

Abstract

Following the initial cation formation, the synthesis of ionic liquids (ILs) often involves an anion-exchange or metathesis reaction. For hydrophobic ILs, this is generally performed through several cross-current contacts of the IL with a fresh salt solution of the desired anion. However, if a large number of contacts is required to attain an adequate conversion, this procedure is not economical because of the large excess of the reagent that is consumed. In this study, the metathesis of an IL, Aliquat 336 or [A336][Cl], to ILs with other anions ([A336][X] with X = HSO4–, Br–, NO3–, I–, and SCN–) was studied in a continuous counter-current mixer-settler setup. McCabe–Thiele diagrams were constructed to estimate the required number of stages for quantitative conversion. Significantly higher IL conversions were achieved, combined with reduced reagent consumption and waste production. This improvement in efficiency was most pronounced for anions placed low in the Hofmeister series, for example, HSO4–, Br–, and NO3–, which are difficult to exchange. The performance of the counter-current experiments was compared with the conventional multistep cross-current batch process by calculating the reaction mass efficiency (RME) and the environmental factor (E-factor). The RMEs of the cross-current experiments were notably smaller, that is, 38–78% of the values observed for the counter-current experiments. The E-factors of the counter-current experiments were a factor of 2.0–6.8 smaller than those of the cross-current experiments. These sustainability metrics indicate a highly efficient reagent use and a considerable, simultaneous decrease in waste production for the counter-current IL metathesis reactions.
Keywords: anion exchange, counter-current, green solvents, ionic liquids, metathesis, solvent extraction
Short abstract
A counter-current flow diagram facilitates the efficient metathesis of ionic liquids and allows for a high conversion with minimal reagent consumption.
Introduction
Ionic liquids (ILs) solely consist of ions and are generally defined as salts with a melting point below 100 °C, although this threshold is arbitrary, and many ILs are liquid at ambient temperatures. Remarkable properties such as a wide liquidus range, intrinsic electrical conductivity, extremely low vapor pressure, and low flammability have made them a promising class of materials and interesting, environmentally friendly alternatives for volatile molecular solvents.1−3 They have been applied as solvents in organic synthesis,4,5 extractants for solvent extraction,6−8 and solvents for the processing of (bio)polymers.9,10 Because of the high variety of the possible cation–anion combinations, ILs can be tailor-made to each application and are, therefore, often referred to as “task-specific” liquids.11 Although different methods exist, the majority of ILs is generally prepared via a two-step synthesis method.2,12,13 The first step constitutes cation formation through the quaternization of a precursor such as an imidazole, amine, phosphine, or pyridine. This is mostly performed using a haloalkane, resulting in the formation of the corresponding halide salt. In the second step, the targeted anion is introduced via an anion-metathesis or anion-exchange reaction:
| 1 |
with Q+ as the cation, Y– as a halide, A– as the targeted anion, and M+ as an alkali metal, Ag+, or NH4+. For hydrophobic ILs, anion metathesis can easily be performed by contacting the IL with an aqueous solution of the desired anion. For this, both the acid and metal or ammonium salts of the anion can be used. The byproducts can subsequently be washed from the IL product using water. When the initial IL is hydrophilic, the same strategy can be used when the formed IL is hydrophobic and separates from the aqueous mixture.14 Otherwise, the use of silver salts is preferred as the byproducts, that is, silver halides precipitate from the solution.15 However, this is a very expensive method, and the final IL is often contaminated by silver impurities. Other methods that have been used for the anion metathesis of ILs include the use of anion-exchange resins and membranes and the use of electrophilic reagents, for example, dimethyl sulfate, and the direct reaction of halide ILs with alkaline salts of the targeted anion.16−20
The efficiency of anion exchange between an IL-containing organic phase and an aqueous phase can be predicted based on the Hofmeister series:21−24
| 2 |
In the Hofmeister series, anions are ranked according to their hydration enthalpy. Anions to the left of the series are characterized by a higher hydration enthalpy and prefer hydration in the aqueous phase over transfer to the organic phase and association with an organic cation, whereas anions to the right of the series are characterized by a lower hydration enthalpy and prefer to reside in the organic phase. Consequently, when an IL of a certain anion is contacted with an aqueous phase containing an anion placed more to the right in the series, the exchange equilibrium is generally favorable and a more or less efficient exchange reaction can be expected to occur. For anions placed more to the left in the series, the exchange equilibrium is generally disfavored, although some exchange can still be expected.
For most research purposes where anion exchange of a hydrophobic IL is required, eq 1 is performed in a multistep batch (or cross-current) process, in which an aliquot of the IL is contacted multiple times with a fresh aqueous salt solution of the desired anion.6,25,26 Often, a significant excess of the reagent is consumed in such processes because of the relatively large number of contacts required to reach an adequate conversion. Likewise, in solvent extraction applications, cross-current processes consume a large amount of the extracting organic phase to obtain a high recovery of an aqueous solute. To this extent, an alternative flow diagram, that is, a counter-current process, is often preferred by industry.27 In such a counter-current process, the phase volumes remain constant, but the two phases are fed at opposite ends of the cascade of contactors. Figure 1 compares cross-current and counter-current flow diagrams for an IL metathesis process. The driving force behind the process, that is, the anion concentration gradient between the two phases, is maximized in the counter-current mode. This can intuitively be understood when considering the first and last contacts of such a process, where a fresh IL is contacted with a largely depleted aqueous phase or an almost completely converted IL is contacted with a fresh salt solution, resulting in the more efficient usage of the salt solution. Consequently, a reduction in both reagent use and waste production can be expected for counter-current operation.
