

ABSTRACT
Endoplasmic reticulum stress (ERS) occurs when cellular demand for protein folding exceeds the capacity of the organelle. Adaptation and cell survival in response to ERS requires a critical contribution by mitochondria and peroxisomes. During ERS responses, mitochondrial respiration increases to ameliorate reactive oxygen species (ROS) accumulation. We now show in yeast that peroxisome abundance also increases to promote an adaptive response. In pox1Δ cells, which are defective in peroxisomal β-oxidation of fatty acids, the respiratory response to ERS is impaired and ROS accrues. However, the respiratory response to ERS is rescued and ROS production is mitigated in pox1Δ cells overexpressing Mpc1, the mitochondrial pyruvate carrier that provides another source of acetyl CoA to fuel the tricarboxylic acid cycle and oxidative phosphorylation. Using proteomics, select mitochondrial proteins were identified that undergo upregulation upon ERS to remodel the respiratory machinery. The abundance of several peroxisome-based proteins was also increased, corroborating the role of peroxisomes in ERS adaptation. Finally, ERS stimulates assembly of respiratory complexes into higher-order supercomplexes, underlying increased electron transfer efficiency. Our results highlight peroxisomal and mitochondrial support for ERS adaptation to favor cell survival.
KEY WORDS: Endoplasmic reticulum, Mitochondria, Stress surviva, Peroxisome, Reactive oxygen species
Summary: Mitochondria and peroxisomes respond to endoplasmic reticulum stress to help promote cell survival by reducing accumulation of reactive oxygen species.
INTRODUCTION
The endoplasmic reticulum (ER) is host to the folding and modification of ∼30% of newly made cellular protein. When demand for these activities exceeds ER capacity, ER stress (ERS) can develop. Severe and prolonged ERS is a threat to cell survival. The ERS response includes the unfolded protein response (UPR), which is initiated by the ERS sensor Ire1 (Senft and Ronai, 2015). In addition, cellular reactions involving multiple organelles participate in protecting against ERS. Mitochondria have a well-known role in apoptosis, but evidence is building that mitochondria also play a critical role in the survival response to ERS. The mitochondrial electron transport chain (ETC) is the major source of production of reactive oxygen species (ROS) (Zhao et al., 2019). Because of potential toxicity, excessive ROS must be constrained (Haynes et al., 2004; Knupp et al., 2019). In both mammalian cells and yeast, limiting ROS while increasing respiration are adaptive responses that help protect cells from death during ERS (Balsa et al., 2019; Bravo et al., 2011; Haynes et al., 2004; Hijazi et al., 2020; Knupp et al., 2019; Urano et al., 2000).
In the yeast Saccharomyces cerevisiae, ERS activates the RTG retrograde signaling pathway, which conveys mitochondrial needs to adjust nuclear gene expression (Hijazi et al., 2020; Liu and Butow, 2006). The activated RTG pathway not only upregulates the tricarboxylic acid (TCA) cycle, but also promotes peroxisomal proliferation, both of which are necessary to optimally drive oxidative phosphorylation (Hijazi et al., 2020; Liu and Butow, 2006). Peroxisome abundance is dynamically regulated by the cellular metabolic state, as peroxisomes host diverse reactions, including β-oxidation of fatty acids and detoxification of hydrogen peroxide (Schrader et al., 2016). Exchange of metabolites between the ER, mitochondria and peroxisomes, reflected in their physical juxtaposition, is critical to maintaining the homeostatic function of these organelles (Cohen et al., 2014). Because β-oxidation of fatty acids occurs solely in peroxisomes in yeast, delivery of fatty acid metabolites is one such requirement for organellar contact.
Notably, the mitochondrial response to ERS occurs in cells exponentially growing in abundant glucose, when a glycolytic metabolism is normally favored (Pfeiffer and Morley, 2014). Because the numbers of mitochondria do not change during adaptive ERS-induced response (Hijazi et al., 2020), metabolic changes coupled with remodeling of the respiratory machinery appear to mediate the response.
In this study, we show the importance of lipid metabolism via peroxisomal fatty acid β-oxidation and organellar contact with mitochondria to fuel a respiratory response during ERS adaptation and cell survival. In addition, we identify changes in the mitochondrial proteome that occur in response to ERS to build a more efficient, less ROS-prone respiratory machinery. We confirm some of the activities and components required to increase respiration and ameliorate ERS-induced ROS, including formation of higher-order mitochondrial respiratory supercomplexes.
RESULTS
Peroxisomal fatty acid β-oxidation is required during adaptation to ERS
Previously, we have reported that activation of RTG signaling occurs in the yeast ERS response (Hijazi et al., 2020). Because RTG signaling is also associated with induction of peroxisomal biogenesis (Epstein et al., 2001), we tested whether peroxisome numbers increase during ERS. Cells were treated for 5 h with 0.5 µg/ml tunicamycin (an inhibitor of N-linked glycosylation that induces protein misfolding and ERS), and peroxisomes were visualized in living cells using fluorescent mNeonGreen-tagged Pex11 (a peroxisomal membrane protein). Indeed, fluorescent peroxisomal puncta were more abundant in tunicamycin-stressed cells by contrast with control cells (Fig. 1A). Importantly, peroxisomal accumulation during ERS response occurs independently of the yeast UPR, as it was similarly observed in ire1Δ cells (Fig. 1A, quantified by flow cytometry in Fig. 1B). Peroxisomal abundance was also increased in response to dithiothreitol (DTT), an independent ER stressor, and this too was not inhibited in ire1Δ cells (Fig. 1B). In rtg1Δ cells, the efficiency of peroxisomal proliferation in response to DTT was not significantly impaired, but the tunicamycin response was reduced (Fig. 1B); these results suggest that the RTG pathway may at least partially play a role in peroxisomal induction by ERS.
Fig. 1.

Peroxisomes ameliorate ERS. (A) Micrographs showing ERS-induced peroxisome increase, visualized using Pex11–mNeonGreen. Exponentially growing wild-type (WT) and ire1Δ cells were treated with or without ER stressor (0.5 µg/ml tunicamycin; tun) for 5 h. Scale bar: 10 µm. (B) Quantitation of ERS-induced peroxisomal increase by flow cytometry of Pex11–mNeonGreen (Pex11-NG)-labeled cells. Wild-type, ire1Δ and rtg1Δ cells were treated with or without 0.5 µg/ml tunicamycin or 1 mM DTT. Pex11–NG fluorescence was quantitated using flow cytometry. The peroxisomal response to ERS was intact in ire1Δ cells but impaired in rtg1Δ mutants. Data are presented as mean±s.e.m. n≥3 using independent colonies. P values are indicated on the graph (Student's t-test, one-tailed, unpaired). (C) Left: western blots showing low levels of Cox2 protein in pox1Δ and pex34Δ cells compared with levels in WT. Cells were treated with or without 0.5 µg/ml tunicamycin for 5 h. Lysates were normalized using total protein content and blotted with anti-Cox2 and anti-Por1 antibodies. Pgk1 is shown as the loading control. Right: densitometric quantitation of the western blots using ImageJ software. Data are representative of three experiments. (D) Increased O2 consumption in response to ERS requires peroxisomal β-oxidation. Exponentially growing cells were treated with or without 0.5 µg/ml tunicamycin for 5 h. Cellular OCR was measured in a high-resolution respirometer. Data are presented as mean±s.e.m. n≥3 using independent colonies. P values are indicated on the graph (Student's t-test, one-tailed, unpaired). At α=0.01, OCR between untreated and treated pex mutants is statistically insignificant. (E) Pex34 is required for adaptation to ERS. Cells were treated with or without a low dose (0.5 µg/ml) or high dose (10 µg/ml) of tunicamycin for 4 h. Adaptation was assayed by treating cells with low dose tunicamycin for 4 h followed by high dose for 4 h (low→high). Cell viability was quantitated by colony forming assay, as described in the Materials and Methods. Data are presented as mean±s.e.m. n=3 with independent colonies.
