

Abstract
Niemann-Pick disease, type C1 is a progressive, lethal, neurodegenerative disorder due to endolysosomal storage of unesterified cholesterol. Cerebellar ataxia, as a result of progressive loss of cerebellar Purkinje neurons, is a major symptom of Nieman-Pick disease, type C1. Comparing single cell RNAseq data from control (Npc1+/+) and mutant (Npc1−/−) mice, we observed significantly decreased expression of Slc1a3 in Npc1−/− astrocytes. Slc1a3 encodes a glutamate transporter (GLAST, EAAT1) which functions to decrease glutamate concentrations in the post synaptic space after neuronal firing. Glutamate is an excitatory neurotransmitter and elevated extracellular levels of glutamate can be neurotoxic. Impaired EAAT1 function underlies type-6 episodic ataxia, a rare disorder with progressive cerebellar dysfunction, thus suggesting that impaired glutamate uptake in Niemann-Pick disease, type C1 could contribute to disease progression. We now show that decreased expression of Slc1a3 in Npc1−/− mice has functional consequences that include decreased surface protein expression and decreased glutamate uptake by Npc1−/− astrocytes. To test whether glutamate neurotoxicity plays a role in Niemann-Pick disease, type C1 progression, we treated NPC1 deficient mice with ceftriaxone and riluzole. Ceftriaxone is a β-lactam antibiotic that is known to upregulate the expression of Slc1a2, an alternative glial glutamate transporter. Although ceftriaxone increased Slc1a2 expression, we did not observe a treatment effect in NPC1 mutant mice. Riluzole is a glutamate receptor antagonist that inhibits postsynaptic glutamate receptor signaling and reduces the release of glutamate. We found that treatment with riluzole increased median survival in Npc1−/− by 12%. Given that riluzole is an approved drug for the treatment of amyotrophic lateral sclerosis, repurposing of this drug may provide a novel therapeutic approach to decrease disease progression in Niemann-Pick disease type, C1 patients.
Keywords: Niemann-Pick disease, type C1, NPC1, Lysosomal Disease, Glutamate-mediated Neurotoxicity, Purkinje Neurons, Riluzole, SLC1A3, EAAT1, GLAST
Introduction
Niemann-Pick disease, type C (NPC) is a rare, lethal lysosomal disease clinically characterized by progressive neurodegeneration. NPC can be caused by mutation of either NPC1 or NPC2; however, most cases are due to impaired NPC1 function [1]. The incidence of NPC1 is estimated to be on the order of 1/100,000–120,000 [1, 2]. The NPC1 protein, in conjunction with NPC2, transports unesterified cholesterol out of the lysosomal lumen to the lysosomal limiting membrane where it becomes bioavailable [3]. Impaired function of either NPC1 or NPC2 results in endolysosomal accumulation of unesterified cholesterol and decreased cholesterol bioavailability for cellular functions. Major clinical symptoms observed in NPC1 are progressive cerebellar ataxia and dementia [4]. The age of onset is heterogeneous, but NPC1 primarily affects children and adolescents with a median age of death of 13 years [5]. Long-term treatment data has shown that miglustat, a glycosphingolipid synthesis inhibitor, delays NPC1 neurological disease progression and increases survival [6–8]. Although miglustat has been approved for treatment of NPC by the European Medicines Agency and other countries, it is not approved in the United States. Currently there are no FDA approved therapies for NPC1, thus there remains a significant unmet medical need for therapeutic interventions to treat patients with NPC1.
Loss of cerebellar Purkinje neurons underlies the progressive cerebellar ataxia that is observed in NPC1 patients. Purkinje neurons integrate extensive excitatory cerebellar neuronal inputs and are the sole output of the cerebellar cortex, via inhibitory GABAergic signaling, to coordinate motor function. Purkinje neurons show differential sensitivity to loss of NPC1 function with aldolase C positive cells being more resistant to loss [9]. Aldolase C positive Purkinje neurons are enriched in posterior lobules and lateral stripes. A characteristic histopathological finding in NPC1 mice is an anterior to posterior progression of Purkinje neuron loss [10, 11]. The pathological mechanisms underlying the differential sensitivity are not known; however, Purkinje neurons are highly susceptible to glutamate-mediated excitotoxicity [12, 13]. Glutamate is an excitatory neurotransmitter and after synaptic release, extracellular glutamate concentrations are decreased by sodium-dependent plasma membrane glutamate transporters (Excitatory Amino Acid Transporters or EAATs). EAAT1 (GLAST) is encoded by SLC1A3 and EAAT4 is encoded by SLC1A6. These glutamate transporters function to maintain low extracellular glutamate levels and play a major role in protecting Purkinje neurons from glutamate-mediated excitotoxicity. In the cerebellum, SLC1A3 is expressed uniformly by glial cells including both Bergmann and radial glia which are closely associated with the soma of Purkinje neurons [14]. SLC1A6 is expressed by Purkinje neurons, with higher SLC1A6 expression in aldolase C positive Purkinje neurons [12, 15]. Glutamate hyperstimulation leads to increase calcium influx, mitochondrial dysfunction and unregulated activation of degradative enzymes that ultimately results in cell death [16, 17].
EAAT1 functions as the primary glutamate transporter in the cerebellum and is expressed by astrocytes [18, 19]. Using single cell transcriptome analysis, we recently observed decreased expression of Slc1a3 in cerebellar astrocytes from NPC1 mutant mice [20] and previous work by Rabenstein et al. [21] showed decreased EAAT1 protein expression in cerebellar tissue from NPC1 mutant mice. Loss of EAAT1 function results in type-6 episodic ataxia [22] and glutamate-mediated neurotoxicity has been proposed to contribute to neurodegeneration in common disorders such as Alzheimer disease and Amyotrophic Lateral Sclerosis [23]. We thus hypothesized that glutamate-mediated neurotoxicity could contribute to Purkinje neuron loss and that therapies designed to reduce glutamate toxicity would have potential benefit in the treatment of NPC1. In this paper we further characterized EAAT1 dysfunction in NPC1 and explored the therapeutic potential of reducing glutamate-mediated neurotoxicity.
Materials and Methods
Mouse models and phenotypic evaluation
Mouse experiments were approved by the NICHD Animal Care and Use Committee. BALB/cNctr-Npc1m1N/J (Npc1+/−) [24] mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Heterozygous Npc1+/− mice were intercrossed to obtain control (Npc1+/+) and mutant (Npc1−/−) littermates. Mice were genotyped by PCR using ear punch DNA and primers listed in Table S1. Water and chow were available ad libitum. An a priori power calculation was performed to guide adequate design of the survival experiment. The mean survival of Balb/c Npc1−/− mice in our colony is 75 ± 4 days. Assuming no change in the experimental control mean and standard deviation (e.g. due to the vehicle or genetic background) and a similar standard deviation in the experimental group we have 95% power to detect an 8–9 day (~12%) increase in survival at p=0.05 using cohorts of 6–8 mice. Additional mice are enrolled to account for experimental variation and to allow for post-hoc determination of gender differences. A humane survival endpoint was defined as hunched posture, reluctance to move, inability to remain upright when moving and weight loss >30% of peak weight. Ledge test, grooming, motor coordination, kyphosis, and hind limb clasping were assessed as previously described [25]. Individuals conducting the phenotypic assessments were blinded as to treatment status.