Figure 1.

Flow diagrams of a cross-current (a) and counter-current (b) IL metathesis process. N represents the total number of contactors or theoretical stages.
Only a few reports of continuous counter-current IL metathesis exist. A process patented by Gaudernack et al. mentions the production of the nitrate analogue of the IL Aliquat 336 by contacting a 1 mol L–1 solution of NH4NO3 with Aliquat 336 dissolved in the diluent Solvesso 100 in an eight-stage counter-current mixer-settler setup.28 Lu et al. reported a similar metathesis of Aliquat 336, where the IL was dissolved in the diluent Naphtha-100.29 Even if the metathesized IL product is to be used as an extractant in a counter-current process, the metathesis reaction is often performed prior in a separate cross-current batch process.30 Counter-current flow diagrams have rarely been considered for the purpose of IL synthesis, despite the various inherent advantages and the fact that the applicability of viscous ILs in mixer-settlers has been successfully shown in several publications.31,32
The present study involves an in-depth study of a counter-current process for the metathesis of ILs. The metathesis of an IL, Aliquat 336, to its bisulfate, bromide, nitrate, iodide, and thiocyanate analogues was studied using a laboratory-scale counter-current mixer-settler setup. The anions were selected based on their relevance for phase-transfer catalysis, solvent extraction research, and metal separations and purifications.25,30,33 Aliquat 336 is a commercially available, hydrophobic IL and is a mixture of different quaternary ammonium chlorides, with trioctylmethylammonium chloride as the main component. The metatheses were performed in continuous counter-current mode as well as in a multistep cross-current batch process. The processes were compared in terms of product conversion and reagent consumption and evaluated through the calculation of efficiency metrics.
Experimental Section
Materials
Aliquat 336 was purchased from Thermo Fisher Scientific (Merelbeke, Belgium). KBr (≥99%) and di-isobutyl ketone (DIBK, >96%) were acquired from Acros Organics (Geel, Belgium). Potassium iodide (KI) (≥99.5%) was bought from Honeywell Riedel de Haën (Seelze, Germany). KSCN (>99%) and KNO3 (>99%) were purchased from Chem-Lab (Zedelgem, Belgium). NaHSO4 (>93%) was acquired from Carl Roth (Karlsruhe, Germany). Water was always of ultrapure quality (Milli-Q water), deionized with a Merck Millipore Milli-Q Reference A+ system. All chemicals were used as received, without any further purification.
Batch Experiments
Batch experiments were performed in 50 mL centrifuge tubes by contacting 15 mL of the organic phase with a certain volume of an aqueous salt solution of the corresponding anion for 60 min using a Kuhner ES-X orbital shaker. To construct the distribution isotherms, the organic-over-aqueous volumetric phase ratio (O/A) was varied between 10 and 0.25. To study the metathesis rate, the system was stirred with a magnetic stirrer at 1000 rpm for varying amounts of time between 1 and 60 min. Afterward, the samples were centrifuged for 30 s at 5000 rpm (Eppendorf centrifuge 5804), and the phases were separated.
Residual Chloride Measurement Using Wavelength Dispersive X-Ray Fluorescence Spectrometry
The degree of metathesis or conversion was determined through the measurement of the residual chloride content of the ILs using wavelength dispersive X-ray fluorescence spectrometry (WDXRF). Measurements were performed on a Bruker S8 Tiger 4 kW WDXRF system equipped with a Rh anode, 50 μm Be filter, PET (pentaerythritol) diffraction crystal, and a gas-flow proportional counter detector. All measurements were performed on the Cl Kα line (2.622 keV) under atmospheric helium with the IL samples in polyethylene cups (XRF Scientific) with a 4 μm polypropylene film (Chemplex) bottom, which were rotated at 0.5 rev/s. The WDXRF system was calibrated with solutions prepared by mixing varying volumes of [A336][Cl] and [A336][X] (with X = HSO4–, Br–, I–, NO3–, or SCN–). More details about the chloride determination by WDXRF can be found in a recent study.34 Optimal measurement parameters were as follows: 10 mL sample volume, 34 mm collimator mask, and 0.23° collimator. Samples were measured after a washing step with Milli-Q water to remove any dissolved salts.
The chloride content is displayed as “conversion” (eq 3) and not as an exact value such as mol L–1 or ppm for two reasons: (1) the quaternary compound concentration is prone to change throughout a metathesis process because of a change in the water content of the IL, and (2) the exact concentration of the quaternary compound in undiluted Aliquat 336 is not exactly known, although the literature reports values of approximately 1.8 mol L–1.35−37
| 3 |
with V([A336][Cl]) and V([A336][X]) being the volumes of water-saturated, pure [A336][Cl] and [A336][X] (X = HSO4–, Br–, I–, NO3–, or SCN–) that can be mixed to obtain an IL with a chloride content that will result in the same WDXRF Cl Kα intensity as the experimental sample with unknown chloride concentration.
