

Abstract
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide. However, there is a lack of adequate means of treatment prognostication for HCC. Pyroptosis is a newly discovered way of programmed cell death. However, the prognostic role of pyroptosis in HCC has not been thoroughly investigated. Here, we generated a novel prognostic signature to evaluate the prognostic value of pyroptosis-related genes (PRGs) using the data from The Cancer Genome Atlas (TCGA) database. The accuracy of the signature was validated using survival analysis through the International Cancer Genome Consortium cohort (n = 231) and the First Affiliated Hospital of Wenzhou Medical University cohort (n = 180). Compared with other clinical factors, the risk score of the signature was found to be associated with better patient outcomes. The enrichment analysis identified multiple pathways related with pyroptosis in HCC. Furthermore, drug sensitivity testing identified six potential chemotherapeutic agents to provide possible treatment avenues. Interestingly, patients with low risk were confirmed to be associated with lower tumor mutation burden (TMB). However, patients at high risk were found to have a higher count of immune cells. Consensus clustering was performed to identify two main molecular subtypes (named clusters A and B) based on the signature. It was found that compared with cluster B, better survival outcomes and lower TMB were observed in cluster A. In conclusion, signature construction and molecular subtype identification of PRGs could be used to predict the prognosis of HCC, which may provide a specific reference for the development of novel biomarkers for HCC treatment.
1. Introduction
The etiology and molecular mechanism of hepatocellular carcinoma (HCC), a significant subtype of liver cancer, remain largely unknown [1]. HCC ranks fourth among the most lethal cancers and lacks appropriate treatment [2]. In the United States, the 5-year survival rate for patients with HCC is approximately 18% [3]. In addition, HCC is a highly heterogeneous disease, which has been documented at interpatient, intertumoral, and intertumoral level [4–6]. Previous studies have indicated that hepatocyte death chronically promotes HCC, but the related molecular mechanism is not well defined [7]. Thus, both the poor prognostic conditions and the unknown molecular mechanisms indicate the urgent need to improve the prognosis of HCC.
Pyroptosis, a type of programmed cell death, manifests as the continuous swelling of cells until the cell membrane ruptures, resulting in the release of intracellular contents, followed by the activation of a strong inflammatory response [8]. As programmed necrosis mediated by gasdermin, pyroptosis is different from other cell death modalities, such as apoptosis and necrosis in the morphological features, occurrence, and regulatory mechanism [9]. Pyroptosis has been reported to take a part in tumor genesis, invasion, and metastasis [10]. Some studies have been found that pyroptosis is widely involved in the occurrence and development of various types of diseases [11, 12] and could inhibit the onset of associated diseases to improve the overall survival (OS) of patients [13]. Pyroptosis has also been confirmed to have strong associations with multiple known biomarkers [14, 15]. A recent study demonstrated the critical regulatory role of pyroptosis in the tumor microenvironment (TME), which provides new therapeutic insights for cancer treatment [16]. Therefore, an in-depth study of pyroptosis may help understand its role in the occurrence and development of cancers including HCC and provide new ideas for the clinical prevention and treatment [17, 18].
The tumor mutation burden (TMB) is the total number of substitution and insertion/deletion mutations that occur/megabase in the exon coding regions of the genes evaluated in one tumor sample. TMB, as a biomarker for high-frequency mutations and neoantigens, plays an important role in the immunotherapy in various cancers [19, 20]. Elevated TMB in tumor cells have more neoantigens, resulting in an increase in antitumor T cells in the TME. Interestingly, patients with high TMB have a higher probability in the response to tumor immunotherapy [21]. Dysregulated TMB has also been reported to be involved in the prognosis of cancers [22]. Different cancer species vary in the expression of TMB [23]. In HCC, the higher of TMB means the worse in the OS of patients [24]. However, whether TMB could serve as a biomarker in HCC is still unclear.
Herein, we constructed a novel prognostic gene signature to explore the prognostic value of pyroptosis-related genes (PRGs) and the relationships with tumor mutation and immunotherapy. Our data suggested that risk score was identified as an independent prognostic factor. Notably, the prognostic prediction of our risk signature was also confirmed by the International Cancer Genome Consortium (ICGC) cohort (n = 231) and the First Affiliated Hospital of Wenzhou Medical University (FAHWMU) cohort (n = 180). Finally, the effects of risk scores and molecular subtypes on TMB and immune filtration were explored to further evaluate the value of the signature in molecular therapy.
2. Materials and Methods
2.1. Data Preparation
The RNA sequencing data and relevant clinical characteristics of 371 HCC patients were downloaded from TCGA database (https://portal.gdc.cancer.gov/). We matched the RNA sequencing data and clinical features according to each patient's ID number and excluded six follow-up tumor samples. Thus, TCGA cohort with 365 HCC patients was finally enrolled as the training cohort. The other 231 patients with HCC, along with their RNA-seq data and clinical features, were obtained from the ICGC database (https://dcc.icgc.org/projects/LIRI-JP/). HCC patients obtained from the ICGC database were derived from a subset of Japanese population with HBV or HCV infection [25]. Thus, the ICGC cohort was used as the testing cohort. The data of TCGA cohort and ICGC cohort were downloaded from public databases; thus, our study followed the public data access policies; there was no ethical relationship involved. The FAHWMU cohort (n = 180) was obtained from the First Affiliated Hospital of Wenzhou Medical University (Wenzhou, China). HCC samples in the FAHWMU cohort were collected from 2010 to 2020, and OS time was used as the main indicator of the survival time. The collection of this cohort was reviewed and approved by the human research ethics committee of the First Affiliated Hospital of Wenzhou Medical University. The patients/participants provided their written informed consent to participate in this study. All the clinical characters of these patients with HCC are shown in Table 1. Meanwhile, 55 pyroptosis-related genes (PRGs) used in this study were extracted from the MsigDB database (http://www.gsea-msigdb.org/gsea/msigdb/) and prior reviews (Table S1) [26–28]. In addition, the data of TMB and immune infiltration of HCC patients in TCGA cohort were also obtained from TCGA database.
Table 1.
The clinical characteristics of TCGA cohort, ICGC cohort and FAHWMU cohort.
| Variables | TCGA cohort (N = 365) | ICGC cohort (N = 231) | FAHWMU cohort (N = 180) |
| Survival status | |||
| OS days (median) | 556 | 780 | 803 |
| Age (median) | 61 | 69 | 64 |
| Gender | |||
| Female | 119 | 61 | 114 |
| Male | 246 | 170 | 66 |
| TNM stage | |||
| Stage I | 170 | 36 | 62 |
| Stage II | 84 | 105 | 50 |
| Stage III | 83 | 71 | 59 |
| Stage IV | 4 | 19 | 9 |
| Unknown | 24 | 0 | 0 |
| Grade | |||
| G1 | 55 | NA | 24 |
| G2 | 175 | NA | 76 |
| G3 | 118 | NA | 64 |
| G4 | 12 | NA | 13 |
| Unknown | 5 | NA | 3 |
2.2. Identification of Prognosis-Related DEPRGs
The Wilcoxon rank sum test was used for the differential analysis to identify differentially expressed pyroptosis-related genes (DEPRGs) between HCC samples and adjacent nontumorous samples (P < 0.05). Then, univariate Cox regression analysis was used to further identify prognosis-related DEPRGs. The expression of each gene (i) was adjusted to log2(i + 1) to increase the accuracy of the Cox regression results. In the univariate Cox regression analysis, FDR < 0.05 was set as the cut-off value. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed using the “http://org.Hs.eg.db” and “enrichplot” package.