Investigational drug administration and sample collection
Riluzole (Selleckchem, Houston, TX, USA) was resuspended in DMSO at 30mg.ml−1 as a 500-fold concentrated stock solution and further diluted in drinking water. Water was replaced every three days. Ceftriaxone (Cayman Chemical, Ann Arbor, MI, USA) was resuspended in PBS at 100 mg.ml−1 and a 100 mg.kg−1 dose was administered daily by intraperitoneal injection. Dosing of investigational agents was initiated after weaning of the animals on day of life ~21. Six mice per group were sacrificed at 7 weeks of age for pathology and RNA extraction, the remaining animals were treated until they reached the humane disease end point described above. Mutant, control, treated and untreated animals were enrolled concurrently, and efforts were made to balance sex distribution.
Cell culture and glutamate uptake
Cell lines, growth conditions and TaqMan Probes are listed in Table S1. Glutamate concentrations in astrocyte culture media were measured using a fluorometric glutamate assay kit (MET-5080, CellBiolabs, San Diego, USA). Media containing 50µM glutamate was added to the cells and aliquots were collected at 0, 1, 3 and 6 hours for measurement of glutamate concentration. The data represents the percent glutamate remaining in the media after each time point normalized to the initial glutamate concentration. Glutamate uptake was considered to be the difference between initial and remaining concentration of glutamate in the culture medium. Results were normalized to the mean uptake by Npc1+/+ astrocytes.
Human tissue
Anonymous post-mortem tissue was obtained from the NICHD Blood and Tissue Bank (http://medschool.umaryland.edu/btbank/) and processed for western blot analysis as previously described [26]. Tissue reference numbers are listed in Table S1.
RNA extraction and qPCR
Total RNA was extracted from frozen cell pellets or cerebellar tissue using TRIzol (Thermo Fischer Scientific, Waltham, MA, USA), followed by RNA purification using Qiagen RNeasy Mini Columns (Qiagen, Hilden, Germany). For gene expression analysis by real-time qPCR, the quality and the quantity of RNA was assessed using a NanoDrop (Thermo Fischer Scientific, Waltham, MA, USA). One microgram of total RNA was reverse-transcribed to obtain cDNA using a High-Capacity cDNA Archive Kit, according to the manufacturer’s instructions (Thermo Fischer Scientific, Waltham, MA, USA). TaqMan assays used are listed in Table S1 and were used according to the manufacturer’s instructions. Amplifications were performed in 96-well plates with an Applied Biosystems 7300 real-time PCR system (Carlsbad, CA, USA). Each sample was analyzed in duplicate, using 50ng of total cDNA per reaction.
Immunofluorescence and Flow Cytometry analysis
Mice were euthanized at 7 weeks by CO2 asphyxiation and transcardially perfused with 30 ml of PBS, pH 7.4. Brain tissue was further post-fixed in 4% paraformaldehyde solution for 24 hours and then cryoprotected in 30% sucrose (Sigma-Aldrich Millipore, St Louis, MO, USA). Frozen parasagittal sections (20μm) of cerebellar tissue were obtained using a cryostat and then floated in PBS with 0.25% Triton X-100 (Sigma-Aldrich Millipore, St Louis, MO, USA). The tissue sections were stained at 4°C overnight with the antibody and dyes listed in Table S1. Images were taken using a Zeiss Axio Observer Z1 microscope fitted with an automated scanning stage, Colibri II LED illumination and Zeiss ZEN2 software using a high-resolution AxioCam MRm camera and a 20× objective. Each fluorophore channel was pseudo-colored in ZEN2, exported as a .czi file, and analyzed using ImageJ [27]. Antibodies and working dilutions are listed in Table S1. We have previously described in detail the gating strategy and cell preparation for FACS analysis of mice cerebella [26]. Mean Fluorescence Intensities (MFI) were determined using FlowJo software v9 (TreeStar, Ashland, OR, USA). Histopathological quantification was adapted from our previous work [28]. In ImageJ, GFAP and CD68 density used the “moments” thresholding method at 95% for the measurement performed on manually drawn cerebellar area. Purkinje cells were counted manually by counting the number of calbindin positive cell bodies with a recognizable dendritic tree or axonal projection remaining within a given cerebellar lobule. The data were expressed as the number of Purkinje cells per 100 μm of Purkinje cell layer: granule cell layer interface. The whole cerebellar area or individual cerebellar lobules were manually identified from the Hoechst 33342 channel images (DNA counterstain) using the default region of interest function of ImageJ.
Statistical analysis
Statistical comparisons were performed using GraphPad Prism 5 software (San Diego, CA, USA). Box and whisker plots display the Tukey method, and direct comparison between 2 groups was performed using the Mann-Whitney test. For experiments using multiple concentrations of a drug, significance was assessed using Kruskal-Wallis One-way ANOVA. For the lifespan estimation, data were plotted in Kaplan-Meier survival curves, using the log rank test for survival analysis, plotting percent survival as a function of time in days. Results were considered significant if the p- value was <0.05. Figures and Tables were created in Microsoft office 2016 (Redmond, WA, USA).
Results
EAAT1/Slc1a3 cerebellar expression and function
Although Slc1a3 expression is not significantly reduced in cerebellar lobules from Npc1−/− mice (Fig. 1a) [11], single cell transcriptome analysis showed Slc1a3 expression was significantly decreased in Npc1−/− cerebellar astrocytes (Fig. 1b)[20]. Western blot analysis confirmed decreased EAAT1 protein expression in cerebellar tissue from Npc1−/− mice (Fig. 2a) and NPC1 patients (Fig. 2b), suggesting that EAAT1 dysregulation may contribute to both mouse and human pathology. In the cerebellum, EAAT1 is expressed primarily by astrocytes. Decreased surface expression of EAAT1 protein on Npc1−/− astrocytes was measured by flow cytometry analysis of cells from mechanically disassociated cerebellar tissue and of cultured astrocytes (Fig. 2c). To assess if the decreased expression of EAAT1 by Npc1−/− astrocytes had functional consequences, we measured glutamate uptake. In comparison to Npc1+/+ astrocytes, cultured Npc1−/− astrocytes deplete exogenous glutamate from the culture medium at a significantly slower rate at 3 (p<0.05) and 6 (p<0.01, two-way ANOVA) hours (Fig. 2d). Decreased expression of EAAT1 protein and functional impairment of glutamate uptake by Npc1−/− astrocytes, is consistent with the hypothesis that glutamate-mediated neurotoxicity contributes to NPC1 neuropathology.
Figure 1.