Because Pox1, the fatty acyl CoA oxidase and first enzyme in β-oxidation, is a target of the RTG pathway (Epstein et al., 2001), we asked whether β-oxidation of fatty acids is necessary for ERS-induced mitochondrial response. Fig. 1C shows that the complex IV subunit Cox2 and the mitochondrial outer membrane protein Por1 were induced in wild-type cells after 5 h incubation with 0.5 µg/ml tunicamycin. Previously, we have reported that Cox2 and Por1 levels are constitutively increased upon constitutive expression of CPY*, a misfolded secretory pathway protein (Hijazi et al., 2020; Knupp et al., 2019), reflecting mitochondrial response to ERS. By contrast, Cox2 protein levels after ERS were consistently reduced in cells lacking Pox1 or Pex34, a membrane contact site protein that helps to transfer β-oxidation intermediates from peroxisomes to mitochondria (Fig. 1C) (Farre et al., 2019; Shai et al., 2018).
Because ERS adaptation requires induction of cellular O2 consumption rate (OCR) even under glucose- and oxygen-replete conditions that normally favor a predominantly glycolytic metabolism (Hijazi et al., 2020; Knupp et al., 2019), the OCR was assayed in pox1Δ, pex34Δ and pex3Δ cells (Fig. 1D; the peroxisomal membrane protein Pex3 is necessary for the targeting of other peroxisomal membrane proteins; Motley et al., 2008). Cellular OCR induction by ERS was reduced in all the peroxisomal mutants examined, by contrast with adapted wild-type cells in which OCR was increased ∼2.5 fold (Fig. 1D). As in wild-type cells, OCR in pox1Δ and pex34Δ mutants was completely abolished by the oxidative phosphorylation inhibitor antimycin A (Fig. S1A), indicating that cellular O2 consumption was entirely attributable to mitochondrial respiration. In the absence of ERS, the maximum cellular OCR in pex34Δ and pox1Δ (determined by addition of the protonophore CCCP) was similar to that in wild-type cells (Fig. S1A), indicating that the capacity of the ETC machinery is unaffected by loss of peroxisomal function. Furthermore, pox1Δ and pex34Δ mutants were all able to grow well in the non-fermentable carbon source glycerol, confirming that these proteins are not necessary for respiration per se (Fig. S1B). However, consistent with a lack of respiratory response to ERS (Fig. 1C,D), pox1Δ and pex34Δ cells could not significantly expand their maximum OCR after tunicamycin treatment; by contrast, the maximum OCR of wild-type cells increased slightly, but significantly, indicating an ERS-mediated augmentation in respiratory capacity (Fig. S1B). These data suggest that β-oxidation and peroxisomes are integrally connected in the adaptive mitochondrial response to ERS. Fatty acid metabolism via peroxisomal β-oxidation is used specifically during ERS response, presumably as a fuel source to stimulate the TCA cycle and oxidative phosphorylation. These relationships are underscored by physical contacts between peroxisomes, mitochondria and ER (Shai et al., 2016).
ERS adaptation confers protection against higher levels of subsequent ERS (Hijazi et al., 2020). To determine whether peroxisomal activity is necessary for ERS adaptation, wild-type and pex34Δ cells were treated directly with a low or high dose of tunicamycin for 4 h or, alternatively, pretreated with a low dose for 4 h followed by the high dose for 4 h. Cell viability was then assessed using a colony plating assay. Fig. 1E shows comparable survival of wild-type and pex34Δ cells after a single dose of ERS. However, whereas wild-type cells could adapt when pre-exposed to a single low dose, thereby better surviving a subsequent high dose, pex34Δ cells were impaired in adaptation, as judged by their loss of viability after the subsequent high dose of ER stressor (Fig. 1E). By using a UPRE-lacZ reporter to assay the ERS response (Cox and Walter, 1996), we found that the inability of pex34Δ cells to adapt to ERS (Fig. 1E) is not caused by impaired ability to mount a UPR (Fig. S2A).
Proteomics reveals select mitochondrial targets of ERS
To identify targets of the mitochondrial response to ERS, we prepared crude mitochondrial fractions from unstressed exponentially growing yeast cells, and from cells treated for 5 h with a low dose of tunicamycin to give cells time to adapt (Hijazi et al., 2020). Sample replicates were analyzed using unbiased liquid chromatography–tandem mass spectrometry (LC-MS/MS) to identify upregulated ERS targets (Table S1). Based on gene ontology information (from the Saccharomyces Genome Database), proteins enriched upon ERS were functionally grouped (Table 1). Several upregulated peroxisomal proteins were found (Table 1), perhaps due to physical links between peroxisomes and mitochondria, corroborating a peroxisomal role in the ERS response. Upregulated peroxisomal proteins included the β-oxidation enzymes Pox1 and Sps19, and fatty acyl CoA synthase (Faa2), although the cells were cultured in glucose-replete and lipid-free synthetic complete (SC) medium. These findings provide a molecular basis for the observed respiratory increase during ERS (Hijazi et al., 2020; Knupp et al., 2019).
Table 1.
Proteins upregulated by ERS identified in proteomics analysis

Numerous components and assembly factors of the TCA cycle and ETC were identified as ERS response targets, including those of respiratory complex II (succinate dehydrogenase) and complex IV (cytochrome c oxidase) (Table 1). The ETC is mostly made up of nuclear-encoded proteins, but several critical components are encoded by mitochondrial DNA. ERS-mediated expansion of the ETC thus requires mitochondrial translation machinery (Table 1). Cox2 is one such ERS-induced, mitochondrial DNA-encoded subunit of complex IV, as revealed by both proteomics (Table 1) and western blotting (Fig. 1C; Tiwari et al., 2002). Proteomics analysis also identified an uncharacterized mitochondrial ribosome-associated protein, Mrx9 (Table 1). Remarkably, Mrx9 has previously been isolated as a high-copy suppressor that can rescue ERS-sensitive mutants (such as ire1Δ cells impaired in the UPR) from cell death (Knupp et al., 2019). Mrx9 is localized to the polypeptide tunnel exit of the mitochondrial ribosome and colocalizes with proteins acting in early assembly steps of ETC subunits (Singh et al., 2020), suggesting a possible role in assisting assembly of new respiratory complexes. In addition, proteomics revealed upregulation of mitochondrial transport machinery to import new proteins from the cytoplasm (Table 1). These data suggest a critical coordination between increased mitochondrial protein synthesis and increased delivery of nuclear-encoded gene products destined for mitochondria during ERS.