2.3. Generation and Validation of the Signature
The LASSO Cox regression analysis was applied to identify hub DEPRGs to minimize the risk of overfitting among the signature [29, 30]. The independent variable in the regression was the normalized expression matrix of candidate prognostic DEPRGs, and the response variables were OS and the status of patients in TCGA cohort. Penalty parameter (λ) for the gene signature was determined by tenfold cross-validation following the minimum criteria (i.e., the value of λ corresponding to the lowest partial likelihood deviance). Thus, a total of five optimal genes were screened, and their relevant coefficients were calculated. Next, we used the following formula to calculate the risk score for each patient:
| (1) |
Based on the median risk score, all HCC patients were separated into high- and low-risk groups. The different OS between high- and low-risk groups was analyzed via the log rank test. The sensitivity and specificity of the signature were evaluated by time-dependent receiver operating characteristic (ROC) analysis.
The hazard proportional model was constructed by employing multivariate Cox regression analysis to determine the independent prognostic factors. A novel nomogram, including the risk score and other three clinical factors (age, gender, and TNM stage), was constructed to explore the proportional hazards assumption of the multivariate Cox model. Next, the calibration curves of the 1st, 2nd, and 3rd years were generated to verify the accuracy of the nomogram. The correlation between risk scores and individual clinical characteristics was analyzed via Wilcoxon rank sum test.
2.4. Function Enrichment Analyses and Drug Sensitivity Test
All DEGs were screened between the high-risk and low-risk groups using the following filter (∣log2FC | ≥1, FDR < 0.05). The GO and KEGG enrichment analyses were performed based on the “clusterProfiler” R package. Estimated by the Gene Set Variation Analysis algorithm, the infiltrating score of 16 immune cells and the activity of 13 immune-related pathways were calculated. The half-maximal inhibitory concentration (IC50) was estimated by R package “pRRophetic” to evaluate the drug sensitivity [31]. The Connectivity Map (CMap) database (https://portals.broadinstitute.org/cmap/) was used to predict potential chemotherapeutic drugs.
2.5. Consensus Clustering Analyses
In the consensus clustering, the cumulative distribution function (CDF) and consistent matrix were used to evaluate the optimal number of subtypes [32]. Thus, two robust subtypes (clusters A and B) were obtained according to the transcription matrix of the five genes in the signature. The Kaplan-Meier survival curves were performed to analyze the OS of subtypes. The correlations between the subtypes, OS status, and risk score were explored using the “ggalluvial” R package.
2.6. Tumor Mutation Burden Correlation
The TMB score of each HCC patient in TCGA cohort was evaluated using the somatic mutation analysis. We constructed correlation scatter and boxplots based on the Pearson correlation analysis to search the effect of risk score on TMB. Waterfall plots regarding high- and low-risk groups were generated by R package “maftools.”
2.7. Immune Infiltration Analysis
The immune infiltration analysis was performed to calculate the correlation coefficient and construct bubble chart. The ESTIMATE algorithm was applied to derive the corresponding immune score, stromal score, and ESTIMATE score [33]. Next, the proportions of 22 immune cell types of HCC patients were calculated via the CIBERSORT algorithm.
2.8. Quantitative Real-Time PCR (qRT-PCR)
The total RNA from the liver tissues of the FAHWMU cohort was extracted using TRIzol reagent. The mRNA was then reverse transcribed into cDNA using ribo SCRIPTTM reverse transcription kit. The expression level of mRNA was calibrated with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The designed primers are shown in Table S2. SYBR Green master mix was added, and real-time PCR was carried out using a 7500 rapid quantitative PCR system (Applied Biosystems, USA). The CT value of each well was recorded, and the relative quantification of the amplified products was performed using the 2−ΔCt method.
2.9. Statistical Analysis
Here, the R version 3.6.1 (http://www.R-project.org) and its appropriate packages were used to perform all statistical analyses. P < 0.05 was considered as the standard of significantly statistical difference. The FDR method was used for multiple testing. Pearson test was used to compare the categorical variables. The overall workflow of this study is shown in Figure 1.
Figure 1.
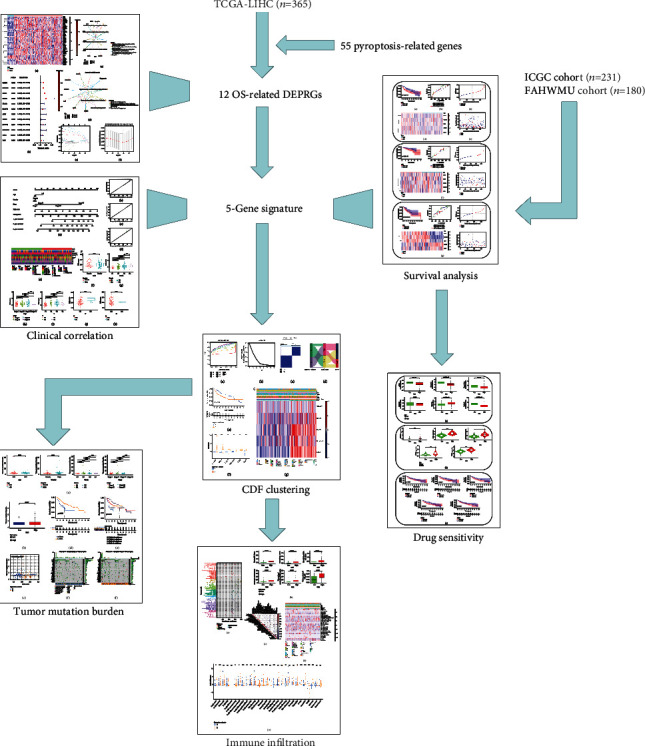
The flow chart of this study.
3. Results
3.1. Twelve Prognosis-Related DEPRGs Were Identified between Adjacent Nontumorous Samples and HCC Samples
The expressions of 55 PRGs were compared between 50 adjacent nontumorous and 365 HCC samples, and 40 DEGs were identified (P < 0.05). Among these, the expressions of 3 genes (IL6, IL1B, and NLRP3) were found to be downregulated in the tumor group while others were upregulated compared to the adjacent nontumorous group. The expression of these DEGs was shown in the heat maps (Figure 2(a)). The univariate Cox regression analyses further identified 12 DEPRGs regarding OS. The DEPRGs with P < 0.05 and hazard ratio (HR) > 1.000 were regarded as prognosis-related DEPRGs (Figure 2(b)). In addition, the GO enrichment analyses revealed that prognosis-related DEPRGs were mainly enriched in the pathways, including activation of cysteine−type endopeptidase activity involved in the apoptotic processes (Figure 2(c), P < 0.05 and Q < 0.05). The KEGG pathway enrichment plots demonstrated enrichment in Kaposi sarcoma-associated herpesvirus infection, necroptosis, and human immunodeficiency virus 1 infection (Figure 2(d), P < 0.05 and Q < 0.05). The overfitting of genes during the signature generation was prevented by LASSO regression analysis and finally identified the optimal five genes (GSDMC, DHX9, CHMP4B, BAK1, and NOD2) (Figures 2(e) and 2(f)).
Figure 2.
