

Abstract
Triple-negative breast cancer (TNBC) is the most aggressive subtype of breast cancer, and it often becomes resistant to paclitaxel (PTX) therapy. Autophagy plays an important cytoprotective role in PTX-induced tumor cell death, and targeting autophagy has been promising for improving the efficacy of tumor chemotherapy in recent years. The aim of the present study was to clarify the mechanism of PTX inducing autophagy in TNBC cells to provide a potential clinical chemotherapy strategy of PTX for TNBC. The present study reported that PTX induced both apoptosis and autophagy in MDA-MB-231 cells and that inhibition of autophagy promoted apoptotic cell death. Furthermore, it was found that forkhead box transcription factor O1 (FOXO1) enhanced PTX-induced autophagy through a transcriptional activation pattern in MDA-MB-231 cells, which was associated with the downstream target genes autophagy related 5, class III phosphoinositide 3-kinase vacuolar protein sorting 34, autophagy related 4B cysteine peptidase, beclin 1 and microtubule associated protein 1 light chain 3β. Knocking down FOXO1 attenuated the survival of MDA-MB-231 cells in response to PTX treatment. These findings may be beneficial for improving the treatment efficacy of PTX and to develop autophagic targeted therapy for TNBC.
Keywords: forkhead box transcription factor O1, autophagy, apoptosis, paclitaxel, triple-negative breast cancer
Introduction
Triple-negative breast cancer (TNBC) is the most aggressive subtype of breast cancer. Higher proliferation rates and a higher incidence of metastases with a lack of targeted therapies have made TNBC an unmet clinical challenge (1). Currently, chemotherapy is regarded as a systemic therapeutic alternative for TNBC, and taxanes such as paclitaxel (PTX) are commonly used as antitumor agents against TNBC (2,3). However, numerous types of TNBC cells, especially the MDA-MB-231 cell line that carry mutant K-Ras and TP53, are highly aggressive and invasive breast cancer cell lines and often escape drug-induced apoptosis, leading to poor therapeutic effects (4,5). It has been reported that autophagy can protect tumor cells from damage and undergoing apoptosis during PTX treatment (6). Therefore, treatment approaches targeting autophagy may provide novel therapeutic value for TNBC.
Autophagy is a classical self-digestive process that maintains cellular homeostasis and ensures cell survival under stress conditions by degrading and recycling damaged organelles and protein aggregates (7). It plays a pivotal role in tumor progression, during early stages of cancer development it prevents tumor initiation, but in fully developed tumors it protects tumor cells from various stressful stimuli (4,8,9). Various chemotherapy drugs induce autophagy by activating different signaling pathways. For example, SKF-96365, a store-operated calcium entry inhibitor, induces cytoprotective autophagy by preventing the release of cytochrome C in colorectal cancer cells, which is mechanistically involved in AKT-related signaling (10). In addition, apatinib-induced autophagy in anaplastic thyroid carcinoma cells is related to the downregulation of phosphorylated (p)-AKT and p-mechanistic target of rapamycin kinase (mTOR) signaling (11). PTX treatment often activates different autophagy-related pathways depending on the tumor type. In A549 lung cancer cells, PTX induces autophagy by regulating the autophagy-related genes, including autophagy related 5 (ATG5) and beclin 1 (BECN1) (12). However, in ovarian cancer, PTX induces autophagy by upregulating the thioredoxin domain-containing protein 17 gene, which shortens the survival of patients (13). However, the key molecule or mechanism associated with PTX-induced autophagy in TNBC cells remains unclear.
Apoptosis is a terminal pathway of cell death and is closely related to morphogenesis during the elimination of aged or harmful cells to maintain adult tissue homeostasis (14). Some crosstalk between autophagy and apoptosis has been reported in tumors; autophagy may be an adaptive stress response prior to apoptotic cell death, either enabling or antagonizing apoptosis (9). Therefore, the relationship between apoptosis and autophagy is critical for tumor targeted therapy.
Given the high invasiveness of the MDA-MB-231 cell line, which is commonly used to investigate the molecular basis of TBNC and for the development of novel therapeutic approaches (15), MDA-MB-231 cells were used in the present study to investigate the molecular mechanism of PTX against TNBC in vitro. The present study assessed whether PTX induced cell apoptosis and autophagy at certain concentrations, whether inhibition of autophagy contributed to PTX-induced apoptosis and the underlying molecular mechanism of PTX. Finally, it was investigated whether inhibition of the molecular mechanism promotes PTX-induced apoptosis and may be a clinical chemotherapy strategy of PTX for TNBC.
Materials and methods
Regents and antibodies
All experimental reagents and antibodies were purchased from Sigma-Aldrich (Merck KGaA) unless otherwise stated. Bafilomycin A1 (Baf A1) was purchased from Sangon Biotech Co., Ltd. The antibodies used in this study included anti-FOXO1 antibody (cat. no. 2880; Cell Signaling Technology, Inc.), anti-p-FOXO1 antibody (cat. no. WL03634; Wanleibio Co., Ltd.) and anti-Lamin AC antibody (cat. no. 10298-1-AP; ProteinTech Group, Inc.).
Cell culture
MDA-MB-231 TNBC cell lines were purchased from The Cell Bank of Type Culture Collection of The Chinese Academy of Sciences and HCC-1937 cell lines were provided by the Kunming Institute of Zoology, Chinese Academy of Sciences. The cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM; cat. no. C11995500BT; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS; cat. no. 10099-141; Gibco; Thermo Fisher Scientific, Inc.), penicillin (100 U/ml) and streptomycin (100 µg/ml) (Invitrogen; Thermo Fisher Scientific, Inc.) in a humidified atmosphere of 5% CO2 at 37°C.