ROS accumulation during ERS
To analyze further the roles of mitochondria and peroxisomes, and some of their resident antioxidants and redox proteins, in the ERS response, ROS accumulation was examined during ERS. ROS (predominantly H2O2) is a byproduct of peroxisomal β-oxidation (Pascual-Ahuir et al., 2017), and ROS (primarily superoxide) is also produced by electron leak from the ETC (Murphy, 2009). Live cells were stained with dihydroethidium (DHE), which becomes oxidized by ROS to generate a fluorescent product (Wojtala et al., 2014). Previously, we have reported that mutant cells that have increased susceptibility to ERS-induced death, such as ire1Δ cells, display intense DHE fluorescence covering the entire cell (Knupp et al., 2019). Such ‘whole-cell ROS’ is virtually undetectable in wild-type cells in the absence of ERS, and this staining pattern is observed in only a very small percentage (∼5%) of the cells after ERS (Knupp et al., 2019). However, in wild-type cells, a low intensity fluorescent staining of ROS was readily observable, and this ROS staining colocalized substantially with mitochondria (labeled on the outer membrane with GFP-tagged Tom70; Fig. S3). Although Tom70–GFP fluorescence levels remained similar, mitochondrial DHE staining was diminished after the wild-type cells had adapted to ERS induced by a low dose of tunicamycin for 5 h (Fig. 2A; Fig. S3), suggesting that ROS reduction accompanies the adaptation. A time course of DHE staining was examined for acute ROS changes in response to ERS. DHE staining remained similar in untreated and wild-type cells treated with ER stressor for up to 150 min. Transition from a mitochondrial pattern of DHE staining to a cytoplasmic pattern occurred by ∼150 min of ERS (Fig. S4).
Fig. 2.

Mitochondrial response to ERS includes amelioration of ROS. (A) ROS amelioration requires superoxide dismutase and catalase activity during ERS. Wild-type (WT), sod1Δ and cta1Δ cells were treated with or without 0.5 µg/ml tunicamycin for 2 h or 5 h. Cells were then stained with DHE (2 µg/ml for 15 min), and fluorescence images (top) were collected at the same exposure and adjusted with Photoshop using the same settings. DIC images are also shown (bottom). In low-exposure images, adjustments were made with Photoshop settings to reduce the fluorescence signal. Scale bar: 10 µm. Images are representative of three experiments. (B) ROS accumulation during ERS is exacerbated in pex34Δ and tsa1Δ cells. Wild-type, pex34Δ and tsa1Δ cells were treated with or without 0.5 µg/ml tunicamycin for 2 h or 5 h, and then stained with DHE as in A. Fluorescence images were collected at the same exposure and adjusted in Photoshop with the same settings. As in A, adjustments were made with Photoshop to reduce to the fluorescence signal in low-exposure images. Scale bar: 10 µm. (C) Quantitation of ROS accumulation shown in B, as described in the Materials and Methods. DHE fluorescence data are presented as mean±s.e.m. n≥38 cells, quantified from at least two distinct fields of view.
As positive controls for ROS accumulation, we examined sod1Δ and cta1Δ cells, which lack superoxide dismutase activity that converts superoxide to H2O2 in mitochondria and cytoplasm (Tank et al., 2018), and catalase A activity that detoxifies H2O2 in peroxisomes and the mitochondrial matrix (Petrova et al., 2004), respectively. By contrast with wild-type cells, DHE fluorescence of untreated and ER-stressed sod1Δ cells was more intense, and by 5 h, ROS accumulation in sod1Δ cells appeared mitochondrially distributed (Fig. 2A). In cta1Δ cells, DHE fluorescence intensity without or after 2 h of ERS was similar to that in wild-type cells. By 5 h of stress, however, fluorescence intensity of cta1Δ cells was quite extreme; to display these images, exposures were adjusted to reduce fluorescence saturation; the same image from 2 h ERS shown at higher intensity is barely visible at reduced intensity (Fig. 2A, images 2 and 3). These results suggest that both Sod1 and Cta1 antioxidants play critical roles to ameliorate ROS during ERS.
ROS accumulation was then examined in pex34Δ cells. Consistent with impaired ERS adaptation in pex34Δ cells (Fig. 1E), ROS accumulation was not mitigated after 5 h incubation with tunicamycin (Fig. 2B). Although readily apparent, fluorescence intensity of DHE-stained, ER-stressed pex34Δ cells was compared quantitatively with that in wild-type cells using ImageJ software (Fig. 2C). To determine the source of ERS-induced ROS accumulation in pex34Δ and pox1Δ cells, cells were treated with the protonophore CCCP to dissipate the mitochondrial membrane potential (MMP) and drive the OCR to maximal levels. Fig. S5 shows that CCCP addition abrogated ERS-induced ROS accumulation in pex34Δ and pox1Δ cells, underscoring that ROS is predominantly produced by mitochondrial respiratory complexes.
Suppression of ROS accumulation in pox1Δ cells by MPC1 overexpression
The inability of pox1Δ and pex mutants to respond to ERS with increased respiration (Fig. 1C) suggests a possible requirement for delivery of metabolites from peroxisomal β-oxidation to fuel the TCA cycle and the ETC. Pyruvate derived from glycolysis is another source of acetyl CoA to drive oxidative phosphorylation; the mitochondrial pyruvate carrier Mpc1 is an important determinant of pyruvate oxidation to acetyl CoA (Schell et al., 2014). Therefore, we asked whether overexpression of Mpc1 can rescue impaired respiratory response to ERS in pox1Δ cells. OCR in response to ERS was measured in wild-type and pox1Δ cells with and without MPC1 overexpression (Fig. 3A). Although OCR induced by tunicamycin in wild-type cells remained similar with or without MPC1 overexpression, pox1Δ cells overexpressing MPC1 responded to ERS with a significant increase in OCR (Fig. 3A). Proteomics analysis revealed upregulation of Mpc1 after tunicamycin treatment, supporting a role for Mpc1 in ERS response (Table 1). These results suggest that both glycolysis and fatty acid β-oxidation participate in energizing the ETC.
Fig. 3.

Rescue of pox1Δ cells by overexpression of MPC1. (A) OCR measured with or without MPC1 overexpression. OCR of wild-type (WT) cells with and without ERS (0.5 µg/ml tunicamycin for 5 h) was unchanged by MPC1 overexpression; OCR was significantly induced by ERS in wild-type cells. Without MPC1 overexpression, OCR was not significantly increased by ERS in pox1Δ cells. In pox1Δ cells with MPC1 overexpression, OCR was induced significantly by ERS. Data are presented as mean±s.e.m. n=3. *P≤0.05; NS, not significant (Student's t-test, one-tailed, unpaired). (B) ROS accumulation in pox1Δ and pex34Δ cells is ameliorated by MPC1 overexpression. WT and mutant cells with or without a HIS3-marked high-copy plasmid bearing MPC1 (MPC1 OE) were treated with or without tunicamycin (0.5 µg/ml tunicamycin for 5 h) and then stained with DHE to detect ROS, as described for Fig. 2A. Fluorescence images were collected at the same exposure and adjusted with Photoshop using the same settings. Scale bar: 10 µm. Images are representative of three experiments. (C) Schematic diagram illustrating the mitochondrial respiratory response to ERS and the requirement for fatty acid β-oxidation contributed by peroxisomes and pyruvate delivered to mitochondria via the Mpc1 carrier.
Because ROS accumulation was increased following ERS in pox1Δ and pex34Δ cells by comparison with wild-type cells (Figs 2B, 3B), the effect of MPC1 overexpression was examined by DHE staining. Remarkably, ROS buildup in pox1Δ and pex34Δ cells was mitigated by MPC1 overexpression (Fig. 3B), suggesting that increased flux into the TCA cycle and ETC is necessary for increased respiration and reduced ROS production. Fig. 3C schematically depicts the contributions of peroxisomal β-oxidation and pyruvate imported into mitochondria to form acetyl CoA during the ERS response.