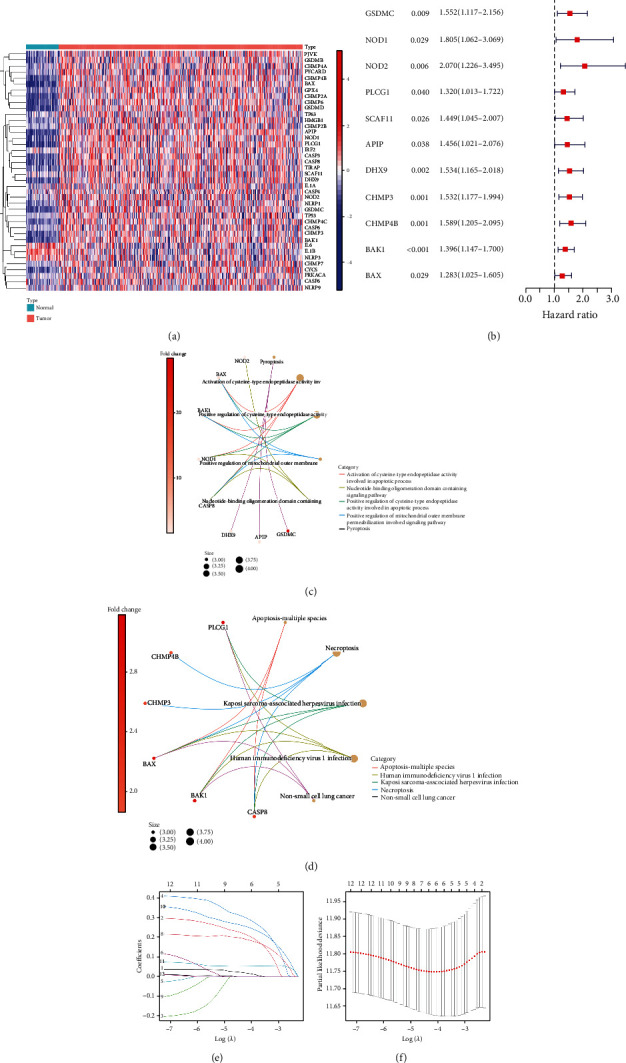
Identification of five optimal prognosis-related DEPRGs. (a) The heat map illustrated the differential expression of 40 PRGs between tumor samples and adjacent normal samples. (b) The Forest plot displayed the univariate Cox regression analyses regarding overall survival, and only DEPRGs with P < 0.05 and HR > 1 were regarded as prognosis-related DEPRGs. (c) CIRC plot for GO function enrichment of 12 prognosis-related DEPRGs. (d) CIRC plot for KEGG pathway enrichment of 12 prognosis-related DEPRGs. (e, f) LASSO regression analyses for screening five optimal PRGs.
3.2. Prognostic Value of the Signature Was Validated in TCGA Cohort and the Extra Validation Cohort
In TCGA cohort, using five optimal genes and the relevant coefficients, the risk score was calculated using the following formula:
| (2) |
According to the median score calculated by the risk score formula, 365 patients were separated into low- and high-risk groups (Figures 3(c) and 3(d)). With an increase in the risk score, there was a gradual decrease in the survival time as well as an increase in the number of patients in death status (Figure 3(e)). Compared to the high-risk group, the low-risk group showed a better OS probability (Figure 3(a), P < 0.05). We found that the area under the ROC curve (AUC) reached 0.729 for 1st year, and the AUC value for 2nd and 3rd years was both >0.600 (Figure 3(b)). HCC patients in the ICGC cohort and the FAHWMU cohort were also divided into high-risk group and low-risk group. The results of the survival analysis were similar to TCGA cohort (Figures 3(f) and 3(g)). Taken together, our results suggest the potential prognostic value of our signature in the prognosis of HCC patients.
Figure 3.
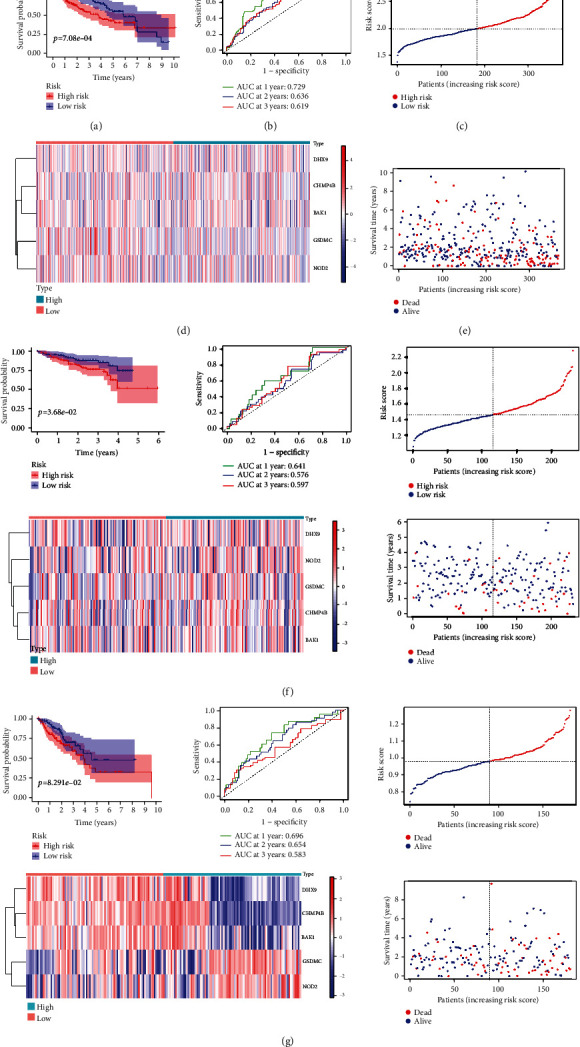
Generation and validation of the signature. (a) K-M survival curve displays the OS of patients between different risk groups in TCGA cohort. (b) Time-dependent ROC curves of the patients in TCGA cohort. (c) Heat map illustrated the expression patterns of five PRGs in TCGA cohort. (d) Distribution of the risk score in TCGA cohort. (e) Survival status scatter plots of patients in TCGA cohort. (f) Survival analysis with respect to the ICGC cohort (n = 231). (g) Survival analysis of the FAHWMU cohort (n = 180).
3.3. Risk Score Was Identified as an Independent Prognostic Factor
In the univariate Cox analysis, the risk score and clinical factors (age, gender, and TNM stage) were significantly correlated with OS (Fig. S1A). Importantly, the risk score was confirmed as an independent predictor for OS in the multivariate Cox model (Fig. S1B). The OS-related nomogram was constructed to test the proportional hazards assumption in the multivariate Cox model (Figure 4(a)). Compared with other clinical factors, risk score had a better effect on the OS in the nomogram. With the integration of the risk score and clinical features (age, gender, and TNM stage), the 1st, 2nd, and 3rd years of OS of patients with HCC could be predicted accurately. The subsequent calibration curves further verified the accuracy of the nomogram (Figures 4(b)–4(d)). The correlations between the risk and clinical characteristics (age, gender, tumor grade, TNM stage, T stage, N stage, and M stage) were shown in the complex heat map (Figure 4(e)). With increasing risk, the correlation between tumor grade and risk score was most significant (P < 0.001). The boxplot further confirmed that there was a significant upward trend of risk score with tumor grade from G1 to G4 (Figure 4(j), G1-G4: P = 0.0071). Additionally, with stage T from T1 to T3, the risk score was also significantly increased (Figure 4(i), T1-T3: P = 0.0094). The results of TNM stage were consistent with the results of stage I to stage III (Figure 4(h), stages I-III, P = 0.0015). Compared to the female patients with HCC, a lower risk score was found in male patients with HCC (Figure 4(f)).
Figure 4.