Cell morphological observation
Exponentially growing MDA-MB-231 and HCC-1937 cells were transferred to 12-well plates at a density of 2×104 cells/well and cultured at 37°C in a 5% CO2 atmosphere. The cells were treated with 0, 10, 20 and 30 nM PTX (cat. no. 48248; MedChemExpress) at 37°C. When the cells reached 60-70% confluency, they were rinsed twice with PBS and supernatant was discarded and cells were used for morphological observation. Images were acquired using an Olympus IX 71 microscope (Olympus Corporation). Next, MDA-MB-231 cells were treated with different concentrations of PTX (0, 0.03, 0.1, 0.3, 1, 3, 10 and 30 nM) at 37°C, 5% CO2 for 24 h to perform cell proliferation assay using cell counting kit (CCK-8) (cat. no. P10330; TransGen Biotech) according to manufacturer's instruction.
Annexin V-FITC/PI staining for apoptosis analysis
For detecting PTX-induced cell apoptosis, MDA-MB-231 cells were treated with 0, 10, 20 and 30 nM PTX at 37°C, 5% CO2 for 24 h. For detecting PTX-induced cell apoptosis under FOXO1 inhibition, MDA-MB-231 cells were transfected with small interfering RNA at 37°C for 7 h, and then were treated with or without 20 nM PTX at 37°C, 5% CO2 for 24 h. The apoptosis assay was performed using an Annexin V-FITC/PI test kit (cat. no. FXP018-100; Beijing 4A Biotech Co., Ltd.), according to the manufacturer's instructions. Flow cytometry analysis was performed using a Beckman CytoFLEX flow cytometer (Beckman Coulter, Inc.). The cells undergoing apoptosis were defined as reported previously (16) early (Annexin V-FITC-positive/PI-negative, Q3) + late apoptotic cells (Annexin V-FITC-positive/PI-positive; Q2).
Immunofluorescence
For detecting LC3, MDA-MB-231 cells were pretreated with Baf A1 (0 and 10 nM) at 37°C, 5% CO2 for 24 h and then treated with or without 20 nM PTX at 37°C, 5% CO2 for 24 h. Cells were treated with 0, 10 and 30 nM PTX combined with or without 5 nM 3-methyladenine (3-MA; 5 nM; a PI3K inhibitor that is a widely used inhibitor of autophagy via its inhibitory effect on class III PI3K) at 37°C, 5% CO2 for 24 h. For detecting FOXO1, MDA-MB-231 cells were treated with 0, 10 and 20 nM PTX at 37°C, 5% CO2 for 24 h. Immunofluorescence analysis of light chain 3 (LC3) and FOXO1 proteins was performed as described in our previous study (17). In brief, the treated cells were fixed in 95% methanol at room temperature for 10 min and blocked in a buffer containing 1% BSA (cat. no. A8020; Beijing Solarbio Science & Technology Co., Ltd.) and 0.1% Triton X-100 at RT for 1 h. Then, the fixed cells were incubated with primary antibodies against LC3B (1:200) and FOXO1 (1:100) at 4°C overnight. Next, the cells were incubated with secondary fluorescence-conjugated antibodies (Green for LC3, 1:200, cat. no. 111-545-003; Red for FOXO1, 1:200, cat. no. 711-605-152, Jackson ImmunoResearch Inc.) at room temperature for 1 h for visualization via laser confocal microscopy (Olympus FV 1000; Olympus Corporation).
Colony formation assay
This assay was performed according to our previous study (18). Briefly, adherent MDA-MB-231 cells (250 cells/plate) were treated with or without 3-MA and/or PTX (0, 10, 30 nM) at 37°C for 72 h and then cultured for 15 days. Thereafter, the cells were fixed with 4% paraformaldehyde (cat. no. P1110; Solarbio) at RT for 30 min and stained with 10% Giemsa (cat. no. G1015; Beijing Solarbio Science & Technology Co., Ltd.) at RT for 15 min. The colonies were washed, air dried, imaged and counted (>100 cells as one colony). Finally, the colony formation ratio was calculated according to the following formula: Colony formation ratio=number of colonies/number of seeded cells ×100%.
Small interfering RNA (siRNA) and transient transfection
MDA-MB-231 cells (2×106 cells/well) were seeded into 96- or 6-well plates. Then, control random siRNA (cat. no. sc-37007; proprietary sequence) or FOXO1-targeted siRNA (cat. no. sc-35382) that included a pool of three sequences are in Table SI (Santa Cruz Biotechnology, Inc.; 100 pmol/well) were transfected into MDA-MB-231 cells using an siRNA transfection reagent (Santa Cruz Biotechnology, Inc.) according to the manufacturer's protocol. The cells were transfected at 37°C for 7 h, washed with PBS and then were immediately treated with 0 and 20 nM PTX at 37°C for 24 h. Then, the cells were immediately collected, and cell lysates were prepared for reverse transcription-quantitative PCR (RT-qPCR) and western blotting. Cells were also used for apoptosis analyses.