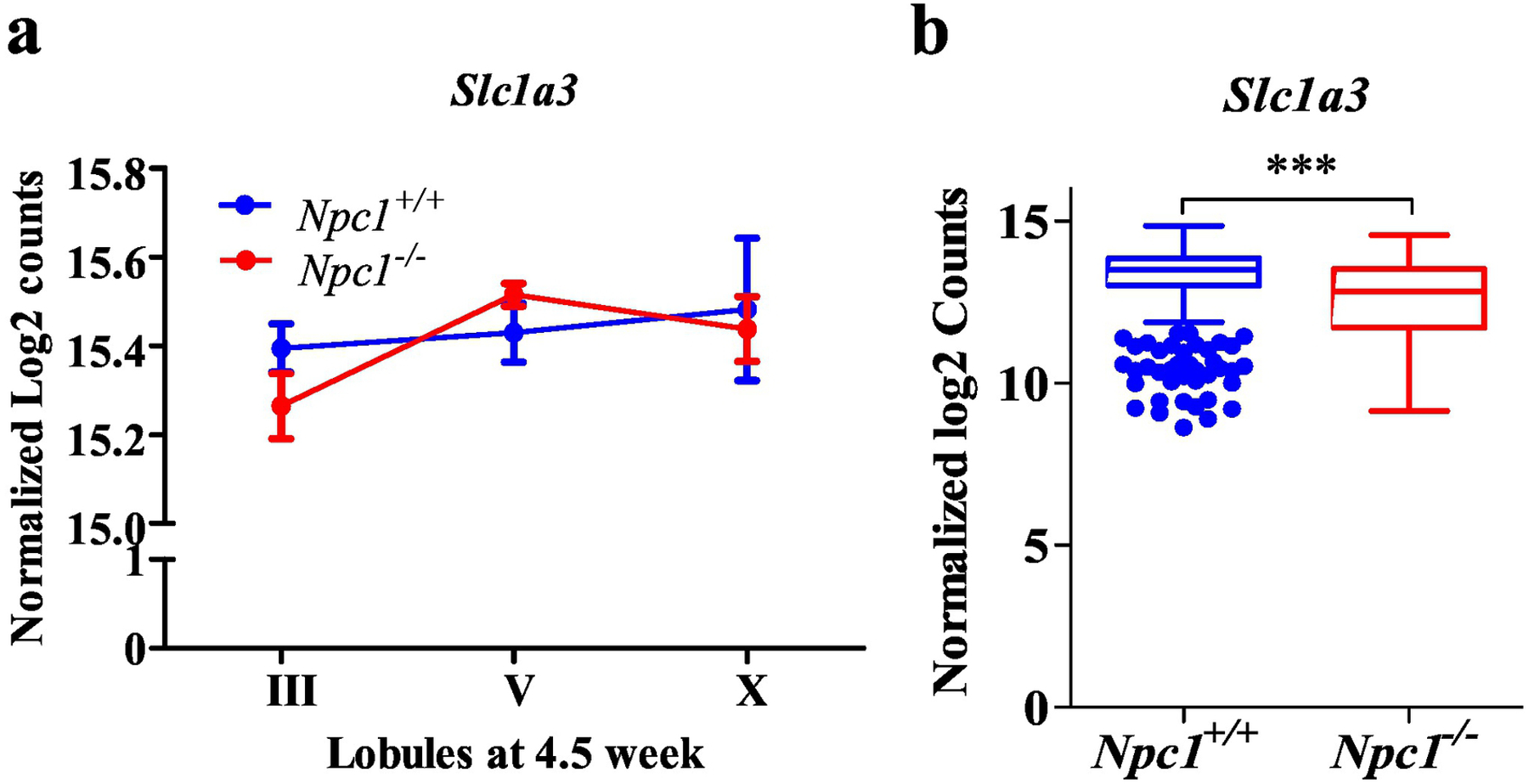
Slc1a3 normalized expression derived from our previously published datasets investigating transcriptional changes in Npc1−/− mouse cerebellar lobules (a) at 4.5 weeks of age [11] (https://porterlab.shinyapps.io/cerebellarlobules/), and by single cell transcriptome analysis (b) of astrocytes from Npc1+/+ (n=435) and Npc1−/− (n=226) mouse cerebellar tissue at 7 weeks of age (SRA accession number: PRJNA606815) [20]. Npc1+/+ (blue). Npc1−/− (red). ***p<0.001
Figure 2.
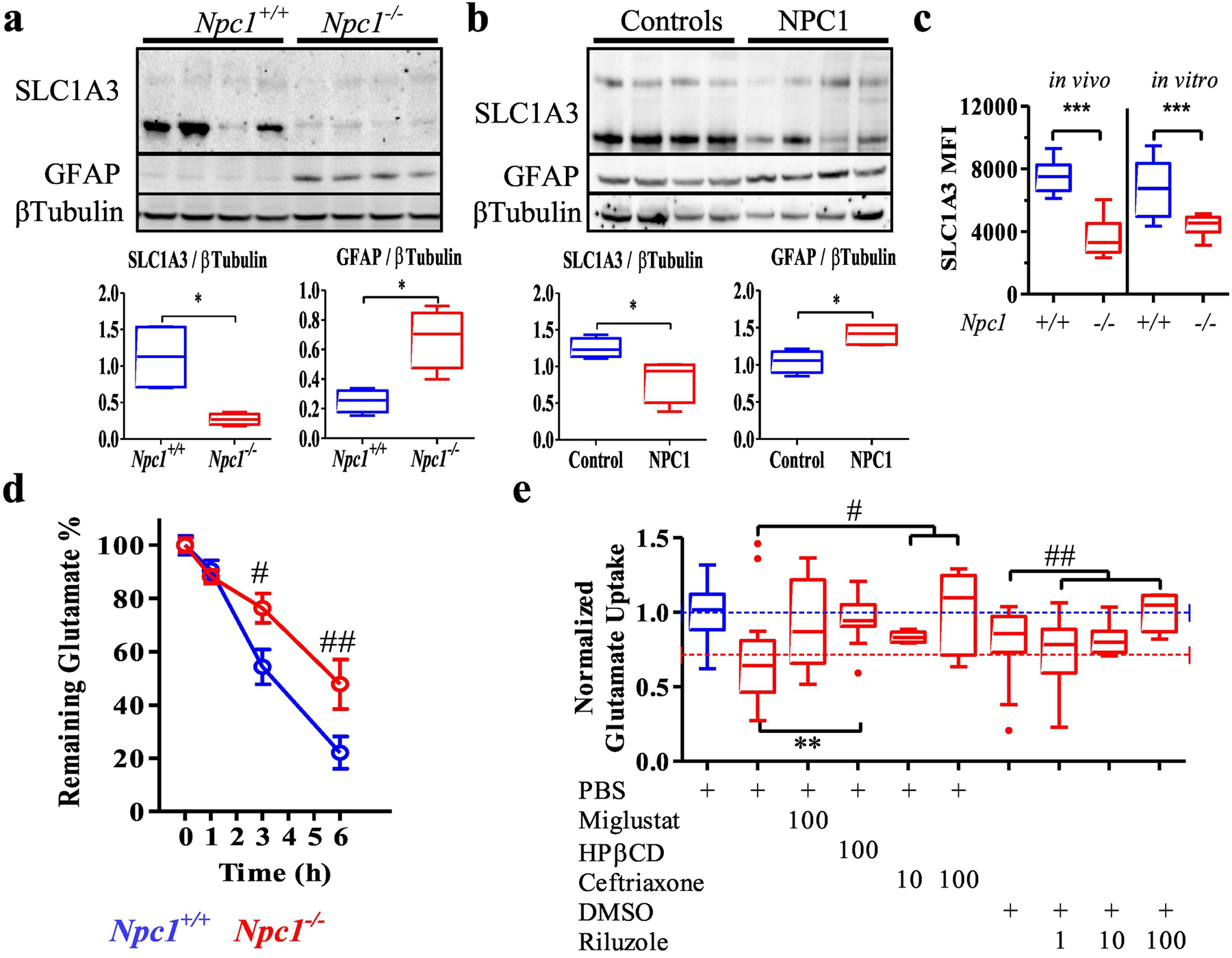
SLC1A3 dysregulation in Niemann-Pick type C. a Western blot analysis of SLC1A3, GFAP and βTubulin expression in 7-week-old mouse cerebellar extracts and densitometric quantification. N=4. b Western blot analysis of SLC1A3, GFAP and βTubulin expression in NPC1 and control human cerebellar extracts and densitometric quantification. N=4 c SLC1A3 surface expression level (Mean Fluorescence Intensity) on astrocytes isolated from 7-week-old cerebellar tissue (in vivo) or primary cerebellar astrocytes cultured (in vitro) from Npc1+/+ and Npc1−/− mice. d Decrease in glutamate levels in the culture media from Npc1+/+ and Npc1−/− primary astrocytes. e Tukey box and whisker plot of glutamate uptake from culture media was significantly (p<0.01) reduced in Npc1−/− astrocytes compared to uptake by Npc1+/+ astrocytes. Data was normalized to Npc1+/+ uptake. The average values for Npc1+/+ (blue) and Npc1−/− (red) are indicated by dashed lines. Glutamate uptake by Npc1−/− astrocytes was increased by treatment with miglustat (p=0.07), HPβCD (p<0.005), ceftriaxone (p<0.05) or riluzole (p<0.005). PBS was the vehicle for miglustat, HPβCD and ceftriaxone. DMSO was the vehicle for riluzole. Drug concentrations are in micromolar. Npc1+/+ (blue), Npc1−/− (red). *p<0.05, **p<0.01 and ***p<0.001 Mann-Whitney, #p<0.05 and ##p<0.01 ANOVA.
In vitro stimulation of astrocyte glutamate uptake
We evaluated the potential of candidate therapeutic agents to increase glutamate uptake in Npc1−/− astrocytes using an in vitro assay. Miglustat and 2-hydroxypropyl-β-cyclodextrin (HPβCD, Kleptose HPB) are two drugs that have been shown to be effective in reducing neurological disease progression in animal models [29–31] and in NPC1 patients [32]. Glutamate uptake significantly decreased by 30% in Npc1−/− astrocytes (p<0.01) relative to Npc1+/+ astrocytes (Fig. 2e). Exposure of Npc1−/− astrocytes to 100µM miglustat increased glutamate uptake from 70% to 92% of control levels (p=0.07); whereas exposure to 100µM HPβCD increased (p<0.005) glutamate uptake to 97% of control (Fig. 2e). These data suggest that drugs which decrease endolysosomal glycosphingolipid (miglustat) or unesterified cholesterol (HPβCD) improve glutamate uptake by Npc1 mutant astrocytes.