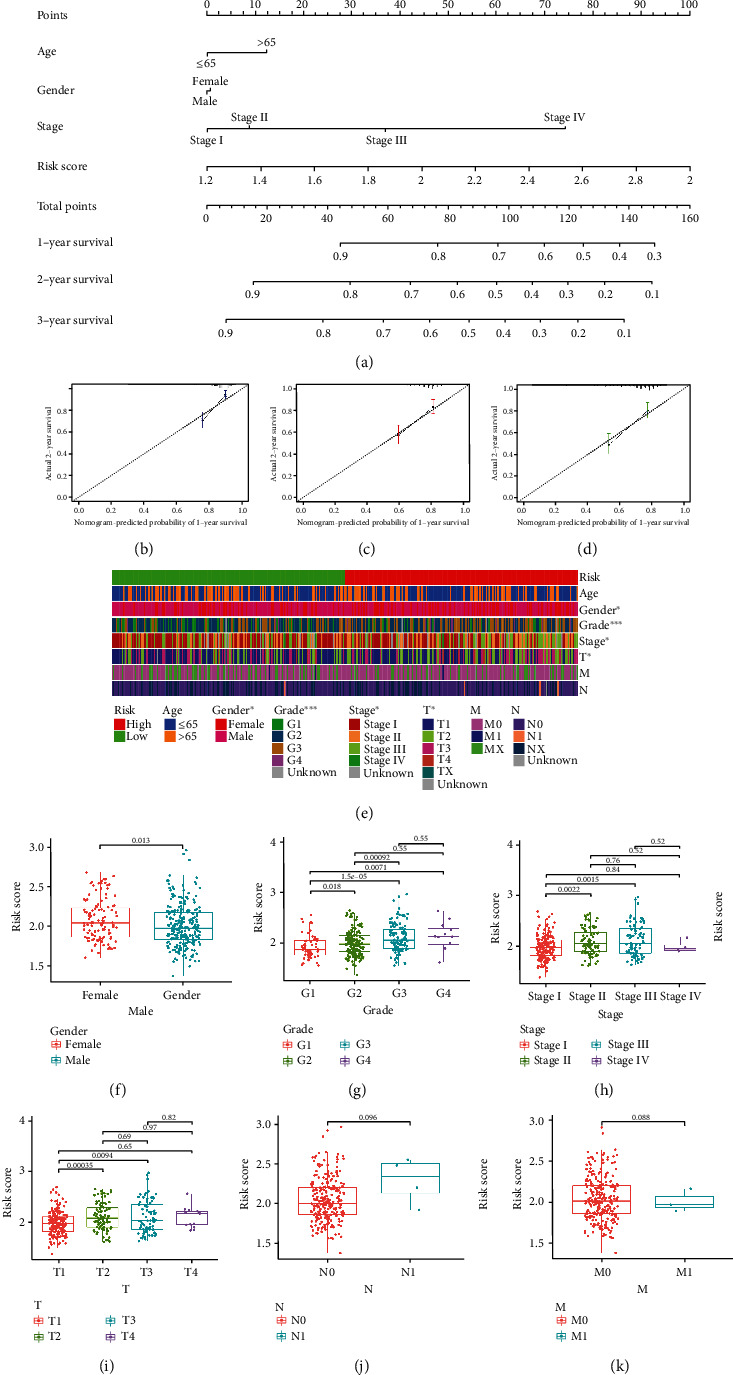
Selection of the independent prognostic factors. (a) The nomogram of the risk score and clinical parameters (age, gender, and TNM stage) in TCGA cohort. (b–d) The calibration curves displayed the accuracy of the nomogram in the 1st, 2nd, and 3rd years. (e) Complex heat map illustrated the correlation between risk and clinical characteristics (∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001). (f–k) Boxplots demonstrated the differences in risk scores across clinical features (F: gender; G: grade; H: TNM stage; I: T stage; J: N stage; K: M stage).
3.4. Functional and the Immunological Activity Analyses
GO function enrichment and KEGG pathway enrichment were performed based on the risk score. In the results of GO enrichment, we found that the DEGs were mainly associated with immune response-activating cell surface receptor signaling pathway and immune response-activating signal transduction (Figure 5(a)), suggesting the involvement of immune infiltration in pyroptosis. KEGG pathway enrichment indicated that DEGs were mainly enriched in human T cell leukemia virus 1 infection, cell cycle, and phagosome (Figure 5(b)). ssGSEA was used to further analyze the scores of immune cells and immune-related pathways between the low- and high-risk groups. The scores of most immune cells (aDCs, DCs, iDCs, Tfh, Th2 cells, and Treg) were significantly increased with the increasing of risk (Figure 5(c), P < 0.001). In addition, the activity of 8 immune pathways in the high-risk group was higher than that in the low-risk group, except for the APC coinhibition, Cytolytic activity, T cell coinhibition, and type I IFN response (Figure 5(d)). Notably, type II IFN response was significantly lower in the high-risk group.
Figure 5.
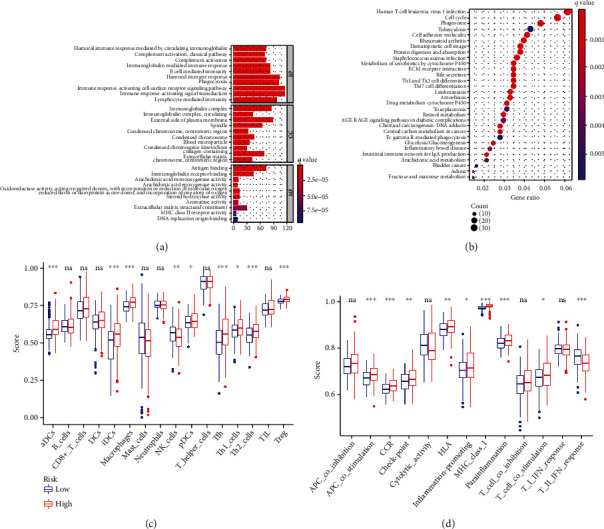
The enrichment analyses of the signature. (a) GO function enrichment of the signature (abscissa: number of DEGs, P < 0.05, Q < 0.05). (b) KEGG pathway enrichment of the signature (abscissa: number of DEGs, P < 0.05, Q < 0.05). (c) Comparison of the enrichment scores of 16 immune cells between different risk groups. (d) Comparison of the enrichment scores of 13 immune-related functions between different risk groups.
3.5. Drug Sensitivity Test Screened Six Potential Chemotherapy Drugs
The boxplots showed the results of drug sensitivity test (Figure 6(a)). By estimating IC50 between the low-risk and high-risk groups, 6 potential chemotherapy drugs were identified. The patients with HCC in the high-risk subtype showed obvious sensitivity to chemotherapy drugs, like ATRA, Bleomycin, Doxorubicin, Etoposide, Nilotinib, and Tipifarnib (all P < 0.05). Moreover, violin plots showed that individual genes in the signature were enhanced in the high-risk group (Figure 6(b)). Additionally, the K-M survival curves of individual genes in the signature showed a better OS in the low-expression group (Figure 6(c)).
Figure 6.