RT-qPCR
The MDA-MB-231 cells treated with 0, 10 and 20 nM PTX at 37°C for 24 h were used for the analysis of mRNA expression of the autophagy-regulated genes. The MDA-MB-231 cells transfected with siRNA at 37°C for 7 h were treated with 0 and 20 nM PTX at 37°C, 5% CO2 for 24 h and used for the analysis of mRNA expression of the autophagy-related genes (ATG). The mRNA expression levels of genes were assessed via qPCR, including the autophagy-regulated genes, sestrin 1 (SESN1), phosphatase and tensin homolog (PTEN), mTOR, ectopic P-granules autophagy protein 5 homolog (EPG5), TSC complex subunit 1 (TSC1), serine/threonine kinase 1 and 11 (AKT and LKB1), FOXO1, lamin A/C (LMNA), autophagy and beclin 1 regulator 1 (AMBRA1), DNA damage regulated autophagy modulator 1 and 2 (DRAM1 and DRAM2), as well as, the ATGs, ATG5, class III phosphoinositide 3-kinase vacuolar protein sorting 34 (VPS34), autophagy related 4B cysteine peptidase (ATG4B), BECN1 and microtubule associated protein 1 light chain 3β (MAP1LC3B). Total RNA was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was synthesized from total RNA using a Prime-Script RT reagent kit (Takara Bio, Inc.) according to the manufacturer's protocol. The obtained cDNA was used as a template for TB Green-based qPCR kit (cat. no. RR820B; Takara Bio, Inc.) on a CFX-96 system (Bio-Rad Laboratories, Inc.). GAPDH was used for normalization. The primer sequences are shown in Table SII. Thermocycling conditions were as follows: Initial denaturation, 95°C for 30 sec followed by 40 cycles of 95°C for 5 sec, 60°C for 30 sec, and 72°C for 30 sec. Finally, the data was analyzed using 2−∆∆Cq method (19).
Western blotting
MDA-MB-231 and HCC-1937 cells were treated with 0, 10, 20 and 30 nM PTX at 37°C, 5% CO2 unless otherwise stated for 24 h. At the end of the designed treatment time, the cells were washed twice with PBS and collected. The cells were lysed in RIPA lysis buffer (cat. no. BB-3201-2; Bestbio) with protease inhibitors (cat. no. D1201-2; TransGen Biotech) at 4°C. Then, the total protein concentrations of the cell lysates were determined using a BCA Protein Assay kit (cat. no. P0009-1 & 2; Beyotime Institute of Biotechnology). The cytosolic and nuclear proteins were extracted using Nuclear and Cytoplasmic Protein Extraction kit (cat. no. P0028-1; Beyotime Institute of Biotechnology) according to manufacturer's instruction. Protein samples (50 µg/lane) were separated via 12% SDS-PAGE and subsequently transferred onto PVDF membranes. The membranes were incubated at RT for 30 min in 5% BSA buffer (Beijing Solarbio Science & Technology Co., Ltd.) with gentle shaking to block non-specific binding before incubation with the diluted primary antibody (Bcl-2, 1:1,000, cat. no. AF6139, Affinity; Bax, 1:1,000, cat. no. WL01637, Wanlei, China; LC3B, 1:2,000, cat. no. L7543; Sigma-Aldrich; Merck KGaA; P62; 1:2,000, cat. no. P0067; Sigma-Aldrich; Merck KGaA; FOXO1, 1:1,000; p-FOXO1, 1:1,000; AKT, 1:1,000, cat. no. 4691S; Cell Signaling Technology, Inc.; p-AKT, 1:1,000, cat. no. 9271S; Cell Signaling Technology, Inc.; Lamin AC, 1:2,000; β-actin, 1:5,000, cat. no. A5441; Sigma-Aldrich; Merck KGaA) overnight at 4°C. Subsequently, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (rabbit IgG, 1:5,000, cat. no. HAF008; mouse IgG, 1:10,000, cat. no. HAF007; R&D Systems) for 90 min at RT. Membranes were washed three times in PBS for 10 min each time. Then, the membranes were treated for 3 min in the dark with reagent from an Easysee Western Blot kit (TransGen Biotech) and visualized using an imaging system (Bio-Rad Laboratories, Inc.). The densitometry of protein bands was performed using Image Lab Software (version 6.1; Bio-Rad Laboratories, Inc.).
Statistical analysis
All numerical data are representative of at least three independent experiments and are presented as the mean ± standard deviation (SD) on the basis of three independent experiments. The data from the different concentrations of PTX were analyzed by one-way ANOVA, while the data from the combined treatment with PTX + Baf A1 or PTX + si-FOXO1 were analyzed by two-way ANOVA followed by a Tukey's post hoc test. The data from PTX and PTX + 3-MA treatment were analyzed using unpaired Student's t-test. Analysis of all numerical data was performed using SPSS version 22 software (IBM Corp.). P<0.05 was considered to indicate a statistically significant difference.
Results
PTX induces cytotoxicity and apoptosis of MDA-MB-231 cells
PTX is a chemotherapeutic agent that promotes the polymerization of tubulin, which disrupts normal microtubule dynamics, leading to cell death (20). To determine the effect of PTX on MDA-MB-231 cell viability, morphological observations, and CCK-8 assays were performed after PTX treatment. Morphological changes revealed that MDA-MB-231 cell death increased with increasing doses of PTX (Fig. 1A). Furthermore, the CCK-8 assay showed that the cell survival rate also decreased with increasing PTX doses, and the half maximal inhibitory concentration (IC50) of PTX fell between 10 and 30 nM (Fig. 1B). These results indicated that PTX induced MDA-MB-231 cell death in a dose-dependent manner.
Figure 1.

PTX exerts cytotoxicity and induces apoptosis in MDA-MB-231 cells. (A) Images of MDA-MB-231 cells exposed to 0, 10, 20 and 30 nM PTX for 24 h (scale bar, 200 µm). (B) Quantification of cell viability after treatment with different doses of PTX. *P<0.05 vs. 0 nM; #P<0.05 vs. 1 nM PTX; $P<0.05 vs. 3 nM PTX; &P<0.05 vs. 10 nM PTX. (C) Effect of different doses of PTX on expression levels of the apoptosis proteins Bcl-2 and Bax, with β-actin as an internal control (left) and semi-quantification of the Bcl-2/Bax ratio (right). (D) Effect of different doses of PTX on the apoptosis of MDA-MB-231 cells. (E) Quantification of the ratio of apoptotic cells in response to different doses of PTX. *P<0.05 vs. 0 nM PTX; #P<0.05 vs. 20 nM PTX. PTX, paclitaxel.