Redox biology during mitochondrial response to ERS
Peroxisome activity plays an important role in restricting mitochondrial ROS accumulation. Moreover, proteomics analysis showed that several members of the peroxiredoxin family of antioxidants that act as regulators of redox signaling and homeostasis were induced by ERS (Table 1) (Finkel, 2011). A role for peroxiredoxins in ROS mitigation during ERS response was examined. Loss of the Tsa1 peroxiredoxin had a major adverse effect on ERS adaptation. Mitochondrial ROS accumulated in tsa1Δ cells during ERS to such an extent that many cells displayed saturating levels of fluorescence, even after decreasing exposure settings to allow comparison of 2 h and 5 h images (Fig. 2B); a comparison of the fluorescence intensity of DHE-stained tsa1Δ cells and wild-type cells is shown in Fig. 2C.
Although tsa1Δ and wild-type cells survived low or high doses of tunicamycin to the same extent, tsa1Δ cells failed to adapt, as their survivability was not improved by a high dose following an initial low dose of stressor (Fig. 4A). Association of impaired ERS adaptation with mitochondrial ROS accumulation in tsa1Δ cells (Fig. 2B) supports the idea that mitigation of ROS production is important for adaptation. Concomitantly, ERS stimulation of mitochondrial OCR was diminished in tsa1Δ mutants (Fig. 4B). Surprisingly, however, in the absence of ERS, constitutive OCR was increased in tsa1Δ cells (Fig. 4B). The OCR phenotype of the tsa1Δ single mutant remained similar when tsa1Δ was combined in double mutants with loss of the homologous Tsa2 peroxiredoxin or mitochondrial peroxiredoxins Ccp1 and Prx1 (Fig. 4B). Like tsa1Δ cells, loss of the Cta1 catalase also resulted in increased basal OCR with impaired respiratory response to ERS (Fig. 4B).
Fig. 4.

Mitochondrial remodeling in response to ERS is redox dependent. (A) Adaptation to ERS. Wild-type (WT) and tsa1Δ cells were treated without and with low dose (0.5 µg/ml) or high dose (10 µg/ml) of tunicamycin for 4 h, or a low dose for 4 h followed by a high dose for 4 h (low→high). Cell viability was determined by colony forming assay, with viability of unstressed wild-type cells expressed as 100%. Data are presented as mean±s.e.m. n=3 with independent colonies. (B) tsa1Δ cells have a constitutively increased OCR with diminished response to ERS. Exponentially growing wild-type, tsa1Δ, tsa1Δ tsa2Δ, tsa1Δ prx1Δ, tsa1Δ ccp1Δ, and cta1Δ cells were treated with or without 0.5 µg/ml tunicamycin for 5 h. Cellular OCR was measured in a high-resolution oxygraph. Data are presented as mean±s.e.m. n=3 with independent colonies. P values are indicated on the graph (Student's t-test, one-tailed, unpaired). (C) MMP is constitutively increased in tsa1Δ and tsa1Δ ccp1Δ cells, with diminished response to ERS. Cells were treated with or without 0.5 µg/ml tunicamycin for 5 h. Live cells were then stained with 5 nM TMRM for 30 min, and fluorescence and DIC images were taken with an Olympus microscope and a Hamamatsu CCD camera. Scale bar: 10 µm. Images are representative of three experiments. (D) Oxidative stress is induced by ERS, as assayed by TRX2-lacZ assay. The indicated cell lines were either untreated (−), treated for 5 h with 0.5 µg/ml tunicamycin (+tun), treated for 5 h with 1 mM DTT (+DTT) or treated for 5 h with 0.5 µM H2O2 (+H2O2). β-galactosidase activity is expressed as µmol/min/mg lysate protein. Data are presented as mean±s.e.m. n≥3. *P≤0.05; ****P≤0.0001; ns, P>0.05 (Student's t-test, one-tailed, unpaired).
Respiratory response to ERS was confirmed by staining with the cell permeant fluorescent dye TMRM in non-quenching mode. Mitochondrial TMRM staining is dependent on the MMP (Perry et al., 2011). In wild-type cells, TMRM staining was almost undetectable in untreated cells, but significantly increased upon ERS, reflecting increased respiration and MMP (Fig. 4C; Knupp et al., 2019). By contrast, TMRM fluorescence was detectable in untreated tsa1Δ and tsa1Δ prx1Δ cells, but increased MMP in response to ERS was diminished compared with that of wild-type cells (Fig. 4C). Constitutively higher TMRM fluorescence in the peroxiredoxin mutants is consistent with their higher constitutive OCR (Fig. 4B). These results suggest that loss of peroxiredoxin activity may impair ERS response by affecting the cellular redox balance (Pascual-Ahuir et al., 2017).
To examine ERS-induced ROS further, transcriptional activation of a thioredoxin reporter, TRX2-lacZ, was used to detect oxidative stress response (OSR). A positive control in Fig. 4D shows that TRX2-lacZ activity was induced ∼5-fold in wild-type cells challenged with 0.5 mM H2O2 for 5 h, in agreement with previous reports (Ross et al., 2000). Strikingly, TRX2-lacZ activity was increased ∼2 fold by ER stressors such as tunicamycin or DTT, suggesting an OSR is activated by an initiating ERS (Harding et al., 2003). The OSR derives from mitochondrial electron transport, as no significant tunicamycin-induced OSR was detected by the TRX2-lacZ reporter in rho0 cells lacking a functional ETC (Fig. 4D). Additionally, ERS-induced OSR was intact in ire1Δ cells, suggesting that the classical UPR is bypassed (Fig. 4D). Interestingly, as assayed with the TRX2-lacZ reporter, ERS-induced OSR was very slightly decreased in pex mutants by comparison with wild-type cells (Fig. S2B); by contrast, ERS-induced DHE staining was moderately increased over wild-type levels in pex34Δ cells (Figs 2B and 3B). The different reporters, TRX2-lacZ induction and DHE staining, likely reflect different sites and kinds of ROS accumulation.
In tsa1Δ cells, oxidative stress was constitutively increased, as evidenced by TRX2 induction (Fig. S6). Further accretion of the OSR in response to tunicamycin was ∼1.4 fold, diminished by comparison with that seen in wild-type cells (Fig. 4D). Although loss of the homologous Tsa2 peroxiredoxin (Wong et al., 2002) did not affect OSR, constitutive and ERS-induced OSR of the tsa1Δ tsa2Δ double mutant was even higher than in tsa1Δ cells, demonstrating a combined effect of Tsa1 and Tsa2 in antioxidant defense (Fig. S6).
In the mitochondrial peroxiredoxin mutants, OSR was unaffected in prx1Δ and ccp1Δ single mutants, and in prx1Δ ccp1Δ cells; however, prx1Δ tsa1Δ and ccp1Δ tsa1Δ double mutants displayed synthetically increased constitutive and ERS-induced OSR (Fig. S6). These results provide genetic evidence that the mitochondrial peroxiredoxins cooperate with the Tsa1 peroxiredoxin to protect cells against ERS.