Using this in vitro assay system, we also evaluated two drugs, ceftriaxone and riluzole, which directly modulate glutamate uptake, a therapeutic approach that has not been studied in NPC1. In addition to EAAT1, astrocytes also express EAAT2 (GLT-1) which, although less prominent in the cerebellum, is the major glutamate transporter in the brain [33]. EAAT2 is encoded for by SLC1A2. In a high throughput screen of FDA-approved drugs Rothstein et al. [34] found that beta-lactam antibiotics stimulate SLC1A2 expression, and that ceftriaxone increased both gene and protein expression in vivo. We thus tested whether ceftriaxone, a brain penetrant β-lactam, could increase glutamate uptake in Npc1−/− astrocytes. Mean glutamate uptake was increased in a dose dependent manner (p<0.05, Kruskal-Wallis One-way ANOVA) to 84% and 102% of control levels when Npc1−/− astrocytes were exposed overnight to either 10 or 100µM ceftriaxone, respectively (Fig. 2e). Riluzole (Rilutek) is a neuroprotective drug that has been approved by the FDA for the treatment of amyotrophic lateral sclerosis. Although the clinical mechanism of action has not been definitively established, riluzole may decrease glutamate neurotoxicity by both inhibiting the presynaptic release of glutamate and noncompetitive inhibition of NMDA receptors [35]. Riluzole has also been shown to increase glutamate uptake by glutamate transporters including EAAT2 and EAAT1 [36–38]. We thus tested the ability of riluzole to increase glutamate uptake by Npc1−/− cerebellar astrocytes. Although the vehicle, DMSO, has been reported to have a beneficial effect on the Npc1−/− cellular phenotype [39], the increase in glutamate uptake in DMSO-treated compared to PBS-treated Npc1−/− astrocytes was not significant (p=0.13). Using the in vitro assay, we observed a dose-dependent restoration (p<0.005, Kruskal-Wallis One-way ANOVA) of glutamate uptake to 74%, 82% and 107% of control levels for 1, 10 and 100µM riluzole, respectively (Fig. 2e).
In vivo efficacy of ceftriaxone
Based on our in vitro study we investigated the potential efficacy of ceftriaxone in vivo. Ceftriaxone (100mg.kg−1 (n=14) or vehicle (PBS, n=15)) was administered daily to NPC1 mutant mice by intraperitoneal injection. Treatment of Npc1−/− mice with ceftriaxone did not delay neurological progression (Fig. 3a) or extend lifespan (Fig. 3b). Of note, throughout the treatment period, both control and mutant ceftriaxone treated mice developed diarrhea and had poor weight gain. This is a known side effect of antibiotic treatment and may have confounded survival data. However, histopathological evaluation showed no significant effect of ceftriaxone treatment on either microgliosis, astrogliosis or Purkinje neuron loss at 7 weeks of age when comparing PBS and ceftriaxone treated Npc1−/− cerebellar tissue (Figs. 3c–e and S1a). In addition, the expression of both Slc1a2 and Slc1a3 was increased in response to ceftriaxone treatment in cerebellar tissue from Npc1+/+ mice. In contrast, only Slc1a2 expression was increased in Npc1−/− mice, whereas Slc1a3 expression was unchanged (Fig. S1b). These data suggest a differential expression pattern for Slc1a3 in the Npc1−/− cerebellum. Consistent with the histopathological result, Gfap expression showed only a trend (p=0.07) toward being decreased in ceftriaxone treated mice (Fig. S1b).
Figure 3.