To further determine whether the observed cell death was caused by PTX inducing the apoptotic pathway, the expression of apoptotic markers was evaluated and the results indicated that the expression levels of the apoptosis markers Bcl-2 and Bax, especially expression levels of Bcl-2, were decreased in a dose-dependent manner (Fig. 1C). Further, PI and Annexin V staining coupled with flow cytometry were also performed and it was observed that PTX exposure resulted in a dose-dependent increase in the apoptosis rate (Fig. 1D and E). Collectively, these results indicated that PTX induced the apoptosis of MDA-MB-231 cells.
PTX induces autophagy in MDA-MB-231 cells
In response to PTX-induced MDA-MB-231 cell apoptosis, the effect of PTX on autophagy was investigated. A significant increase in LC3-II/LC3-I and a decrease in P62 in response to 30 nM PTX was observed (Fig. 2A-C), indicating an increased level of autophagy. Considering that the IC50 value of PTX was close to 20 nM, it was speculated whether 20 nM PTX could induce autophagy. Therefore, autophagic flux levels in MDA-MB-231 cells treated with 20 nM PTX were assessed after blocking autophagosome-lysosome fusion and degradation with Baf A1. Immunoblotting results revealed a significant increase in LC3 puncta in the presence of PTX + Baf A1 (Fig. 2D and E), suggesting that 20 nM PTX induced increased autophagic flux. Taken together, 20 and 30 nM PTX induced autophagy, while simultaneously inducing apoptosis in MDA-MB-231 cells.
Figure 2.

PTX induces autophagy in MDA-MB-231 cells. (A) Protein expression levels of the autophagy markers P62, LC3-I and LC3-II at different doses of PTX. β-actin was used as an internal control. The semi-quantification of (B) P62 (*P<0.05 vs. 0 nM PTX), and (C) LC3-II/LC3-I protein expression (*P<0.05 vs. 0 nM PTX). (D) Representative images of LC3 puncta. Cells were stained with antibodies against LC3 (green), and nuclei were stained with DAPI (blue). Scale bar, 20 µm. (E) Quantification of LC3 puncta (**P<0.01 vs. 0 nM PTX; ##P<0.01 vs. 20 nM PTX). PTX, paclitaxel; LC3, light chain 3; Baf A1, bafilomycin A1.
In addition, another TNBC cell line (HCC-1937) were also treated with 0, 10, 20 and 30 nM PTX and it was observed that HCC-1937 cells were more sensitive to PTX treatment than MDA-MB-231 cells because more HCC-1937 cells died compared with MDA-MB-231 cells at the same dose of PTX (Fig. S1A). A significant decrease in the Bcl-2/Bax ratio (Fig. S1B and C) and a significant increase in the LC3-II/LC3-I ratio (Fig. S1F and G) was found, although no change in P62 protein expression (Fig. S1D and E) was found at 30 nM PTX, suggesting that PTX-induced apoptosis and autophagy in TNBC cells is a common phenomenon.
Inhibition of autophagy increases PTX-induced apoptotic cell death in MDA-MB-231 cells
Given that PTX enabled the simultaneous induction of apoptosis and autophagy in MDA-MB-231 cells, it was next investigated whether inhibition of autophagy contributed to improving PTX-induced apoptotic cell death. After treatment with PTX + 3-MA, inhibition of autophagy was clearly observed, including a significant decrease in the LC3-II/LC3-I ratio at 10 and 30 nM PTX (Fig. 3A and B) and an increase in P62 expression at 30 nM (Fig. 3A and C). In addition, LC3 puncta also markedly decreased at 10 and 30 nM PTX after treatment with 3-MA (Fig. 3D). Furthermore, the formation of cell colonies was effectively inhibited at 10 and 30 nM PTX + 3-MA (Fig. 3E and F). These results indicated that inhibition of autophagy with 3-MA promoted PTX-induced apoptosis in MDA-MB-231 cells.
Figure 3.

Inhibition of autophagy with 3-MA promotes PTX-induced apoptotic cell death. (A) Effect of 3-MA on protein expression levels of the autophagy markers P62, LC3-I and LC3-II. β-actin was used as an internal control. Semi-quantification of the (B) LC3-II/LC3-I ratio and (C) P62 protein expression. (D) Representative images of LC3 puncta after treatment with or without 3-MA and/or PTX. (E) Detection of cell viability by colony formation assay. (F) Quantification of the cellular colony formation rate shown in E. Data are presented as the means ± SD of three independent experiments. *P<0.05 and **P<0.01; ns, non-significant; PTX, paclitaxel; LC3, light chain 3; 3-MA, 3-methyladenine.
