

ABSTRACT
Because the majority of bacterial species divide by binary fission, and do not have distinguishable somatic and germline cells, they could be considered to be immortal. However, bacteria ‘age’ due to damage to vital cell components such as DNA and proteins. DNA damage can often be repaired using efficient DNA repair mechanisms. However, many proteins have a functional ‘shelf life’; some are short lived, while others are relatively stable. Specific degradation processes are built into the life span of proteins whose activities are required to fulfil a specific function during a prescribed period of time (e.g. cell cycle, differentiation process, stress response). In addition, proteins that are irreparably damaged or that have come to the end of their functional life span need to be removed by quality control proteases. Other proteases are involved in performing a variety of specific functions that can be broadly divided into three categories: processing, regulation and feeding. This review presents a systematic account of the proteases of Bacillus subtilis and their activities. It reviews the proteases found in, or associated with, the cytoplasm, the cell membrane, the cell wall and the external milieu. Where known, the impacts of the deletion of particular proteases are discussed, particularly in relation to industrial applications.
Keywords: protein quality control, proteostasis, protein processing, protein folding, degradation
This review systematically discusses the crucial cellular roles of the intracellular, membrane, cell wall and extracellular proteases of Bacillus subtilis.
INTRODUCTION
Five catalytic categories of proteases have been recognized in bacteria: the serine, threonine, cysteine and aspartic proteases, and metalloproteases in which metal ions play a central role in catalysis (Barrett et al. 1998). However, Bacillus subtilis only encodes serine, cysteine, aspartic and metallo-type proteases. The proteases of B. subtilis perform a variety of specific functions that can be divided into three broad categories: (i) quality control—the removal of misfolded or truncated proteins; (ii) regulation—the activation or degradation of proteins that have transitory functions; and (iii) feeding—the degradation of cellular and environmental proteins as a source of amino acids and peptides. In some cases, these functions and the proteins involved overlap, as is best seen in the processing of aberrant of misfolded proteins and the turnover of ribosomal proteins during stationary phase. It is important to understand that cellular proteins are not immortal but have a useful ‘shelf life’, after which they are targeted for degradation. To this end, some proteins have a functionally short half-life, while others are relatively stable. Specified degradation processes are built into the life cycle of some proteins whose activities are required to fulfil a specific function during a prescribed period of time (e.g. ComK: Turgay et al. 1998). Other proteins may be damaged by reactive oxygen or nitrogen species that are either generated endogenously by oxidative respiration or exogenously from compounds in the environment.
This review presents a systematic review of the proteases and their activities in B. subtilis, from the inside to the outside of the cell. It reviews the proteases found in, or associated with, the cytoplasm, the cell membrane, the cell wall and the external milieu. However, it does not discuss proteases associated with the differential process leading to the formation of endospores or the peptidases responsible for cell wall synthesis and turnover. Where known, the impact of the deletion of particular proteases are discussed. An excellent review, specifically of regulated proteolysis in B. subtilis, was published by Molière and Turgay (2013).
CELLULAR PROTEOSTASIS NETWORK
To avoid widespread misfolding, nascent protein chains are directed to a cellular proteostasis network, consisting of chaperones and protease, that maintains a balanced proteome of correctly folded proteins. However, because proteins are structurally dynamic, endogenous and exogenous stresses can lead to misfolding and other forms of damage. Consequently, the proteostasis network also has the role of constantly surveying the existing proteome to help maintain protein homeostasis (Cho et al. 2015; Santra et al. 2017). The proteostasis network is composed of a combination of decapping enzymes, chaperones and proteases (Balchin et al. 2016) and therefore functions from the cradle to the grave.
For newly born proteins, the cell has first to identify their final destinations. During protein synthesis, individual aminoacyl residues are added to the growing nascent peptide chain within the peptidyl transferase centre (PTC) and emerge from the ribosome via the nascent chain exit channel and exit port. The exit channel is ∼100 Å in length and can accommodate a peptide of 20–30 aminoacyl residues. While in the exit channel, folding is constrained, although it is wide enough (10–20 Å) to accommodate some secondary structures such as α-helices (Balchin et al. 2016). In the case of cytoplasmic proteins, the formation of tertiary structure begins towards the distal end of the channel. The exit port itself can accommodate small single-domain proteins (<50 residues) that are able to fold to completion before emerging fully from the ribosome. In the case of multi-domain proteins, their N-terminal domains can fold immediately outside the exit port, in most cases assisted by chaperones such as the ribosome-associated trigger factor (TF) (Stoller et al. 1995; Kaiser et al. 2006; Holtkamp et al. 2015).
The N-terminal residues of membrane and secreted proteins are recognized early in nascent peptide chain synthesis and direct their cognate proteins to the signal recognition particle (SRP) pathway (membrane proteins) or the Sec or twin-arginine transporter (Tat)-mediated secretion pathways (Blaudeck et al. 2001; Janda et al. 2010; Oh et al. 2011). The SRP pathway brings about translational arrest and subsequent cotranslational translocation of their protein cargo directly into the membrane, aided by the YidC (YqjG) insertase/chaperone protein. SecA-dependent secretory preproteins are posttranslationally targeted to the membrane translocase in an essentially non-folded secretion competent state, only finally folding into their native configuration at the trans side of the cytoplasmic membrane (Harwood and Cranenburg 2008). Substrates of the Tat pathway are, however, folded and in some cases (e.g. Fe-S cluster proteins) assembled prior to translocation, presumably aided by cytoplasmic chaperones (Goosens et al. 2013).
In the case of cytoplasmic proteins, the rate of folding is considerably faster than the rate of translation and, as a result, folding is effectively cotranslational (O'Brien et al. 2011). Trigger factor (∼50 kDa), an abundant ATP-independent chaperone with peptidyl-prolyl isomerase (PPIase) activity, engages its target proteins when they are ∼110 amino acids in length (Oh et al. 2011). This means that TF does not engage the membrane and secretory proteins, which are identified much earlier by their cognate recognition factors. TF has an elongated three-domain structure that binds to hydrophobic stretches in the nascent chains of most cytoplasmic proteins. TF slows the rate of cotranslational folding but increases the yield of authentically folded substrates (Kaiser et al. 2011; Cabrita et al. 2016). There is also evidence that the presence of infrequently used codons plays an important role in the process of cotranslational folding by stalling or slowing the rate of translation at key points in nascent chain synthesis to allow the individual domains of multi-domain proteins to fold before the peptide of the next domain emerges from the exit port (Yu et al. 2015). Controlling the rate of synthesis and cotranslational folding therefore helps prevent illegitimate inter-domain interactions and misfolding.
For the majority of proteins that are destined for the cytoplasm, but which are too large to fold within ribosome exit tunnel, chaperones of the proteostasis network (e.g. DnaK(JE) and GroES/EL) interact with the nascent polypeptide chains as they emerge from the ribosome and guide them along a productive folding pathway (Hartl et al. 2011). Misfolding occurs when the so-called off-pathway nascent proteins fail to be recognized by the folding pathway. Off-pathway proteins can be the result from endogenous or exogenous stresses (e.g. heat shock, over production, high osmolarity, etc.) or, in the case of heterologous proteins, because they are perceived as ‘foreign’ and not recognized by the chaperone systems. A major function of the proteostasis network is therefore to prevent aberrant and often irreversible interactions from forming and to remove them if they do (Hartl et al. 2011).
Chaperones can promote protein folding by partitioning non-native states. ATP binding and hydrolysis is used to switch between folding intermediates that are detected by the presence of surface-exposed hydrophobic amino acid residues (Hartl et al. 2011). The interaction between folding intermediates and chaperones helps to block proteins from aggregating and reduces the concentration of aggregation-prone molecules. Chaperones do this by facilitating authentic folding that ensures that hydrophobic residues are located within the structure of the protein (Dobson 2004; Balchin et al. 2016).
About a third of proteins that cannot fold spontaneously or with the help of TF are triaged and directed to either the DnaK/DnaJ/GrpE or GroES/GroEL chaperone systems (Santra et al. 2017). DnaK is an abundant protein that helps facilitate cotranslational folding, by reducing nonproductive inter-domain interactions, a main cause of aggregate formation. DnaK can either release its substrates, allowing them to refold spontaneously, or retain them in a folding competent state before passing them on to other chaperons. DnaK acts together with DnaJ and GrpE also forming an ATPase cycle (as described for GroEL/ES later).
GroEL is a multi-subunit chaperone that forms a nanocage with two cavities (Hayer-Hartl et al. 2016). Each cavity consists of a heptameric ring of ∼57 kDa subunits. GroES forms a dome-shaped heptameric ring of ∼10 kDa subunits that binds to each end of the GroEL cylinder, forming a cage that encapsulates the substrate protein. Extensive conformational changes that both enlarges the ring cavity and alters its physical properties from hydrophobic to hydrophilic allow the substrate protein to fold, driven by intramolecular rather than intermolecular interactions. Ultimately, ATP hydrolysis brings about allosteric changes that lead to the dissociation of GroES and the release of authentically folded protein. Proteins that are still incompletely folded or misfolded may be rebound and subjected to additional folding cycles. If these are unsuccessful, the aberrant proteins can be removed by proteolysis.
PROTEIN RECYCLING
Protein degradation is essential for the maintenance of protein homeostasis, and the amino acids that are generated by degradation are, for the most part, reused for the synthesis of new proteins. This recycling process functions even during bacterial growth and, as a result, protein synthesis and degradation are in a dynamic equilibrium. However, when bacteria have consumed the available amino acids and other nitrogen sources in the culture medium, protein turnover supports the synthesis of new proteins that allows the cell to reconfigure its proteome to resist stress or to differentiate into specific cell types (e.g. endospores, motile cells, etc.). Consequently, protein turnover is critical for the maintenance of viability during conditions of nutrient and other stresses.
The ability to recycle the component subunits that are present in otherwise stable macromolecules plays an important role in cell survival. For example, ribosomes are normally extremely stable macromolecular complexes but, under conditions that lead to marked reductions in cell growth rate, the need for large numbers of functional ribosomes decreases and they become substrates for degradation and recycling (Okamura et al. 1973; Cohen and Kaplan 1977; Zundel et al. 2009; Akanuma et al. 2016). This is significant because ribosomes are the largest single source of cellular proteins and, consequently, of recyclable amino acids. As a result, under nutrient or other stress conditions, entry into stationary phase is characterized by a rapid decline in the number of active ribosomes, to the level that is required for the maintenance of cell viability (Basturea, Zundel and Deutscher 2011; Piir et al. 2011).
The decline in the number of ribosomes also coincides with the appearance of translationally inactive 100S ribosomal particles, referred to a disomes. Disomes result from the dimerization of 70S ribosomes and, being devoid of translational activity, are often referred to as ‘hibernating ribosomes’ (Yoshida and Wada 2014; Shcherbakova et al. 2015). In Escherichia coli, the dimerization of 70S ribosomes to form the 100S ribosome is directed by two proteins, a short form of the ribosome modulation factor (SRMF) and the hibernation promoting factor (Yoshida and Wada 2014; Gohara and Yap 2018). SRMF is found widely throughout the γ-proteobacteria, but less so in other groups of bacteria. Instead, bacteria such as B. subtilis have a long form of the ribosome modulation factor that catalyses this interaction (Beckert et al. 2017). Mutants that are unable to form or dissociate 100S ribosomes are more susceptible to early death and consequently both the formation and dissociation of 100S ribosomes require tight temporal regulation to maintain viability.
In addition to disome formation, the absolute number of ribosomes decreases rapidly during stationary phase, leaving a relatively small number of translating 70S ribosomes. The trigger for ribosome degradation is the increase in free 30S and 50S ribosome subunits, resulting from the reduced translational activity during nutrient starvation (Zundel et al. 2009). The free ribosome subunits, with their exposed RNA interfaces, are substrates for existing ribonucleases, since the synthesis of new genes is not a requirement for degradation to proceed (Zundel et al. 2009). In B. subtilis, while RNase II is responsible for rRNA precursor maturation (DiChiara et al. 2016), RNase Y is responsible for rRNA degradation (Lehnik-Habrink et al. 2012; Segev et al. 2012). The ribosomal proteins released following rRNA degradation are themselves degraded by cellular proteases, as discussed later.
Degrons
Degrons, proteolysis signal tags, have been identified at the N-terminus, C-terminus and internal positions of bacterial proteins. Degrons can either be incorporated within a native protein sequence to specify the protein's turnover rate or generated de novo, either as result of proteolytic cleavage or during the premature release of a nascent polypeptide chain from the ribosome. Proteins with degrons are generally degraded by ATP-dependent proteases (see later). FtsH recognizes degrons directly. However, Clp-like proteases use adaptor proteins to expand the classes of substrates they recognize, as in the case of the ClpX adaptor protein, which binds directly to the ‘ALAA’ motif located at the C-terminal degron of SsrA-processed polypeptides (Moore and Sauer 2007).
Protein stability is also determined by specific amino acid residue at the extreme N-terminus of a protein (so-called N-degrons), with fMet and Leu degrons being particular degradation signals in bacteria (Dougan et al. 2010; Varshavsky 2019). In the case of the fMet degron, this is normally removed shortly after a nascent peptide chain emerges from the ribosomal exit tunnel. The nascent chain's first interaction is with peptide deformylase (PDF) and methionine aminopeptidase (MAP), enzymes that oversee the removal of the N-terminal f-Met. This occur once the chain is ∼40 amino acids in length or at least very shortly after. The fMet degron is designed to identify certain non-native N-terminal sequences or structures, either because they do not collapse rapidly enough in the non-native cellular environment, or collapse into globules that impede deformylation of the N-terminal fMet by PDF. Proteins that retain the fMet residue are then subject to proteolysis, probably by the FtsH, Lon or ClpP-containing proteases (Sauer and Baker 2011; Piatkov et al. 2015).
Ribosomal stalling and the degradation of truncated polypeptides
Ribosomes read and decode the genetic information encoded by messenger RNA in the form of codons until a stop codon is reached. Stop codons not only signal the end of the protein-coding sequence but also serve as the binding sites for factors that promote the release of the nascent polypeptide and the recycling of the ribosome subunits for further rounds of translation. Messenger RNA molecules that lack appropriate termination signals, due to premature transcription termination, transcription errors or the presence of rare codons, are unable to bind release factors. This results in the accumulation of stalled ribosomes that potentially leads to:
a significant loss of translational capacity, due to the sequestering of stalled ribosomes;
the presence of aberrant mRNA molecules that, if not promptly removed, could engage the ribosomal machinery in additional futile translation cycles; and
the presence of aberrant protein products that, if released, might be deleterious for the cell.