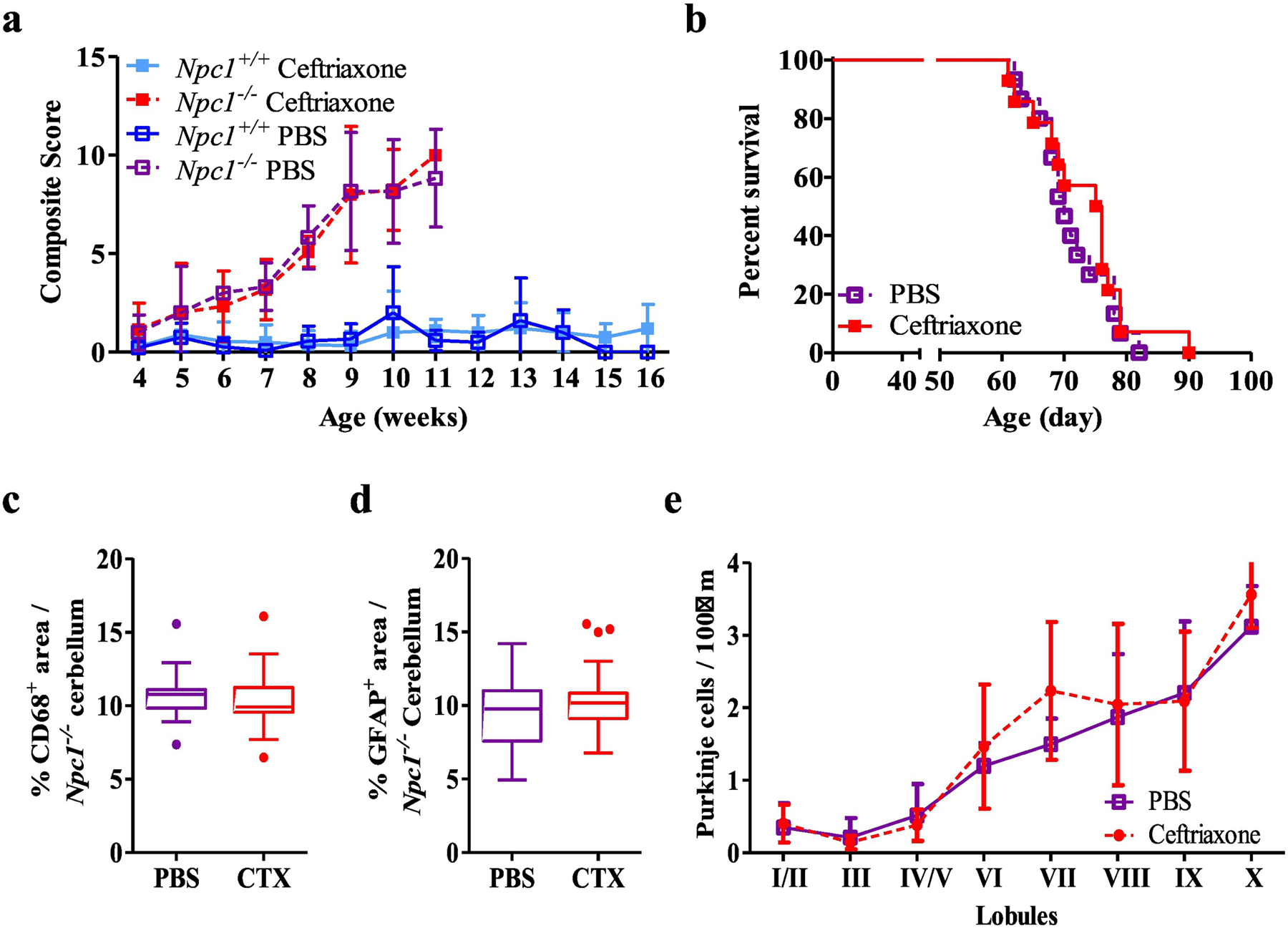
In vivo evaluation of ceftriaxone. a NPC1 disease composite score evaluation between 4 and 16 weeks for vehicle (PBS, initial n=9 and 23) and ceftriaxone (100 mg.kg−1, initial n=13 and 15) treated Npc1−/− and Npc1+/+ mice. Initial n values are for Npc1−/− and Npc1+/+ respectively). These numbers become truncated as the experiment progresses. b Kaplan-Meier survival analysis of PBS (n=15) and ceftriaxone (n=14) treated Npc1−/− mice. c Immunofluorescence quantification of CD68 in cerebellar tissue from 7-week-old PBS or ceftriaxone treated Npc1−/− mice. d Immunofluorescence quantification of GFAP in cerebellar tissue from 7-week-old PBS or ceftriaxone treated Npc1−/− mice. e Cerebellar lobule Purkinje neuron density per 100µm in 7-week-old of PBS or ceftriaxone treated Npc1−/− mice. Purkinje neurons were identified by immunostaining for calbindin. N≥6, Npc1−/− PBS (purple), Npc1−/− ceftriaxone (red), Npc1+/+ PBS (dark blue), Npc1+/+ ceftriaxone (light blue).
Riluzole delays neurological disease progression in Npc1−/− mice
Based on our in vitro data, we also evaluated the therapeutic potential of riluzole to treat NPC1. To test this, we treated Npc1−/− mice with 60µg.ml−1 riluzole added to the drinking water starting at 3 weeks of age. Analysis of cerebellar tissue from 7-week-old mice confirmed that riluzole increased expression of Slc1a3 by 2.1- and 1.7-fold over vehicle control levels in Npc1+/+ (p=0.005) and Npc1−/− mice (p=0.002), respectively (Fig. S2b). Histopathological evaluation of cerebellar tissue showed that riluzole did not decrease microglial activation characterized by CD68+ staining, but significantly (p<0.001) decreased astrogliosis as characterized by GFAP+ staining (Fig. 4 c,d and Fig. S2b). Treatment with riluzole also resulted in preservation of Calbindin D positive Purkinje neurons (Figs. 4e and S2a). Consistent with improved histopathology, we observed that riluzole treatment (n=13) delayed NPC1 disease progression (Fig. 4a) and significantly (p=0.02) increased survival compared to vehicle (DMSO, n=20) treated Npc1−/− mice (Fig. 4b). Median survival was increased 12% from 73 to 82 days. 2-hydroxypropyl-β-cyclodextrin (HPβCD) has been shown to significantly increase survival of Npc1−/− mice, thus we explored whether combined riluzole/HPβCD therapy would be of benefit. No significant (p=0.63) survival difference was observed comparing HPβCD (median 106 days, n=13) and HPβCD + riluzole (median 108 days, n=12) treatment (Fig. 4b).
Figure 4.
