleicher RL, Carroll MD, Ford ES, Lacher DA. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr.2009;90:1252–1263. [DOI] [PubMed] [Google Scholar]
- 69.Isola G, Polizzi A, Muraglie S, Leonardi R, Lo Giudice A. Assessment of Vitamin C and Antioxidant Profiles in Saliva and Serum in Patients with Periodontitis and Ischemic Heart Disease. Nutrients.2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cahill LE, El-Sohemy A. Haptoglobin genotype modifies the association between dietary vitamin C and serum ascorbic acid deficiency. Am J Clin Nutr.2010;92:1494–1500. [DOI] [PubMed] [Google Scholar]
- 71.Lima de Araujo L, Maciel Barbosa J, Gomes Ribeiro AP, Oliveira dos Santos AC, Pedrosa F. Nutritional status, dietary intake and serum levels of vitamin C upon diagnosis of cancer in children and adolescents. Nutr Hosp.2012;27:496–503. [DOI] [PubMed] [Google Scholar]
- 72.Na N, Delanghe JR, Taes YE, Torck M, Baeyens WR, Ouyang J. Serum vitamin C concentration is influenced by haptoglobin polymorphism and iron status in Chinese. Clin Chim Acta.2006;365:319–324. [DOI] [PubMed] [Google Scholar]
- 73.Lykkesfeldt J, Tveden-Nyborg P. The Pharmacokinetics of Vitamin C. Nutrients.2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Evans RM, Currie L, Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br J Nutr.1982;47:473–482. [DOI] [PubMed] [Google Scholar]
- 75.Agathocleous M, et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature.2017;549:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nuclear Zhitkovich A. and Cytoplasmic Functions of Vitamin C. Chem Res Toxicol.2020;33:2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilson JX. Regulation of vitamin C transport. Annu Rev Nutr.2005;25:105–125. [DOI] [PubMed] [Google Scholar]
- 78.Kennel KB, Burmeister J, Schneider M, Taylor CT. The PHD1 oxygen sensor in health and disease. J Physiol.2018;596:3899–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prolyl Myllyharju J. 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann Med.2008;40:402–417. [DOI] [PubMed] [Google Scholar]
- 80.Young JI, Zuchner S, Wang G. Regulation of the Epigenome by Vitamin C. Annu Rev Nutr.2015;35:545–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yue X, Rao A. TET family dioxygenases and the TET activator vitamin C in immune responses and cancer. Blood.2020;136:1394–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li W, Maeda N, Beck MA. Vitamin C deficiency increases the lung pathology of influenza virus-infected gulo−/− mice. J Nutr.2006;136:2611–2616. [DOI] [PubMed] [Google Scholar]
- 83.Kim Y, et al. Vitamin C Is an Essential Factor on the Anti-viral Immune Responses through the Production of Interferon-alpha/beta at the Initial Stage of Influenza A Virus (H3N2) Infection. Immune Netw.2013;13:70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hemann EA, Green R, Turnbull JB, Langlois RA, Savan R, Gale M, Jr. Interferon-lambda modulates dendritic cells to facilitate T cell immunity during infection with influenza A virus. Nature immunology.2019;20:1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bae S, et al. In vivo consequence of vitamin C insufficiency in liver injury: vitamin C ameliorates T-cell-mediated acute liver injury in gulo(−/−) mice. Antioxid Redox Signal.2013;19:2040–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Magri A, et al. High-dose vitamin C enhances cancer immunotherapy. Science translational medicine.2020;12. [DOI] [PubMed] [Google Scholar]
- 87.Kuhn SO, Meissner K, Mayes LM, Bartels K. Vitamin C in sepsis. Curr Opin Anaesthesiol.2018;31:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Klimant E, Wright H, Rubin D, Seely D, Markman M. Intravenous vitamin C in the supportive care of cancer patients: a review and rational approach. Curr Oncol.2018;25:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luchtel RA, et al. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proceedings of the National Academy of Sciences of the United States of America.2020;117:1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Noh K, et al. Mega-dose Vitamin C modulates T cell functions in Balb/c mice only when administered during T cell activation. Immunol Lett.2005;98:63–72. [DOI] [PubMed] [Google Scholar]