Because stalled ribosomes and their peptide products reduce the translational capacity of the cell and are ultimately lethal (Bengtson and Joazeiro 2010), bacteria have evolved two widely conserved mechanisms that mediate subunit recycling, template mRNA degradation and nascent peptide chain proteolysis (Moore and Sauer 2007; Janssen and Hayes 2012; Defenouillère and Fromont-Racine 2017; Joazeiro 2017). These are the trans-translation system, mediated by transfer messenger RNA (tmRNA) and SmpB, and the stop-codon-independent peptide release, mediated by the RqcH/YabO system (Fig. 1).
Figure 1.
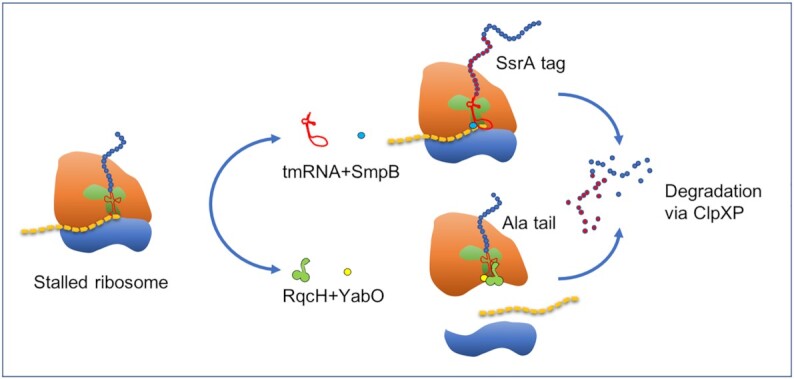
The processing and release of nascent peptide chains from stalled bacterial ribosomes in B. subtilis. The SmpB/tmRNA system tags the nascent peptide chain with a 15-residue-long peptide degron, while the RqcH/YabO system generates an alanine tail. Both C-terminal extensions target the resulting peptides for degradation by ClpXP.
The SmpB/tmRNA system consists of the SmpB protein and the RNA product of the ssrA gene that encodes a hybrid transfer-messenger (tm)RNA molecule (Fig. 1). The tmRNA molecule contains both a tRNA‐like structure at its 5′ end and an internal reading frame encoding a 15-residue-long peptide ‘tag’ at its 3′ end (Karzai et al. 2000; Wiegert and Schumann 2001). The SmpB/tmRNA system recognizes and rescues stalled ribosomes by removing the nascent peptide from the ribosome, allowing the aberrant mRNA to be released and its subunits to dissociated to reinitiate translational activity. It does this by shifting the translation of the nascent peptide from the defective mRNA molecule to the coding sequence encoded by the messenger component of tmRNA. During this process, the tmRNA binds to the empty ‘A’ site in the PTC of ribosomes stalled at the 3′ end of aberrant mRNAs. The alanine-charged tRNA moiety of tmRNA is then covalently bonded to the C-terminus of the incomplete nascent-polypeptide, while the open reading frame in its mRNA moiety allows the ribosome to resume translation, to terminate appropriately and be released. The released peptide has an in-frame C-terminal peptide proteolysis tag (i.e. degron) and, consequentially, the aberrant mRNA molecules and their aberrant/incomplete polypeptide product are targeted for degradation (Moore and Sauer 2007; Janssen and Hayes 2012). In B. subtilis, the translated tmRNA open reading frame encodes a 15-residue-long peptide degron (AGKTNSFNQNVALAA), the so-called SsrA tag, that targets the peptide to the ClpXP protease (Dulebohn et al. 2007; Moore and Sauer 2007; Janssen and Hayes 2012).
While the SmpB/tmRNA system is unique to eubacteria, the ribosome-associated protein quality control (RQC) pathway is present in all domains of life (Bradmann and Hegde 2016; Lytvynenko et al. 2019). The RQC pathway provides an alternative proteolytic pathway in which stalled ribosomes are split by as yet unknown rescue factors, producing 50S subunits that remain obstructed with peptidyl-tRNA (Burroughs and Aravind 2014). Recent work on B. subtilis has shown that RqcH and YabO are functional components of the RQC pathway that sense the incomplete peptide, allowing charged tRNAAla to tag the nascent chains with an untemplated C-terminal Ala tail (Lytvynenko et al. 2019; Crowe-McAuliffe et al. 2020). YabO detects 50S-peptidyl-tRNA complexes by binding to peptidyl-tRNA at the P-site, freeing the A-site for RqcH to deliver Ala-tRNAAla. Cycles of YabO binding and dissociation drive the processivity of alanine tailing (Crowe-McAuliffe et al. 2020), and the resulting homopolymeric Ala peptide functions as a degron proteolysis tag, targeting the peptide to the ClpXP protease for degradation (Fig. 1).
INTRACELLULAR PROTEASES
In B. subtilis, 14 proteases have been identified as being located and active in the cytoplasm of vegetative cells (Table 1). Only one of these proteases, namely Prp responsible for processing ribosomal protein L27, is essential (Wall et al. 2017). However, defective mutants in some of the other proteases exhibit phenotypic traits that are likely to affect growth and productivity. The intracellular proteases include: the AAA+ (ATPases associated with diverse cellular activities) proteolytic machines that are key components of the cellular proteostasis network; proteases such as LonA and IspA that are induced in response to stresses that damage proteins (e.g. heat, oxidative stress) and repressed in the presence of branched chain amino acids; proteases that perform specific regulatory or processing functions. Each of these proteases and their known activities are described later.
Table 1.
Intracellular proteases of B. subtilis.
| Protein | Activity | Location | Regulon | Protein family | Koo mutantsa |
|---|---|---|---|---|---|
| AprX | Alkaline serine protease | Intracellular | LexA, SigA | Peptidase S8 | BKE/BKK17260 |
| ClpC | AAA unfoldase, ATPase subunit of the ClpC-ClpP protease, directs proteins phosphorylated on arginine residues to ClpP | Intracellular—colocalization with ClpP | CtsR, SigM, SigA, SigB, SigF, Spx | ClpA/ClpB | BKE/BKK00860 |
| ClpE | AAA unfoldase, ATPase subunit of the ClpE-ClpP protease (class III stress gene) | Intracellular—colocalization with ClpP | CtsR, SigA | ClpA/ClpB | BKE/BKK13700 |
| ClpP | ATP-dependent Clp serine protease proteolytic subunit (class III heat-shock protein) | Intracellular—colocalization with ClpX | Cts, SigA, SigB | Peptidase S14 | BKE/BKK34540 |
| ClpQ | Two-component ATP-dependent serine protease | Intracellular—colocalization with ClpY | CodY, SigA | Peptidase T1B | BKE/BKK16150 |
| ClpX | AAA unfoldase, ATP-dependent Clp protease, ATP-binding subunit (class III heat-shock protein) | Intracellular—colocalization with ClpP | Cts, SigA, SigB | ClpX chaperone family | BKE/BKK28220 |
| ClpY | Two-component ATP-dependent protease, ATPase subunit | Intracellular—colocalization with ClpQ | CodY, SigA | ClpX chaperone family | BKE/BKK16160 |
| ImmA | A site-specific metalloprotease that degrades ImmR | Intracellular | ImmR | Unknown | BKE/BKK04810 |
| IspA | Serine protease—a major component of the degradome | Intracellular | CodY, SigA | peptidase S8 | BKE/BKK13190 |
| LonA | Class III heat-shock ATP-dependent serine protease | Intracellular—nucleoid | CtsR, SigA | Peptidase S16 | BKE/BKK28200 |
| LonB | Lon-like ATP-dependent serine protease involved sporulation | Intracellular | SigF | Peptidase S16 | BKE/BKK28210 |
| MlpA | Metallopeptidase, involved in regulation of protease gene expression | Intracellular | Not known | Peptidase M16 | BKE/BKK16710 |
| Prp | Cysteine protease—N-terminal cleavage of ribosomal protein L27 | Intracellular | Stringent response | Unknown | None |
| YpwA | Carboxypeptidase, metalloprotease | Intracellular | Not known | peptidase M32 | BKE/BKK22080 |
Koo et al. (2017).
AprX serine protease
AprX (48 kDa) is an intracellular serine protease with a peptidase S8 domain that shows 33% identity to AprE (subtilisin E), the major extracellular protease of B. subtilis (Valbuzzi et al. 1999). AprX is not essential for either growth or sporulation. The aprX gene is 1326 bp in length and transcription analysis indicates that it is expressed in stationary phase. Unlike that of aprE, expression of aprX is not dependent on transition state regulators such as DegU, DegQ, AbrB, SinR and Hpr, but is instead a member of the LexA regulon, induced as part of the SOS response. It is therefore likely to be involved in the degradation of oxidatively damaged proteins. AprX has been detected in the culture medium at late stationary phase, presumably the result of autolysis. Consequently, deletion of the gene encoding AprX reportedly reduces the degradation of secreted heterologous protein at the later stages of cultivation (Kodama et al. 2007).
Clp proteases
Bacillus subtilis encodes a number of Hsp100 family proteases, namely Clp (ClpC, ClpE, ClpP, ClpQ, ClpX and ClpY), Lon (LonA and LonB) and FtsH (Hayer-Hartl et al. 2016; Olivares et al. 2016, 2017). The Clp proteases are discussed in detail in this section while the Lon and FtsH proteases are discussed in separate sections later.
Clp-controlled proteolysis plays a significant role in the B. subtilis cellular proteostasis network, particularly under stress conditions (Ogura and Wilkinson 2001; Erzberger and Berger 2006; Sauer and Baker 2011). Clp proteases form large barrel-shaped hetero-oligomeric complexes that disassemble and then degrade damaged, misfolded or aggregated proteins (Weber-Ban et al. 1999; Wickner et al. 1999). The complexes consist of a hexameric ATPase unfoldase component and a proteolytic component. The ATPase components are members of the AAA+ (ATPases associated with various cellular activities) protein family (Neuwald et al. 1999).
Clp-mediated proteolysis is irreversible and therefore a combination of sequence tags (e.g. degrons), adapter proteins and Clp-complex architecture ensure the specificity of this degradation machinery. The specific sequence tags and/or adaptor proteins are necessary for the recognition, selection and preparation of substrate proteins for degradation by the AAA+ protease complexes. The synthesis and activity of adaptor proteins can be regulated by a variety of mechanisms and input signals. For example, adaptor protein activity can be controlled by sequestration, proteolysis, posttranslational modification or by anti-adaptor proteins (Kirstein et al. 2009; Sauer and Baker 2011; Battesti and Gottesman 2013; Kuhlmann and Chien 2017; Yeom et al. 2017). In B. subtilis, MecA, YpbH and McsB are adaptor proteins for ClpC, YjbH and CmpA for ClpX, and SmiA for LonA (Kirstein et al. 2009; Mukherjee et al. 2015; Tan et al. 2015; Elsholz et al. 2017). The adaptor proteins of ClpC not only recognize substrate proteins, but also facilitate the activation of the ClpC hexamer, which in turn facilitates the subsequent formation of the functional protease complex. In the absence of their substrates, these adaptor proteins are themselves degraded, which leads to the inactivation of ClpC–ClpP proteolytic complex. This regulatory mechanism is designed to curb the activity of the ClpCP protease when potential substrates are not present (Kirstein et al. 2006). A specific targeting mechanism for directing proteins to the ClpC–ClpP complex has been identified in B. subtilis (Schmidt et al. 2014; Trentini et al. 2016). This involves the phosphorylation of arginine residues by the McsB kinase. The docking site for phosphoarginine is located in the amino-terminal domain of the ClpC ATPase. Phosphoarginine therefore functions as a bona fide degradation tag for the ClpC–ClpP protease, a tagging system that is widely distributed across Gram-positive bacteria.
The unfoldase and protease components of the Clp complexes work together in the presence of a suitable substrate. An exposed segment of the substrate, often the degron tag, binds to an axial pore in the unfoldase domain of the complex. Because the axial entry pore is too narrow, even for small protein, the substrate needs to be dis-assembled and spooled into the unfoldase compartment as a more or less unstructured peptide strand (Fig. 2). This dis-assembly is mediated by the ATPase components (ClpC, ClpE, ClpX or ClpY) that use the mechanical energy generated by ATP binding and hydrolysis to facilitate the translocation of the substrate into the unfoldase compartment and thereafter the protease compartment (Horwich et al. 1999; Sauer and Baker 2011). Proteolysis is carried out by the partner protease, ClpP or ClpQ. ClpP colocalizes with ClpC, ClpE or ClpX, while ClpQ colocalizes with ClpY (Gottesman 1996; Grimaud et al. 1998; Wickner et al. 1999; Singh et al. 2000; Hartl et al. 2011; Mogk et al. 2011). In some instances, the unfoldase components, uncoupled from their cognate protease component, can function together with refolding chaperones to disassembly misfolded or aggregated proteins to mediate their refolding (Sauer and Baker 2011).
Figure 2.
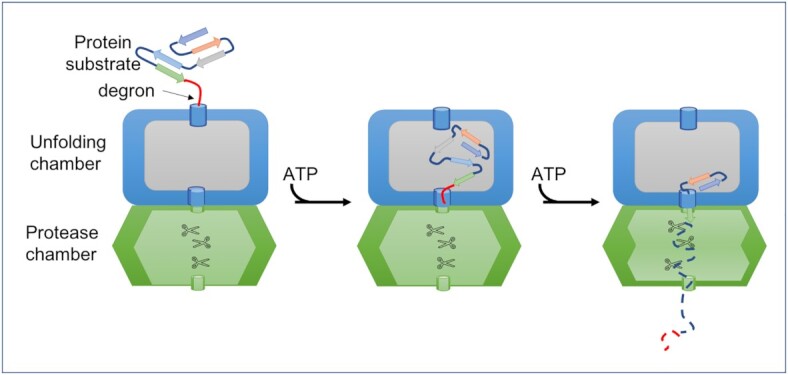
The Clp complex consists of unfolding and protease chambers that function together in the presence of a suitable substrate. The substrate, often with a degron tag, binds to the axial pore in the unfoldase domain and is spooled into the unfoldase compartment by the ATPase components (ClpC, ClpE, ClpX or ClpY), using energy from ATP binding and hydrolysis. From here, the unfolded substrate is channelled to the protease chamber where proteolysis is carried out by the partner protease, ClpP or ClpQ.