PTX increases FOXO1 expression in MDA-MB-231 cells
Autophagy is regulated by autophagy-regulated genes, such as the FOXO1, PTEN, LKB1, mTOR, SESN1, EPG5, TSC1, AKT, LMNA, AMBRA1 and DRAM1 genes, which are essential for autophagy signaling pathways (21–23). The mRNA levels of these genes were measured via RT-qPCR and it was found that 20 nM PTX induced a 2.7-fold increase in FOXO1 mRNA in MDA-MB-231 cells (Fig. 4A). Therefore, the role of FOXO1 in 20 nM PTX-treated MDA-MB-231 cells was the focus of subsequent assays. First, FOXO1 and p-FOXO1 protein expression in response to PTX treatment was assessed. The results showed that PTX induced a significant decline in the p-FOXO1/FOXO1 ratio in a dose-dependent manner, suggesting that FOXO1 rather than p-FOXO1 played an important role in PTX-induced autophagy in MDA-MB-231 cells (Fig. 4B and C). In addition, the upstream inhibitor of FOXO1, p-ATK, exhibited a significantly downregulated pattern (Fig. 4D and E). Thus, it was speculated that the decreased p-FOXO1/FOXO1 ratio was related to decreased p-AKT levels. Since FOXO1 is a transcription factor, it was assessed whether FOXO1 exerted a transcriptional function in PTX-mediated autophagy of MDA-MB-231 cells. Therefore, the localization of FOXO1 was further detected via western blotting and immunofluorescence. The results showed that FOXO1 was primarily distributed in the nucleus, further confirming that FOXO1 exerted its autophagy-regulated function in a transcriptional activation pattern (Fig. 5A and B).
Figure 4.

Autophagy regulatory pathway induction by PTX in MDA-MB-231 cells. (A) mRNA expression levels of autophagy signaling pathway-related genes in response to treatment with 10 and 20 nM PTX. GAPDH was used as an internal control. (*P<0.05 vs. 0 nM PTX; #P<0.05 vs. 10 nM PTX). (B) Protein expression of FOXO1 and p-FOXO1 and (C) semi-quantification of the p-FOXO1/FOXO1 ratio (*P<0.05 vs. 0 nM PTX; #P<0.05 vs. 10 nM PTX). (D) Protein expression of AKT and p-AKT and (E) semi-quantification of the p-AKT/AKT ratio (*P<0.05 vs. 0 nM PTX). β-actin was used as an internal control. Data are presented as the means ± SD of three independent experiments. PTX, paclitaxel; FOXO1, forkhead box transcription factor O1; p-, phosphorylated; SESN1, sestrin 1; PTEN, phosphatase and tensin homolog; mTOR, mechanistic target of rapamycin kinase; EPG5, ectopic P-granules autophagy protein 5 homolog; TSC1, TSC complex subunit 1; AKT, serine/threonine kinase 1; LKB1, serine/threonine kinase 11; LMNA, lamin A/C; AMBRA1, autophagy and beclin 1 regulator 1; DRAM1, DNA damage regulated autophagy modulator 1; DRAM2, DNA damage regulated autophagy modulator 2.
Figure 5.

PTX induces increased nuclear FOXO1 in MDA-MB-231 cells. (A) Localization of FOXO1 by western blotting. Lamin AC, a nucleus-specific marker, and β-actin were used as internal controls. (B) Localization of FOXO1 was detected by immunofluorescence staining (scale bar, 20 µm). PTX, paclitaxel; FOXO1, forkhead box transcription factor O1.
FOXO1 is required for PTX-induced autophagy in MDA-MB-231 cells
To investigate the function of FOXO1 in PTX-induced autophagy in MDA-MB-231 cells, FOXO1 expression was knocked down using siRNA. At the protein level, the expression of FOXO1 was successfully inhibited after treatment with siRNA (Fig. 6A and B). Knocking down the FOXO1 gene led to significantly decreased LC3II and significantly increased P62 protein expression levels in PTX-treated MDA-MB-231 cells, suggesting reduced autophagy levels (Fig. 6A, C and D). A number of autophagy-related genes, such as ATG5, VPS34 (PI3KC3), ATG4B, BECN1 and MAP1LC3B, are transcriptionally regulated by FOXO1 (24). In this study, it was also found that these genes were suppressed in response to FOXO1 knockdown in PTX-treated MDA-MB-231 cells (Fig. 6E). Importantly, knockdown of FOXO1 enhanced PTX-induced apoptotic cell death (Fig. 6F and G). These results illustrated that FOXO1 played a role in PTX-induced autophagy in MDA-MB-231 cells and that targeting FOXO1 may improve the efficacy of PTX in TNBC therapy.
Figure 6.

Knockdown of FOXO1 attenuates PTX-induced autophagy and promotes apoptotic cell death in MDA-MB-231 cells. (A) Protein expression of the autophagy markers P62, LC3-I and LC3-II after treatment with si-FOXO1, with β-actin as an internal control. Semi-quantification of (B) FOXO1, (C) P62 and (D) LC3-II protein expression. (E) The mRNA expression levels of FOXO1 and its downstream target genes after treatment with siRNA and/or PTX. (F) The effect of FOXO1 knockdown on PTX-induced apoptosis was analyzed by flow cytometry. (G) Quantification of the ratio of apoptotic cells after the knockdown of FOXO1. PTX was used at a concentration of 20 nM. Data are presented as the means ± SD of three independent experiments. *P<0.05 vs. 0 nM PTX; **P<0.01 vs. 0 nM PTX; #P<0.05 vs. 20 nM PTX + si-Con; ##P<0.01 vs. 20 nM PTX + si-Con. PTX, paclitaxel; FOXO1, forkhead box transcription factor O1; LC3, light chain 3; si, small interfering; ATG5, autophagy related 5; VPS34, class III phosphoinositide 3-kinase vacuolar protein sorting 34; ATG4B, autophagy related 4B cysteine peptidase; BECN1, beclin 1; MAP1LC3B, microtubule associated protein 1 light chain 3β.