Mixer-Settler Setup and the Experimental Procedure
Mixer-settler experiments were performed using a Rousselet Robatel MD UX 1.1 system fabricated using polytetrafluoroethylene (PTFE). A top view of a four-stage cascade is shown in Figure 2, and several of such cascades can be coupled if more than four stages are required. One mixer-settler unit consists of a mixing chamber in which the aqueous and organic phases are mixed using a motored impeller and a settling chamber, where the phases disengage under gravity. The settling chambers are fitted with a viewing window to allow for easy monitoring of the phase disengagement and the phase ratio. Coalescence plates were used to aid the phase disengagement. The effective volumes of each mixing and settling chambers were 35 and 143 mL, respectively. Both aqueous and organic phases were pumped using a Masterflex L/S precision variable-speed console drive equipped with an L/S 16 polycarbonate or an easy-load pump head. Tygon Fuel & Lubricant tubing was used for organics, while Norprene or Versilon tubing was used for aqueous phases, all with an internal diameter of 3.1 mm (Masterflex).
Figure 2.

(a) Top view of a four-stage mixer-settler battery. (1) Mixing chamber; (2) settling chamber with PTFE coalescence plates; (3) adjustable weir for interface-level control; (4) organic phase inlet; (5) organic phase outlet; (6) aqueous phase inlet; (7) aqueous phase outlet; and (8) recycling channel (not used in the present study). (b) Schematic representation of the four-stage setup with the yellow line as the path of the organic phase and the blue line as the path of the aqueous phase.
At the start of the experiment, the mixing and settling chambers were, respectively, filled with the aqueous and organic phases in a ratio corresponding to the applied flow rate ratio. Next, the pumps and stirrers were started. The pumps were calibrated to the correct flow rate prior to starting the experiment, and the stirrers were operated at 1000 rpm. Subsequently, the height of the weirs was adjusted until the correct settling chamber phase ratio and a stable flow were achieved. Samples of the IL product were taken periodically from the organic outlet, and the settling chambers were sampled when the experiment was terminated.
Process Efficiency Calculations
The reaction mass efficiency (RME, eq 4) and the environmental factor (E-factor, eq 5) were used to evaluate the efficiency of the metathesis process in terms of reagent use and waste production, respectively.
| 4 |
| 5 |
The RME relates the mass of the IL product, that is, m([A336][X]), to the mass of the used reagents, that is, m([A336][Cl]) and m(MX) with MX as the used salt of the desired anion. The formula can be rearranged to incorporate molar masses (MM), the product conversion (i.e., yield) and whether or not an excess of reagent is used. The latter is found in both the volume and concentration of the salt solution, V(MX) and C(MX), respectively. For the concentration of [A336][Cl], that is, C([A336][Cl]), it was assumed that the quaternary compound concentration in the undiluted IL is 1.8 mol L–1.35−37 An IL metathesis process will, however, never reach an RME of 1 because of the loss of a certain amount of mass in the form of a salt of the initial anion, that is, MCl (with M = K+ or Na+ for the current experiments). The RME also does not incorporate the consumption of solvents or diluents such as water and DIBK. An E-factor, which relates the total mass of waste produced, m(waste), to the mass of the product, is able to account for both shortcomings. Solvent recovery and recycling can also be accounted for in the calculations.
Results and Discussion
Single-Stage Batch and Cross-Current Metatheses
To create a basis of comparison for the counter-current experiments, single-stage and cross-current experiments were performed. In order to avoid viscosity-related issues, that is, inefficient mixing and unstable flow rates, DIBK was used as a diluent during all experiments. The choice of diluent is discussed extensively in the Supporting Information.
In a first series batch experiment, the influence of the initial aqueous anion concentration on the degree of IL conversion was studied. Figure 3 shows the conversion of 70 wt % [A336][Cl] in DIBK as a function of the aqueous anion, that is, HSO4–, Br–, NO3–, I–, and SCN–, concentration. The initial aqueous anion concentrations were varied between 0.1 and 2.5 mol L–1, and an organic-to-aqueous volumetric phase ratio (O/A) of 1 was used. The data clearly adhere to the Hofmeister series, with HSO4– being the least and SCN– being the most efficiently exchanged for the original organic chloride ion. The formation of [A336][SCN] is very efficient, and only a slight aqueous excess of thiocyanate ions is required for the quantitative exchange of the Cl– ions in the organic phase, approximately 1.22 mol L–1 for the 70 wt % [A336][Cl] dilution, and to achieve a conversion of >0.990. The formation of [A336][I] is slightly less efficient, and a quantitative conversion is reached at 1.5 mol L–1 of iodide, which corresponds to an aqueous excess of approximately 0.3 mol L–1 of iodide ions. At lower initial aqueous NO3– and Br– concentrations, that is, up to 0.5 mol L–1, the conversion profiles of [A336][NO3] and [A336][Br] match those of [A336][SCN] and [A336][I], and the conversion seems to proceed equally efficient with a complete removal of the respective anions from the aqueous phase. However, subsequent conversion proceeds with more difficulty, as can be seen by the deviation from linearity. A quantitative conversion of [A336][Cl] into [A336][NO3] or [A336][Br] could not be achieved under the experimental conditions used, indicating that a significant aqueous excess of reagent would be required. The conversion into [A336][HSO4] was found to be the most difficult and a maximum conversion of about 0.550 was achieved at the highest initial aqueous HSO4– concentration.
Figure 3.