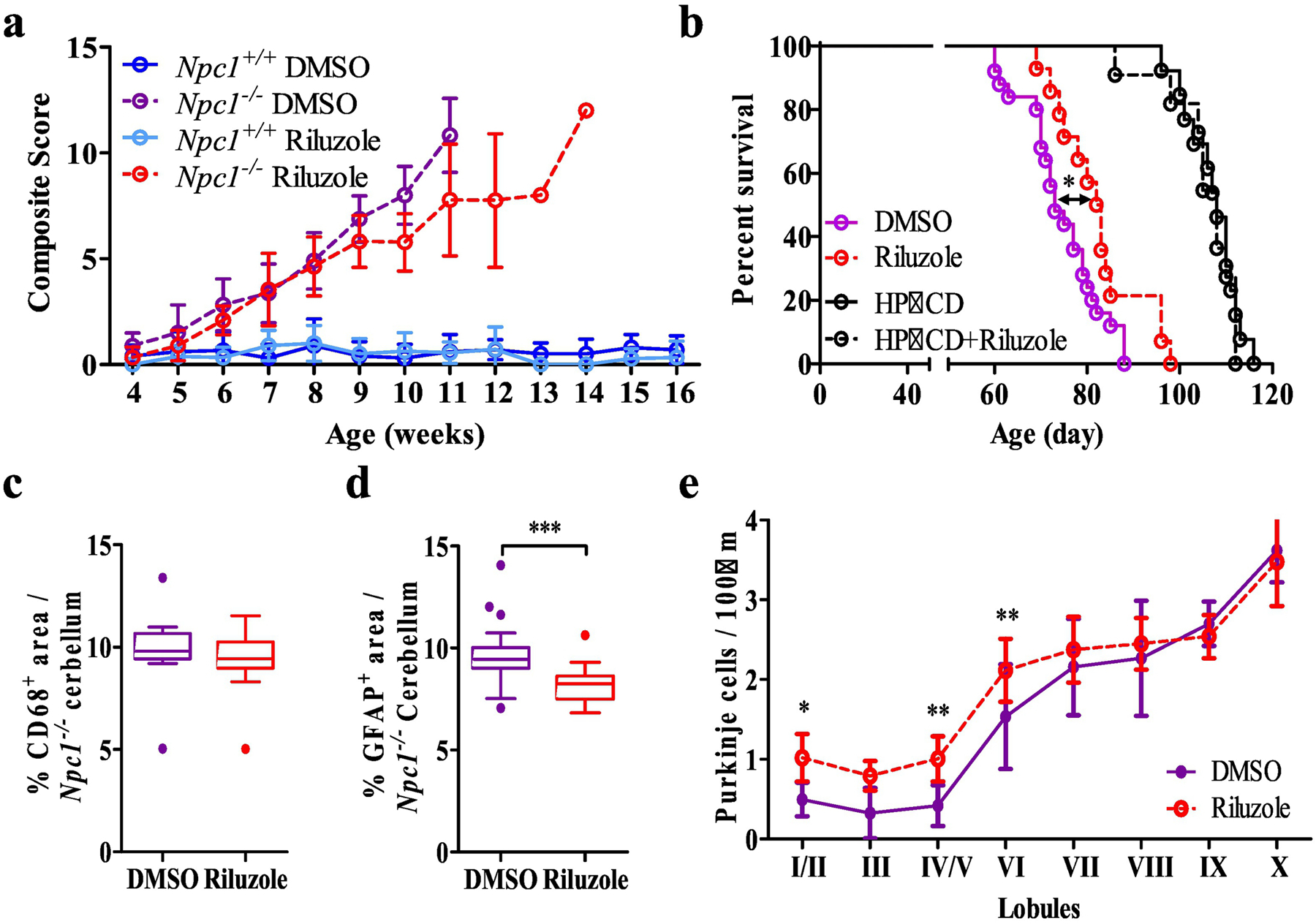
In vivo evaluation of riluzole. a NPC1 disease composite score evaluation between 4 and 16 weeks of age with DMSO (initial n=16 and 14) and riluzole treated (60µg.ml−1 in the drinking water, initial n=12 and 18) Npc1−/− and Npc1+/+ mice. Initial n values are for Npc1−/− and Npc1+/+ respectively). These numbers become truncated as the experiment progresses. b Kaplan-Meier survival analysis of DMSO (n=20), riluzole (n=14), HPβCD (n=13) and HPβCD + riluzole (n=12) treated Npc1−/− mice. c Immunofluorescence quantification of cerebellar CD68 staining in 7-week-old of DMSO or riluzole treated Npc1−/− mice. d Immunofluorescence quantification of cerebellar GFAP staining in 7-week-old of DMSO or riluzole treated Npc1−/− mice. e Cerebellar lobule Purkinje cell density per 100µm in 7-week-old of PBS or ceftriaxone treated Npc1−/− mice. Purkinje neurons were identified by immunostaining for calbindin. N≥6, Npc1−/− DMSO (purple), Npc1−/− riluzole (red), Npc1+/+ DMSO (blue), Npc1+/+ Riluzole (light blue). *p<0.05, **p<0.01 and ***p<0.001.
Discussion
Glutamate mediated excitotoxicity has been proposed to be a pathological mechanism contributing to neurodegeneration in multiple disorders including amyotrophic lateral sclerosis, Huntington disease and Alzheimer disease [23]. We previously noted decreased Slc1a3 expression in cerebellar astrocytes from Npc1−/− mice [20]. These data were consistent with work by Rabenstein et al. [21] who showed reduced protein levels of EAAT1, EAAT2 and EAAT4 in cerebellar tissue from Npc1−/− mice. Based on these results we considered that the reduced levels of these glutamate transporters contribute to glutamate-mediated excitotoxicity and neurodegeneration in NPC1. We hypothesized that increasing their level and function would have a beneficial effect in treating NPC1. To test this hypothesis, we evaluated the therapeutic benefit of both ceftriaxone and riluzole in Npc1−/− mice.
Ceftriaxone is a beta-lactam antibiotic that has been shown to increase Slc1a2 expression and corresponding EAAT2 activity in vivo [34]. Although ceftriaxone treatment of Npc1+/+ and Npc1−/− mice increased expression of Slc1a2, we did not observe any histopathological, phenotypic or survival benefit in ceftriaxone treated NPC1 mutant mice. There are several possible explanations for this negative result. EAAT2 is the major glutamate transporter in the brain; however, it plays less of a role in the cerebellum where EAAT1 plays a major role [33]. Thus, upregulation of EAAT2 may be less effective for disorders such as NPC1 where cerebellar Purkinje neuron loss is a major pathological feature. Our data also show that ceftriaxone induced expression of Slc1a2 appears to be blunted in the NPC1 mutant cerebellar tissue relative to controls. Ceftriaxone induced expression of Slc1a2 is mediated by an NFκB binding site [40], thus it is possible that the prominent neuroinflammation and neurodegeneration in the mutant mouse cerebellar tissue alters NFκB signaling. Alternatively, although we confirmed that ceftriaxone increased glutamate uptake by Npc1−/− astrocytes in vitro and observed increased, but blunted, Slc1a2 expression in cerebellar tissue from treated mice, we did not establish a corresponding increase in expression of EAAT2 protein. We did observe increased expression of Slc1a3 in cerebellar tissue from ceftriaxone treated Npc1+/+ mice. This contrasts with a lack of increased EAAT1 protein in hippocampal tissue from ceftriaxone treated mice [34]. To our knowledge this observation has not been previously reported; however, given that we did not observe increased expression of Slc1a3 in the Npc1−/− mutant mice, this observation was not pursued further in the context of this study.