Clp proteins have important roles in many cellular processes, including stress responses, competence, sporulation, motility, swarming and biofilm formation. Consequently, mutations in the genes encoding the Clp proteins show a variety of phenotypes: clpC mutants reduce sporulation efficiency, show delayed entry into sporulation, reduced stress tolerance (Krüger et al. 1994; Turgay et al. 1997; Turgay et al. 2001; Meeske et al. 2016; Molière et al. 2016); clpP and clpX mutants are nonmotile and show increased thermotolerance due to the increased stability of Spx and thus increased expression of trxA; clpQ and clpY show impaired swarming motility (Runde et al. 2014; Molière et al. 2016). Deletion of the ClpXP complex improved the expression levels of signal peptidases SipS, SipT, SipV and Lps, leading to overproduction of the B. amyloliquefaciens α-amylase, AmyQ, in B. subtilis (Pummi et al. 2002).
ImmA metalloprotease
The mobile genetic element ICEBs1 is an integrative and conjugative transposon found in B. subtilis. ICEBs1 gene expression is repressed by ImmR, an Xre-type repressor of the immR-immA-int operon (Bose et al. 2008; Bose and Grossman 2011). ImmR repression is relieved by is cleavage by ImmA, a 19 kDa metalloprotease that specifically targets this repressor. ImmA-mediated proteolysis of ImmR, leading to derepression of ICEBs1 gene expression, can be increased by: (i) increasing the amount of ImmA and (ii) increasing the specific activity of ImmA, as both RapI and RecA appear to do by an as yet unknown mechanism (Bose and Grossman 2011). There is little evidence that deletions in ImmA increase protein production.
IspA serine protease
IspA is a 34 kDa intracellular member of the subtilisin family of serine proteases. As a member of the CodY regulon, its expression is repressed during growth in the presence of branched chain amino acids (Barbieri et al. 2015). Originally thought to be essential for sporulation, it is now thought that IspA plays a key role in precise protein processing when the cell enters the stationary phase (Lee et al. 2004). A 2D gel electrophoresis analysis of the intracellular proteome of lspA mutant indicates that, among others, ClpC, EF-Tu, SpoIIE and XkdX are physiologically relevant substrates of this protease (Lee et al. 2004).
There is little evidence that deletions in ispA increase recombinant protein production per se, except for a report that a multiply deleted strain DB431, with deficiencies in the aprE, npr, epr, mpr, ispA and bpr genes leading to an approximate doubling of the concentration of the single chain antibody scFv compared with the wild-type strain (Lakowitz et al. 2018). However, it is not possible to ascribe this increase specifically to a lack of IspA. It should be noted that in the literature, the gene name ispA has also been used for farnesyl pyrophosphate synthase, involved in the methyl erythritol phosphate pathway, and for geranyltransferase, involved in peptidoglycan synthesis (synonym yqiD).
LonA/B ATP-dependent serine proteases
The Lon paralogues, LonA and LonB, are hexameric AAA+ proteases, in which the ATPase domain and the protease domain are present on a single polypeptide chain (cf. the two-component Clp proteases). The proteolytic domains of LonA and LonB are serine–lysine hydrolases, however, the intramolecular environments in which catalytic sites are located are different, due to the presence of additional domains. LonA has an additional domain at the N terminus while the additional domain in LonB is within the AAA+ module. The extended N‐terminal region of LonA includes a domain referred to as HI(CC) (helical inserted with a coiled-coil fragment). The HI(CC) domain is formed exclusively by α‐helices and reminiscent of the structure of the H1 domain of the first AAA+ module of ClpB chaperones (Rotanova et al. 2019).
LonA is an 86 kDa adaptor regulated class III heat-shock ATP-dependent serine protease, a member of the CtsR regulon. LonA does not appear to have a significant role in the degradation of misfolded proteins (Riethdorf et al. 1994; Schmidt et al. 1994; Serrano et al. 2001), except perhaps under heat stress. This is reflected in its different cellular location compared with Clp proteases (Simmons et al. 2008).
LonA is involved in the switch from swimming to swarming motility on surfaces, which is accompanied by the hyperflagellation of the swarming cells (Kearns 2010). The transcriptional activator SwrA determines the number of flagella in B. subtilis cells (Mukherjee and Kearns 2014) and the transition from swimming to swarming is controlled by the regulated proteolysis of SwrA, which in swimming cells is targeted for degradation by LonA and its adaptor protein SmiA (Mukherjee et al. 2015). As a result, mutants in lonA show the presence of pre-differentiated swarmer cells in liquid medium.
LonA is not upregulated in cells overproducing an insoluble heterologous protein, namely PorA, an outer membrane protein from Neisseria meningitidis (Jürgen et al. 2001), and there is little evidence in the literature that deletions in lonA increase heterologous protein production.
LonB is a member of the SigF regulon and is localized to the forespore membrane early in sporulation and to the whole forespore during later stages of the process (Serrano et al. 2001; Simmons et al. 2008).
MlpA metalloprotease
MlpA is a 46 kDa metallopeptidase, a member of the peptidase M16 family. There is evidence that MlpA is involved in the regulation of extracellular protease gene expression, since inactivation of the mlpA gene results in a fivefold increase in the level of proteolytic activity in the growth medium (Bolhuis et al. 2000). Expression of the aprE promoter was strongly increased in a ΔmlpA mutant, indicating that MlpA acts to negatively regulate the expression aprE, independently of DegU, a key regulator known to influence aprE transcription (Smith 1993). MlpA may function either by binding to the upstream sequences of the aprE gene, thereby acting as a repressor or by modulating the activity of a transcriptional regulator of aprE through proteolysis. However, the absence of a helix-turn-helix motif in MlpA makes the latter more likely. This potentially interesting observation appears not to have been followed up. While the ΔmlpA mutant increases extracellular protease activity, and is therefore likely to reduce the recovery of some extracellular products, there is currently no evidence that the upregulation of mlpA improves heterologous protein production.
Prp cysteine protease
Prp is a 12 kDa cysteine protease, required for N-terminal posttranslational cleavage of ribosomal protein L27. Prp is widely conserved among bacteria encoding the L27 N-terminal extension (Wall et al. 2017). Prp is regulated as part of the stringent response regulon that responds to nutrient starvation. Prp is reported to be essential in Staphylococcus aureus (Wall et al. 2017) and likely to be essential in B. subtilis since it was not possible to generate a prp knockout mutant (Koo et al. 2017).
YpwA metalloprotease
YpwA is a 58 kDa M32 family peptidase with carboxypeptidase activity (Lee et al. 2009). It releases a C-terminal amino acid from proteins with broad specificity, excepting for X-Pro (Lee et al. 2009). There is little evidence in the literature that deletions in ypwA increase heterologous protein production.
MEMBRANE PROTEASES
In B. subtilis, 22 proteases have been identified as being active in the cell membrane of vegetative cells (Table 2). None are essential for growth and there is little evidence that the deletion of membrane or membrane-associated proteases impacts on the production of heterologous protein, except for the HtrA-like serine proteases.
Table 2.
Cell membrane and membrane-associated proteases of B. subtilis.
| Protein | Activity | Location | Regulon | Protein family | Koo mutantsa |
|---|---|---|---|---|---|
| ComC | Prepilin processing aspartic protease—ComGC, ComGD, ComGE, ComGG | Cell membrane | ComK, SigA | Peptidase A24 | BKE/BKK28070 |
| CtpA | Carboxy-terminal processing serine protease—YneA, DNA damage checkpoint recovery | Cell membrane—extracellular protease domain | Not known | Peptidase s41a | BKE/BKK19590 |
| DdcP | Carboxy-terminal processing serine protease—YneA, DNA damage checkpoint recovery | Cell membrane—extracellular protease domain | Not known | Peptidase S16 | BKE/BKK15050 |
| FtsH | ATP-dependent metalloprotease—Spo0E, Spo0M, EzrA, SopoIIE | Cell membrane | SigM, TilS, SigA, HprT, SigF | AAA ATPase | BKE/BKK00690 |
| HtpX | Stress-responsive membrane metalloprotease | Cell membrane | YkrK, Rok | Peptidase M48B | BKE/BKK13490 |
| HtrA | Secretion stress-responsive quality control serine protease | Cell membrane—extracellular protease domain | CssR, SigA | Peptidase S1C | BKE/BKK12900 |
| HtrB | Secretion stress-responsive quality control serine protease | Cell membrane—extracellular protease domain | CssR, SigA | Peptidase S1C | BKE/BKK33000 |
| HtrC | Wall stress quality control serine protease | Cell membrane—extracellular protease domain | WalR, SigG | Peptidase S1C | BKE/BKK40360 |
| LspA | Type II signal peptidase | Cell membrane—extracellular peptidase domain | Not known | Peptidase A8 | BKE/BKK15450 |
| PrsW | Cleave RsiW in the presence of antimicrobial peptides | Cell membrane | Not known | Peptidase PrsW | BKE/BKK22940 |
| RasP | Intramembrane metalloprotease, cleaves—FtsL, RsiV, RsiW & released signal peptides | Cell membrane | Not known | Peptidase M50B | BKE/BKK16560 |
| SipS | Type I signal peptidase | Cell membrane—extracellular peptidase domain | SigA | Peptidase S26 | BKE/BKK23310 |
| SipT | Type I signal peptidase | Cell membrane—extracellular peptidase domain | DegU | Peptidase S26 | BKE/BKK14410 |
| SipU | Type I signal peptidase | Cell membrane—extracellular peptidase domain | Not known | Peptidase S26 | BKE/BKK04010 |
| SipV | Type I signal peptidase | Cell membrane—extracellular peptidase domain | LexA | Peptidase S26 | BKE/BKK10490 |
| SipW | Type I signal peptidase | Cell membrane—extracellular peptidase domain | AbrB, SinR, RemA, LutR, SigA | Peptidase S26B | BKE/BKK24630 |
| SpoIVFB | Metalloprotease processing proSigK to the active SigK | Mother cell membrane | SpoIIID, SigE | Peptidase M50B | BKE/BKK27970 |
| SppA | Signal peptide serine peptidase, cleaves signal peptides remnants within the membrane | Cell membrane | SigW | Peptidase S49 | BKE/BKK29530 |
| YhfN | Metalloprotease | Cell membrane | Not known | Peptidase M48B | BKE/BKK10290 |
| YpuD | Intramembrane metalloprotease | Cell membrane | LexA, SigB, SigM | Not known | BKE/BKK23300 |
| YqgP/GluP | Serine peptidase | Cell membrane | PhoP | Peptidase S54 | BKE/BKK24870 |
| YwhC | Site-2 metalloprotease putatively involved in intramembrane proteolysis (RIP) | Cell membrane | Not known | Peptidase M50B | BKE/BKK37530 |
Koo et al. (2017).
ComC aspartic protease
ComC is a 26 kDa A24 family aspartic protease with prepilin leader peptidase and N-methyltransferase activities. ComC is a member of the large ComK regulon that regulates competence development (Mann et al. 2013). It is a late competence gene and is required for the processing and translocation of type IV prepilin-like competence proteins: ComGC, the main pilin component and ComGD, ComGE and ComGG, the minor components (Mann et al. 2013). ComC cleaves a -Gly-|-Phe- bond to release an N-terminal, basic peptide of 5–8 residues from the prepilin, and then N-methylates the resulting N-terminal phenylalanine group, using S-adenosyl-l-methionine as the methyl donor (Chen et al. 2006). A second membrane located enzyme, BdbDC, generates an intramolecular disulfide bond in ComGC prior to pilin assembly. There is no evidence that deletions in comC affect heterologous protein production.
CtpA, CtpB and DdcP serine proteases
CtpA (51 kDa peptidase S41a), CtpB (53 kDa, peptidase S41a) and DdcP (39 kDa, peptidase S16) are carboxy-terminal processing serine proteases. Although unrelated, CtpA and DdcP are both DNA damage checkpoint recovery proteases. They are membrane-associated proteins with an extracellular protease domain. Deletion of the genes encoding CtpA and DdcP leads to the accumulation of the DNA damage checkpoint protein YneA, a small membrane bound inhibitor that delays cell division and thereby provides a time frame during which damaged chromosomes are repaired (Burby et al. 2018, 2019). CtpA and CtpB specifically recognize a C-terminal tripeptide, X-Y-Z, in which X is preferably Ala or Leu, Y is preferably Ala or Tyr, and Z is preferably Ala. However, it cleaves at a variable distance from the C-terminus. In contrast, DdcP (synonym YlbL) contains a transmembrane domain, a Lon protease domain and a PDZ domain (Burby et al. 2018).
CtpB, a paralogue of CtpA, is located in the intracellular space between the mother cell and the forespore. CtpB is a member of the SigE and SigG regulons and is responsible for the cleavage and inactivation of SpoIVFA, an inhibitor of the metalloprotease SpoIVFB responsible for activating the sporulation specific sigma factor, SigK (Sun et al. 2021). There is no evidence that deletions in ctpA, ctpB or ddcP affect heterologous protein production.
FtsH ATP-dependent metalloprotease
FtsH is a 71 kDa membrane-associated FtsH AAA+ ATPase family metalloprotease that forms a hexameric barrel-like complex with ATPase and protease domains. FtsH is involved in cell division, sporulation initiation, biofilm formation, cell envelope stress and heat shock (class III). FtsH appears to be directly involved in protein quality control, since deletion of ftsH causes pleiotropic effects, such as salt stress, defective biofilms and heat sensitivity (Deuerling et al. 1995, 1997). FtsH has also been shown to degrade tmRNA-tagged peptides (Herman et al. 1998; Dulebohn et al. 2007) The FtsH protease is restricted to protein substrates that are locally available and have low thermodynamic stability and, as a result, FtsH appears to have a narrower specificity than ClpXP and ClpAP (Herman et al. 2003). FtsH recognizes an N-terminal degradation tag on the SpoIIE phosphatase, involved in the control of Sigma F activity. It is the differential degradation of SpoIIE in the mother cell but not the fore spore that leads the accumulation of this protein in the latter compartment.
The secretion of bulk exoproteins was severely impaired in a ftsH null mutant after entry into stationary phase. While the activity of α‐amylase increased upon entry into stationary phase in both the wild type and the ftsH mutant, the activity of subtilisin (AprE) was prevented at the level of transcription, presumably due in part to the failure to synthesize appropriate amounts of Spo0A in the ftsH null mutant (Deuerling et al. 1997).