ERS promotes supercomplex formation
During ERS adaptation, the mitochondrial respiratory machinery is remodeled to become more efficient in order to reduce ROS accumulation and promote cell survivability (Hijazi et al., 2020; Knupp et al., 2019). To address the underlying molecular mechanism by which the ETC can achieve increased efficiency during ERS adaptation, we examined the assembly of respiratory chain complexes into higher-order structures called supercomplexes (den Brave and Becker, 2020; Genova and Lenaz, 2015). In yeast, the predominant supercomplexes are formed between complex III and complex IV, corresponding to III2+IV1 and III2+IV2 stoichiometries (Chen et al., 2012). A mitochondrial fraction was prepared from exponentially growing wild-type cells untreated or treated with tunicamycin for 5 h; mitochondria were solubilized with digitonin and analyzed using blue native PAGE. As shown in Fig. 5, western blotting with anti-Qcr7 and anti-Cox2 antibodies enabled detection of mitochondrial supercomplexes containing complex III and complex IV, respectively. Without ERS, two bands were detected with anti-Qcr7 antibody that had molecular masses consistent with a III2+IV1 trimer (∼780 kDa) and a possible III2 dimer (∼640 kDa). After tunicamycin treatment, another band of higher molecular mass appeared in the Qcr7 blot, corresponding to III2+IV2 tetramer (∼850 kDa), indicating supercomplex formation. Abundance of the tetrameric supercomplex was also increased by the presence of the misfolded ER-retained protein CPY* (Fig. 5, right panel). The Cox2 blot also showed two bands, representing both the trimer and tetramer, but after tunicamycin treatment, more tetramer accumulated (Fig. 5). Consistently, supercomplex formation was not induced in response to ERS in pex34Δ and cta1Δ cells (Fig. 5), as they were unable to increase their OCR (Figs 1D and 4B) and accumulated ROS (Fig. 2). These results are consistent with a report showing that ERS stimulates supercomplex assembly in mammalian cells (Balsa et al., 2019). Our results suggest that supercomplex formation is the molecular basis for ERS adaptation.
Fig. 5.
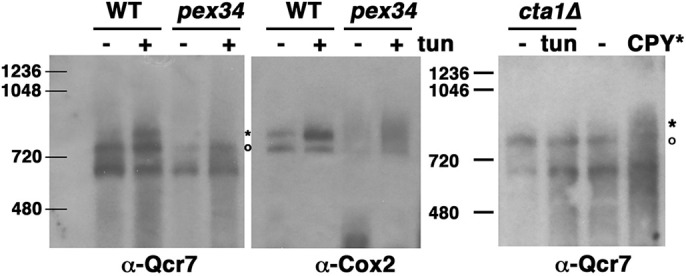
Supercomplex assembly during ERS. Abundance of ETC supercomplexes is increased by ERS. Mitochondria were prepared from wild-type (WT), pex34Δ and cta1Δ cells treated with or without tunicamycin (0.5 µg/ml; tun) for 5 h, and from cells with or without constitutive expression of CPY*, as described in the Materials and Methods, and then frozen. Cells constitutively expressing CPY* were not treated with tunicamycin. For analysis on blue native PAGE gels, mitochondria were thawed, solubilized with digitonin and run on a 3–12% gradient gel, then transferred to PVDF membrane, as described in the Materials and Methods. Supercomplexes were analyzed by western blotting with anti-Cox2 and anti-Qcr7 antibodies. Asterisks indicate a III2+IV2 tetramer; circles indicate a III2IV1 trimer. Positions of molecular mass markers (kDa) are shown. Images are representative of three experiments.
DISCUSSION
In this study, we suggest that peroxisomal β-oxidation of fatty acids is necessary to provide fuel for increased respiration via enhanced TCA cycle and ETC activity that accompanies ERS adaptation. Our hypothesis is supported by ERS-induced increase in peroxisome abundance (Fig. 1) (Epstein et al., 2001). Also participating in ERS adaptation, peroxisomal citrate synthase (Cit2) converts acetyl CoA to citrate for transport to mitochondria and consumption by the TCA cycle (Hijazi et al., 2020); CIT2 is transcriptionally activated by ERS (Tank et al., 2018). In yeast, β-oxidation occurs exclusively in peroxisomes (Hiltunen et al., 2003); the pox1Δ mutant, impaired in β-oxidation, and pex34Δ cells, impaired in peroxisomal–mitochondrial contacts and transfer of fatty acid metabolites (Shai et al., 2018), are defective in respiratory response and ERS adaptation (by contrast, UPR induction and OSR in the mutants appear relatively unaffected; Fig. S2). In addition to peroxisomal β-oxidation, pyruvate from the glycolytic pathway serves as another source of acetyl CoA, as proteomics analysis shows that the mitochondrial pyruvate carrier proteins Mpc1 and Mpc3 are upregulated by ERS (Table 1). Moreover, MPC1 overexpression can ameliorate the impaired respiratory response to ERS and ROS accumulation in pox1Δ and pex3Δ cells (Fig. 3A,B). In addition to providing fuel for respiration, acetyl CoA abundance has been suggested to regulate ETC assembly (Nowinski et al., 2020; Van Vranken et al., 2018); if so, increased acetyl CoA production in response to ERS may act as a signal to initiate ETC remodeling.
The ETC is the major source of cellular ROS (Murphy, 2009). We propose that during adaptive response to ERS, ROS production is held in check by remodeling of the respiratory machinery to increase the rate and efficiency of electron transfer in the ETC (Hijazi et al., 2020; Knupp et al., 2019). Respiratory complexes of the ETC are proposed to exist in a dynamic equilibrium as free complexes and supercomplexes. Although the physiological role of supercomplexes remains unresolved, supercomplex formation is induced by ERS in yeast and mammalian cells (Fig. 5) (Balsa et al., 2019). Cryo-electron microscopy analysis of yeast supercomplexes III2+IV and III2+IV2 has revealed that the supercomplexes mediate decreased distance for electron transfer from complex III to complex IV, suggesting more efficient electron transfer (Rathore et al., 2019). Recent evidence also suggests that by increasing electron transfer efficiency, supercomplex formation is necessary for competitive cellular fitness (Berndtsson et al., 2020). Although a previous report of ERS-induced supercomplex formation in mammalian cells did not address its functional significance (Balsa et al., 2019), our results in yeast show that increased supercomplex assembly is linked to ERS adaptation (Fig. 5). Importantly, the changes underlying adaptation result in reduced mitochondrial ROS accumulation (Fig. 2).
We hypothesize that respiratory supercomplex assembly to mitigate mitochondrial ROS production is a molecular basis of ERS adaptation. Increased mitochondrial cristae density is associated with ERS response (Balsa et al., 2019) – which is consistent with mitochondrial cristae morphology modulating oxidative phosphorylation machinery and bioenergetics (Cogliati et al., 2016) – possibly reflecting a means for increased packing of respiratory complexes within the inner mitochondrial membrane. Further work is necessary to identify the signal produced by ERS that leads to supercomplex formation. Potentially, supercomplex formation could be driven by the local concentration of the ETC complex components, or peroxisome-based alterations in the lipid environment of the mitochondrial inner membrane.
Here, we identify the contributions of peroxiredoxins, Cu-Zn superoxide dismutase and Cta1 catalase activity in ROS mitigation during ERS (Fig. 2). Because β-oxidation is a ROS-producing activity, Cta1 activity is transcriptionally activated when β-oxidation is upregulated by the RTG pathway (Chelstowska and Butow, 1995). Cytoplasmic Tsa1 has a role in controlling mitochondrially derived ROS because mitochondrial H2O2 can readily diffuse into the cytosol. Tsa1 also mitigates mitochondrial ROS promoted by protein aggregates that damage mitochondria (Hanzen et al., 2016; Weids and Grant, 2014). Loss of Tsa1 and Cta1 results in constitutively higher oxidative stress and is linked to constitutively higher O2 consumption (Fig. 4B; Fig. S6). The relationship between oxidative stress and O2 consumption in tsa1Δ and cta1Δ cells is not currently understood, although oxidative stress has been reported to disturb respiratory activity and promote ROS production (Tiwari et al., 2002).