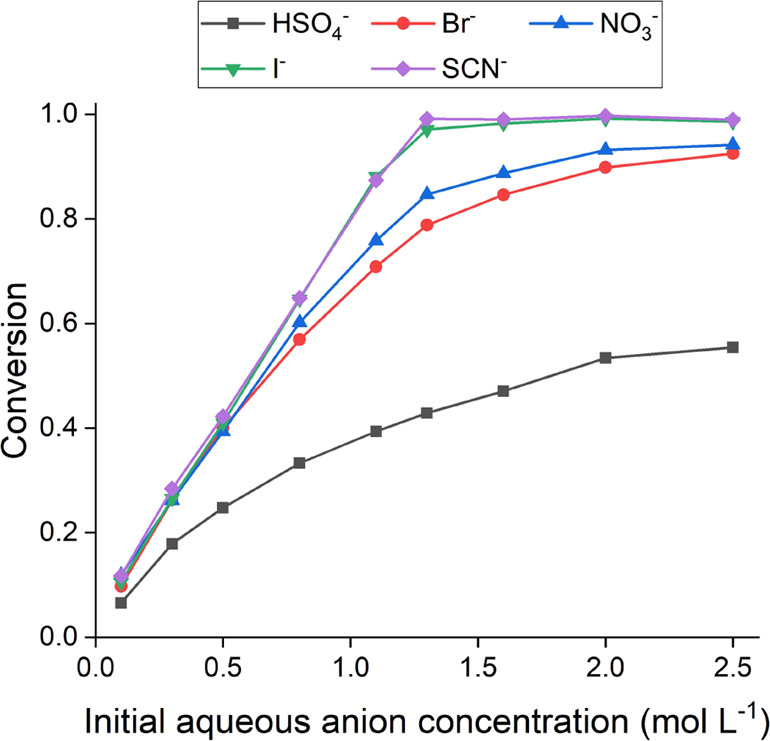
Conversion of [A336][Cl] 70 wt % in DIBK to [A336][X] (X = HSO4–, Br–, NO3–, I– or SCN–) as a function of the initial aqueous anion concentration. Conditions: 0.1–2.5 mol L–1 MX (X = HSO4–, NO3–, Br–, I–, and SCN–), room temperature, O/A = 1, 60 min.
The data in Figure 3 indicate that only for [A336][SCN] and [A336][I], a fully converted product (conversion >0.990) can be achieved in a single-stage batch process using moderate aqueous anion concentrations. Because of the position of SCN– and I– on the right side of the Hofmeister series, only a small excess of reagent is required to do so. For [A336][NO3], [A336][Br], and [A336][HSO4], a fully converted product cannot be achieved in a single-stage batch process, even at elevated aqueous anion concentrations. Multiple contacts with an aqueous salt solution are thus required.
Cross-current experiments were performed where the IL was contacted multiple times with a fresh salt solution. Two experimental conditions were chosen, that is, aqueous anion concentrations of 1.3 and 2.0 mol L–1. Working with O/A = 1, 1.3 mol L–1 of the reagent can be considered as near-equimolar conditions because for the employed 70 wt % [A336][Cl] dilution, the organic quaternary compound concentration is about 1.22 mol L–1. The use of 2.0 mol L–1 of the reagent is a significant excess compared to the amount of quaternary compound present. The use of these two different conditions highlights the increased process efficiency of a counter-current setup (vide infra). The results of the cross-current batch experiments are summarized in Table 1 for both anion concentrations (1.3 and 2.0 mol L–1) and O/A = 1. The experiment was terminated after eight consecutive steps or as soon as a conversion of 0.990 was reached. As expected, complete conversion (>0.990) to [A336][SCN] was achieved, for both experimental conditions after only a single contact (Figure 3). The conversion to [A336][I] was slightly less efficient and, under near-equimolar conditions, the conversion settled at 0.971 after the first contact. The second contact with a fresh 1.3 mol L–1 KI solution increased the conversion to >0.990. When using a more significant excess of the reagent (i.e., 2.0 mol L–1), a full conversion could be achieved in a single step. However, more important are the results for conversion to [A336][HSO4], [A336][Br], and [A336][NO3], as these systems require multiple contacts to achieve adequate conversions. Using a 1.3 mol L–1 solution, four and three contacts are required for [A336][Br] and [A336][NO3], respectively, in order to reach a quantitative conversion. For a 2.0 mol L–1 solution, this number is reduced to two contacts in both cases. The conversion to [A336][HSO4] proved to be significantly more difficult. Using a 1.3 mol L–1 solution, the conversion only reached a value of 0.985 after eight contacts. With a 2.0 mol L–1 solution, a fully converted IL was obtained after seven steps.
Table 1. Conversion of [A336][Cl] 70 wt % in DIBK to [A336][X] (X = HSO4–, Br–, NO3–, I–, and SCN–) for a Multistage Batch Process and Two Aqueous Anion Concentrations.