- 91.Maeng HG, et al. Vitamin C enters mouse T cells as dehydroascorbic acid in vitro and does not recapitulate in vivo vitamin C effects. Immunobiology.2009;214:311–320. [DOI] [PubMed] [Google Scholar]
- 92.Jeong YJ, et al. Vitamin C-treated murine bone marrow-derived dendritic cells preferentially drive naive T cells into Th1 cells by increased IL-12 secretions. Cell Immunol.2011;266:192–199. [DOI] [PubMed] [Google Scholar]
- 93.Jeong YJ, et al. Vitamin C treatment of mouse bone marrow-derived dendritic cells enhanced CD8(+) memory T cell production capacity of these cells in vivo. Immunobiology.2014;219:554–564. [DOI] [PubMed] [Google Scholar]
- 94.Sasidharan Nair V, Song MH, Oh KI. Vitamin C Facilitates Demethylation of the Foxp3 Enhancer in a Tet-Dependent Manner. Journal of immunology.2016;196:2119–2131. [DOI] [PubMed] [Google Scholar]
- 95.Nikolouli E, et al. Alloantigen-Induced Regulatory T Cells Generated in Presence of Vitamin C Display Enhanced Stability of Foxp3 Expression and Promote Skin Allograft Acceptance. Frontiers in immunology.2017;8:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oyarce K, Campos-Mora M, Gajardo-Carrasco T, Pino-Lagos K. Vitamin C Fosters the In Vivo Differentiation of Peripheral CD4(+) Foxp3(−) T Cells into CD4(+) Foxp3(+) Regulatory T Cells but Impairs Their Ability to Prolong Skin Allograft Survival. Frontiers in immunology.2018;9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hwang JH, et al. Suppressive effects of vitamin C-treated induced-regulatory T cells on heart allograft rejection under vitamin C-deficient or -sufficient conditions. PloS one.2021;16:e0246967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee Chong T, Ahearn EL, Cimmino L. Reprogramming the Epigenome With Vitamin C. Front Cell Dev Biol.2019;7:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brabson JP, Leesang T, Mohammad S, Cimmino L. Epigenetic Regulation of Genomic Stability by Vitamin C. Frontiers in genetics.2021;12:675780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Das AB, Smith-Diaz CC, Vissers MCM. Emerging epigenetic therapeutics for myeloid leukemia: modulating demethylase activity with ascorbate. Haematologica.2021;106:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Esteban MA, et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell stem cell.2010;6:71–79. [DOI] [PubMed] [Google Scholar]
- 102.Pera MF. Epigenetics, vitamin supplements and cellular reprogramming. Nature genetics.2013;45:1412–1413. [DOI] [PubMed] [Google Scholar]
- 103.Blaschke K, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature.2013;500:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yin R, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. Journal of the American Chemical Society.2013;135:10396–10403. [DOI] [PubMed] [Google Scholar]
- 105.Wang T, et al. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell stem cell.2011;9:575–587. [DOI] [PubMed] [Google Scholar]
- 106.Ebata KT, et al. Vitamin C induces specific demethylation of H3K9me2 in mouse embryonic stem cells via Kdm3a/b. Epigenetics Chromatin.2017;10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen J, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nature genetics.2013;45:34–42. [DOI] [PubMed] [Google Scholar]
- 108.Manning J, et al. Vitamin C promotes maturation of T-cells. Antioxid Redox Signal.2013;19:2054–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Song MH, Nair VS, Oh KI. Vitamin C enhances the expression of IL17 in a Jmjd2-dependent manner. BMB Rep.2017;50:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yue X, Samaniego-Castruita D, Gonzalez-Avalos E, Li X, Barwick BG, Rao A. Whole-genome analysis of TET dioxygenase function in regulatory T cells. EMBO reports.2021:e52716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Someya K, et al. Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. International immunology.2017;29:365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Floess S, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol.2007;5:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Polansky JK, et al. DNA methylation controls Foxp3 gene expression. European journal of immunology.2008;38:1654–1663. [DOI] [PubMed] [Google Scholar]