The location of FtsH at the cytoplasmic membrane means that it is well suited to the degradation of membrane proteins and, in E. coli at least, three quarters of the substrates identified by in vivo trapping are located in this membrane (Arends et al. 2016). A good example is that of SecY, a component of the SecYEG protein translocase. When the concentration of SecY was significantly higher than that of SecE or when SecE is under expressed, the fraction of the SecY that is not interacted with SecE is rapidly degraded. The role of FtsH in the degradation of SecY has been identified by both in vivo substrate-trapping and mutation analysis. Overproduction of SecY in a ftsH mutant affected both cell growth and protein export, indicating that the removal of uncomplexed SecY is important for optimal protein export and membrane the integrity (Kihara et al. 1995). However, it has not been established experimentally whether FtsH similarly controls the level of SecY in B. subtilis.
HtpX metalloprotease
HtpX is a 33 kDa peptidase M48B family metalloprotease that has been implicated in membrane protein quality control in response to stress. Expression of the htpX gene is negatively controlled by both Rok and the novel transcriptional regulator, YkrK. However, its induction in response to heat stress appears to be due to transient negative control mediated by SigB in a Rok- and YkrK-independent manner (Lin et al. 2012). The activity of HtpX partially overlap with that of FtsH (Sakoh et al. 2005; Marciniak et al. 2012). While the absence of FtsH or HtpX alone does not impair the viability at high temperatures, the absence of both FtsH and HtpX caused a severe growth defect. This triple negative control at high temperatures may help to avoid uncontrolled and potentially detrimental over synthesis of HtpX (Lin et al. 2012). While heat stress could be a factor in industrial fermenters, there is no evidence in the literature that deletions in htpX affect heterologous protein production.
HtrA-like (HtrA, HtrB, HtrC) serine proteases
Bacillus subtilis encodes three peptidase S1C family HtrA-like serine proteases, namely HtrA (48 kDa), HtrB (49 kDa) and HtrC (43 kDa). All are membrane anchored with their protease domains located on the trans side of the membrane. HtrA and HtrB are protein quality control proteases that scan secretory proteins at the membrane/wall interface and in the wall for structural authenticity (Darmon et al. 2002; Pohl and Harwood 2010). Misfolded or slowly folding proteins are rapidly degraded to prevent interference with cell-wall growth (Jensen et al. 2000; Harwood and Cranenburgh 2008). Although the genes encoding these proteases can be deleted individually without major effects on cell physiology, strains in which both the htrA and htrB genes are deleted exhibited a significant reduction in viability and are prone to accumulating suppressor mutations (Darmon et al. 2002). However, the htrA/htrB double mutant is more stable in the absence of the seven extracellular ‘feeding proteases’ (see later), for reasons that are not fully understood (Pohl et al. 2013). The importance of the HtrA and HtrB became apparent when the expression of htrC was monitored. The absence of these proteases leads to a marked increase in htrC expression, indicating that the accumulation of misfolded secretory proteins at the membrane–wall interface interferes with cell-wall biosynthesis and activates the wall stress-induced WalR operon, in which htrC is located (Hyyryläinen et al. 2001; Noone et al. 2001; Pohl et al. 2013).
Increased levels of HtrA and HtrB produced are produced at the end of exponential growth, possibly because they are predicted to have chaperone activity (Antelmann et al. 2003), although this has yet to be experimentally confirmed. However, the production of these proteases is regulated by the CssR/CssS two-component signal transduction system in response to secretion stress and other stresses such as heat stress and the presence of slowly folding or misfolded proteins. For example, when the native xylanase (XynA) and the Geobacillus stearothermophilus α-amylase (AmyM) are overexpressed in B. subtilis from identical expression vectors, the htrA gene was upregulated in the latter but not the former (Cruz 2016; Ploss et al. 2016). It is not clear why the production of an α-amylase from a related species causes stress in B. subtilis under conditions when the native protein does not. A possible explanation is that the kinetics of α-amylase folding is slow following membrane translocation and that the CssR-CssS regulatory pathway responds rapidly to the presence of hydrophobic residues that would not normally be exposed at the surface of an authentically folded protein (Stephenson et al. 2000). Transcriptionally linking green fluorescent protein (GFP) (or similar reporters) to the htrA promoter and inserting on to the chromosome at an ectopic location provides a useful reporter system for identifying heterologous secretory proteins that are not well tolerated by the B. subtilis secretion pathway.
HtrC is a member of the WalR and Sigma G regulons. It is anchored to the cytoplasmic membrane by a single domain, with the protease moiety on the trans side of the membrane. HtrC is also found at the inner spore membrane where it is responsible for the processing of the spore protein YpeB to a stable form during the early stages of germination. The stable incorporation of SleB, a spore cortex-specific lytic enzyme, requires YpeB, and evidence suggests that the two proteins interact within the dormant spore to stabilize each other. In the absence of HtrC, YpeB fails to stabilize during spore formation when its interaction partner was missing, indicating that other proteases are involved in their degradation during sporulation (Bernhards et al. 2015). The htrC gene is also part of the walR-walK-walH-walI-walJ-htrC operon and is therefore also likely to be induced in response to cell wall stress sensor system, WalRK, as was predicted in the case of the htrA/htrB double mutation mentioned earlier (Pohl et al. 2013).
In summary, decreasing the level of HtrA‐type quality control proteases generally result in growth defects that seem to outweigh any potential benefits to enhancing protein secretion. This is presumably because misfolded protein that accumulate at the membrane/cell wall interface due to the absence of quality control are likely to interfere with cell wall biosynthesis and therefore growth, resulting on the one hand in increased cell lysis and, on the other, in the accumulation of suppressors (Darmon et al. 2002).
LspA type II signal peptidase
LspA is a type II signal peptidase responsible for the removal of signal peptides from lipid-modified preproteins (Prágai et al. 1997; Tjalsma et al. 1999). LspA is a 17.3 kDa A8 family aspartate peptidase with four membrane-spanning α-helices and a β-cradle located at the trans side of the membrane. It has a number of conserved aspartate and asparagine residues, with Asp111 and Asp129 likely to form the active site catalytic dyad (Vogeley et al. 2016). Lsp recognizes a conserved tetrapeptide recognition site, the Lipobox, at the C-termini of lipoprotein signal peptides (Leu-Ala/Ser-Gly/AlaßCys). The g·Leu–g·Ile–g·Ser tripeptide of globomycin mimics the first three residues of the LspA recognition site, inhibiting its catalytic activity. Cleavage generates a Cys residue at the N terminus of the mature protein that is diacyl modified to facilitate attachment to the trans side of the membrane.
PrsW regulatory metalloprotease
PrsW is a 26k Da zinc-dependent metalloprotease responsible for the specific intramembrane proteolytic (RIP) cleavage of the anti-sigma factor RsiW, that in turn moderates the activity of the extracytoplasmic function (ECF) sigma factor W (SigW). The resulting induction of the SigW regulon allows the cell to adapt to the presence of membrane active peptides such as polymyxin and D-cycloserine. PrsW-regulated intramembrane proteolysis (RIP) removes 40 amino acids by cleaving between residues Ala168 and Ser169 of the extracytoplasmic domain of RsiW (Heinrich, Hein and Wiegert 2009; Heinrich and Wiegert2009). Subsequent degradation takes place in the cytoplasm by Clp peptidases. PrsW does not show obvious similarities to other protease family proteins and instead shows similarities to proteins in the Clusters of Orthologous Group (COG) 1266, that share conserved sequence motifs with the eukaryotic type II CAAX prenyl endopeptidase family proteins (Ellermeier and Losick 2006), themselves part of a superfamily of membrane‐embedded metalloproteases (MEM‐superfamily).
RasP regulatory metalloprotease
RasP is a 47 kDa regulatory metalloprotease that cleaves FtsL (Bramkamp et al. 2006), involved in cell division, and anti-sigma factors RsiV and RsiW (Ellermeier et al. 2006). Removing the anti-sigma activities of RsiV and RsiW means that the cells are able to conduct SigV- and SigW-activated RIP. RasP also appears to have a role in controlling the amounts of the quality control proteases HtrA and HtrB and in the degradation of signal peptides following their release by signal peptide peptidases (Park and Schumann 2015). RasP mutants are unable to activate SigW, and therefore have defects in cell division.
The deletion of the gene encoding RasP led to elevated levels of FtsL, HtrA and HtrB, but compromised the production of a number of other membrane proteins (Bramkamp et al. 2006; Zweers et al. 2009). The rasP deletion mutant also affected the processing of the AmyQ α-amylase of Bacillus amyloliquefaciens, AmyE of B. subtilis, AmyL of Bacillus licheniformis and the serine protease BPN’ of B. amyloliquefaciens (Heinrich et al. 2008, 2009; Neef et al. 2017). However, enhanced expression of rasP overcomes the negative effects associated with the secretion of Properase (Danisco US Inc, Palo Alto, Ca, US), a subtilisin variant of Bacillus clausii, and an engineered α-amylase from Paenibacillus curdlanolyticus that belongs to the AmyAc family—in the latter case, boosting production up to 10-fold (Neef et al. 2017). It therefore appears that in some contexts, manipulating the expression of RasP can benefit the production of certain heterologous proteins.
SipS,T,U,V,W type I signal peptidases
Strains of B. subtilis encode a number of type I signal peptidases, some of which are plasmid encoded. However, B. subtilis strain 168 encodes five type I signal peptidases, namely SipS (21kDa), SipT (22kDa), SipU (21kDa), SipV (19kDa) and SipW (21kDa), all of which are chromosomally encoded and members of the peptidase S26 protein family. Their role is to cleave the signal peptides of secretory preproteins to facilitate the release of the mature protein from the membrane. They do this by recognizing an Ala-X-Ala motif at the signal peptidase recognition site, located at positions −3 to −1 relative to the start of the mature protein. While SipS and SipT, SipU and SipV are P-type signal peptidases, SipW is atypical, and more related to ER (endoplasmic reticulum) type signal peptidases (Tjalsma et al. 1998). Although P-type I signal peptidases are serine proteases, they are unusual in having a Ser-Lys dyad (Ser43 and Lys83 in SipS) at the active site, rather than the more usual Ser-His-Asp catalytic triad (Ekici et al. 2008).
SipS and SipT, SipU and SipV have overlapping substrate specificities and are not individually essential. However, cell lacking both SipS and SipT are not viable. SipW is more substrate specific and is discussed separately later. SipS and SipT are regarded as the major signal peptidases and are highly expressed under all conditions. SipS is a member of the SigA regulon and is upregulated in minimal medium during glucose starved stationary phase. SipT is a member of the DegU regulon, but is actually slightly down regulated in minimal medium during glucose starvation. SipU and SipV are minor signal peptidases and their expression is both lower and more variable. SipU is a member of the SigA regulon and is down regulated during sporulation and glucose starvation. SipV is a member of the LexA regulon and its expression is increased in the presence of hydrogen peroxide and diamide.
The ER-type SipW is a member of the RemA, Spo0A, AbrB, LutR and SinR regulons. Site directed mutagenesis indicates that the triad Ser47, His87 and Asp106 is essential for catalytic activity (Tjalsma et al. 2000). Unlike the P-type signal peptidases, SipW is highly substrate specific. It is transcribed as part of the tapA-sipW-tasA operon, and catalytically active SipW is required for the incorporation of mature TasA into spores (Erskine et al. 2018). Moreover, SipW is a bifunctional protein since, in addition to its role in TasA translocation, it was found to activate biofilm matrix genes specifically when cells were on a solid surface. This activity requires the presence of the C-terminal twenty amino acid residues that, unlike the peptidase domain, is located on the cis side of the cytoplasmic membrane (Terra et al. 2012).
SpoIVFB
SpoIVFB is a spore mother cell intramembrane metalloprotease, a peptidase M50B family protein. It has a cystathionine β-synthase domain that regulates access to its active site via an adenine-based nucleotide. SpoIVFB is recruited to the outer forespore membrane where it is responsible for activating the sporulation-specific sigma factor, SigK, by removing its propeptide (Lu et al. 1995; Sun et al. 2021). SpoIVFB is controlled at the both the transcriptional and translational level; it is a member of the SpoIIID and SigE regulons and part of a complex regulatory network that ensures its activity is tightly controlled. SpoIVFB forms a ternary complex with SpoIVFA and BofA in which the role of SpoIVFA is to help recruit BofA to the complex that, in turn, inhibits the proteolytic activity of SpoIVFB. Consequently, degradation of BofA by CtpB (see earlier) activates SpoIVFB (Rudner, Fawcett and Losick 1999; Rudner and Losick 2002).
SppA serine protease
SppA is a 37 kDa peptidase M48B family protein that cleaves the remnant signal peptides within the membrane following signal peptidase activity. SppA synthesis is controlled by the ECF sigma factor, SigW, in response to cell envelope stress. SppA is essential for the efficient translocation and processing of secretory proteins. In a recent study, both nattokinase and α-amylase were used to evaluate the functional activity of SppA in Bacillus licheniformis. Significant decreases in the concentrations of these proteins were observed in a sppA-deficient strain, while the extracellular yields of these proteins were increased in a strain overexpressing SppA (Cai et al. 2017).
YhfN metalloprotease
YhfN is a 49 kDa peptidase M48B family metalloprotease. TMHMM predicts YhfN to be a membrane protein with two loops located on the trans side of the membrane. There is no relevant information about the regulation or activity of this protease in the published literature.
YqgP (GlpG, GluP) serine peptidase
YqgP (synonyms GlpG and GluP) is a 56 kDa intramembrane serine protease. It is a member of a group of rhomboid proteases that cleave type-1 transmembrane domains using a catalytic dyad composed of serine and histidine residues located on separate transmembrane domains. The yqgP gene is present in an operon with yqgQ and glcK, the latter encoding glucokinase. In the single study that looks at the activity of this protein (Mesak et al. 2004), YqgP appears to have a role in glucose transport, sporulation and cell division. The effects on glucose transport are minor and could be due to polarity, while those on sporulation are also minor and could be due to the cell division defect. However, the effect on cell division itself appears to be significant since the ΔyqgP mutants are filamentous (Mesak et al. 2004).
YqgP was recently shown to interact with FtsH and cleave MgtE, a high‐affinity magnesium transporter. This cleavage takes place under conditions of low magnesium and high manganese or zinc, protecting the cells from Mn2+/Zn2+ toxicity (Began et al. 2020). It also presents MgtE or its cleavage products to FtsH for more extensive cleavage: while the proteolytic activity of YqgP is not needed for this activity, its unoccupied active site is essential. There is no evidence that deletions in yqgP affect heterologous protein production.