Riluzole is a neuroprotective drug which inhibits the release of presynaptic glutamate and facilitates glutamate uptake by excitatory amino acid transporters [35, 41] and riluzole has been shown to protect Purkinje neurons in a rat model of cerebellar ataxia [42]. Riluzole has been approved by the FDA for the treatment of amyotrophic lateral sclerosis and has been studied in children with obsessive compulsive disorder [43]. This raises the possibility that riluzole could be repurposed for the treatment of individuals with NPC1. In this study we found that treatment of Npc1−/− mice with riluzole decreased astrogliosis, delayed Purkinje neuron loss, improved the neurological phenotype and increased survival. The histopathological analysis is notable in that riluzole did not appear to impact neuroinflammation which occurs early in the disease [11, 20, 26], but rather had a more pronounced effect on astrogliosis which appears to occur later in the NPC1 pathological cascade [44]. Indeed, the disease severity score began to diverge after 8 weeks (Fig. 4a) as reduced disease severity scores were observed in the treated mice. This is coincidental with the timing of increased astrogliosis during normal disease progression and suggests a reduced progression of the disease in the riluzole treated mice due to the reduced astrogliosis. This apparent later effect of riluzole on the NPC1 pathological cascade may explain the lack of an observed benefit when combined with HPβCD. HPβCD has been shown to have very early effects on NPC1 histopathology. In this paper we only explored the potential of riluzole to complement an optimized dose of HPβCD initiated in presymptomatic animals. These data do not exclude the possibility that this drug combination could show benefit under other experimental paradigms. The observation that riluzole appears to have more of an impact on astrogliosis also suggests that combining riluzole with an anti-inflammatory drug which suppresses microgliosis may be of benefit.
We postulate that the beneficial effect of riluzole in Npc1−/− mice is due to decreased glutamate-mediated neurotoxicity. This conclusion is based on riluzole’s reported mechanism of action, increased in vitro glutamate uptake by Npc1−/− astrocytes treated with riluzole and increased expression of Slc1a3 in cerebellar tissue from riluzole treated Npc1−/− mice. In addition to the work presented in this paper, multiple studies suggest a disturbance of glutamate signaling in Npc1 mutant mice. Analogous to our finding of decreased cerebellar astrocyte EAAT1 protein expression, Byun et al. [45] reported decreased protein expression of EAAT3 (EAAC1, SLC1A1) in hippocampal tissue from Npc1−/− mice. Work by both D’Arcangelo et al. [46] and Feng et al. [47] indicate impaired AMPA receptor internalization. These observations provide additional evidence of impaired glutamate homeostasis in NPC1 and support our conclusion that glutamate-mediated neurotoxicity contributes to NPC1 neuropathology.
Conclusions
The data reported in this paper support the conclusion that glutamate mediated neurotoxicity likely contributes to NPC1 neuropathology and demonstrate that treatment with riluzole has beneficial effects in the NPC1 mouse model. Given that riluzole is an approved drug for the treatment of amyotrophic lateral sclerosis, repurposing of this drug, or use of other agents which reduce glutamate-mediated neurotoxicity, may provide a novel therapeutic approach to decrease disease progression in Niemann-Pick disease type C1 patients.
Supplementary Material
Figure S1. a Immunofluorescence analysis of CD68, GFAP and Calbindin staining of 7-week-old of PBS or ceftriaxone treated Npc1−/− mice cerebellar tissue. b Relative transcription levels of Slc1a2, Slc1a3 and Gfap to β-tubulin (Tubb) in 7-week-old cerebellar tissue from Npc1−/− PBS (purple), Npc1−/− ceftriaxone (red), Npc1+/+ PBS (dark blue), Npc1+/+ ceftriaxone (light blue) groups. N=6. Mann-Whitney test, ** p<0.01.
Figure S2. a Immunofluorescence analysis of CD68, GFAP and Calbindin staining of 7-week-old of vehicle (DMSO) or riluzole treated Npc1−/− mice cerebellar tissue. b Relative transcription levels of Slc1a2, Slc1a3 and Gfap to β-tubulin (Tubb) in 7-week-old cerebellar tissue from DMSO (purple), Npc1−/− riluzole (red), Npc1+/+ DMSO (blue), Npc1+/+ riluzole (light blue) cohorts. N=6. Mann-Whitney test, ** p<0.01.
Table S1 Materials and software.
Acknowledgements
Not applicable
Funding
This work was supported by the intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health. This work was also supported by a grant from the Ara Parseghian Medical Research Foundation.
List of Abbreviations
- CALB
Calbindin
- CTX
Ceftriaxone
- DMSO
Dimethyl Sulfoxide
- EAAT
Excitatory Amino Acid Transporters
- FDA
Federal Drug Administration
- GFAP
Glial Fibrillary Acidic Protein
- HPβCD
2-hydroxypropyl-β-cyclodextrin
- MFI
Mean Fluorescent Intensity
- NICHD
Eunice Kennedy Shriver National Institute of Child Health and Human Development
- NPC
Niemann-Pick Disease, type C
- NPC1
Niemann-Pick Disease, type C1
- NPC2
Niemann-Pick Disease, type C2
- PBS
Phosphate Buffered Saline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations
Mouse experiments were approved by the NICHD Animal Care and Use Committee. All data generated or analyzed during this study are included in the published article and supplementary information files. The authors declare that they have no financial or non-financial competing interests.