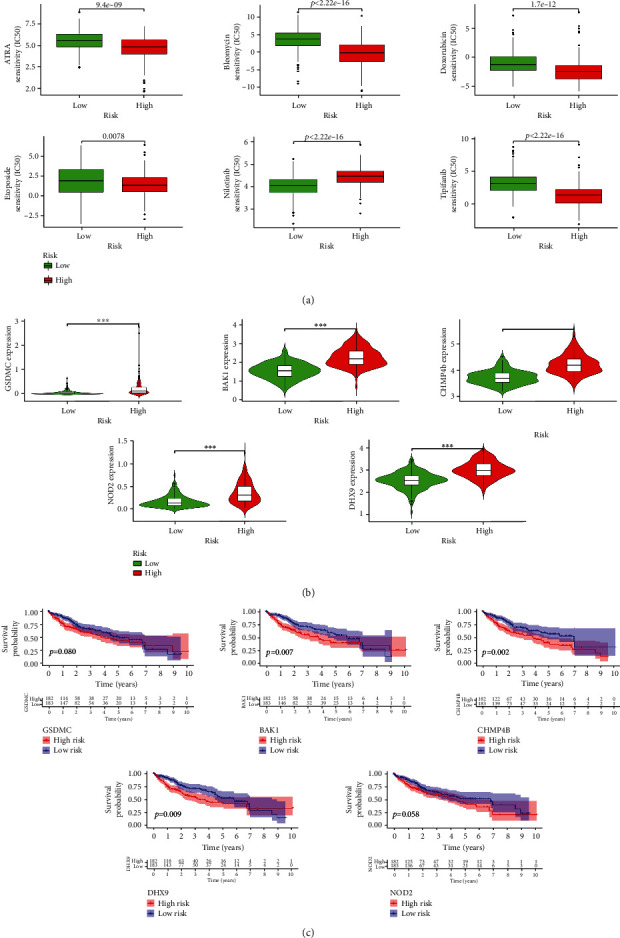
Drug sensitivity of the signature. (a) Boxplots displayed the estimated IC50 of different potential drug sensitivity (ATRA, Bleomycin, Doxorubicin, Etoposide, Nilotinib, and Tipifarnib) between low- and high-risk groups (all P < 0.05). (b) Violin plots presented significant differences in the expression of individual genes across the signature in high- and low-risk groups (∗∗∗P < 0.001). (c) K-M survival curves illustrated that the lower expression of individual genes across signature had better OS (P < 0.1).
3.6. Two Main Subtypes Were Divided Based on the Consensus Clustering Analysis
According to the k value selected by the highest cophenetic correlation coefficient, we divided all patients with HCC into two main subtypes (clusters A and B) (Figures 7(a)–7(c)). Compared to cluster A, patients in cluster B had higher risk scores and worse OS (Figures 7(d) and 7(e)). Moreover, all the expressions of individual genes were found to be higher in cluster B (Figure 7(f), P < 0.001). Combined with other clinical characteristics (age, gender, grade, TNM stage, T stage, N stage, and M stage) and risk score, the complex cluster-based heat map was constructed (Figure 7(g)).
Figure 7.
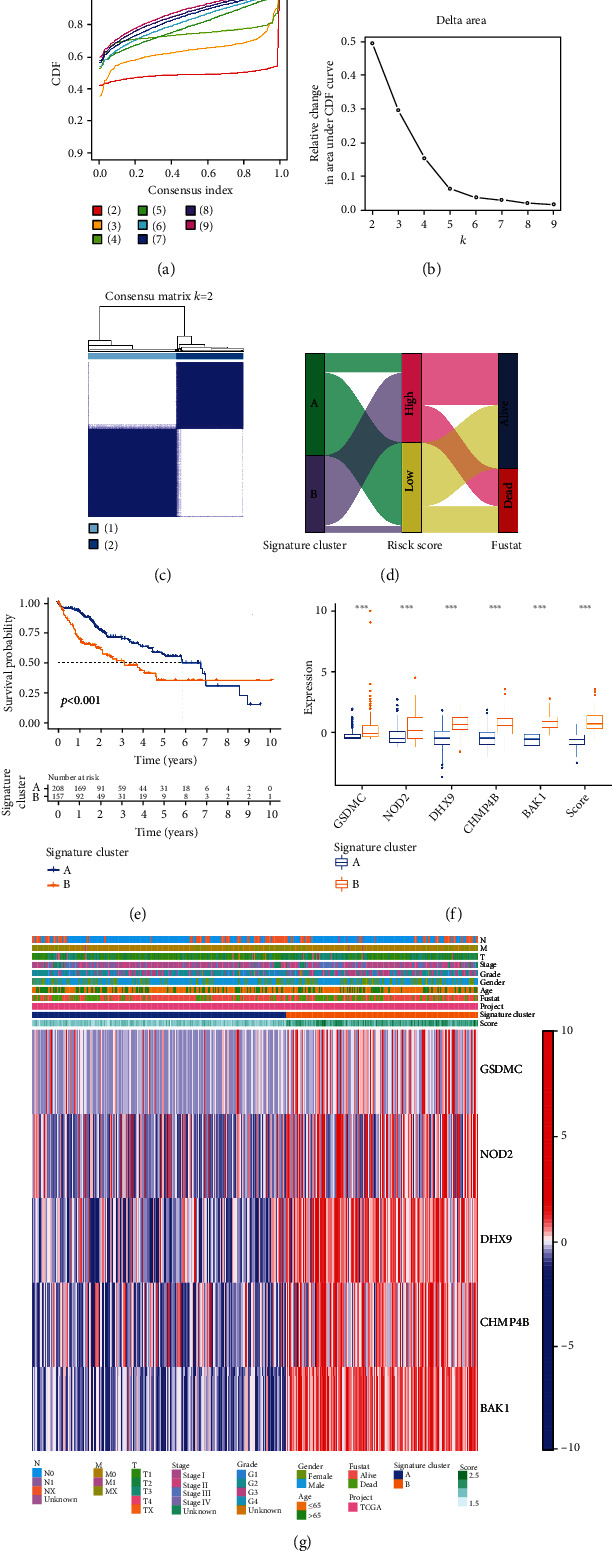
Clustering analyses of the signature. (a, b) Cumulative distribution function based on the signature indicated that the optimal number of subtypes was 2. (c) Concordance matrix of subtypes. (d) ggalluvial of two clusters displayed the correlation between clusters, risk, and survival status. (e) K-M survival curve of the two clusters. (f) Expression of individual genes across signature and risk score between cluster A and cluster B. (g) Complex heat map illustrated the expression patterns between cluster A and cluster B.
3.7. The Potential Correlation between Signature and Tumor Mutation
The association between clinical features and TMB is shown in Figure 8(a). We found that patients with male and >65 y had a higher TMB value. TMB values were obviously increased with tumor grade from G1 to G4 and TNM stage from stages I to II. With an increase in risk score, TMB values were additionally increased (Figure 8(b), P = 0.047). Unfortunately, the correlation between TMB value and risk score was not significant (Figure 8(c), R = 0.019, P = 0.72). Our results showed that the higher level of TMB was observed in cluster A. Interestingly, K-M survival curves showed that the combination of higher TMB value and higher risk score was associated with worse OS (Figures 8(d) and 8(e), P < 0.001). Additionally, waterfall plots revealed that the mutation profiles of patients with HCC was lower in the low-risk group (Figures 8(f) and 8(g)).
Figure 8.