YwhC metalloprotease
YwhC is a 25 kDa site-2 peptidase M50B family metalloprotease. TMHMM predicts YwhC to be a membrane protein with three loops located on the trans side of the membrane and two on the cis side. There is no relevant information on this protease in the published literature.
Extracellular proteases
Bacillus subtilis and its close relatives are major producers of industrial enzymes, among which alkaline serine proteases are of major commercial significance (Priest 1977; Harwood 1992). The main class of alkaline serine proteases isolated from B. subtilis are known as subtilisins, although slightly different versions of this protease have been purified from different isolates of B. subtilis (e.g. Subtilisin Novo, Subtilisin BPN', Subtilisin Carlsberg). These enzymes were first purified in the 1960s and their catalytic activities and structures have since been studied extensively. Indeed, this group of enzymes was used as model proteins for the development of protein engineering techniques, with the objective of designing engineered variants with improved stability and catalytic activity under a range of commercially relevant environments (Thomas et al. 1985; Bryan 2000). The main drivers were the development of so-called biological detergents for the laundry market and the observation that the native enzyme was rapidly inactivated by detergents at the high temperatures involved. Proteases can also be classified on the basis of: (i) pH, (ii) substrate specificity, (iii) similarity in action to well-characterized enzymes such as trypsin, chymotrypsin and elastase and (iv) active site amino acid residue and catalytic mechanism.
Bacillus subtilis encodes eight extracellular proteases, none of which are essential for growth or viability (Table 3). Five of these enzymes are serine proteases, the remaining three are metalloproteases. AprE (subtilisin) and NprE are the most abundant proteases and are found in the culture medium during stationary phase where they contribute >95% of the extracellular proteolytic activity of B. subtilis. The remaining five extracellular proteases (Bpr, Epr, Mpr, NprB, Vpr) and single wall-associated protease (WprA), are responsible for most of the remaining activity, together with intracellular proteases release as a result of cell lysis.
Table 3.
The extracellular and wall-associated proteases of B. subtilis.
| Protein | Activity | Location | Regulon | Protein family | Koo mutantsa |
|---|---|---|---|---|---|
| AprE | Major serine protease subtilisin E—feeding protease | Extracellular | SinR, ScoC, AbrB DegU, CodY, SigA | Peptidase S8 | BKE/BKK10300 |
| Bpr | Minor serine bacillopeptidase F | Extracellular | DegU | Peptidase S8 | BKE/BKK15300 |
| Epr | Minor serine protease—control of swarming motility | Extracellular | SinR, SocC, Spo0A, SigD, DegU | Peptidase S8 | BKE/BKK38400 |
| Mpr | Minor metalloprotease | Extracellular | CodY | Peptidase S1B | BKE/BKK02240 |
| NprB | Minor Neutral metalloprotease B—facilitates the YIT toxin | Extracellular | DegU | Peptidase M4 | BKE/BKK11100 |
| NprE | Major neutral metalloprotease—feeding protease | Extracellular | SooC, AbrB, CodY | Peptidase M4 | BKE/BKK14700 |
| Vpr | Minor serine protease—processing protease | Extracellular | CodY, PhoP, LexA, DnaA, SigH | Peptidase S8 | BKE/BKK38090 |
| WprA | Serine protease—quality control protease | Extracellular cell wall associated | CcpA, YvrHb, SigA | Peptidase S8 | BKE/BKK10770 |
Koo et al. 2017.
AprE alkaline serine protease
AprE (39 kDa), or subtilisin, is the major extracellular protease produced by B. subtilis and is the protease that has been most widely exploited commercially. Its regulation is extraordinarily complex since it is regulated as part of the SinR, ScoC, AbrB, DegU, CodY and SigA regulons (Barbieri et al. 2016). A discussion of its complex regulation is beyond the scope of the current report, except to say that, like a number of other extracellular enzymes, its expression is upregulated in the DegUhy mutant of the DegS-DegU two-component system (Olmos et al. 1997; Mäder et al. 2002; Cairns, Hobley and Stanely-Wall 2014). The DegS-DegU system is responsible for regulating a number of transition phase processes in addition to extracellular degradative enzymes, including competence development and the switch between swimming and biofilm formation. The DegUhy mutation stabilizes the phosphorylated form of the response regulator (DegU-P), and the resulting overproduction of certain extracellular degradative enzymes is widely used in their commercial production.
While the native AprE enzyme was of commercial value for the degradation of proteinaceous stains, its susceptibility to detergents and temperature limited its commercial application. As a result, variants of the enzyme were screened for improved operational characteristics. The resulting structure/activity studies on the variants resulted in the use of AprE as a model for the development the tools for engineering proteins with improved characteristics (e.g. temperature tolerance, detergent resistance and modified substrate specificity; Bryan 2000). AprE was the first B. subtilis protease gene to be cloned and sequenced (Stahl and Ferrari 1984; Wong et al. 1984; Wong and Doi 1986). The primary translation product of the aprE gene is a 381 residue prepropeptide, consisting of a 29-residue signal peptide, a 77-residue propeptide and a 275-residue mature protein. The Class I propeptide of AprE is essential for its rapid posttranslocational folding (Yabuta et al. 2002). It functions by overcoming the large kinetic barriers in the productive folding pathway and is a potent inhibitor of the enzyme's activity (Yabuta et al. 2001). During the translocation of the preproprotein across the membrane, the signal peptide is removed in the usual manner by a Type I signal peptidase, during or immediately following translocation. The propeptide then accelerates post-translocational folding by stabilizing an intermediate complex that provides the nucleus for folding (Gallagher et al. 1995; Wang et al. 1998). Once proAprE is folded, the propeptide temporally inhibits its proteolytic activity (Fu et al. 2000). Full subtilisin activity is only achieved after proteolytic self-cleavage and the subsequent degradation of the propeptide (Yabuta et al. 2001). In the absence of the propeptide, the protein is trapped in a molten globular-like intermediate folding state (Wang et al. 1998). Propeptide catalysed folding and propeptide removal are necessary for subtilisin to pass through the cell wall, presumably because exposed hydrophobic residues can interact with cell wall components (Power et al. 1986). Although propeptides are intrinsic intramolecular chaperones, they can be provided extrinsically to catalyse the folding of their cognate mature protein in vitro in both an intra and intermolecular fashion.
Subtilisin E has two calcium binding sites, the high-affinity Ca1 site and the low-affinity Ca2 site. The Ca1 site is conserved in various subtilisin-like proteases and is important for stability. This site is not formed in Pro-subtilisin E, because the structural rearrangement of the N-terminal region of the subtilisin domain upon autoprocessing is necessary for the formation of this site. As a result, Pro-subtilisin E is not fully folded (Vévodová et al. 2010; Uehara et al. 2013).
Bpr serine bacillopeptidase F
Bpr, referred to as bacillopeptidase F, is initially synthesized as a 154 kDa prepropeptide. However, the details of the synthesis and processing of the B. subtilis version of this enzyme in the literature are ambiguous, with reports of two forms (33 000 and 50 000 kDa) being secreted by B. subtilis 168 after the end of exponential growth. Initial DNA sequence analysis suggested bacillopeptidase F is synthesized as a prepropeptide of 96 kDa and processed at both the amino and carboxyl termini to generate variants with molecular masses that range from 80 to 40 kDa (Sloma et al. 1990). A clearer picture arises from studies on the bacillopeptidase F from Bacillus amyloliquefaciens, a close relative of B. subtilis (Kwon et al. 2011). The B. amyloliquefaciens bpr gene encodes a 1431 residue prepropeptide with a calculated mass of 155 kDa, with a 71% identity to the similarly sized primary product of the B. subtilis enzyme. The first 30 residues are predicted to encode a signal peptide and the following 166 residues a propeptide. The expected mass of the mature protein was 133 kDa. However, when the gene was cloned and expressed in a strain of B. subtilis lacking bpr and five other extracellular protease genes, proteins of 90, 55 and 40 kDa were detected in the culture medium, suggestive of further processing at both the N- and C-termini of the protein (Kwon et al. 2011). It is not clear if some or all of these bpr products have protease activity and their precise roles are unknown excepting for their contribution to feeding protease activity.
Bpr is a member of the DegU regulon and the regulatory region of bpr contains three direct repeats of a DegU-binding consensus sequence. Like a number of other extracellular proteases, Bpr transcription of the bpr promoter is upregulated in a DegUhy mutant and, over time, during biofilm formation (Veening et al. 2008; Marlow et al. 2014).
Epr serine protease
Epr is a minor extracellular serine protease with a mass of 70 kDa. It is a member of the SinR, ScoC, Spo0A, DegU and SigD regulons and is suggested to play a role in the DegU-mediated control of swarming motility. However, more recent evidence suggests that Epr is not exclusive to this role and several other extracellular proteases appear to perform the same function (Connelly et al. 2004). The epr gene encodes a primary product of 645 amino acids that is partially homologous to both subtilisin (AprE) and the major intracellular serine protease (IspA). Deletion analysis indicates that the C-terminal 240 amino acids of Epr are not necessary for activity and this is consistent with finding of active forms of the enzyme with apparent molecular masses of 34 and 40 kDa. The C-terminal region exhibits several unusual features, including a high abundance of lysine residues and the presence of a partially homologous sequence of 44 amino acids that is directly repeated five times (Sloma et al. 1988). This suggests that at least a portion of the synthesized Epr is retained in the negatively charged cell wall.
Epr is involved in cell-to-cell communication. Bacillus subtilis uses peptide signals for communication, such as the Phr pentapeptides that are secreted with short ‘pro’ domains that are cleaved to produce the active signalling peptide. One such Phr pentapeptide is the competence and sporulation factor (CSF), which is formed from the inactive proCSF precursor by cleavage with Epr, as well as AprE and another minor serine protease, Vpr. The processed form of CSF is a secreted ERGMT pentapeptide that is transported into the cell by an oligopeptide permease when the peptide reaches a critical concentration in the environment. Once in the cytoplasm, CSF stimulates the quorum response and sporulation by antagonizing RapC and RapB activity (Lanigan-Gerdes et al. 2007). Finally, Epr, together with WprA, are responsible for the degradation of FlgM, a SigD specific anti-sigma factor that is secreted from the cell using the flagellar export apparatus (Calvo and Kearns 2015).
Mpr metalloprotease
Mpr is a 34 kDa metalloprotease and a negatively regulated member of the CodY regulon. Mpr is synthesized as a prepropeptide (32 kDa) that is subsequently processed to the active form (28 kDa) by removal of the signal and pro-peptide moieties. While many B. subtilis extracellular proteases appear to be activated by autoprocessing, Mpr is an exception. This is due to its high substrate specificity for a glutamate residue as a P1 cleavage site and the absence of this residue at the Mpr propeptide cleavage site (Park et al. 2004). Analysis of Mpr processing, using defined protease-deficient mutants, indicates that bacillopeptidase F (Bpr) is required for pro-Mpr processing. Pro-Mpr remains unprocessed in a bpr-deficient mutant and its glutamate-specific proteolytic activity is not activated.
NprB neutral metalloprotease
NprB, neutral metalloprotease B, is a 37 kDa metalloprotease with a role in biofilm-associated toxin sensitivity. It is a paralogue of NprE but produced at lower concentrations. NprB is synthesized as an 89 kDa prepropeptide with a 28-residue signal peptide and a 195-residue propeptide. NprB is a member of the DegU regulon.
Bacillus subtilis produces a biofilm-associated toxin (YIT), the product of the yitM gene, and an antitoxin, the product of the yitQ gene. The YIT toxin attacks toxin-sensitive competitor cells by passing through the protective barriers of the biofilm and does so with the assistance of NprB (Kobayashi and Ikemoto 2019). YIT toxin resistance is mediated by a combination of YitQ and SigW and in their absence, but in the presence of NprB, YIT inhibits biofilm formation. The role of NprB in this process is not clear, since it is not involved in YIT toxin production and the toxin is not a NprB substrate. Instead, since the biofilm matrix is responsible for the increased tolerance of its cells to antibiotics and toxins, it is suggested that NprB facilitates the migration of the YIT toxin through the polysaccharide/protein biofilm matrix. It is likely to do this by degrading a proteinaceous component of the biofilm, comprising the fibrous TasA protein and the hydrophobin-like BslA protein that forms a hydrophobic coating around the biofilm (Brandani et al. 2015; Arnaouteli et al. 2021). Since fibres of TasA are generally resistant to extracellular proteases (Erskine et al. 2018), it is likely that NprE facilitates the permeation of the YIT toxin through the biofilm matrix of toxin-sensitive cells by degradation of BslA. This is, in part at least, confirmed by the absence of a requirement for NprB in a ΔbslA mutant (Kobayashi and Ikemoto 2019).
NprE neutral metalloprotease A
NprE is a 56 kDa metalloprotease and a paralogue of NrpB. It differs from NprB mainly on the basis of is regulation, being repressed as a member of the ScoB, AbrB and CodY regulons (Barbieri et al. 2016). NprE is produced as a prepropeptide precursor with a 27-residue signal peptide and a 194-residue propeptide, meaning that the mature protein is 300 residues in length. NprE is second only to AprE as one of the major proteases found in the stationary phase culture medium. Deletions in the genes encoding NprE and AprE reduce the protease activity of the culture medium by 95% (Karamura and Doi 1984) and these genes are routinely inactivated in industrial production strains. The nprE gene is monocistronic, and consequently it is frequently used for the insertion of expression cassettes encoding heterologous proteins.
Vpr serine protease
Vpr is a minor extracellular protease with an initial mass of 85 kDa. PreproVpr is 806 amino acids in length, with a 28-residue signal sequence and a 132-residue propeptide. The Vpr protein has a predicted molecular weight of 68 kDa with a long C-terminal region; however, the mature protein isolated from the culture medium has an apparent molecular weight of 28 kDa, suggesting that Vpr undergoes maturation by C-terminal processing (Sloma et al. 1991). Vpr is a member of the CodY, PhoP, LexA and DnaA regulons, indicating that it is induced under specific environmental conditions such as phosphate starvation (Allenby et al. 2005). The vpr gene is expressed from a SigH promoter and repressed by CodY. In a CodY mutant, Vpr expression increased 30- to 50-fold and, as a result, becomes a major component of the extracellular proteome (Barbieri et al. 2015).