References
- 1.Vanier MT, Niemann-Pick disease type C. Orphanet J Rare Dis, 2010. 5: p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wassif CA, et al. , High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet Med, 2016. 18(1): p. 41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfeffer SR, NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J Biol Chem, 2019. 294(5): p. 1706–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crocker AC and Farber S, Niemann-Pick disease: a review of eighteen patients. Medicine (Baltimore), 1958. 37(1): p. 1–95. [DOI] [PubMed] [Google Scholar]
- 5.Bianconi SE, et al. , Evaluation of age of death in Niemann-Pick disease, type C: Utility of disease support group websites to understand natural history. Mol Genet Metab, 2019. 126(4): p. 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patterson MC, et al. , Long-term survival outcomes of patients with Niemann-Pick disease type C receiving miglustat treatment: A large retrospective observational study. J Inherit Metab Dis, 2020. [DOI] [PMC free article] [PubMed]
- 7.Patterson MC, et al. , Treatment outcomes following continuous miglustat therapy in patients with Niemann-Pick disease Type C: a final report of the NPC Registry. Orphanet J Rare Dis, 2020. 15(1): p. 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solomon BI, et al. , Association of Miglustat With Swallowing Outcomes in Niemann-Pick Disease, Type C1. JAMA Neurol, 2020. [DOI] [PMC free article] [PubMed]
- 9.Sarna JR and Hawkes R, Patterned Purkinje cell death in the cerebellum. Prog Neurobiol, 2003. 70(6): p. 473–507. [DOI] [PubMed] [Google Scholar]
- 10.Higashi Y, et al. , Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol, 1993. 85(2): p. 175–84. [DOI] [PubMed] [Google Scholar]
- 11.Martin KB, et al. , Identification of Novel Pathways Associated with Patterned Cerebellar Purkinje Neuron Degeneration in Niemann-Pick Disease, Type C1. Int J Mol Sci, 2019. 21(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perkins EM, et al. , Loss of cerebellar glutamate transporters EAAT4 and GLAST differentially affects the spontaneous firing pattern and survival of Purkinje cells. Hum Mol Genet, 2018. 27(15): p. 2614–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slemmer JE, De Zeeuw CI, and Weber JT, Don’t get too excited: mechanisms of glutamate-mediated Purkinje cell death. Prog Brain Res, 2005. 148: p. 367–90. [DOI] [PubMed] [Google Scholar]
- 14.Miyazaki T, et al. , Glutamate transporter GLAST controls synaptic wrapping by Bergmann glia and ensures proper wiring of Purkinje cells. Proc Natl Acad Sci U S A, 2017. 114(28): p. 7438–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagao S, Kwak S, and Kanazawa I, EAAT4, a glutamate transporter with properties of a chloride channel, is predominantly localized in Purkinje cell dendrites, and forms parasagittal compartments in rat cerebellum. Neuroscience, 1997. 78(4): p. 929–33. [DOI] [PubMed] [Google Scholar]
- 16.Farooqui AA and Horrocks LA, Involvement of glutamate receptors, lipases, and phospholipases in long-term potentiation and neurodegeneration. J Neurosci Res, 1994. 38(1): p. 6–11. [DOI] [PubMed] [Google Scholar]
- 17.Lau A and Tymianski M, Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch, 2010. 460(2): p. 525–42. [DOI] [PubMed] [Google Scholar]
- 18.Arriza JL, et al. , Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci, 1994. 14(9): p. 5559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Lozada Z, Guillem AM, and Robinson MB, Transcriptional Regulation of Glutamate Transporters: From Extracellular Signals to Transcription Factors. Adv Pharmacol, 2016. 76: p. 103–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cougnoux A, et al. , Single Cell Transcriptome Analysis of Niemann-Pick Disease, Type C1 Cerebella. Int J Mol Sci, 2020. 21(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabenstein M, et al. , Impact of Reduced Cerebellar EAAT Expression on Purkinje Cell Firing Pattern of NPC1-deficient Mice. Sci Rep, 2018. 8(1): p. 3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi KD, et al. , Late-onset episodic ataxia associated with SLC1A3 mutation. J Hum Genet, 2017. 62(3): p. 443–446. [DOI] [PubMed] [Google Scholar]
- 23.Lewerenz J and Maher P, Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front Neurosci, 2015. 9: p. 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loftus SK, et al. , Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science, 1997. 277(5323): p. 232–5. [DOI] [PubMed] [Google Scholar]
- 25.Guyenet SJ, et al. , A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp, 2010(39). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cougnoux A, et al. , Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum Mol Genet, 2018. 27(12): p. 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schindelin J, et al. , Fiji: an open-source platform for biological-image analysis. Nat Methods, 2012. 9(7): p. 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cougnoux A, et al. , Necroptosis inhibition as a therapy for Niemann-Pick disease, type C1: Inhibition of RIP kinases and combination therapy with 2-hydroxypropyl-beta-cyclodextrin. Mol Genet Metab, 2018. 125(4): p. 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davidson CD, et al. , Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One, 2009. 4(9): p. e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu B, et al. , Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc Natl Acad Sci U S A, 2009. 106(7): p. 2377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vite CH, et al. , Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci Transl Med, 2015. 7(276): p. 276ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ory DS, et al. , Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial. Lancet, 2017. 390(10104): p. 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim K, et al. , Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol, 2011. 226(10): p. 2484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothstein JD, et al. , Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature, 2005. 433(7021): p. 73–7. [DOI] [PubMed] [Google Scholar]
- 35.Doble A, The pharmacology and mechanism of action of riluzole. Neurology, 1996. 47(6 Suppl 4): p. S233–41. [DOI] [PubMed] [Google Scholar]
- 36.Fumagalli E, et al. , Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol, 2008. 578(2–3): p. 171–6. [DOI] [PubMed] [Google Scholar]
- 37.Carbone M, Duty S, and Rattray M, Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem Int, 2012. 60(1): p. 31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dall’Igna OP, et al. , Riluzole increases glutamate uptake by cultured C6 astroglial cells. Int J Dev Neurosci, 2013. 31(7): p. 482–6. [DOI] [PubMed] [Google Scholar]
- 39.Blanchette Mackie EJ, et al. , Type C Niemann-Pick disease: dimethyl sulfoxide moderates abnormal LDL-cholesterol processing in mutant fibroblasts. Biochim Biophys Acta, 1989. 1006(2): p. 219–26. [DOI] [PubMed] [Google Scholar]
- 40.Lee SG, et al. , Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem, 2008. 283(19): p. 13116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azbill RD, Mu X, and Springer JE, Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res, 2000. 871(2): p. 175–80. [DOI] [PubMed] [Google Scholar]
- 42.Janahmadi M, et al. , Co-treatment with riluzole, a neuroprotective drug, ameliorates the 3-acetylpyridine-induced neurotoxicity in cerebellar Purkinje neurones of rats: behavioural and electrophysiological evidence. Neurotoxicology, 2009. 30(3): p. 393–402. [DOI] [PubMed] [Google Scholar]
- 43.Grant P, et al. , Riluzole Serum Concentration in Pediatric Patients Treated for Obsessive-Compulsive Disorder. J Clin Psychopharmacol, 2017. 37(6): p. 713–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pressey SN, et al. , Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol Dis, 2012. 45(3): p. 1086–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byun K, et al. , Alteration of the glutamate and GABA transporters in the hippocampus of the Niemann-Pick disease, type C mouse using proteomic analysis. Proteomics, 2006. 6(4): p. 1230–6. [DOI] [PubMed] [Google Scholar]
- 46.D’Arcangelo G, et al. , Glutamatergic neurotransmission in a mouse model of Niemann-Pick type C disease. Brain Res, 2011. 1396: p. 11–9. [DOI] [PubMed] [Google Scholar]
- 47.Feng X, et al. , Stimulation of mGluR1/5 Improves Defective Internalization of AMPA Receptors in NPC1 Mutant Mouse. Cereb Cortex, 2020. 30(3): p. 1465–1480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. a Immunofluorescence analysis of CD68, GFAP and Calbindin staining of 7-week-old of PBS or ceftriaxone treated Npc1−/− mice cerebellar tissue. b Relative transcription levels of Slc1a2, Slc1a3 and Gfap to β-tubulin (Tubb) in 7-week-old cerebellar tissue from Npc1−/− PBS (purple), Npc1−/− ceftriaxone (red), Npc1+/+ PBS (dark blue), Npc1+/+ ceftriaxone (light blue) groups. N=6. Mann-Whitney test, ** p<0.01.
Figure S2. a Immunofluorescence analysis of CD68, GFAP and Calbindin staining of 7-week-old of vehicle (DMSO) or riluzole treated Npc1−/− mice cerebellar tissue. b Relative transcription levels of Slc1a2, Slc1a3 and Gfap to β-tubulin (Tubb) in 7-week-old cerebellar tissue from DMSO (purple), Npc1−/− riluzole (red), Npc1+/+ DMSO (blue), Npc1+/+ riluzole (light blue) cohorts. N=6. Mann-Whitney test, ** p<0.01.
Table S1 Materials and software.