Discussion
Although PTX has been widely used for the treatment of various solid tumors, including ovarian, lung and breast cancer (25), its chemotherapeutic efficacy varies among different types of cancer, particularly in cases where resistance has been developed against it. Previously, it has also been found that TNBC frequently acquires resistance against PTX through different regulatory pathways (26,27). For example, PTX can trigger breast cancer type 1 (BRCA1)-IRIS expression, a product of the oncogene BRCA1, which enhances AKT-related signaling and results in a PTX resistance phenotype in TNBC (28). Another study demonstrated that aurora kinase A can also promote PTX resistance in TNBC by stabilizing FOXM1 (3). Therefore, targeting these pathways or inhibiting tumor-associated factors may be helpful for improving PTX efficacy in TNBC (29–33). Additionally, it has been demonstrated that inhibition of autophagy enhances PTX-induced cell death in TNBC MDA-MB-231 cells with PTX resistance (34). Consistently, inhibition of autophagy also contributed to promoting PTX-induced apoptosis in MDA-MB-231 cells in the present study. Elevated autophagic flux was implicated in the significant increase in nuclear FOXO1. Furthermore, knockdown of FOXO1 enhanced PTX-induced apoptosis. These findings may provide a potential anticancer target and be important for improving PTX efficacy in TNBC treatment.
Autophagy and apoptosis are two antagonistic and interconnected molecular mechanisms of various cellular stresses. Autophagy, a prosurvival regulatory process, often attenuates apoptotic cell death (35,36). PTX, as a broad-spectrum anticancer agent, often induces cytotoxic apoptosis or inhibits autophagy (37,38). However, it has also been reported that PTX induces autophagy in tumor cells (39), which might have an adverse effect on PTX efficacy. Another previous study indicated that PTX induces both autophagy and apoptosis in several cancer cells. The upregulated levels of autophagy are related to the autophagosome-regulatory genes ATG5 and BECN1. After 3-MA treatment or BECN1 knockdown, the tumor cell response switches from autophagic to apoptotic (12). Similarly, the current study also demonstrated that autophagy was induced in MDA-MB-231 cells accompanied by PTX-induced apoptosis, and autophagic inhibition with 3-MA enhanced apoptotic cell death. In addition, to assess that whether PTX generally induced apoptosis and autophagy in TNBC cells, another TNBC cell line, HCC-1937, was treated with PTX (Fig. S1) and the downregulated value of Bcl-2/Bax and the upregulated value of LC3-II/LC3-I showed that PTX induced apoptosis and autophagy (Fig. S1B-G). Therefore, we speculated that PTX treatment in different TNBC cells would have similar effects. However, HCC-1937 cells seemed to be more sensitive to PTX treatment than MDA-MB-231 cells (Fig. S1A). Therefore, HCC-1937 cells were not selected for subsequent assays.
FOXO1 is a representative member of the forkhead transcription factor (FOX) family, which plays a crucial role in the inhibition of tumor proliferation and the induction of cellular responses (7). In the present study, it was found that FOXO1 played an important role in PTX-induced autophagy in MDA-MB-231 cells. The increase in FOXO1 was accompanied by attenuated AKT1 phosphorylation, suggesting that PTX induces elevated FOXO1 expression by inhibiting AKT1-related signaling. Previously, it was also determined that p-ATK1 catalyzed the phosphorylation of FOXO1, resulting in the loss of transcriptional activity and translocation from the nucleus to the cytoplasm (40). However, in the current study, it was observed that FOXO1 accumulated in the nucleus rather than translocating to the cytoplasm. Therefore, the expression of core autophagy-related genes that are transcriptionally regulated by FOXO1 were analyzed. The results revealed that ATG5, BECN1 and MAP1LC3B were significantly upregulated in response to PTX treatment, consistent with previous reports that nuclear FOXO1 transcriptionally activates ATG5 and BECN1 to promote autophagy (41). After inhibition of FOXO1 in combination with PTX treatment, ATG5, VPS34, ATG4B, BECN1 and MAP1LC3B were markedly downregulated. These findings suggested that FOXO1-mediated autophagy in PTX-treated MDA-MB-231 cells regulates its downstream target genes through transactivation, which is different than cytosolic FOXO1 inducing autophagy by binding to the ATG7 gene (42). In addition, FOXO1 transcriptionally inhibits mTOR activity by increasing SENSE3 expression to promote autophagy (43). Importantly, in the present study autophagy was inhibited after FOXO1 knockdown; at the same time, the rate of apoptotic MDA-MB-231 cell death induced by PTX was increased. This finding was consistent with the concept that regulation of apoptosis by autophagy enhances cancer therapy (9) and provides a potential therapeutic target for TNBC. However, the current study only explored the in vitro role of FOXO1 in MDA-MB-231 cells, thus whether PTX treatment would lead to elevated FOXO1 expression in vivo is still unclear. Therefore, we are creating a tumor-bearing mouse model by injecting MDA-MB-231 cells into nude mice. In future studies, we will treat these mice with PTX and investigate whether FOXO1 plays the same role in vivo. Clinically, it has been reported that FOXO1 expression is not observed in tissues from patients with TNBC (44). Therefore, it is important to determine whether the tissues of patients with TNBC treated with PTX exhibit increased FOXO1 expression, which may provide novel insight into the clinicopathological significance.
In summary, PTX induced both autophagy and apoptosis, and inhibition of autophagy promoted apoptosis in MDA-MB-231 cells. Furthermore, elevated FOXO1 was associated with increased autophagic flux. Knocking down FOXO1 enhanced PTX-induced cell death, which might be important for improving PTX efficacy in TNBC (Fig. 7).
Figure 7.