ROS is known to have hormetic effects such that low ROS levels promote signaling while ROS at high levels is linked to cell death (Finkel, 2011). One can speculate that signaling involved in the mitochondrial response during ERS may depend on a net change in ROS levels. Thus, an imbalance of ROS in mutants such as tsa1Δ and cta1Δ might contribute to deficient signaling to elicit an adaptive response. Moreover, there are likely other ways by which cellular redox conditions may affect mitochondrial response to ERS, such as oxidative mitochondrial import via the Mia40 pathway (Bihlmaier et al., 2007; Mordas and Tokatlidis, 2015).
We conclude that a series of changes beyond the classical UPR, including peroxisomal support of mitochondrial respiratory efficiency, and regulation of redox metabolism, are required for ERS adaptation and cell survival. It is likely that these same basic adaptive survival mechanisms have developed in all eukaryotes, and these pathways will play an important role in the disease pathogenesis linked to chronic ERS.
MATERIALS AND METHODS
Yeast media and strains
Yeast were grown in standard synthetic complete (SC) medium with 2% glucose (Sherman et al., 1986). Yeast transformations were by lithium acetate protocol (Gietz et al., 1992). Yeast strains were in the BY4742 or BY4741 background. Deletion strains were confirmed by PCR. Double mutants were constructed by mating single mutants, followed by tetrad dissection and selection of the G418+ mutant from a nonparental ditype tetrad.
Protein lysates, western blotting, enzyme assays and cell viability assays
Exponentially growing cells were pelleted by centrifugation and frozen in liquid nitrogen. Lysate was prepared by vortexing cells with glass beads in the presence of protease inhibitors, as described previously (Chang and Slayman, 1991). Protein content of lysates was determined by Bradford assay (Bradford, 1976). Cell lysates were analyzed by SDS–PAGE and western blotting. Monoclonal antibodies against yeast Cox2 (1:3000 dilution; ab110271) and Por1 (1:3000 dilution; ab110326) are from Abcam (UK). Anti-PGK1 antibody is from Invitrogen (1:106 dilution; 459250).
To overexpress MPC1, cells were transformed with pAB423-GPD-MPC1-dsRed, a HIS3-marked 2 µ plasmid (Timon-Gomez et al., 2013; a gift from Markus Proft, Instituto de Biomedicina de Valencia, Valencia, Spain).
To detect oxidative stress response and the UPR, cells were transformed with the URA3-marked centromeric plasmid bearing TRX2-lacZ (Coleman et al., 1999; a gift from Scott Moye-Rowley, University of Iowa, Iowa City, IA) and pJC104 (Cox and Walter, 1996), a URA3-marked high-copy plasmid bearing a UPRE-lacZ reporter (a gift from Peter Walter, University of California, San Francisco, CA), respectively. Exponentially growing cell transformants were harvested by freezing in liquid nitrogen. Cell lysate was prepared, and β-galactosidase activity was assayed as described previously (Rose et al., 1990) and expressed as µmol/min/mg lysate protein.
Cell viability was determined by colony plating assay. After 5 h treatment with tunicamycin (Sigma-Aldrich), cells were normalized to give an equivalent OD600/ml and then plated on SC plates at 30°C. To assay adaptation to ERS, cells were treated with a low dose of tunicamycin (0.5 μg/ml) for 4 h, followed by addition of high dose tunicamycin (10 μg/ml) for 4 h. Cells were then serially diluted in SC medium and plated for viability assay. After a 2 d incubation at 30°C, colonies were counted and expressed as a percentage of colonies formed by untreated cells. Controls from all strains ranged from 253 to 361 colonies, expressed as 100% viability; n=2 for assays of pex34Δ cells; n=3 for assays of tsa1Δ cells.
Cell staining with fluorescent dyes, microscopy and flow cytometry
For detecting ROS, mid-log cells were incubated with dihydroethidium (DHE; MilliporeSigma) for 15 min. To preserve nutrient status, cells were incubated in SC medium, but DHE concentration was then increased to 30 µg/ml. Live cells were washed once and resuspended in phosphate-buffered saline (PBS) before visualizing with an Olympus fluorescence microscope. Images were collected at the same exposure with a Hamamatsu CCD camera and adjusted with Adobe Photoshop software at the same settings.
Fluorescence images were quantitated using NIH ImageJ software. Cell boundaries were identified in a differential interference contrast (DIC) image, then fluorescence within these boundaries was measured from the corresponding DHE image after background subtraction. n≥38 cells were quantified from at least two distinct fields of view.
For measuring mitochondrial membrane potential (MMP) in non-quenching mode, live cells were stained with TMRM (5 nM; Sigma-Aldrich) for 30 min, washed and then visualized by fluorescence and DIC microscopy. Images were collected with an Olympus fluorescence microscope and Hamamatsu CCD camera. Fluorescence images were taken at the same exposure and adjusted using the same settings in Adobe Photoshop. Peroxisomes were visualized using a BY4741 strain with integrated Pex11–mNeonGreen::HIS3 (a gift from Ewald Hettema, University of Sheffield, UK).
For flow cytometry, cells were grown to exponential phase in SC medium and treated with drugs as described. Cells (2 OD600) were collected while growing in log phase, and resuspended in 1 ml phosphate-buffered saline. Cells were then analyzed in triplicate using 200 μl of resuspended cells per well in a Nunclon 96-well flat-bottom plate with an Intellicyt iQue Screener Plus flow cytometer equipped with violet, blue and red (VBR) lasers. Filters used for analysis of GFP/NG levels were BL-1 (530/30) and BL-2 (572/28). Once flow cytometric data were obtained, they were then gated for cells on the ForeCyt Standard Edition 6.2 software to exclude debris based on forward scatter height (FSC-H) and 90-degree side scatter (SSC-H). Singlets were then isolated using FSC-H and forward scatter area (FSC-A). Final singlet count typically fell around 100,000. Overall median fluorescence of singlets for each experiment was taken as an average of triplicate values for the median BL1-H of singlets. Experimental data was then compiled for each experimental group.
Oxygen consumption, mitochondrial isolation and blue native PAGE
Cellular O2 flux was determined by measuring the fall in O2 concentration in a sealed oxygraph. For whole-cell oxygen consumption, exponentially growing cells were pelleted and resuspended in medium at 20 OD600/ml. Cells were then added into a high-resolution Oroboros Oxygraph 2K chamber containing the same medium at a concentration of 2 OD600/ml, and O2 flux was followed at 25°C. Where indicated, antimycin (4 µM) and CCCP (5 μM) (both Sigma-Aldrich) were added.
Mitochondria were isolated as described previously (Meisinger et al., 2006) except the homogenization step was omitted; instead, spheroplasts were vortexed for 30 s in water, and then diluted with an equal volume of homogenization buffer.
Blue native PAGE analysis was essentially as described previously (Wittig et al., 2006). Mitochondrial concentration was determined by Bradford assay (Bradford, 1976). Frozen mitochondria were solubilized with digitonin (Sigma-Aldrich) at a ratio of 0.15 protein:digitonin. Mitochondrial proteins were analyzed on 3–12% gradient blue native gels (Invitrogen). NativeMark unstained protein standards (Invitrogen) were run in order to approximate supercomplex sizes. Gels were run at 100 V for 15 min, then the current was set to 8 mA for 3 h at 4°C. Gels were then transferred to PVDF membranes at 4°C. Before blotting with antibodies, membranes were blocked with 5% milk in Tris-saline buffer. Western blots were visualized using primary antibodies followed by peroxidase-conjugated secondary antibodies and chemiluminescence detection. Anti-Qcr7 was a gift from Martin Ott (University of Stockholm, Sweden). Anti-Cox2 antibody was from Abcam.