| anion X | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| number of contacts with a 1.3 mol L–1 aqueous solution | ||||||||
| HSO4– | 0.439 | 0.665 | 0.799 | 0.874 | 0.927 | 0.956 | 0.973 | 0.985 |
| Br– | 0.793 | 0.973 | 0.989 | >0.990 | ||||
| NO3– | 0.847 | 0.986 | >0.990 | |||||
| I– | 0.971 | >0.990 | ||||||
| SCN– | >0.990 | |||||||
| number of contacts with a 2.0 mol L–1 aqueous solution | ||||||||
| HSO4– | 0.525 | 0.762 | 0.872 | 0.935 | 0.967 | 0.983 | >0.990 | |
| Br– | 0.895 | >0.990 | ||||||
| NO3– | 0.933 | >0.990 | ||||||
| I– | 0.990 | |||||||
| SCN– | >0.990 | |||||||
The results of the above batch experiments indicate that the conversion to [A336][SCN] is very efficient, even for a single contact and at near-equimolar conditions. A counter-current process will thus not provide any benefit in terms of reagent use or product conversion. To a large extent, the same is true for [A336][I]. Here, a counter-current process can only be considered at near-equimolar conditions, where a quantitative conversion is not quite reached after a single contact. However, the main interest for the application of a counter-current process to the metathesis of ILs lies with the anions positioned on the left side of the Hofmeister series, for example, HSO4–, NO3–, and Br–. For such systems, a significant excess of the reagent is generally consumed because of the multiple contacts and/or high reagent concentrations that are required to achieve high conversions (cfr. Figure 3 and Table 1).
McCabe–Thiele Diagrams
Prior to the continuous counter-current experiments, McCabe–Thiele diagrams were constructed for the HSO4–, Br–, NO3–, and I– systems. This was performed to determine the feasibility of the process and to estimate the required number of stages to achieve a quantitatively converted IL. The diagrams were constructed by considering the metathesis process as a removal or stripping of chloride from the organic phase. The required equilibrium data were collected by varying the O/A phase ratio. Because of the efficient conversion to [A336][I], only the near-equimolar condition of 1.3 mol L–1 was examined. Conversely, for conversion to [A336][HSO4], which proved to be more difficult to achieve, only the 2.0 mol L–1 experimental condition was used. For conversion to [A336][Br] and [A336][NO3], both experimental conditions were applied. The McCabe–Thiele plots using an operating line for an O/A phase ratio of 1 are shown in Figure 4.
Figure 4.

McCabe–Thiele diagrams for the conversion of [A336][Cl] 70 wt % in DIBK to [A336][X]: (a) X = HSO4–; (b) X = Br–; (c) X = NO3–; and (d) X = I–. Conditions: 1.3 and 2 mol L–1 MX, room temperature, O/A = 10–0.25, 60 min.
Because of the similar position of Cl– and HSO4– in the Hofmeister series, the equilibrium line of the HSO4– system is positioned closely to the O/A = 1 operating line.24 Consequently, a large number (>14) of counter-current stages would be required to achieve an organic Cl– concentration of near-zero, or a fully converted product. Through the manipulation of the O/A ratio, this number can be significantly reduced, for example, 5 stages for O/A = 1/2. However, this would also result in a doubling in reagent consumption, whereas the aim of the current study was to minimize the reagent consumption by working in counter-current mode. An O/A ratio of 1 therefore remains preferred. In the bromide system, three stages would be required when working under near-equimolar conditions (1.3 mol L–1) while two stages would suffice for the 2.0 mol L–1 condition. In both cases, one additional step might be necessary to expel the last traces of chloride from the organic phase. For the nitrate system, three and two stages would be required for both conditions, respectively. In the iodide system, the equilibrium line is angular in shape, resulting in only two stages being required for a full conversion under near-equimolar conditions.
Metathesis Reactions in Continuous Counter-Current Mode
To determine the required flow rates and residence times for the continuous experiments, the rate of the metathesis process was studied first. The conversion of [A336][Cl] 70 wt % in DIBK to [A336][X] (X = HSO4–, Br–, NO3–, I–, and SCN–) as a function of contact time is shown in Figure 5. The experiments were performed with O/A = 1 and both experimental conditions (initial aqueous salt concentrations of 1.3 and 2.0 mol L–1). The results indicate that a contact time of 5 min is sufficient to reach equilibrium values for all anion systems. There seems to be no correlation between the rate of conversion and the position of the anions in the Hofmeister series, that is, anions placed on the right side of the series do not necessarily result in a fast metathesis. It was, however, noticed that the higher the equilibrium conversion, the longer it takes to reach that equilibrium position. For example, under near-equimolar conditions (1.3 mol L–1 salt solution), [A336][HSO4] reaches its equilibrium conversion of 0.439 within 1 min, while [A336][SCN] requires about 5 min to reach its equilibrium conversion of >0.990.
Figure 5.
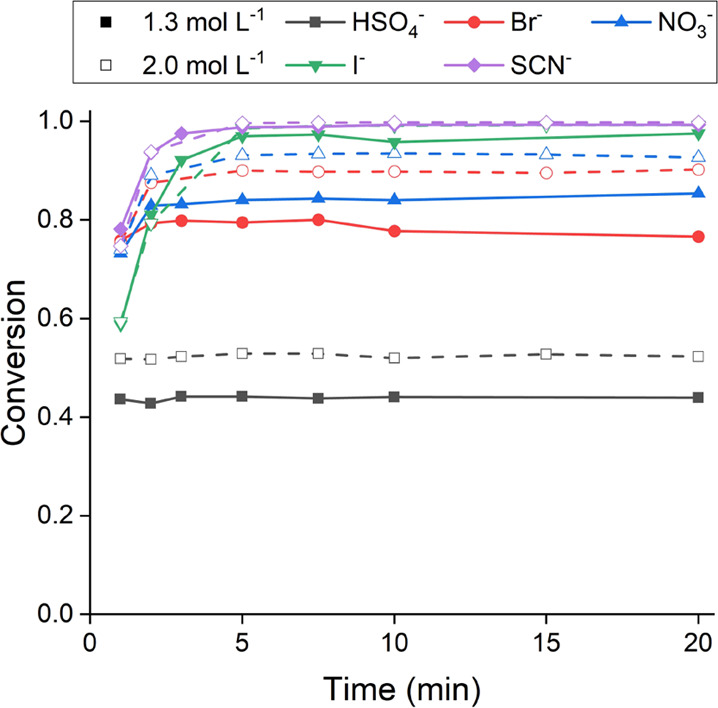
Conversion of [A336][Cl] 70 wt % in DIBK to [A336][X] (X = HSO4–, Br–, NO3–, I–, and SCN–) in batch as a function of time. Conditions: 1000 rpm, 1.3, and 2.0 mol L–1 MX (X = HSO4–, Br–, NO3–, I–, and SCN–), room temperature, and O/A = 1.