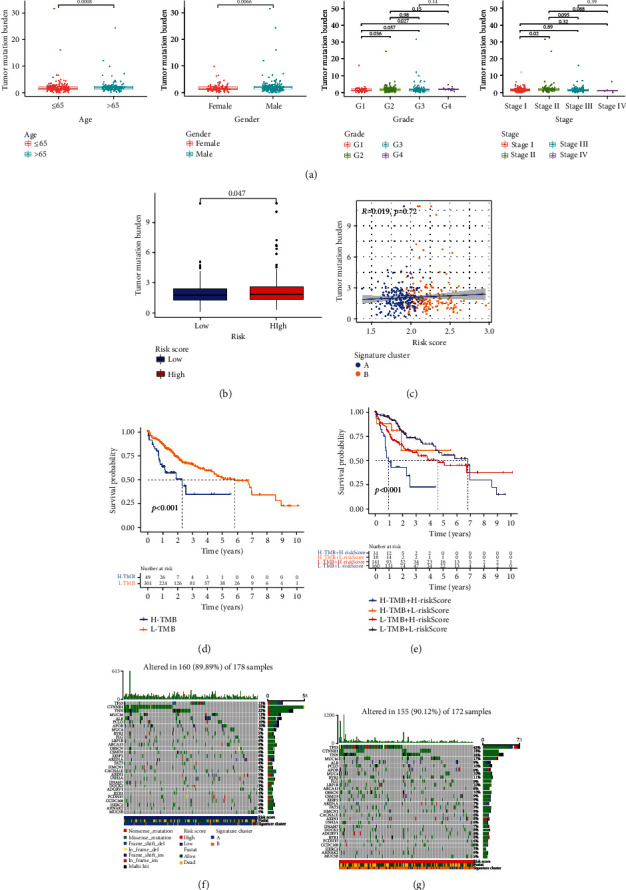
Tumor mutation burden analyses. (a) Boxplots showed the differences in TMB value among different clinical characteristics (age: P = 0.0008, gender: P = 0.0066, and G1-G4: P = 0.027). (b) Boxplot illustrated that the TMB value was significantly higher in the high-risk group (P = 0.047). (c) Scatter plot of correlations between the TMB value and the risk score. (d) K-M survival curve of the high-TMB group and the low-TMB group. (e) K-M survival curve of the comprehensive analyses between TMB and risk score. (f) Comparison of mutation frequencies in the low-risk group. (g) Comparison of mutation frequencies in the high-risk group.
3.8. Significant Correlation between Immune Infiltration and Signature
Next, positive correlations were found between immune infiltration and risk score (Figure 9(a)). Boxplots showed that the contents of 6 immune cells were significantly higher in all the high-risk groups (Figure 9(b), all P < 0.001). Based on the ESTIMATE and CIBERSOFT algorithms, the proportions of 22 immune cell types in patients with HCC, the relevant ESTIMATE scores were calculated (Figure 9(c)). We found that the coefficients of immune cells including T cells and macrophages were significant. The complex heat map revealed the expression patterns of clinical features and the proportions of 22 immune cell types (Figure 9(d)). The boxplot showed the differences in scale of fraction of different immune cells in two clusters (Figure 9(e)).
Figure 9.
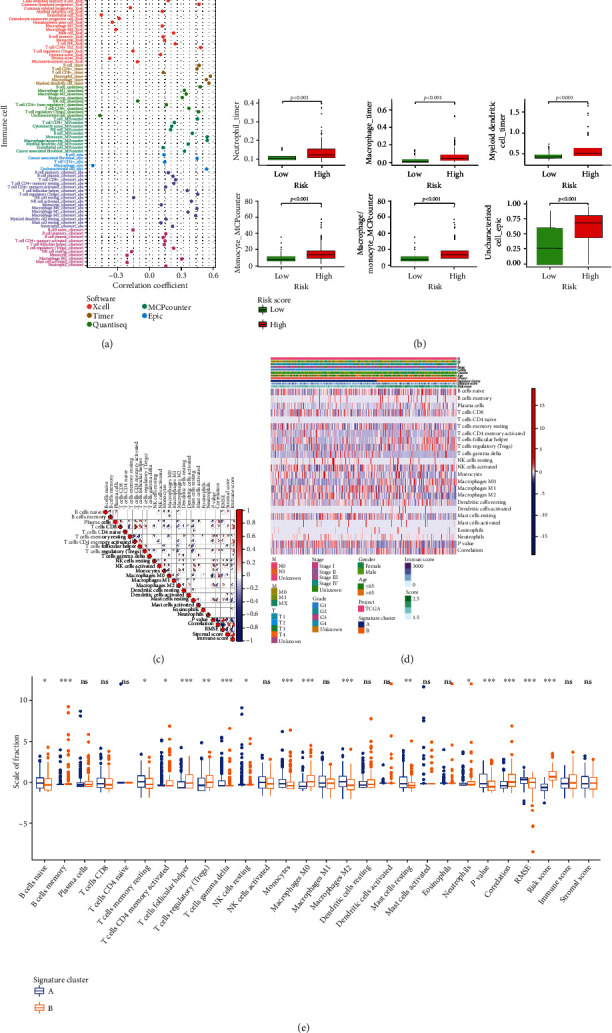
Immune correlation analyses and immune differential analyses of three clusters in the HCC patients. (a) Correlation coefficients between different immune cells and risk score. (b) Boxplots showed the content of immune cells with high correlation coefficients is significantly higher in the high-risk group. (c) The correlation plot of immune cells in TCGA cohort. (d) Complex heat map displayed the association between the expression levels of immune cells and clinical features in the HCC patients. (e) The differential analyses between immune cells and the scale of fraction for cluster A and cluster B.
4. Discussion
Recent studies have identified pyroptosis as a new form of programmed cell death, which plays an essential role in tumor development and treatment mechanisms [8]. Pyroptosis has been found to play a crucial role in various cancers, such as non-small-cell lung cancer and head and neck cancer [34, 35]. Currently, the pyroptosis-related prognostic signature has been constructed in ovarian cancer and gastric cancer, with an excellent prognostic potential [36, 37]. Thus, targeting PRGs may be a promising therapeutic strategy for HCC. However, comprehensive analysis of PRGs for prognosis prediction and targeted therapy in the patients with HCC still remains unclear. In the present study, we aimed to construct a novel prognostic risk signature and identify potential molecular subtypes to better predict the prognosis in HCC. The signature, which was validated by the ICGC cohort and the FAHWMU cohort, contributed to accurate prediction of the OS in patients with HCC. In addition, the high-risk patients identified by this signature were confirmed to be associated with higher TMB, drug sensitivity, and tumor immune cell content. Molecular subtypes (clusters A and B) were identified based on the signature. Further studies revealed that compared with cluster B, better survival outcomes and lower TMB were observed in cluster A. All these results suggest that this signature could serve as a new biomarker to improve the prognosis of HCC.
DHX9, CHMP4B, BAK1, NOD2, and GSDMC were the PRGs included in the prognostic signature. Wang et al. found that DHX9 could interact with CDK6 to promote the growth of HCC [38]. Elevated expression of CHMP4B has been found to play a key role in accelerating cell proliferation and resistance to doxorubicin in HCC [39]. In addition, elevated expression of BAK1 could exacerbate pyroptosis and further aggravate the invasion of HCC [40]. Hepatic NOD2, a well-characterized intracellular PRR of the NOD-like receptor (NLR) family, has been shown to promote hepatocarcinogenesis [41]. In addition, GSDMC, metabolized by α-ketoglutarate and mediated through caspase-8, results in pyroptosis [42]. Overall, increasing studies have confirmed the roles of these genes (DHX9, CHMP4B, BAK1, NOD2, and GSDMC) in HCC.
Previously, Hage et al. found that pyroptosis in macrophages mediates natural killer cell cytotoxicity against HCC [43]. In the present study, the effect of this signature on immune infiltration was also explored. Moreover, there was a positive correlation between macrophages under the TIMER database and risk score. The relative content of macrophages under the TIMER database was elevated in the high-risk group. It is known that drug therapy is crucial for the treatment of HCC [44, 45]. Previous studies have identified Doxorubicin as an effective drug to inhibit HCC via the regulation of apoptosis [46]. Herein, Doxorubicin was also confirmed as a potential drug against HCC, with higher drug sensitivity in the high-risk group. TMB, as a novel biomarker, has been intensively studied in precision medicine for HCC [47, 48]. Xu et al. found that TMB is positively correlated with clinical features in HCC [49]. Liu et al. found that LRP1B mutations are associated with higher TMB and poor prognosis in patients with HCC [50]. We also analyzed the correlation between TMB and the signature. Clearly, there was an obviously lower TMB value in the low-risk group, suggesting a potential correlation between TMB value and the signature.