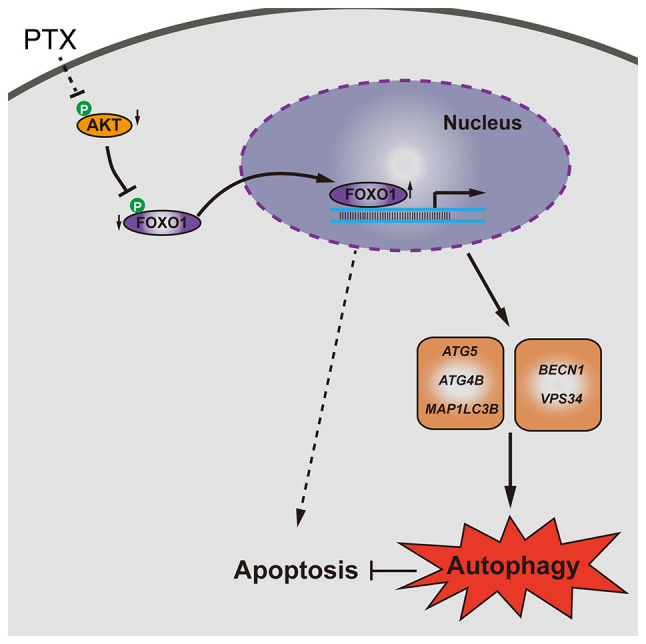
Schematic model of FOXO1-mediated autophagy and apoptosis induced by PTX in MDA-MB-231 cells. PTX, paclitaxel; FOXO1, forkhead box transcription factor O1; p-, phosphorylated; ATG5, autophagy related 5; MAP1LC3B, microtubule associated protein 1 light chain 3β; BECN1, beclin 1; VPS34, class III phosphoinositide 3-kinase vacuolar protein sorting 34; ATG4B, autophagy related 4B cysteine peptidase.
Supplementary Material
Acknowledgements
Not applicable.
Funding Statement
This study was supported by the National Key R&D Program of China (grant no. 2019YFA0110700) and National Natural Science Foundation of China (grant no. 81872452).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
HYZ and HJW conceived and designed the study. KX, WZ, AX, ZX and DJ performed the cell culture and morphological observation. KX, WZ and AX performed the apoptotic assay and immunofluorescence. ZX, DZ, HZ and YQ performed the PCR and western blotting assays. KX, MAJ and HYZ analyzed the data. KX, WZ and AX organized the data. KX, WZ, MAJ and HYZ wrote the manuscript. HYZ and HJW proofread the manuscript and confirm the authenticity of all the raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mustacchi G, De Laurentiis M. The role of taxanes in triple-negative breast cancer: Literature review. Drug Des Devel Ther. 2015;9:4303–4318. doi: 10.2147/DDDT.S86105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang N, Wang C, Wang J, Wang Z, Huang D, Yan M, Kamran M, Liu Q, Xu B. Aurora kinase A stabilizes FOXM1 to enhance paclitaxel resistance in triple-negative breast cancer. J Cell Mol Med. 2019;23:6442–6453. doi: 10.1111/jcmm.14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4:466–479. doi: 10.1158/2159-8290.CD-13-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu CC, Chan ML, Chen WY, Tsai CY, Chang FR, Wu YC. Pristimerin induces caspase-dependent apoptosis in MDA-MB-231 cells via direct effects on mitochondria. Mol Cancer Ther. 2005;4:1277–1285. doi: 10.1158/1535-7163.MCT-05-0027. [DOI] [PubMed] [Google Scholar]
- 6.Wu Q, Wu W, Fu B, Shi L, Wang X, Kuca K. JNK signaling in cancer cell survival. Med Res Rev. 2019;39:2082–2104. doi: 10.1002/med.21574. [DOI] [PubMed] [Google Scholar]
- 7.Xing YQ, Li A, Yang Y, Li XX, Zhang LN, Guo HC. The regulation of FOXO1 and its role in disease progression. Life Sci. 2018;193:124–131. doi: 10.1016/j.lfs.2017.11.030. [DOI] [PubMed] [Google Scholar]
- 8.Cheon SY, Kim H, Rubinsztein DC, Lee JE. Autophagy, cellular aging and age-related human diseases. Exp Neurobiol. 2019;28:643–647. doi: 10.5607/en.2019.28.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tompkins KD, Thorburn A. Regulation of apoptosis by autophagy to enhance cancer therapy. Yale J Biol Med. 2019;92:707–718. [PMC free article] [PubMed] [Google Scholar]
- 10.Jing Z, Sui X, Yao J, Xie J, Jiang L, Zhou Y, Pan H, Han W. SKF-96365 activates cytoprotective autophagy to delay apoptosis in colorectal cancer cells through inhibition of the calcium/CaMKIIγ/AKT-mediated pathway. Cancer Lett. 2016;372:226–238. doi: 10.1016/j.canlet.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng H, Cheng X, Kuang J, Chen L, Yuen S, Shi M, Liang J, Shen B, Jin Z, Yan J, Qiu W. Apatinib-induced protective autophagy and apoptosis through the AKT-mTOR pathway in anaplastic thyroid cancer. Cell Death Dis. 2018;9:1030. doi: 10.1038/s41419-018-1054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xi G, Hu X, Wu B, Jiang H, Young CYF, Pang Y, Yuan H. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett. 2011;307:141–148. doi: 10.1016/j.canlet.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 13.Zhang SF, Wang XY, Fu ZQ, Peng QH, Zhang JY, Ye F, Fu YF, Zhou CY, Lu WG, Cheng XD, Xie X. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy. 2015;11:225–238. doi: 10.1080/15548627.2014.998931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. 2018;36:489–517. doi: 10.1146/annurev-immunol-042617-053010. [DOI] [PubMed] [Google Scholar]
- 15.Mohammed F, Rashid-Doubell F, Taha S, Cassidy S, Fredericks S. Effects of curcumin complexes on MDA-MB-231 breast cancer cell proliferation. Int J Oncol. 2020;57:445–455. doi: 10.3892/ijo.2020.5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiao D, Cheng W, Zhang X, Zhang Y, Guo J, Li Z, Shi D, Xiong Z, Qing Y, Jamal MA, et al. Improving porcine SCNT efficiency by selecting donor cells size. Cell Cycle. 2021;20:2264–2277. doi: 10.1080/15384101.2021.1980983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu W, Qu H, Xu K, Jia B, Li H, Du Y, Liu G, Wei HJ, Zhao HY. Differences in the starvation-induced autophagy response in MDA-MB-231 and MCF-7 breast cancer cells. Anim Cells Syst (Seoul) 2017;21:190–198. doi: 10.1080/19768354.2017.1330763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lv C, Qu H, Zhu W, Xu K, Xu A, Jia B, Qing Y, Li H, Wei HJ, Zhao HY. Low-dose paclitaxel inhibits tumor cell growth by regulating glutaminolysis in colorectal carcinoma cells. Front Pharmacol. 2017;8:244. doi: 10.3389/fphar.2017.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Mekhail TM, Markman M. Paclitaxel in cancer therapy. Expert Opin Pharmacother. 2002;3:755–766. doi: 10.1517/14656566.3.6.755. [DOI] [PubMed] [Google Scholar]