- 114.Prinz W, Bloch J, Gilich G, Mitchell G. A systematic study of the effect of vitamin C supplementation on the humoral immune response in ascorbate-dependent mammals. I. The antibody response to sheep red blood cells (a T-dependent antigen) in guinea pigs. Int J Vitam Nutr Res.1980;50:294–300. [PubMed] [Google Scholar]
- 115.Feigen GA, et al. Enhancement of antibody production and protection against systemic anaphylaxis by large doses of vitamin C. Res Commun Chem Pathol Pharmacol.1982;38:313–333. [PubMed] [Google Scholar]
- 116.Vallance S Relationships between ascorbic acid and serum proteins of the immune system. Br Med J.1977;2:437–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Prinz W, Bortz R, Bregin B, Hersch M. The effect of ascorbic acid supplementation on some parameters of the human immunological defence system. Int J Vitam Nutr Res.1977;47:248–257. [PubMed] [Google Scholar]
- 118.Anderson R, Oosthuizen R, Maritz R, Theron A, Van Rensburg AJ. The effects of increasing weekly doses of ascorbate on certain cellular and humoral immune functions in normal volunteers. Am J Clin Nutr.1980;33:71–76. [DOI] [PubMed] [Google Scholar]
- 119.Kennes B, Dumont I, Brohee D, Hubert C, Neve P. Effect of vitamin C supplements on cell-mediated immunity in old people. Gerontology.1983;29:305–310. [DOI] [PubMed] [Google Scholar]
- 120.Ichiyama K, et al. Promotion of IL-4- and IL-5-dependent differentiation of anti-mu-primed B cells by ascorbic acid 2-glucoside. Immunol Lett.2009;122:219–226. [DOI] [PubMed] [Google Scholar]
- 121.Tanaka M, Muto N, Gohda E, Yamamoto I. Enhancement by ascorbic acid 2-glucoside or repeated additions of ascorbate of mitogen-induced IgM and IgG productions by human peripheral blood lymphocytes. Jpn J Pharmacol.1994;66:451–456. [DOI] [PubMed] [Google Scholar]
- 122.Nojima T, et al. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nature communications.2011;2:465. [DOI] [PubMed] [Google Scholar]
- 123.Camaschella C Iron deficiency. Blood.2019;133:30–39. [DOI] [PubMed] [Google Scholar]
- 124.Drakesmith H, et al. Vaccine efficacy and iron deficiency: an intertwined pair? Lancet Haematol.2021;8:e666–e669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nature reviews Immunology.2015;15:500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stoffel NU, et al. Iron Deficiency Anemia at Time of Vaccination Predicts Decreased Vaccine Response and Iron Supplementation at Time of Vaccination Increases Humoral Vaccine Response: A Birth Cohort Study and a Randomized Trial Follow-Up Study in Kenyan Infants. Frontiers in immunology.2020;11:1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jabara HH, et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nature genetics.2016;48:74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Muckenthaler MU, Rivella S, Hentze MW, Galy B. A Red Carpet for Iron Metabolism. Cell.2017;168:344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cronin SJF, Woolf CJ, Weiss G, Penninger JM. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front Mol Biosci.2019;6:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bradley JM, Le Brun NE, Moore GR. Ferritins: furnishing proteins with iron. J Biol Inorg Chem.2016;21:13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee TS, Shiao MS, Pan CC, Chau LY. Iron-deficient diet reduces atherosclerotic lesions in apoE-deficient mice. Circulation.1999;99:1222–1229. [DOI] [PubMed] [Google Scholar]