Proteomics
Mitochondrial fractions were prepared from untreated and tunicamycin-treated cells (0.5 µg/ml for 5 h), solubilized with RIPA buffer (Thermo Fisher Scientific). The samples, normalized to protein content (75 µg), were proteolyzed and labeled with TMT-6-plex (Thermo Fisher Scientific) for analysis by LC-MS/MS, essentially by following the manufacturer's protocol. Further details are as described in Tank et al. (2018). TMT labeling and LC-MS/MS was done by the Proteomics Resource Facility in the Department of Pathology at the University of Michigan.
Supplementary Material
Acknowledgements
We thank Dan Beard and Fran Van Den Bergh (University of Michigan) for sharing their expertise and oxygraph, and Ewald Hettema (University of Sheffield, UK), Scott Moye-Rowley (University of Iowa), Markus Proft (Instituto de Biomedicina de Valencia) and Martin Ott (Stockholm University) for plasmids, antibodies and yeast strains.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.C.; Methodology: M.O.; Investigation: I.H., E.W., M.O., S.P.; Resources: A.C.; Writing - original draft: A.C.; Writing - review & editing: I.H., E.W., A.C.; Funding acquisition: A.C.
Funding
This work was supported by funds from the University of Michigan Protein Folding Disease Initiative (to A.C.) and the National Institutes of Health (R21 AG058862 to A.C). Deposited in PMC for release after 12 months.
Peer review history
The peer review history is available online at https://journals.biologists.com/jcs/article-lookup/doi/10.1242/jcs.259254.
References
- Balsa, E., Soustek, M. S., Thomas, A., Cogliati, S., Garcia-Poyatos, C., Martin-Garcia, E., Jedrychowski, M., Gygi, S. P., Enriquez, J. A. and Puigserver, P. (2019). ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK-eIF2alpha axis. Mol. Cell 74, 877-890.e876. 10.1016/j.molcel.2019.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndtsson, J., Aufschnaiter, A., Rathore, S., Marin-Buera, L., Dawitz, H., Diessl, J., Kohler, V., Barrientos, A., Buttner, S., Fontanesi, F.et al. (2020). Respiratory supercomplexes enhance electron transport by decreasing cytochrome c diffusion distance. EMBO Rep. 21, e51015. 10.15252/embr.202051015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihlmaier, K., Mesecke, N., Terziyska, N., Bien, M., Hell, K. and Herrmann, J. M. (2007). The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol. 179, 389-395. 10.1083/jcb.200707123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford, M. (1976). A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248-254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- Bravo, R., Vicencio, J. M., Parra, V., Troncoso, R., Munoz, J. P., Bui, M., Quiroga, C., Rodriguez, A. E., Verdejo, H. E., Ferreira, J.et al. (2011). Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 124, 2143-2152. 10.1242/jcs.080762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, A. and Slayman, C. W. (1991). Maturation of the yeast plasma membrane [H+]ATPase involves phosphorylation during intracellular transport. J. Cell Biol. 115, 289-295. 10.1083/jcb.115.2.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelstowska, A. and Butow, R. A. (1995). RTG genes in yeast that function in communication between mitochondria and the nucleus are also required for expression of genes encoding peroxisomal proteins. J. Biol. Chem. 270, 18141-18146. 10.1074/jbc.270.30.18141 [DOI] [PubMed] [Google Scholar]
- Chen, Y. C., Taylor, E. B., Dephoure, N., Heo, J. M., Tonhato, A., Papandreou, I., Nath, N., Denko, N. C., Gygi, S. P. and Rutter, J. (2012). Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 15, 348-360. 10.1016/j.cmet.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati, S., Enriquez, J. A. and Scorrano, L. (2016). Mitochondrial cristae: where beauty meets functionality. Trends Biochem. Sci. 41, 261-273. 10.1016/j.tibs.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Cohen, Y., Klug, Y. A., Dimitrov, L., Erez, Z., Chuartzman, S. G., Elinger, D., Yofe, I., Soliman, K., Gartner, J., Thoms, S.et al. (2014). Peroxisomes are juxtaposed to strategic sites on mitochondria. Mol. Biosyst. 10, 1742-1748. 10.1039/C4MB00001C [DOI] [PubMed] [Google Scholar]
- Coleman, S. T., Epping, E. A., Steggerda, S. M. and Moye-Rowley, W. S. (1999). Yap1p activates gene transcription in an oxidant-specific fashion. Mol. Cell. Biol. 19, 8302-8313. 10.1128/MCB.19.12.8302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, J. S. and Walter, P. (1996). A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391-404. 10.1016/S0092-8674(00)81360-4 [DOI] [PubMed] [Google Scholar]
- den Brave, F. and Becker, T. (2020). Supercomplex formation boosts respiration. EMBO Rep. 21, e51830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, C. B., Waddle, J. A., Hale, W., Dave, V., Thornton, J., Macatee, T. L., Garner, H. R. and Butow, R. A. (2001). Genome-wide responses to mitochondrial function. Mol. Biol. Cell 12, 297-308. 10.1091/mbc.12.2.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre, J. C., Mahalingam, S. S., Proietto, M. and Subramani, S. (2019). Peroxisome biogenesis, membrane contact sites, and quality control. EMBO Rep. 20, e46864. 10.15252/embr.201846864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7-15. 10.1083/jcb.201102095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco, L. V. R., Bremner, L. and Barros, M. H. (2020). Human mitochondrial pathologies of the respiratory chain and ATP synthase: contributions from studies of saccharomyces cerevisiae. Life (Basel) 10, 304. 10.3390/life10110304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genova, M. L. and Lenaz, G. (2015). The Interplay Between Respiratory Supercomplexes and ROS in Aging. Antioxid Redox Signal. 23, 208-238. 10.1089/ars.2014.6214 [DOI] [PubMed] [Google Scholar]
- Gietz, D., St. Jean, A., Woods, R. A. and Schiestl, R. H. (1992). Improved method for high efficiency transformation of intact yeast cells. Nucl. Acids Res. 20, 1425. 10.1093/nar/20.6.1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzen, S., Vielfort, K., Yang, J., Roger, F., Andersson, V., Zamarbide-Fores, S., Andersson, R., Malm, L., Palais, G., Biteau, B.et al. (2016). Lifespan control by redox-dependent recruitment of chaperones to misfolded proteins. Cell 166, 140-151. 10.1016/j.cell.2016.05.006 [DOI] [PubMed] [Google Scholar]
- Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., Sadri, N., Yun, C., Popko, B., Paules, R.et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619-633. 10.1016/S1097-2765(03)00105-9 [DOI] [PubMed] [Google Scholar]
- Haynes, C. M., Titus, E. A. and Cooper, A. A. (2004). Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 15, 767-776. 10.1016/j.molcel.2004.08.025 [DOI] [PubMed] [Google Scholar]
- Hijazi, I., Knupp, J. and Chang, A. (2020). Retrograde signaling mediates an adapative survival response to endoplasmic reticulum stress in Saccharomyces cerevisiae. J. Cell Science 133, jcs241539. 10.1242/jcs.241539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen, J. K., Mursula, A. M., Rottensteiner, H., Wierenga, R. K., Kastaniotis, A. J. and Gurvitz, A. (2003). The biochemistry of peroxisomal β-oxidation in the yeastSaccharomyces cerevisiae. FEMS Microbiol. Rev. 27, 35-64. 10.1016/S0168-6445(03)00017-2 [DOI] [PubMed] [Google Scholar]