There are several advantages in this study. First, the prognostic signature could accurately predict the OS for patients with HCC. In addition, the signature is significantly correlated with immune infiltration and TMB, suggesting its biomarker potential in HCC. The prognosis prediction of the signature is further confirmed by the FAHWMU cohort, suggesting its good prognostic prediction ability. More clinical samples are needed to validate the reliability of HCC prognostic value of this signature.
In conclusion, a novel prognostic PRG-signature is constructed for better prediction of prognosis in HCC, which may provide new insights into the treatment of HCC. In addition, this signature is closely associated with TMB and immune infiltration.
Acknowledgments
The project was supported by the National College Students Innovation and Entrepreneurship Training Program (No. 202110343028).
Abbreviations
- HCC:
Hepatocellular carcinoma
- TCGA:
The Cancer Genome Atlas
- ICGC:
International Cancer Genome Consortium
- PRGs:
Pyroptosis-related genes
- LASSO:
Least absolute shrinkage and selection operator
- DEPRGs:
Differentially expressed pyroptosis-related genes
- PDEPRGs:
Prognostic differentially expressed pyroptosis-related genes
- FDR:
False-discovery rate
- OS:
Overall survival
- AUC:
Area under the curve
- ROC:
Receiver operating characteristic
- ssGSEA:
Single-sample gene set enrichment analysis
- K-M:
Kaplan-Meier
- GO:
Gene Ontology
- KEGG:
Kyoto Encyclopedia of Genes and Genomes
- CDF:
Cumulative distribution function
- TME:
Tumor microenvironment
- FAHWMU:
The First Affiliated Hospital of Wenzhou Medical University.
Contributor Information
Boyu Lv, Email: 13780109835@163.com.
Zhengping Yu, Email: yzpwk@126.com.
Changyong Lin, Email: linchangyong111@126.com.
Data Availability
The data and materials can be obtained by contacting the corresponding author.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Ji Chen and Changyong Lin designed the study; Zhengping Yu, Boyu Lv, and Suhui Yu obtained and assembled data; Qiqi Tao and Zhichao Lang wrote the manuscript; Yuxiang Gao and Yan Jin sorted out all the figures and tables; Xiaoqi Li and Yajing Wang revised the manuscript; Yuxiao Zhang performed the literature search. The manuscript was edited by all authors, who have approved the final version for submission. Ji Chen, Qiqi Tao, Zhichao Lang, and Yuxiang Gao contributed equally to this work.
Supplementary Materials
Figure S1: univariate Cox analysis and multivariate Cox analysis containing risk score and clinical factors. Table S1: 55 pyroptosis-related genes used in this study. Table S2: list of primer sequences.
References
- 1.Yang J., Hainaut P., Gores G., Amadou A., Plymoth A., Roberts L. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nature Reviews. Gastroenterology & Hepatology. . 2019;16(10):589–604. doi: 10.1038/s41575-019-0186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Llovet J., Kelley R., Villanueva A., et al. Hepatocellular carcinoma. Nature Reviews Disease Primers . 2021;7(1) doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A., Ward E., Johnson C., et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. JNCI: Journal of the National Cancer Institute . 2017;109(9) doi: 10.1093/jnci/djx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoshida Y., Nijman S., Kobayashi M., et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Research . 2009;69(18):7385–7392. doi: 10.1158/0008-5472.Can-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nault J., Villanueva A. Intratumor molecular and phenotypic diversity in hepatocellular carcinoma. Clinical Cancer Research . 2015;21(8):1786–1788. doi: 10.1158/1078-0432.Ccr-14-2602. [DOI] [PubMed] [Google Scholar]
- 6.Torrecilla S., Sia D., Harrington A., et al. Trunk mutational events present minimal intra- and inter-tumoral heterogeneity in hepatocellular carcinoma. Journal of Hepatology . 2017;67(6):1222–1231. doi: 10.1016/j.jhep.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Boege Y., Malehmir M., Healy M. E., et al. A dual role of caspase-8 in triggering and sensing proliferation-associated DNA damage, a key determinant of liver cancer development. Cancer Cell . 2017;32(3):342–359.e10. doi: 10.1016/j.ccell.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovacs S., Miao E. A. Gasdermins: effectors of pyroptosis. Trends in Cell Biology . 2017;27(9):673–684. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broz P., Pelegrin P., Shao F. The gasdermins, a protein family executing cell death and inflammation. Nature Reviews. Immunology . 2020;20(3):143–157. doi: 10.1038/s41577-019-0228-2. [DOI] [PubMed] [Google Scholar]
- 10.Kolb R., Liu G.-H., Janowski A. M., Sutterwala F. S., Zhang W. Inflammasomes in cancer: a double-edged sword. Protein & Cell . 2014;5(1):12–20. doi: 10.1007/s13238-013-0001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang X., Feng Z., Jiang Y., et al. Nlrp3VSIG4 mediates transcriptional inhibition of and β in macrophages. Science Advances . 2019;5(1, article eaau7426) doi: 10.1126/sciadv.aau7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mamik M., Power C. Inflammasomes in neurological diseases: emerging pathogenic and therapeutic concepts. Brain : A Journal Of Neurology . 2017;140(9):2273–2285. doi: 10.1093/brain/awx133. [DOI] [PubMed] [Google Scholar]
- 13.Zhang F., Zhou B., Yan Z., et al. 6-Gingerol attenuates macrophages pyroptosis via the inhibition of MAPK signaling pathways and predicts a good prognosis in sepsis. Cytokine . 2020;125, article 154854 doi: 10.1016/j.cyto.2019.154854. [DOI] [PubMed] [Google Scholar]
- 14.Ma Y., Pitt J., Li Q., Yang H. The renaissance of anti-neoplastic immunity from tumor cell demise. Immunological Reviews . 2017;280(1):194–206. doi: 10.1111/imr.12586. [DOI] [PubMed] [Google Scholar]
- 15.Gerhauser I., Li L., Li D., et al. Dynamic changes and molecular analysis of cell death in the spinal cord of SJL mice infected with the BeAn strain of Theiler's murine encephalomyelitis virus. Apoptosis : An International Journal On Programmed Cell Death . 2018;23(2):170–186. doi: 10.1007/s10495-018-1448-9. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y., Zhen W., Wang Y., Song S., Zhang H. NaSO nanoparticles trigger antitumor immunotherapy through reactive oxygen species storm and surge of tumor osmolarity. Journal of the American Chemical Society. . 2020;142(52):21751–21757. doi: 10.1021/jacs.0c09482. [DOI] [PubMed] [Google Scholar]
- 17.Tonnus W., Belavgeni A., Beuschlein F., et al. The role of regulated necrosis in endocrine diseases. Nature reviews. Endocrinology . 2021;17(8):497–510. doi: 10.1038/s41574-021-00499-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koren E., Fuchs Y. Modes of regulated cell death in cancer. Cancer Discovery . 2021;11(2):245–265. doi: 10.1158/2159-8290.Cd-20-0789. [DOI] [PubMed] [Google Scholar]
- 19.Chen J., Esteghamat N., Kim E., et al. PD-1 blockade in a liver transplant recipient with microsatellite unstable metastatic colorectal cancer and hepatic impairment. Journal of the National Comprehensive Cancer Network . 2019;17(9):1026–1030. doi: 10.6004/jnccn.2019.7328. [DOI] [PubMed] [Google Scholar]
- 20.le D. T., Durham J., Smith K., et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science . 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hainesworth J. Researchers Strive to Refine TMB. Cancer Discovery . 2021;11(6):p. 1314. doi: 10.1158/2159-8290.CD-ND2021-0107. [DOI] [PubMed] [Google Scholar]