Continuous counter-current metatheses were performed for the HSO4–, Br–, NO3–, and I– systems. The experiments were performed with a total flow rate of 6 mL min–1 so that the residence time in each mixing chamber (35 mL) was approximately 6 min, sufficiently long to reach the equilibrium (cf. Figure 5). The O/A flow rate ratio was always maintained at 1. Figure 6 shows a working four-stage mixer-settler battery with mounted stirrer motors and connected pumps and tubing. As with the McCabe–Thiele diagrams, only the 1.3 mol L–1 condition was applied for the [A336][I] conversion, while for [A336][HSO4], only the 2.0 mol L–1 condition was used. For [A336][Br] and [A336][NO3], both experimental conditions were applied. Based on the results shown in Figure 4, the number of stages was calculated. A value slightly higher than the minimal number of stages resulting from the McCabe–Thiele simulation was chosen. Hence, a four-stage setup (cfr. Figure 6) was used for the [A336][I], [A336][Br], and [A336][NO3] systems, while for [A336][HSO4], the maximum number of available stages, that is, 12, was used.
Figure 6.

Operational four-stage mixer-settler setup with mounted stirrer motors and connected pumps and tubing.
The conversion of the IL product, which exits the last stage of the setup, is shown in Figure 7 as a function of the total run time of the experiment. Horizontal lines represent equilibrium values for single-stage batch experiments under the same conditions (cfr. Table 1). In general, the conversion of the product increases rapidly with time and exceeds the values that are obtained in a single-stage batch process. A relatively stable output is reached after three residence times, that is, 6 h for the four-stage experiments and 18 h for the twelve-stage experiment. From this equilibrium onward, the process can be run for extended periods of time for the production of large amounts of a converted IL product with a consistent purity.
Figure 7.

Product conversion and conversion per stage of [A336][Cl] 70 wt %wt % in DIBK to [A336][X] as a function of the total run time of a continuous multistage mixer-settler experiment. (a) X = HSO4–; (b) X = Br–; (c) X = NO3–; and (d) X = I–. Conditions: 1000 rpm, 1.3, and 2.0 mol L–1 MX, room temperature, O/A = 1, and a total flow rate of 6 mL min–1.
The most significant improvement in terms of final product conversion was obtained for [A336][HSO4], that is, a conversion of 0.980 compared to 0.525 under the same conditions (2.0 mol L–1 and O/A = 1) in a single-stage batch process. For [A336][NO3] and [A336][Br], the counter-current process resulted in a practically completely converted product. A conversion of 0.993 was achieved for both ILs under near-equimolar (1.3 mol L–1) conditions, whereas in batch, values of about 0.800 are obtained. With a more significant excess of reagent (2.0 mol L–1), the final product conversions increased slightly to 0.996 and 0.995, respectively, compared to values of around 0.900 for the batch experiments. These results also indicate that there is practically no difference between both experimental conditions, that is, the same product conversion is achieved within the same timeframe. Moreover, provided that a sufficient number of stages is available, there is no significant added value in terms of product conversion by increasing the reagent concentration over the equimolar amount. For [A336][I], only a minor improvement was observed, with a final conversion of 0.982 under near-equimolar conditions, compared to 0.971. The final product conversion and residual chloride contents of the IL products are summarized in Table 2. Chloride concentrations are provided for both the diluted ILs, that is, 70 wt % in DIBK and the undiluted IL, which can be obtained after the evaporation of DIBK (Supporting Information). For the calculation of the residual chloride concentration, the assumption was made that the quaternary compound concentration in the undiluted IL is 1.8 mol L–1 (vide supra).
Table 2. Final Product Conversion and Residual Chloride Concentrations (ppm) for the Counter-Current Mixer-Settler Metathesis of [A336][Cl] 70 wt % in DIBK to [A336][X] (X = HSO4–, Br–, NO3–, I–, and SCN–).
| conversion |
residual
Cl in diluted IL (ppm) |
residual
Cl in undiluted IL (ppm) |
||||
|---|---|---|---|---|---|---|
| aq. X concentration (mol L–1) | 1.3 | 2.0 | 1.3 | 2.0 | 1.3 | 2.0 |
| [A336][HSO4] | n.d.a | 0.980 | n.d. | 868 | n.d. | 1273 |
| [A336][Br] | 0.993 | 0.995 | 304 | 217 | 445 | 318 |
| [A336][NO3] | 0.993 | 0.996 | 304 | 174 | 445 | 255 |
| [A336][I] | 0.982 | n.d. | 781 | n.d. | 1145 | n.d. |
not determined.