- 132.Grant SM, Wiesinger JA, Beard JL, Cantorna MT. Iron-deficient mice fail to develop autoimmune encephalomyelitis. J Nutr.2003;133:2635–2638. [DOI] [PubMed] [Google Scholar]
- 133.Pedchenko TV, LeVine SM. Desferrioxamine suppresses experimental allergic encephalomyelitis induced by MBP in SJL mice. J Neuroimmunol.1998;84:188–197. [DOI] [PubMed] [Google Scholar]
- 134.Jiang Y, et al. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nature communications.2019;10:2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pattanapanyasat K, Hoy TG. Expression of cell surface transferrin receptor and intracellular ferritin after in vitro stimulation of peripheral blood T lymphocytes. Eur J Haematol.1991;47:140–145. [DOI] [PubMed] [Google Scholar]
- 136.Neckers LM, Yenokida G, James SP. The role of the transferrin receptor in human B lymphocyte activation. Journal of immunology.1984;133:2437–2441. [PubMed] [Google Scholar]
- 137.Neckers LM, Cossman J. Transferrin receptor induction in mitogen-stimulated human T lymphocytes is required for DNA synthesis and cell division and is regulated by interleukin 2. Proceedings of the National Academy of Sciences of the United States of America.1983;80:3494–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Thorson JA, Smith KM, Gomez F, Naumann PW, Kemp JD. Role of iron in T cell activation: TH1 clones differ from TH2 clones in their sensitivity to inhibition of DNA synthesis caused by IgG Mabs against the transferrin receptor and the iron chelator deferoxamine. Cell Immunol.1991;134:126–137. [DOI] [PubMed] [Google Scholar]
- 139.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nature genetics.1999;21:396–399. [DOI] [PubMed] [Google Scholar]
- 140.Ned RM, Swat W, Andrews NC. Transferrin receptor 1 is differentially required in lymphocyte development. Blood.2003;102:3711–3718. [DOI] [PubMed] [Google Scholar]
- 141.Vanoaica L, Richman L, Jaworski M, Darshan D, Luther SA, Kuhn LC. Conditional deletion of ferritin h in mice reduces B and T lymphocyte populations. PloS one.2014;9:e89270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang Z, et al. Iron Drives T Helper Cell Pathogenicity by Promoting RNA-Binding Protein PCBP1-Mediated Proinflammatory Cytokine Production. Immunity.2018;49:80–92 e87. [DOI] [PubMed] [Google Scholar]
- 143.Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ. 2-Oxoglutarate-Dependent Oxygenases. Annual review of biochemistry.2018;87:585–620. [DOI] [PubMed] [Google Scholar]
- 144.Johansson C, et al. The roles of Jumonji-type oxygenases in human disease. Epigenomics.2014;6:89–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Price MJ, Scharer CD, Kania AK, Randall TD, Boss JM. Conserved Epigenetic Programming and Enhanced Heme Metabolism Drive Memory B Cell Reactivation. Journal of immunology.2021;206:1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Erkelens MN, Mebius RE. Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends in immunology.2017;38:168–180. [DOI] [PubMed] [Google Scholar]
- 147.Larange A, Cheroutre H. Retinoic Acid and Retinoic Acid Receptors as Pleiotropic Modulators of the Immune System. Annual review of immunology.2016;34:369–394. [DOI] [PubMed] [Google Scholar]
- 148.Martens PJ, Gysemans C, Verstuyf A, Mathieu AC. Vitamin D’s Effect on Immune Function. Nutrients.2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lio CWJ, Huang SCC. Circles of Life: linking metabolic and epigenetic cycles to immunity. Immunology.2020;161:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lyon P, Strippoli V, Fang B, Cimmino L. B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease. Nutrients.2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wu CH, Huang TC, Lin BF. Folate deficiency affects dendritic cell function and subsequent T helper cell differentiation. J Nutr Biochem.2017;41:65–72. [DOI] [PubMed] [Google Scholar]
- 152.Sales VM, Ferguson-Smith AC, Patti ME. Epigenetic Mechanisms of Transmission of Metabolic Disease across Generations. Cell Metab.2017;25:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]