- 22.Gu W., Wang N., Gu W., et al. Molecular gene mutation profiles, TMB and the impact of prognosis in Caucasians and east Asian patients with lung adenocarcinoma. Translational Lung Cancer Research . 2020;9(3):629–638. doi: 10.21037/tlcr-20-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samstein R., Lee C., Shoushtari A., et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nature Genetics . 2019;51(2):202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shrestha R., Prithviraj P., Anaka M., et al. Monitoring immune checkpoint regulators as predictive biomarkers in hepatocellular carcinoma. Frontiers in Oncology . 2018;8:p. 269. doi: 10.3389/fonc.2018.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujimoto A., Furuta M., Totoki Y., et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nature Genetics . 2016;48(5):500–509. doi: 10.1038/ng.3547. [DOI] [PubMed] [Google Scholar]
- 26.Karki R., Kanneganti T. D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nature Reviews. Cancer . 2019;19(4):197–214. doi: 10.1038/s41568-019-0123-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia X., Wang X., Cheng Z., et al. The role of pyroptosis in cancer: pro-cancer or pro-"host"? Cell Death & Disease . 2019;10(9):p. 650. doi: 10.1038/s41419-019-1883-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang B., Yin Q. AIM2 inflammasome activation and regulation: a structural perspective. Journal of Structural Biology . 2017;200(3):279–282. doi: 10.1016/j.jsb.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simon N., Friedman J., Hastie T., Tibshirani R. Regularization paths for Cox's proportional hazards model via coordinate descent. Journal of Statistical Software . 2011;39(5):1–13. doi: 10.18637/jss.v039.i05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tibshirani R. The lasso method for variable selection in the Cox model. Statistics in Medicine . 1997;16(4):385–395. doi: 10.1002/(sici)1097-0258(19970228)16:4<385::aid-sim380>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 31.Geeleher P., Cox N., Huang R. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One . 2014;9(9, article e107468) doi: 10.1371/journal.pone.0107468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaujoux R., Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics . 2010;11(1) doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshihara K., Shahmoradgoli M., Martínez E., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nature Communications . 2013;4(1):p. 2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan R., Zhao W., Wang Q., et al. Cucurbitacin B inhibits non-small cell lung cancer in vivo and in vitro by triggering TLR4/NLRP3/GSDMD-dependent pyroptosis. Pharmacological Research . 2021;170:p. 105748. doi: 10.1016/j.phrs.2021.105748. [DOI] [PubMed] [Google Scholar]
- 35.Cai J., Yi M., Tan Y., et al. Natural product triptolide induces GSDME-mediated pyroptosis in head and neck cancer through suppressing mitochondrial hexokinase-IotaIota. Journal of Experimental & Clinical Cancer Research . 2021;40(1):p. 190. doi: 10.1186/s13046-021-01995-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shao W., Yang Z., Fu Y., et al. The pyroptosis-related signature predicts prognosis and indicates immune microenvironment infiltration in gastric cancer. Frontiers In Cell And Developmental Biology . 2021;9, article 676485 doi: 10.3389/fcell.2021.676485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye Y., Dai Q., Qi H. A novel defined pyroptosis-related gene signature for predicting the prognosis of ovarian cancer. Cell Death Discovery . 2021;7(1):p. 71. doi: 10.1038/s41420-021-00451-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y., Liu J., Yang J., et al. Lnc-UCID promotes G1/S transition and hepatoma growth by preventing DHX9-mediated CDK6 down-regulation. Hepatology . 2019;70(1):259–275. doi: 10.1002/hep.30613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu B., Jiang D., Chen Y., et al. High CHMP4B expression is associated with accelerated cell proliferation and resistance to doxorubicin in hepatocellular carcinoma. Tumour Biology : The Journal Of The International Society For Oncodevelopmental Biology And Medicine . 2015;36(4):2569–2581. doi: 10.1007/s13277-014-2873-1. [DOI] [PubMed] [Google Scholar]
- 40.Hikita H., Kodama T., Shimizu S., et al. Bak deficiency inhibits liver carcinogenesis: a causal link between apoptosis and carcinogenesis. Journal Of Hepatology . 2012;57(1):92–100. doi: 10.1016/j.jhep.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 41.Zhou Y., Hu L., Tang W., et al. Hepatic NOD2 promotes hepatocarcinogenesis via a RIP2-mediated proinflammatory response and a novel nuclear autophagy-mediated DNA damage mechanism. Journal Of Hematology & Oncology . 2021;14(1):p. 9. doi: 10.1186/s13045-020-01028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J., Zhou B., Sun R., et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Research . 2021;31(9):980–997. doi: 10.1038/s41422-021-00506-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hage C., Hoves S., Strauss L., et al. Sorafenib induces pyroptosis in macrophages and triggers natural killer cell-mediated cytotoxicity against hepatocellular carcinoma. Hepatology . 2019;70(4):1280–1297. doi: 10.1002/hep.30666. [DOI] [PubMed] [Google Scholar]
- 44.Arechederra M., Bazai S. K., Abdouni A., et al. ADAMTSL5 is an epigenetically activated gene underlying tumorigenesis and drug resistance in hepatocellular carcinoma. Journal of Hepatology . 2021;74(4):893–906. doi: 10.1016/j.jhep.2020.11.008. [DOI] [PubMed] [Google Scholar]
- 45.Hsu Y., Yip T., Ho H., et al. Development of a scoring system to predict hepatocellular carcinoma in Asians on antivirals for chronic hepatitis B. Journal Of Hepatology . 2018;69(2):278–285. doi: 10.1016/j.jhep.2018.02.032. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y., Liu L., Zhou Y., et al. CKLF1 enhances inflammation-mediated carcinogenesis and prevents doxorubicin-induced apoptosis via IL6/STAT3 signaling in HCC. Clinical Cancer Research : An Official Journal Of The American Association For Cancer Research . 2019;25(13):4141–4154. doi: 10.1158/1078-0432.Ccr-18-3510. [DOI] [PubMed] [Google Scholar]
- 47.Mauriello A., Zeuli R., Cavalluzzo B., et al. High somatic mutation and neoantigen burden do not correlate with decreased progression-free survival in HCC patients not undergoing immunotherapy. Cancers . 2019;11(12):p. 1824. doi: 10.3390/cancers11121824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng H., Zhang Y., Zhou Z., et al. Intergrated analysis of ELMO1, serves as a link between tumour mutation burden and epithelial-mesenchymal transition in hepatocellular carcinoma. eBioMedicine . 2019;46:105–118. doi: 10.1016/j.ebiom.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Q., Xu H., Deng R., et al. Multi-omics analysis reveals prognostic value of tumor mutation burden in hepatocellular carcinoma. Cancer Cell International . 2021;21(1):p. 342. doi: 10.1186/s12935-021-02049-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu F., Hou W., Liang J., Zhu L., Luo C. LRP1B mutation: a novel independent prognostic factor and a predictive tumor mutation burden in hepatocellular carcinoma. Journal of Cancer . 2021;12(13):4039–4048. doi: 10.7150/jca.53124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: univariate Cox analysis and multivariate Cox analysis containing risk score and clinical factors. Table S1: 55 pyroptosis-related genes used in this study. Table S2: list of primer sequences.
Data Availability Statement
The data and materials can be obtained by contacting the corresponding author.