- 21.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–542. doi: 10.1038/nrc.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting autophagy in cancer. Cancer. 2018;124:3307–3318. doi: 10.1002/cncr.31335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Füllgrabe J, Ghislat G, Cho DH, Rubinsztein DC. Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci. 2016;129:3059–3066. doi: 10.1242/jcs.188920. [DOI] [PubMed] [Google Scholar]
- 25.Weaver BA. How taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25:2677–2681. doi: 10.1091/mbc.e14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of taxol resistance related to microtubules. Oncogene. 2003;22:7280–7295. doi: 10.1038/sj.onc.1206934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yusuf R, Duan Z, Lamendola D, Penson R, Seiden M. Paclitaxel resistance: Molecular mechanisms and pharmacologic manipulation. Curr Cancer Drug Targets. 2003;3:1–19. doi: 10.2174/1568009033333754. [DOI] [PubMed] [Google Scholar]
- 28.Blanchard Z, Paul BT, Craft B, ElShamy WM. BRCA1-IRIS inactivation overcomes paclitaxel resistance in triple negative breast cancers. Breast Cancer Res. 2015;17:5. doi: 10.1186/s13058-014-0512-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, Stanford J, Cook RS, Arteaga CL. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123:1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wee ZN, Yatim SMJ, Kohlbauer VK, Feng M, Goh JY, Bao Y, Lee PL, Zhang S, Wang PP, Lim E, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun. 2015;6:8746. doi: 10.1038/ncomms9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sha L, Zhang Y, Wang W, Sui X, Liu SK, Wang T, Zhang H. MiR-18a upregulation decreases dicer expression and confers paclitaxel resistance in triple negative breast cancer. Eur Rev Med Pharmacol Sci. 2016;20:2201–2208. [PubMed] [Google Scholar]
- 32.Yuan Z, Jiang H, Zhu X, Liu X, Li J. Ginsenoside Rg3 promotes cytotoxicity of Paclitaxel through inhibiting NF-κB signaling and regulating Bax/Bcl-2 expression on triple-negative breast cancer. Biomed Pharmacother. 2017;89:227–232. doi: 10.1016/j.biopha.2017.02.038. [DOI] [PubMed] [Google Scholar]
- 33.Zhou YF, Sun Q, Zhang YJ, Wang GM, He B, Qi T, Zhou Y, Li XW, Li S, He L. Targeted inhibition of notch1 gene enhances the killing effects of paclitaxel on triple negative breast cancer cells. Asian Pac J Trop Med. 2017;10:179–183. doi: 10.1016/j.apjtm.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Wen J, Yeo S, Wang C, Chen S, Sun S, Haas MA, Tu W, Jin F, Guan JL. Autophagy inhibition re-sensitizes pulse stimulation-selected paclitaxel-resistant triple negative breast cancer cells to chemotherapy-induced apoptosis. Breast Cancer Res Treat. 2015;149:619–629. doi: 10.1007/s10549-015-3283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang K. Autophagy and apoptosis in liver injury. Cell Cycle. 2015;14:1631–1642. doi: 10.1080/15384101.2015.1038685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veldhoen R, Banman S, Hemmerling D, Odsen R, Simmen T, Simmonds AJ, Underhill DA, Goping IS. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene. 2013;32:736–746. doi: 10.1038/onc.2012.92. [DOI] [PubMed] [Google Scholar]
- 38.Xie S, Ogden A, Aneja R, Zhou J. Microtubule-binding proteins as promising biomarkers of paclitaxel sensitivity in cancer chemotherapy. Med Res Rev. 2016;36:300–312. doi: 10.1002/med.21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eum KH, Lee M. Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem. 2011;348:61–68. doi: 10.1007/s11010-010-0638-8. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, Liao W, Yang J, Ma K, Li X, Wang Y, Wang D, Wang L, Zhang Y, Yin Y, et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy. 2012;8:1712–1723. doi: 10.4161/auto.21830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu P, Das M, Reilly J, Davi RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011;25:310–322. doi: 10.1101/gad.1984311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12:665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Ng S, Wang J, Zhou J, Tan SH, Yang N, Lin Q, Xia D, Shen HM. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy. 2015;11:629–642. doi: 10.1080/15548627.2015.1023981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rehman A, Kim Y, Kim H, Sim J, Ahn H, Chung MS, Shin SJ, Jang K. FOXO3a expression is associated with lymph node metastasis and poor disease-free survival in triple-negative breast cancer. J Clin Pathol. 2018;71:806–813. doi: 10.1136/jclinpath-2018-205052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.