Vpr, together with Epr, is responsible for the specific processing of TapA, involved in TasA biofilm fibre formation (Earl et al. 2020) by processing of two quorum sensing peptides, CSF and PhrA (Lanigan-Gerdes et al. 2007); Vpr, together with WprA, is also involved in the activation of the peptide antibiotic (lantibiotic), subtilin (Corvey et al. 2003).
WprA serine protease
WprA is a wall-associated serine protease. The WprA protease domain has 28.5% identity to subtilisin and displays a broad substrate specificity. WprA and subtilisin A have similar pH profiles, showing optimal activity near pH 7.5 for substrates with Met, Gln or Lys residues at P1. The primary product of the wprA gene is a 96-kDa prepropeptide that is processed into two previously identified cell wall proteins, namely, CWBP52 and CWBP23. The processing of the WprA precursor during secretion accompanies the targeting of CWBP23 propeptide and CWBP52 protease to the cell wall (Margot and Karamata 1996; Stephenson and Harwood 1998). This processing is analogous to the maturation of another B. subtilis cell-wall-bound protein, namely the WapA, involved in cell contact-dependent growth inhibition (Koskiniemi et al. 2013).
Deletion of the wprA gene increases the yield of α‐amylase production by ∼50% (Stephenson and Harwood 1998). It is proposed that WprA, like that of the HtrA-like membrane-associated proteases, is a quality control protease, clearing misfolded or slowly folding secretory proteins from the cell membrane/wall interface and the wall itself, thereby avoiding interference with cell wall synthesis and cell elongation (Sarvas et al. 2004; Harwood and Cranenburgh 2008). WprA also appears to have a role in controlling the levels of extracytoplasmic protein folding catalysts (e.g. PrsA, HtrA and HtrB) and autolysins (Stephenson et al. 1999; Krishnappa et al. 2014). Together with Epr, WprA is also responsible for the degradation of FlgM, a SigD specific anti-sigma factor that is secreted from the cell via the Type 3 flagellar export apparatus (Calvo and Kearns 2015).
Engineering extracellular protease-deficient strains
Generally speaking, the native extracellular proteins synthesized by B. subtilis, when correctly folded (i.e. not subjected to heat or other stresses during synthesis), are little affected by the presence of the eight extracellular proteases. It is assumed that their coevolution has avoided the presence of surface-exposed protease-sensitive sites. There are exceptions, and these relate to the activities of specific proteases that control or process specific co-expressed extracellular proteins (e.g. degradation of BslA by NprB, the processing of TapA by Vpr and the processing of CSF and PhrA by Epr and Vpr). With heterologous proteins, the situation is highly variable and dependent on the characteristics of the protein itself. For example, in its native host, B. licheniformis, virtually 100% of the synthesized α-amylase AmyL is recovered in the culture medium. However, when this enzyme is synthesized in B. subtilis strain 168, only ∼25% of the initially synthesized enzyme is recovered in the culture medium, where it is stable in the presence of the extracellular proteases (Stephenson and Harwood 1998; Stephenson et al. 1998; Jensen et al. 2000). When engineered versions of AmyL were generated, the amount of synthesized enzyme that was recovered in the culture medium was even lower (∼5%), but was similarly stable in the presence of the extracellular proteases. It was shown that AmyL and its variants were successfully secreted but were rapidly degraded at the membrane/cell wall interface by the wall and membrane located quality control proteases WprA, HtrA and HtrB. A combination of experimental evidence indicated that it was their rate of folding post-translocation that was the main factor involved in their degradation (Stephenson et al. 1998). Factors that influence the rate of folding, such as the upregulation of PrsA (Kontinen and Sarvas 1993) and the absence teichoic acid alanylation (Hyyryläinen et al. 2000), both improve the recovery of certain heterologous proteins from the culture medium.
In many cases, however, the presence of so many, often abundant, proteases in the culture medium has limited the use of B. subtilis for the production of heterologous proteins. Given that B. subtilis has a secretion capacity that can lead to the accumulation of a target protein at levels in excess of 20 g/L of culture, numerous research groups and companies have developed strains in which some or all of these proteases have been deleted. One of the pioneers of protease-negative strains was Alan Sloma and colleagues (Sloma et al. 1989). In subsequent years, various protease-deficient strains have been developed and a full account of these is beyond the scope of this report. Some of the developed extracellular protease negative mutants have been generated by classical mutation approaches (Wu et al. 1991; Jeong et al. 2018), others by using precise nucleotide to nucleotide precision techniques (Pohl et al. 2013; Zhao et al. 2019). While deleting extracellular proteases can facilitate increases in the yields of heterologous proteins, it should be borne in mind that in addition to their roles in degrading environmental proteins as a source of carbon and nitrogen, some of these proteases play roles in quality control, cell signalling and protein processing (Krishnappa et al. 2013; Pohl et al. 2013).
SUMMARY
Bacillus subtilis encodes a wide range of proteases and these are located within the cytoplasm, the cell membrane, the cell wall and the external milieu. In most cases, these proteases are not essential, but their deletion is likely to affect the ability of this bacterium to compete in its natural environment. However, the deletion of these proteases often has less impact on this bacterium's growth characteristics in the laboratory or when designed specifically to improve the yield of secreted heterologous proteins. In many cases, individual proteases perform specific processing functions that, for example, help to control specific aspects of metabolism and cellular behaviour. Others of these proteases are important for protein quality control, degrading misfolded protein that might otherwise interfere with cell growth and physiology. As a result, there are still no ideal strains for producing and secreting all types of heterologous proteins, and suitable strains still need to be evaluated on a case-by-case basis. Bottlenecks in secretory protein production can occur at all stages in the secretion pathway from intracellular misfolding, translocase blockage and rapid degradation at the membrane/cell wall interface and in the culture medium—the latter not only by extracellular proteases but also cytoplasmic and membrane proteases released by cell lysis, particularly in post-exponential cultures.
Contributor Information
Colin R Harwood, Centre for Bacterial Cell Biology, Biosciences Institute, Newcastle University, Newcastle upon Tyne NE2 4AX, UK.
Yoshimi Kikuchi, Research Institute for Bioscience Products & Fine Chemicals, Ajinomoto Co., Inc., Kawasaki 210-8681, Japan.
ACKNOWLEDGEMENTS
The authors are grateful for the information on the genes of B. subtilis strain 168 contained in the SubtiWiki website (Zhu and Stulke 2018), and to Nicola Stanley-Wall and Thibault Rosazza for critically reading parts of the manuscript.
FUNDING
This work was supported by Newcastle University and Ajinomoto Co., Inc.
Conflict of Interest
None declared.
REFERENCES
- Akanuma G, Kazo Y, Tagami Ket al. . Ribosome dimerization is essential for the efficient regrowth of Bacillus subtilis. Microbiology. 2016;162:448–58. [DOI] [PubMed] [Google Scholar]
- Allenby NEE, O'Connor N, Prágai Zet al. . Genome-wide transcriptional analysis of the phosphate starvation stimulon of Bacillus subtilis. J Bacteriol. 2005;187:8063–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antelmann H, Darmon E, Noone Det al. . The extracellular proteome of Bacillus subtilis under secretion stress conditions. Mol Molec. 2003;49:143–56. [DOI] [PubMed] [Google Scholar]
- Arends J, Thomanek N, Kuhlmann Ket al. . In vivo trapping of FtsH substrates by label-free quantitative proteomics. Proteomics. 2016;16:3161–72. [DOI] [PubMed] [Google Scholar]
- Arnaouteli S, Bamford NC, Stanley-Wall NRet al. . Bacillus subtilis biofilm formation and social interactions. Nat Rev Microbiol. 2021;19:600–14. [DOI] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354. [DOI] [PubMed] [Google Scholar]
- Barbieri G, Albertini AM, Ferrari Eet al. . Interplay of CodY and ScoC in the regulation of major extracellular protease genes of Bacillus subtilis. J Bacteriol. 2016;198:907–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri G, Voigt B, Albrecht Det al. . CodY regulates expression of the Bacillus subtilis extracellular proteases Vpr and Mpr. J Bacteriol. 2015;197:1423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AJ, Rawlings ND, Woessner JF (eds). Handbook of Proteolytic Enzymes. London: Academic Press, 1998. [Google Scholar]
- Basturea GN, Zundel MA, Deutscher MP. Degradation of ribosomal RNA during starvation: comparison to quality control during steady-state growth and a role for RNase PH. RNA. 2011;17:338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battesti A, Gottesman S. Roles of adaptor proteins in regulation of bacterial proteolysis. Curr Opin Microbiol. 2013:16:140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckert B, Abdelshahid M, Schafer Het al. . Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. EMBO J. 2017;36:2061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Began J, Cordier B, Březinová Jet al. . Rhomboid intramembrane protease YqgP licenses bacterial membrane protein quality control as adaptor of FtsH AAA protease. EMBO J. 2020;39:e102935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CA. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhards CB, Chen Y, Toutkoushian Het al. . HtrC is involved in proteolysis of YpeB during germination of Bacillus anthracis and Bacillus subtilis spores. J Bacteriol. 2015;197:326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaudeck N, Sprenger G, Freudi Ret al. . Specificity of signal peptide recognition in Tat-dependent bacterial protein translocation. J Bacteriol. 2001;183:604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhuis A, Koetje E, Dubois J-Yet al. . Did the mitochondrial processing peptidase evolve from a eubacterial regulator of gene expression?. Mol Biol Evol. 2000;17:198–201. [DOI] [PubMed] [Google Scholar]
- Bose B, Auchtung JM, Lee CAet al. . A conserved anti-repressor controls horizontal gene transfer by proteolysis. Mol Microbiol. 2008;70:570–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose B, Grossman AD. Regulation of horizontal gene transfer in Bacillus subtilis by activation of a conserved site-specific protease. J Bacteriol. 2011;193;22–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradmann O, Hegde RS. Ribosome-associated protein quality control. Nat Struct Mol Biol. 2016;23:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramkamp M, Weston L, Daniel RAet al. . Regulated intramembrane proteolysis of FtsL protein and the control of cell division in Bacillus subtilis. Mol Microbiol. 2006;62:580–91. [DOI] [PubMed] [Google Scholar]
- Brandani GB, Schor M, Morris Ret al. . The bacterial hydrophobin BslA is a switchable ellipsoidal Janus nanocolloid. Langmuir. 2015;31:11558–63. [DOI] [PubMed] [Google Scholar]
- Bryan PN. Protein engineering of subtilisin. Bioch Biophys Acta. 2000;1543:203–22. [DOI] [PubMed] [Google Scholar]
- Burby PE, Simmons ZW, Schroeder JWet al. . Discovery of a dual protease mechanism that promotes DNA damage checkpoint recovery. PLoS Genet. 2018;14:e1007512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burby PE, Simmons ZW, Simmons LA. DdcA antagonizes a bacterial DNA damage checkpoint. Mol Microbiol. 2019;111:237–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Aravind L. Analysis of two domains with novel RNA-processing activities throws light on the complex evolution of ribosomal RNA biogenesis. Front Genet. 2014;5:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita LD, Cassaignau AME, Launay HMMet al. . A structural ensemble of a ribosome-nascent chain complex during cotranslational protein folding. Nat Struct Mol Biol. 2016;23:278–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Wang H, He Pet al. . A novel strategy to improve protein secretion via overexpression of the SppA signal peptide peptidase in Bacillus licheniformis. Microb Cell Fact. 2017;16:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns LS, Hobley L, Stanely-Wall NR. Biofilm formation by Bacillus subtilis: new insights into regulatory strategies and assembly mechanisms. Mol Microbiol. 2014;93:587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo RA, Kearns DB. FlgM is secreted by the flagellar export apparatus in Bacillus subtilis. J Bacteriol. 2015;197:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Provvedi R, Dubnau D. A macromolecular complex formed by a pilin-like protein in competent Bacillus subtilis. J Biol Chem. 2006;281:21720–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho T, Zhang X, Pobre KFRet al. . Individual and collective contributions of chaperoning and degradation to protein homeostasis in Escherichia coli. Cell Rep. 2015;11:321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L, Kaplan R. Accumulation of nucleotides by starved Escherichia coli cells as a probe for the involvement of ribonucleases in ribonucleic acid degradation. J Bacteriol. 1977;129:651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly MB, Young GM, Sloma A. Extracellular proteolytic activity plays a central role in swarming motility in Bacillus subtilis. J Bacteriol. 2004;186:4159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvey C, Stein T, Dusterhus Set al. . Activation of subtilin precursors by Bacillus subtilis extracellular serine proteases subtilisin (AprE), WprA, and Vpr. Biochem Biophys Res Commun. 2003;304:48–54. [DOI] [PubMed] [Google Scholar]
- Crowe-McAuliffe C, Takada H, Murina Vet al. . Structural basis for bacterial ribosome-associated quality control by RqcH and RqcP. Molecular Cell. 2021;81:115–26. [DOI] [PubMed] [Google Scholar]
- Cruz RAL. A comparative study of native and heterologous enzyme production in Bacillus subtilis. Ph.D. Thesis. Newcastle University. 2016. [Google Scholar]
- Darmon E, Noone D, Masson Aet al. . A novel class of heat and secretion stress-responsive genes is controlled by the autoregulated CssRS two-component system of Bacillus subtilis. J Bacteriol. 2002;184:5661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defenouillère Q, Fromont-Racine M. The ribosome-bound quality control complex: from aberrant peptide clearance to proteostasis maintenance. Curr Genet. 2017;63:997–1005. [DOI] [PubMed] [Google Scholar]
- Deuerling E, Mogk A, Richter Cet al. . The ftsH gene of Bacillus subtilis is involved in major cellular processes such as sporulation, stress adaptation and secretion. Mol Microbiol. 1997;23:921–33. [DOI] [PubMed] [Google Scholar]