- Knupp, J., Arvan, P. and Chang, A. (2019). Increased mitochondrial respiration promotes survival from endoplasmic reticulum stress. Cell Death Differ. 26, 487-501. 10.1038/s41418-018-0133-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. and Butow, R. A. (2006). Mitochondrial retrograde signaling. Annu. Rev. Genet. 40, 159-185. 10.1146/annurev.genet.40.110405.090613 [DOI] [PubMed] [Google Scholar]
- Meisinger, C., Pfanner, N. and Truscott, K. N. (2006). Isolation of yeast mitochondria. Methods Mol. Biol. 313, 33-39. [DOI] [PubMed] [Google Scholar]
- Mordas, A. and Tokatlidis, K. (2015). The MIA pathway: a key regulator of mitochondrial oxidative protein folding and biogenesis. Acc. Chem. Res. 48, 2191-2199. 10.1021/acs.accounts.5b00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley, A. M., Ward, G. P. and Hettema, E. H. (2008). Dnm1p-dependent peroxisome fission requires Caf4p, Mdv1p and Fis1p. J. Cell Sci. 121, 1633-1640. 10.1242/jcs.026344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J 417, 1-13. 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowinski, S. M., Solmonson, A., Rusin, S. F., Maschek, J. A., Bensard, C. L., Fogarty, S., Jeong, M. Y., Lettlova, S., Berg, J. A., Morgan, J. T.et al. (2020). Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. Elife 9, e58041. 10.7554/eLife.58041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Ahuir, A., Manzanares-Estreder, S. and Proft, M. (2017). Pro- and antioxidant functions of the peroxisome-mitochondria connection and its impact on aging and disease. Oxid. Med. Cell Longev. 2017, 9860841. 10.1155/2017/9860841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, S. W., Norman, J. P., Barbieri, J., Brown, E. B. and Gelbard, H. A. (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques 50, 98-115. 10.2144/000113610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova, V. Y., Drescher, D., Kujumdzieva, A. V. and Schmitt, M. J. (2004). Dual targeting of yeast catalase A to peroxisomes and mitochondria. Biochem. J. 380, 393-400. 10.1042/bj20040042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer, T. and Morley, A. (2014). An evolutionary perspective on the Crabtree effect. Front. Mol. Biosci. 1, 17. 10.3389/fmolb.2014.00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore, S., Berndtsson, J., Marin-Buera, L., Conrad, J., Carroni, M., Brzezinski, P. and Ott, M. (2019). Cryo-EM structure of the yeast respiratory supercomplex. Nat. Struct. Mol. Biol. 26, 50-57. 10.1038/s41594-018-0169-7 [DOI] [PubMed] [Google Scholar]
- Rose, M. D., Winston, F. and Hieter, P. (1990). Methods in Yeast Genetics: a Laboratory Manual, p. 198. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Ross, S. J., Findlay, V. J., Malakasi, P. and Morgan, B. A. (2000). Thioredoxin peroxidase is required for the transcriptional response to oxidative stress in budding yeast. Mol. Biol. Cell 11, 2631-2642. 10.1091/mbc.11.8.2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell, J. C., Olson, K. A., Jiang, L., Hawkins, A. J., Van Vranken, J. G., Xie, J., Egnatchik, R. A., Earl, E. G., DeBerardinis, R. J. and Rutter, J. (2014). A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 56, 400-413. 10.1016/j.molcel.2014.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader, M., Costello, J. L., Godinho, L. F., Azadi, A. S. and Islinger, M. (2016). Proliferation and fission of peroxisomes - An update. Biochim. Biophys. Acta 1863, 971-983. 10.1016/j.bbamcr.2015.09.024 [DOI] [PubMed] [Google Scholar]
- Senft, D. and Ronai, Z. A. (2015). UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40, 141-148. 10.1016/j.tibs.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai, N., Schuldiner, M. and Zalckvar, E. (2016). No peroxisome is an island - Peroxisome contact sites. Biochim. Biophys. Acta 1863, 1061-1069. 10.1016/j.bbamcr.2015.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai, N., Yifrach, E., van Roermund, C. W. T., Cohen, N., Bibi, C., IJlst, L., Cavellini, L., Meurisse, J., Schuster, R., Zada, L.et al. (2018). Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 9, 1761. 10.1038/s41467-018-03957-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., Hicks, J. B. and Fink, G. R. (1986). Methods in Yeast Genetics: a Laboratory Manual, pp. 523-585. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Singh, A. P., Salvatori, R., Aftab, W., Aufschnaiter, A., Carlstrom, A., Forne, I., Imhof, A. and Ott, M. (2020). Molecular connectivity of mitochondrial gene expression and OXPHOS biogenesis. Mol. Cell 79, 1051-1065.e1010. 10.1016/j.molcel.2020.07.024 [DOI] [PubMed] [Google Scholar]
- Tank, E. M., Figueroa-Romero, C., Hinder, L. M., Bedi, K., Archbold, H. C., Li, X., Weskamp, K., Safren, N., Paez-Colasante, X., Pacut, C.et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 9, 2845. 10.1038/s41467-018-05049-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timon-Gomez, A., Proft, M. and Pascual-Ahuir, A. (2013). Differential regulation of mitochondrial pyruvate carrier genes modulates respiratory capacity and stress tolerance in yeast. PLoS ONE 8, e79405. 10.1371/journal.pone.0079405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari, B. S., Belenghi, B. and Levine, A. (2002). Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 128, 1271-1281. 10.1104/pp.010999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P. and Ron, D. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664-666. 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- Van Vranken, J. G., Nowinski, S. M., Clowers, K. J., Jeong, M.-Y., Ouyang, Y., Berg, J. A., Gygi, J. P., Gygi, S. P., Winge, D. R. and Rutter, J. (2018). ACP acylation is an acetyl-CoA-dependent modification required for electron transport chain assembly. Mol. Cell 71, 567-580. 10.1016/j.molcel.2018.06.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weids, A. J. and Grant, C. M. (2014). The yeast peroxiredoxin Tsa1 protects against protein-aggregate-induced oxidative stress. J. Cell Sci. 127, 1327-1335. 10.1242/jcs.144022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig, I., Braun, H. P. and Schagger, H. (2006). Blue native PAGE. Nat. Protoc. 1, 418-428. 10.1038/nprot.2006.62 [DOI] [PubMed] [Google Scholar]
- Wojtala, A., Bonora, M., Malinska, D., Pinton, P., Duszynski, J. and Wieckowski, M. R. (2014). Methods to monitor ROS production by fluorescence microscopy and fluorometry. Methods Enzymol. 542, 243-262. 10.1016/B978-0-12-416618-9.00013-3 [DOI] [PubMed] [Google Scholar]
- Wong, C. M., Zhou, Y., Ng, R. W., Kung Hf, H. F. and Jin, D. Y. (2002). Cooperation of yeast peroxiredoxins Tsa1p and Tsa2p in the cellular defense against oxidative and nitrosative stress. J. Biol. Chem. 277, 5385-5394. 10.1074/jbc.M106846200 [DOI] [PubMed] [Google Scholar]
- Zhao, R.-Z., Jiang, S., Zhang, L. and Yu, Z.-B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 44, 3-15. 10.3892/ijmm.2019.4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.