Figure 7 also shows the progression of the conversion through the various counter-current stages which, overall, is in accordance with the McCabe–Thiele simulations shown in Figure 4. The HSO4– system shows three larger steps, after which the steps become progressively smaller because of the convergence of the equilibrium and operating lines. For [A336][NO3] and [A336][Br], high conversions are already achieved after three stages, and the fourth stage might be omitted. The difference between both experimental conditions manifests itself mostly in the first and second stages. For [A336][I], all four stages were required, which was not expected based on Figure 4.
Process Evaluation: Efficiency and Sustainability Metrics
Based on the increased conversions compared to batch experiments, the results described above indicate that counter-current processes can facilitate IL metathesis with a minimal reagent consumption. The maximization of the overall driving force for metathesis can be understood on a qualitative basis. In the first stage of the process, the fresh [A336][Cl] is contacted with a nearly depleted salt solution. This corresponds with the leftmost section of Figure 3. Based on the fact that the slope of the curve is the steepest under these conditions, an efficient consumption of the last traces of the salt solution can be expected. On the other hand, in the last stage of the process, a nearly completely converted IL is contacted with a fresh salt solution, resulting in the more efficient expulsion of the last traces of the initial anion, and this corresponds with the information shown in the rightmost section of Figure 3, where it is shown that the highest conversions are obtained for high aqueous anion concentrations.
A more quantitative description of this increased process efficiency can be made with process metrics such as the RME and the E-factor. In Figure 8, the RME of the mixer-settler experiments, based on the final product conversion, is shown and compared to the values of a multistep cross-current batch process (Table 1). Values for both experimental conditions, that is, 1.3 and 2.0 mol L–1 salt solutions, are shown. Solid horizontal lines indicate the RMEs obtained in the mixer-settler experiments, while theoretical maxima are indicated as horizontal dotted lines. The crosses indicate on which step of the cross-current process a product conversion equal to or higher than the mixer-settler process was achieved and thus indicate which step should be compared to the mixer-settler value. Similarly, E-factors are shown in Figure 9. Here, the arbitrary assumption was made that 90 wt % of the diluent and water can be recovered and recycled. The considered waste thus includes excess reagent, produced chloride salts, 10 wt % of the used DIBK, and 10 wt % of the used water.
Figure 8.

Reaction mass efficiencies (RME) of the mixer-settler experiments compared to a multistep cross-current batch process for the conversion of [A336][Cl] 70 wt % in DIBK to [A336][X]. (a) X = HSO4–; (b) X = Br–; (c) X = NO3–; (d) X = I–. Solid horizontal lines indicate the RMEs obtained in the mixer-settler experiments while theoretical maxima are indicated as horizontal dotted lines. Crosses indicate on which step of the cross-current process a product conversion was achieved equal to or higher than that for the mixer-settler process.
Figure 9.

E-factors of the mixer-settler experiments compared with a multistep cross-current batch process under the assumption of 90 wt % DIBK and water recovery for the conversion of [A336][Cl] 70 wt % in DIBK to [A336][X]. (a) X = HSO4–; (b) X = Br–; (c) X = NO3–; and (d) X = I–. Solid horizontal lines indicate the E-factors obtained in the mixer-settler experiments. Crosses indicate on which step of the cross-current process a product conversion was achieved equal to or higher than that for the mixer-settler process.
Overall, the RME of the mixer-settler metatheses closely approaches the theoretical maximum, while the values for the cross-current batch experiments are significantly lower and decrease with an increasing number of stages because of the increasing reagent consumption. In the HSO4– system, six cross-current steps using a 2.0 mol L–1 solution are required in order to achieve a similar conversion to the 12-stage mixer-settler experiment (0.980). As fresh reagent is consumed in each step of the cross-current process, the RME is only 38% of the value obtained for the counter-current experiment. For the conversion of [A336][Br], two or four cross-current steps are required to achieve a similar product conversion (>0.990) as for the counter-current experiments using a 2.0 or 1.3 mol L–1 solution, respectively. In terms of RME, the cross-current processes result in values that are only 75 or 58% of the values achievable by working in counter-current conditions. For [A336][NO3], similar conclusions can be made with values of 78 or 70% of the counter-current values. For [A336][I], the RME of the cross-current process was 77% of the value of the counter-current process. These results confirm that a counter-current metathesis process is most beneficial for anions placed lower in the Hofmeister series, as for those systems, a more significant increase in RME is possible. In addition, the data of the different experimental conditions, that is, the 1.3 and 2.0 mol L–1 experiments, demonstrate that the potential benefit in RME is most significant for the lowest reagent concentration. In other words, provided that sufficient stages are available for a high conversion, it is most beneficial to work at near-stoichiometric conditions.
Concurrent with this reduction in reagent consumption, a reduction in waste production can be expected, which is reflected by the E-factors shown in Figure 9. The mixer-settler experiments show a relatively low E-factor, whereas the E-factor of the cross-current experiments increases significantly with the number of stages. For the conversion to [A336][HSO4], the E-factor could be reduced by a factor of 6.8 by working in counter-current mode. For the bromide system, a reduction by a factor of 2.1 or 4.3 was achieved using a 2.0 or 1.3 mol L–1 salt solution, respectively. For the nitrate system, which is positioned slightly higher in the Hofmeister series than Br–, this was reduced to 2.0 and 3.1. Finally, for the iodide system, the counter-current process resulted in a reduction in the E-factor with a factor of 2.3.