- Deuerling E, Paeslack B, Schumann W. The ftsH gene of Bacillus subtilis is transiently induced after osmotic and temperature upshift. J Bacteriol. 1995;177:4105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiChiara JM, Liu B, Figaro Set al. . Mapping of internal monophosphate 5′ ends of Bacillus subtilis messenger RNAs and ribosomal RNAs in wild-type and ribonuclease-mutant strains. Nucleic Acids Res. 2016;44:3373–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CM. Principles of protein folding, misfolding and aggregation. Cell Devel Biol. 2004;15:3–16. [DOI] [PubMed] [Google Scholar]
- Dougan DA, Truscott KN, Zeth K. The bacterial N-end rule pathway: expect the unexpected. Mol Microbiol. 2010;76:545–58. [DOI] [PubMed] [Google Scholar]
- Dulebohn D, Choy J, Sundermeier T. Trans-translation: the tmRNA-mediated surveillance mechanism for ribosome rescue, directed protein degradation, and nonstop mRNA decay. Biochemistry. 2007;46:4681–93. [DOI] [PubMed] [Google Scholar]
- Earl C, Arnaouteli S, Bamford NC. The majority of the matrix protein TapA is dispensable for Bacillus subtilis colony biofilm architecture. Mol Microbiol. 2020;114:929–33. [DOI] [PubMed] [Google Scholar]
- Ekici OD, Paetzel M, Dalbey RE. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008;17:2023–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellermeier CD, Hobbs EC, Gonzalez-Pastor JE. A three-protein signaling pathway governing immunity to a bacterial cannibalism toxin. Cell. 2006;124:549–59. [DOI] [PubMed] [Google Scholar]
- Ellermeier CD, Losick R. Evidence for a novel protease governing regulated intramembrane proteolysis and resistance to antimicrobial peptides in Bacillus subtilis. Genes Dev. 2006;20:1911–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsholz AKW, Birk MS, Charpentier E. Functional diversity of AAA+ protease complexes in Bacillus subtilis. Front Mol Biosci. 2017;4:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erskine E, Morris R, Schor Met al. . Formation of functional, non-amyloidogenic fibres by recombinant Bacillus subtilis TasA. Mol Microbiol. 2018;110:897–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. [DOI] [PubMed] [Google Scholar]
- Fu X, Inouye M, Shinde U. Folding pathway mediated by an intramolecular chaperone. J Biol Chem. 2000;275:16871–8. [DOI] [PubMed] [Google Scholar]
- Gallagher T, Gilliland G, Wang Let al. . The prosegment-subtilisin BPN´ complex: crystal structure of a specific ‘foldase’. Structure. 1995;3:907–14. [DOI] [PubMed] [Google Scholar]
- Gohara DW, Yap MF. Survival of the drowsiest: the hibernating 100S ribosome in bacterial stress management. Curr Genet. 2018;64:753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosens V, Otto A, Glasner Cet al. . Novel Twin-arginine translocation pathway-dependent phenotypes of Bacillus subtilis unveiled by quantitative proteomics. J Proteome Res. 2013;12:796–807. [DOI] [PubMed] [Google Scholar]
- Gottesman S. Proteases and their targets in Escherichia coli. Annu Rev Genet. 1996;30:465–506. [DOI] [PubMed] [Google Scholar]
- Grimaud R, Kessel M, Beuron Fet al. . Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. J Biol Chem. 1998;273:12476–81. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–32. [DOI] [PubMed] [Google Scholar]
- Harwood CR, Cranenburgh RM. Bacillus protein secretion: an unfolding story. Trends Microbiol. 2008;16:73–9. [DOI] [PubMed] [Google Scholar]
- Harwood CR. Bacillus subtilis: molecular biological and industrial workhorse, Trends Biotechnol. 1992;10:247–56. [DOI] [PubMed] [Google Scholar]
- Hayer-Hartl M, Bracher A, Hartl FU. The GroEL-GroES chaperone machine: a nano-cage for protein folding. Trends Biochem Sci. 2016;41:62–76. [DOI] [PubMed] [Google Scholar]
- Heinrich J, Hein K, Wiegert T. Two proteolytic modules are involved in regulated intramembrane proteolysis of Bacillus subtilis RsiW. Mol Microbiol. 2009;74:1412–26. [DOI] [PubMed] [Google Scholar]
- Heinrich J, Lundén T, Kontinen VPet al. . The Bacillus subtilis ABC transporter EcsAB influences intramembrane proteolysis through RasP. Microbiology. 2008;154:1989–97. [DOI] [PubMed] [Google Scholar]
- Heinrich J, Wiegert T. Regulated intramembrane proteolysis in the control of extracytoplasmic function sigma factors, Res Microbiol. 2009;160:696–703. [DOI] [PubMed] [Google Scholar]
- Herman C, Prakash S, Lu CZet al. . Lack of a robust unfoldase activity confers a unique level of substrate specificity to the universal AAA protease FtsH. Mol Cell. 2003;11:659–69. [DOI] [PubMed] [Google Scholar]
- Herman C, Thevenet D, Bouloc Pet al. . Degradation of carboxy-terminal-tagged cytoplasmic proteins by the Escherichia coli protease HflB (FtsH). Genes Dev. 1998;12:1348–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkamp W, Kokic G, Jäger Met al. . Cotranslational protein folding on the ribosome monitored in real time. Science. 2015;350:1104–7. [DOI] [PubMed] [Google Scholar]
- Horwich AL, Weber-Ban EU, Finley D. Chaperone rings in protein folding and degradation. Proc Natl Acad Sci USA. 1999;96:11033–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyyryläinen H-l, Bolhuis A, Darmon Eet al. . A novel two-component regulatory system in Bacillus subtilis for the survival of severe secretion stress. Mol Microbiol. 2001;41:1159–72. [DOI] [PubMed] [Google Scholar]
- Hyyryläinen H-L, Vitikainen M, Thwaite Jet al. . D-Alanine substitution of teichoic acids as a modulator of protein folding and stability at the cytoplasmic membrane/cell wall interface of Bacillus subtilis. J Biol Chem. 2000;275:26696–703. [DOI] [PubMed] [Google Scholar]
- Janda C, Li J, Oubridge Cet al. . Recognition of a signal peptide by the signal recognition particle. Nature. 2010;465:507–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen BD, Hayes CS. The tmRNA ribosome-rescue system. Adv Prot Chem Struct Biol. 2012;86:151–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CL, Stephenson K, Jørgensen STet al. . Cell-associated degradation affects yield of secreted engineered and heterologous proteins in the Bacillus subtilis expression system. Microbiology. 2000;146:2583–94. [DOI] [PubMed] [Google Scholar]
- Jeong H, Jeong D E, Park SHet al. . Complete genome sequence of Bacillus subtilis strain WB800N, an extracellular protease-deficient derivative of strain 168. Microbiol Res Ann. 2018;7:e01380–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joazeiro CAP. Ribosomal stalling during translation: providing substrates for ribosome-associated protein quality control. Annu Rev Cell Dev Biol. 2017;33:343–68. [DOI] [PubMed] [Google Scholar]
- Jürgen B, Hanschke R, Sarvas Met al. . Proteome and transcriptome based analysis of Bacillus subtilis cells overproducing an insoluble heterologous protein. App Microbiol Biotech. 2001;55:326–32. [DOI] [PubMed] [Google Scholar]
- Kaiser CM, Chang HC, Agashe VRet al. . Real-time observation of trigger factor function on translating ribosomes. Nature. 2006;444:455–60. [DOI] [PubMed] [Google Scholar]
- Kaiser CM, Goldman DH, Chodera JD. The ribosome modulates nascent protein folding. Science. 2011;334:1723–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karzai AW, Roche ED, Sauer RT. The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nat Struct Biol. 2000;7:449–55. [DOI] [PubMed] [Google Scholar]
- Kawamura F, Doi RH. Construction of a Bacillus subtilis double mutant deficient in extracellular alkaline and neutral proteases. J Bacteriol. 1984;160:442–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns DB. A field guide to bacterial swarming motility. Nat Rev Microbiol. 2010;8:634–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Akiyama Y, Ito K. FtsH is required for proteolytic elimination of uncomplexed forms of SecY, an essential protein translocase subunit. Proc Natl Acad Sci USA. 1995;92:4532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J, Moliere N, Dougan DAet al. . Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol. 2009;7:589–99. [DOI] [PubMed] [Google Scholar]
- Kirstein J, Schlothauer T, Dougan DAet al. . Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J. 2006;25:1481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Ikemoto Y. Biofilm-associated toxin and extracellular protease cooperatively suppress competitors in Bacillus subtilis biofilms. PLoS Genet. 2019;15:e1008232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama T, Endo K, Sawada Ket al. . Bacillus subtilis AprX involved in degradation of a heterologous protein during the late stationary growth phase. J Biosci Bioeng. 2007;104:135–43. [DOI] [PubMed] [Google Scholar]
- Kontinen V, Sarvas M. The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol Microbiol. 1993;8:727–37. [DOI] [PubMed] [Google Scholar]
- Koo BM, Kritikos G, Farelli JDet al. . Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst. 2017;4:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiniemi S, Lamoureux JG, Nikolzkakis KCet al. . Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci USA. 2013;110;7032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnappa L, Dreisbach A, Otto Aet al. . Extracytoplasmic proteases determining the cleavage and release of secreted proteins, lipoproteins, and membrane proteins in Bacillus subtilis. J Proteome Res. 2013;12:4101–10. [DOI] [PubMed] [Google Scholar]
- Krishnappa L, Monteferrantem CG, Neef Jet al. . Degradation of extracytoplasmic catalysts for protein folding in Bacillus subtilis. Appl Environ Microbiol. 2014;80:1463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E, Völker U, Hecker M. Stress induction of clpC in Bacillus subtilis and its involvement in stress tolerance. J Bacteriol. 1994;176:3360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann NJ, Chien P. Selective adaptor dependent protein degradation in bacteria. Curr Opin Microbiol. 2017;36:118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon G-H, Park J-Y, Kim J-Set al. . Cloning and expression of a bpr gene encoding Bacillopeptidase F from Bacillus amyloliquefaciens CH86-1. J Microbiol Biotechnol. 2011;21:515–8. [DOI] [PubMed] [Google Scholar]
- Lakowitz A, Godard T, Biedendieck Ret al. . Mini review: recombinant production of tailored bio-pharmaceuticals indifferent Bacillus strains and future perspectives. Eur J Pharm Biopharm. 2018;126:27–39. [DOI] [PubMed] [Google Scholar]
- Lanigan-Gerdes S, Dooley AN, Faull KFet al. . Identification of subtilisin, Epr and Vpr as enzymes that produce CSF, an extracellular signalling peptide of Bacillus subtilis. Mol Microbiol. 2007;65:1321–33. [DOI] [PubMed] [Google Scholar]
- Lee AY, Park SG, Kho CWet al. . Identification of the degradome of Isp-1, a major intracellular serine protease of Bacillus subtilis, by two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization-time of flight analysis. Proteomics. 2004;4:3437–45. [DOI] [PubMed] [Google Scholar]
- Lee MM, Isaza CE, White JDet al. . Insight into the substrate length restriction of M32 carboxypeptidases: characterization of two distinct subfamilies. Proteins: Struct Funct Bioinf. 2009;77:647–57. [DOI] [PubMed] [Google Scholar]
- Lehnik-Habrink M, Lewis RJ, Mäder Uet al. . RNA degradation in Bacillus subtilis: an interplay of essential endo- and exoribonucleases. Mol Microbiol. 2012;84:1005–17. [DOI] [PubMed] [Google Scholar]
- Lin TH, Huang SC, Shaw GC. Reexamining transcriptional regulation of the Bacillus subtilis htpX gene and the ykrK gene, encoding a novel type of transcriptional regulator, and redefining the YkrK operator. J Bacteriol. 2012;194:6758–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Cutting S, Kroos L. Sporulation protein SpoIVFB from Bacillus subtilis enhances processing of the sigma factor precursor Pro-sigma K in the absence of other sporulation gene products. J Bacteriol. 1995;177:1082–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytvynenko I, Paternoga H, Thrun Aet al. . Alanine tails signal proteolysis in bacterial ribosome-associated quality control. Cell. 2019;178:76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäder U, Antelmann H, Buder Yet al. . Bacillus subtilis functional genomics: genome-wide analysis of the DegS-DegU regulon by transcriptomics and proteomics. Mol Genet Genomics. 2002;268:455–67. [DOI] [PubMed] [Google Scholar]
- Mann JM, Carabetta VJ, Cristea IMet al. . Complex formation and processing of the minor transformation pilins of Bacillus subtilis. Mol Microbiol. 2013;90:1201–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak BC, Trip H, van-der Veek PJet al. . Comparative transcriptional analysis of Bacillus subtilis cells overproducing either secreted proteins, lipoproteins or membrane proteins. Microb Cell Fact. 2012;11:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margot P, Karamata D. The wprA gene of Bacillus subtilis 168, expressed during exponential growth, encodes a cell-wall-associated protease. Microbiology. 1996;142:3437–44. [DOI] [PubMed] [Google Scholar]
- Marlow VL, Cianfanelli FR, Porter Met al. . The prevalence and origin of exoprotease-producing cells in the Bacillus subtilis biofilm. Microbiology. 2014;160:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske AJ, Rodrigues CD, Brady Jet al. . High-throughput genetic screens identify a large and diverse collection of new sporulation genes in Bacillus subtilis. PLoS Biol. 2016;14:e1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesak LR, Mesak FM, Dahl MK. Expression of a novel gene gluP is essential for normal Bacillus subtilis cell division and contributes to glucose transport. BMC Microbiol. 2004;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Huber D, Bukau B. Integrating protein homeostasis strategies in prokaryotes. Cold Spring Harb Persp Biol. 2011;3:a004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molière N, Hoßmann J, Schäfer Het al. . Role of Hsp100/Clp protease complexes in controlling the regulation of motility in Bacillus subtilis. Front Microbiol. 2016;16:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molière N, Turgay K. General and regulatory proteolysis in Bacillus subtilis. Subcell Biochem. 2013;66:73–103. [DOI] [PubMed] [Google Scholar]
- Moore SD, Sauer RT. The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem. 2007;76:101–24. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Bree AC, Liu Jet al. . Adaptor-mediated Lon proteolysis restricts Bacillus subtilis hyperflagellation. Proc Natl Acad Sci USA. 2015;112:250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Kearns DB. The structure and regulation of flagella in Bacillus subtilis. Annu Rev Genet. 2014;48:319–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef J, Bongiorni C, Goosens VJet al. . Intramembrane protease RasP boosts protein production in Bacillus. Microb Cell Fact. 2017;16:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JLet al. . AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- Noone D, Howell A, Collery Ret al. . YkdA and YvtA, HtrA-like serine proteases in Bacillus subtilis, engage in negative autoregulation and reciprocal cross-regulation of ykdA and yvtA gene expression. J Bacteriol. 2001;183:654–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien EP, Christodoulou J, Vendruscolo Met al. . New scenarios of protein folding can occur on the ribosome. J Am Chem Soc. 2011;133:513–26. [DOI] [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure-diverse function. Genes Cells. 2001;6:575–97. [DOI] [PubMed] [Google Scholar]
- Oh E, Becker AH, Sandikci Aet al. . Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell. 2011;147:1295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura S, Maruyama HB, Yanagita T. Ribosome degradation and degradation products in starved Escherichia coli. VI. Prolonged culture during glucose starvation. J Biochem. 1973;73:915–22. [DOI] [PubMed] [Google Scholar]
- Olivares AO, Baker TA, Sauer RT. Mechanistic insights into bacterial AAA+ proteases and proteins remodelling machines. Nat Rev Microbiol. 2016;14:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares AO, Kotamarthi HC, Stein BJet al. . Effect or directional pulling on mechanical protein degradation by ATP-dependent proteolytic machines. Proc Natl Acad Sci USA. 2017;114:E6306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos J, De Anda R, Ferrari Eet al. . Effects of the sinR and degU32 (Hy) mutations on the regulation of the aprE gene in Bacillus subtilis. Mol Gen Genet. 1997;253:562–7. [DOI] [PubMed] [Google Scholar]
- Park CH, Lee SJ, Lee SGet al. . Hetero- and autoprocessing of the extracellular metalloprotease (Mpr) in Bacillus subtilis. J Bacteriol. 2004;186:6457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Schumann W. Optimization of the secretion pathway for heterologous proteins in Bacillus subtilis. Biotechnol Bioprocess Eng. 2015;20:623–33. [Google Scholar]
- Piatkov KI, Vu TT, Hwang CSet al. . Formyl-methionine as a degradation signal at the N-termini of bacterial proteins. Microbial Cell. 2015;2:376–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piir K, Paier A, Liiv Aet al. . Ribosome degradation in growing bacteria. EMBO Rep. 2011;12:458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploss TN, Reilman E, Monteferrante CGet al. . Homogeneity and heterogeneity in amylase production by Bacillus subtilis under different growth conditions. Microb Cell Fact. 2016;15:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl S, Bhavsar G, Hulme Jet al. . Proteomic analysis of Bacillus subtilis strains engineered for improved production of heterologous proteins. Proteomics. 2013;13:3298–308. [DOI] [PubMed] [Google Scholar]
- Pohl S, Harwood CR. Heterologous protein secretion by Bacillus species from the cradle to the grave. Adv App Microbiol. 2010;73:1–25. [DOI] [PubMed] [Google Scholar]
- Power SD, Adams RM, Wells JA. Secretion and autoproteolytic maturation of subtilisin. Proc Natl Acad Sci USA. 1986;83:3096–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prágai Z, Tjalsma H, Bolhuis Aet al. . The signal peptidase II (Isp) gene of Bacillus subtilis. Microbiology. 1997;143:1327–33. [DOI] [PubMed] [Google Scholar]
- Priest FG. Extracellular enzyme synthesis in the genus Bacillus. Bacteriol Rev. 1977;41:711–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pummi T, Leskela S, Wahlstrom Eet al. . ClpXP protease regulates the signal peptide cleavage of secretory preproteins in Bacillus subtilis with a mechanism distinct from that of the Ecs ABC transporter. J Bacteriol. 2002;184:1010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethdorf S, Volker U, Gerth Uet al. . Cloning, nucleotide sequence, and expression of the Bacillus subtilis lon gene. J Bacteriol. 1994;176:6518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotanova TV, Andrianova AG, Kudzhaev AMet al. . New insights into structural and functional relationships between LonA proteases and ClpB chaperones. FEBS Open Bio. 2019;9:1536–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner DZ, Fawcett P, Losick R. A family of membrane-embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc Natl Acad Sci USA. 1999;96:14765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner DZ, Losick R. A sporulation membrane protein tethers the pro-sigmaK processing enzyme to its inhibitor and dictates its subcellular localization. Genes Dev. 2002;16:1007–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runde S, Molière N, Heinz Aet al. . The role of thiol oxidative stress response in heat-induced protein aggregate formation during thermotolerance in Bacillus subtilis. Mol Microbiol. 2014;91:1036–52. [DOI] [PubMed] [Google Scholar]
- Sakoh M, Ito K, Akiyama Y. Proteolytic activity of HtpX, a membrane-bound and stress-controlled protease from Escherichia coli. J Biol Chem. 2005;280:33305–10. [DOI] [PubMed] [Google Scholar]
- Santra M, Farrell DW, Dill KA. Bacterial proteostasis balances energy and chaperone utilization efficiently. Proc Natl Acad Sci USA. 2017;114:E2654–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarvas M, Harwood CR, Bron Set al. . Post-translocational folding of secretory proteins in Gram positive bacteria. Biochim Biophys Acta. 2004;1694:311–27. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Trentini DB, Spiess Set al. . Quantitative phosphoproteomics reveals the role of protein arginine phosphorylation in the bacterial stress response. Mol Cell Proteomics. 2014;13:537–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Decatur AL, Rather PNet al. . Bacillus subtilis Lon protease prevents inappropriate transcription of genes under the control of the sporulation transcription factor sigma G. J Bacteriol. 1994;176:6528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev E, Smith Y, Ben-Yehuda S. RNA dynamics in aging bacterial spores. Cell. 2012;148:139–49. [DOI] [PubMed] [Google Scholar]
- Serrano M, Hovel S, Moran CPet al. . Forespore-specific transcription of the lonB gene during sporulation in Bacillus subtilis. J Bacteriol. 2001;183:2995–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova K, Nakayama H, Shimamoto N. Role of 100S ribosomes in bacterial decay period. Genes Cells. 2015;20:789–801. [DOI] [PubMed] [Google Scholar]
- Simmons LA, Grossman AD, Walker GC. Clp and Lon proteases occupy distinct subcellular positions in Bacillus subtilis. J Bacteriol. 2008;190:6758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Grimaud R, Hoskins JRet al. . Unfolding and internalization of proteins by the ATP-dependent pro- teases ClpXP and ClpAP. Proc Natl Acad Sci USA. 2000;97:8898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloma A, Ally A, Ally Det al. . Gene encoding a minor extracellular protease in Bacillus subtilis. J Bacteriol. 1988;170:5557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloma A, Rufo GA, Rudolph CFet al. . Bacillopeptidase F of Bacillus subtilis: purification of the protein and cloning of the gene. J Bacteriol. 1990;172:1470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloma A, Rufo GA, Rudolph CFet al. . Cloning and deletion of the genes for three minor extracellular proteases of Bacillus subtilis. In: Zukowski MM, Ganesan AT, Hoch JA (eds.) Genetics and Biotechnology of Bacilli, Vol. 3. San Diego, CA: Academic Press, Inc, 1989, 295–302. [Google Scholar]
- Sloma A, Rufo GA, Theriault Ket al. . Cloning and characterization of the gene for an additional extracellular serine protease of Bacillus subtilis. J Bacteriol. 1991;173:6889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I. Regulatory proteins that control late-growth development. In:Sonenshein AL, JA Hoch, Losick R. (eds). Bacillus Subtilis and Other Gram-Positive Bacteria: Biochemistry, Physiology and Molecular Genetics. Washington, DC: ASM, 1993, 785–800. [Google Scholar]
- Stahl ML, Ferrari E. Replacement of the Bacillus subtilis subtilisin structural gene with an in vitro-derived deletion mutation. J Bacteriol. 1984;158:411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson K, Bron S, Harwood CR. Cellular lysis in Bacillus subtilis; the effect of multiple extracellular protease deficiencies. Lett Appl Microbiol. 1999;29:141–5. [Google Scholar]
- Stephenson K, Harwood CR, Petit-Glatron M-Fet al. . The influence of protein folding on the secretion of α-amylases from Bacillus subtilis. FEBS Lett. 1998;430:385–9. [DOI] [PubMed] [Google Scholar]
- Stephenson K, Harwood CR. The influence of a cell-wall-associated protease on the production of α-amylase by Bacillus subtilis. Appl Environ Microbiol. 1998;64:2875–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson K, Jensen C, Jørgensen STet al. . The influence of secretory protein charge on late stages of secretion from the Gram-positive bacterium Bacillus subtilis. Biochem J. 2000;350:31–9. [PMC free article] [PubMed] [Google Scholar]
- Stoller G, Rucknagel KP, Nierhaus KHet al. . A ribosome-associated peptidyl-prolyl cis/trans isomerase identified as the trigger factor. EMBO J. 1995;14:4939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Yang M, Jiang Let al. . Regulation of pro-σK activation: a key checkpoint in Bacillus subtilis sporulation. Environ Microbiol. 2021;23:2366–73. [DOI] [PubMed] [Google Scholar]
- Tan IS, Weiss CA, Popham DLet al. . A quality-control mechanism removes unfit cells from a population of sporulating bacteria. Dev Cell. 2015;34:682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terre R, Stanley-Wall NR, Cao Get al. . Identification of Bacillus subtilis SipW as a bifunctional signal peptidase that controls surface-adhered biofilm formation. J Bacteriol. 2012;194:2781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PG, Russell AJ, Fersht AR. Tailoring the pH dependence of enzyme catalysis using protein engineering. Nature. 1985;318:375–6. [Google Scholar]
- Tjalsma H, Bolhuis A, van Roosmalen MLet al. . Functional analysis of the secretory precursor processing machinery of Bacillus subtilis: identification of a eubacterial homolog of archaeal and eukaryotic signal peptidases. Genes Dev. 1998;12: 2318–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma H, Stöver AG, Driks Aet al. . Conserved serine and histidine residues are critical for activity of the ER-type signal peptidase SipW of Bacillus subtilis. J Biol Chem. 2000;275:25102–8. [DOI] [PubMed] [Google Scholar]
- Tjalsma H, Zanen G, Venema Get al. . The potential active site of the lipoprotein-specific (type II) signal peptidase of Bacillus subtilis. J Biol Chem. 1999;274:28191–7. [DOI] [PubMed] [Google Scholar]
- Trentini DB, Suskiewicz MJ, Heuck Aet al. . Arginine phosphorylation marks proteins for degradation by a Clp protease. Nature. 2016;539:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K, Hahn J, Burghoorn Jet al. . Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 1998;17:6730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K, Hamoen LW, Venema Get al. . Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev. 1997;11:119–28. [DOI] [PubMed] [Google Scholar]
- Turgay K, Persuh M, Hahn Jet al. . Roles of the two ClpC ATP binding sites in the regulation of competence and the stress response. Mol Microbiol. 2001;42:717–27. [DOI] [PubMed] [Google Scholar]
- Uehara R, Angkawidjaja C, Koga Y. Formation of the high-affinity calcium binding site in prosubtilisin E with the insertion sequence IS1 of Pro-Tk-subtilisin. Biochemistry. 2013;52:9080–8. [DOI] [PubMed] [Google Scholar]
- Valbuzzi A, Farrari E, Albertini AM. A novel member of the subtilisin-like protease family from Bacillus subtilis. Microbiology. 1999;145:3121–7. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. N-degron and C-degron pathways of protein degradation. Proc Natl Acad Sci USA. 2019;116:358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening J-W, Igoshin OA, Eijlander RTet al. . Transient heterogeneity in extracellular protease production in Bacillus subtilis. Mol Syst Biol. 2008;4:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vévodová J, Gamble M, Künze Get al. . Crystal structure of an intracellular subtilisin reveals novel structural features unique to this subtilisin family. Structure. 2010;18:744–55. [DOI] [PubMed] [Google Scholar]
- Vogeley L, Arnaout TE, Bailey Jet al. . Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science. 2016;351:876–80. [DOI] [PubMed] [Google Scholar]
- Wall EA, Johnson AL, Peterson SLet al. . Structural modeling and functional analysis of the essential ribosomal processing protease Prp from Staphylococcus aureus. Mol Microbiol. 2017;104:520–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ruan B, Ruvinov Set al. . Engineering the independent folding of the subtilisin BPN′ pro-domain: correlation of pro-domain stability with the rate of subtilisin folding. Biochemistry. 1998;37:3165–71. [DOI] [PubMed] [Google Scholar]
- Weber-Ban EU, Reid BG, Miranker ADet al. . Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature. 1999;401:90–3. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–93. [DOI] [PubMed] [Google Scholar]
- Wiegert T, Schumann W. SsrA-mediated tagging in Bacillus subtilis. J Bacteriol. 2001;183:3885–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SL, Doi RH. Determination of the signal peptidase cleavage site in the preprosubtilisin of Bacillus subtilis. J Biol Chem. 1986;261:10176–81. [PubMed] [Google Scholar]
- Wong SL, Price CW, Goldfarb DSet al. . The subtilisin E gene of Bacillus subtilis is transcribed from a sigma 37 promoter in vivo. Proc Natl Acad Sci USA. 1984;81:1184–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XC, Lee W, Tran Let al. . Engineering a Bacillus subtilis expression-secretion system with a strain deficient in six extracellular proteases. J Bacteriol. 1991;173:4952–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuta Y, Subbian E, Takagi Het al. . Folding pathway mediated by an intramolecular chaperone: dissecting conformational changes coincident with autoprocessing and the role of Ca2+ in subtilisin maturation. J Biochem (Tokyo). 2002;131:31–7. [DOI] [PubMed] [Google Scholar]
- Yabuta Y, Takagi H, Inouye Met al. . Folding pathway mediated by an intramolecular chaperone. Propeptide release modulates activation precision of prosubtilisin. J Biol Chem. 2001;276:44427–34. [DOI] [PubMed] [Google Scholar]
- Yeom J, Wayne KJ, Groisman EA. Sequestration from protease adaptor confers differential stability to protease substrate. Mol Cell. 2017;66:234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Wada A. The 100S ribosome: ribosomal hibernation induced by stress. Wiley Interdiscip Rev RNA. 2014;5:723–32. [DOI] [PubMed] [Google Scholar]
- Yu C-H, Dang Y, Zhou Zet al. . Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell. 2015;59:744–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Ye B, Zhang Qet al. . Construction of second-generation protease-deficient hosts of Bacillus subtilis for secretion of foreign proteins. Biotechnol Bioeng. 2019;116:2052–60. [DOI] [PubMed] [Google Scholar]
- Zhu B, Stülke J. SubtiWiki in 2018: from genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res. 2018;46:D743–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundel MA, Basturea GN, Deutscher MP. Initiation of ribosome degradation during starvation in Escherichia coli. RNA. 2009;15:977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweers JC, Wiegert T, van Dijl JM. Stress-responsive systems set specific limits to the overproduction of membrane proteins in Bacillus subtilis. Appl Environ Microbiol. 2009;75:7356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]