Conclusions
The continuous counter-current conversion of an IL, [A336][Cl], to [A336][X] (with X = HSO4–, Br–, NO3–, I–, and SCN–) by a metathesis reaction was studied, and the results were compared with a cross-current multistep batch process, which is typically used for this purpose. It was shown that the use of a counter-current flow setup allows for significant improvements in product conversion, reagent consumption, and waste production because of a maximization of the driving force, that is, the concentration gradient. Because the anions placed at the right side of the Hofmeister series, that is, SCN– and I–, already showed highly efficient conversions in single-stage batch experiments, the possible benefit of counter-current operation remained limited. However, for anions placed more to the left in the Hofmeister series, such as HSO4–, Br–, and NO3–, the counter-current operation showed considerable potential because higher conversions could be obtained with reduced amounts of the reagent. Provided that a sufficient number of counter-current stages is available, quantitatively converted ILs can be attained using a stoichiometric amount of the reagent. The obtained counter-current data matched quite well with the McCabe–Thiele diagrams, and the improved process efficiency was demonstrated by the calculation of the RME and the E-factor. RMEs of the cross-current experiments varied between 38 and 78% of the values of the counter-current experiments and were thus significantly lower. In terms of the E-factor, the counter-current operation allowed values to be reduced by a factor between 2.0 and 6.8.
Acknowledgments
The research was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program: Grant Agreement 694078 (SOLCRIMET). The authors also thank the KU Leuven for financial support (project C24/18/042, ISOMER).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssuschemeng.1c06873.
Choice of the diluent and the diluent content based on physical properties (settling rates and viscosity); diluent removal from the ILs and recycling through vacuum evaporation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Seddon K. R. Ionic Liquids for Clean Technology. J. Chem. Technol. Biotechnol. 1997, 68, 351–356. . [DOI] [Google Scholar]
- Wasserscheid P.; Keim W. Ionic Liquids - New “Solutions” for Transition Metal Catalysis. Angew. Chem., Int. Ed. 2000, 39, 3772–3789. . [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Savoy A. W. Ionic Liquids Synthesis and Applications: An Overview. J. Mol. Liq. 2020, 297, 112038 10.1016/j.molliq.2019.112038. [DOI] [Google Scholar]
- Wasserscheid P.; Welton T.. Ionic Liquids in Synthesis; John Wiley & Sons: Hoboken, 2008, pp. 369–567 [Google Scholar]
- Welton T. Ionic Liquids in Catalysis. Coord. Chem. Rev. 2004, 248, 2459–2477. 10.1016/j.ccr.2004.04.015. [DOI] [Google Scholar]
- Riaño S.; Binnemans K. Extraction and Separation of Neodymium and Dysprosium from Used NdFeB Magnets: An Application of Ionic Liquids in Solvent Extraction towards the Recycling of Magnets. Green Chem. 2015, 17, 2931–2942. 10.1039/C5GC00230C. [DOI] [Google Scholar]
- Wellens S.; Thijs B.; Binnemans K. An Environmentally Friendlier Approach to Hydrometallurgy: Highly Selective Separation of Cobalt from Nickel by Solvent Extraction with Undiluted Phosphonium Ionic Liquids. Green Chem. 2012, 14, 1657–1665. 10.1039/C2GC35246J. [DOI] [Google Scholar]
- Van den Bossche A.; Vereycken W.; Vander Hoogerstraete T.; Dehaen W.; Binnemans K. Recovery of Gallium, Indium, and Arsenic from Semiconductors Using Tribromide Ionic Liquids. ACS Sustainable Chem. Eng. 2019, 7, 14451–14459. 10.1021/acssuschemeng.9b01724. [DOI] [Google Scholar]
- Winters J.; Dehaen W.; Binnemans K. Solvation Structure of Poly-m-Phenyleneisophthalamide (PMIA) in Ionic Liquids. Phys. Chem. Chem. Phys. 2019, 21, 4053–4062. 10.1039/C8CP07041E. [DOI] [PubMed] [Google Scholar]
- Swatloski R. P.; Spear S. K.; Holbrey J. D.; Rogers R. D. Dissolution of Cellose with Ionic Liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. 10.1021/ja025790m. [DOI] [PubMed] [Google Scholar]
- Davis H. J. Task-Specific Ionic Liquids. Chem. Lett. 2004, 33, 1072–1077. 10.1246/cl.2004.1072. [DOI] [Google Scholar]
- Welton T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2084. 10.1021/cr980032t. [DOI] [PubMed] [Google Scholar]
- Holbrey J. D.; Reichert W. M.; Swatloski R. P.; Broker G. A.; Pitner W. R.; Seddon K. R.; Rogers R. D. Efficient, Halide Free Synthesis of New, Low Cost Ionic Liquids: 1, 3-Dialkylimidazolium Salts Containing Methyl- and Ethyl-Sulfate Anions. Green Chem. 2002, 4, 407–413. 10.1039/B204469B. [DOI] [Google Scholar]
- Billard I.; Moutiers G.; Labet A.; El Azzi A.; Gaillard C.; Mariet C.; Lützenkirchen K. Stability of Divalent Europium in an Ionic Liquid: Spectroscopic Investigations in 1-Methyl-3-Butylimidazolium Hexafluorophosphate. Inorg. Chem. 2003, 42, 1726–1733. 10.1021/ic0260318. [DOI] [PubMed] [Google Scholar]
- Wilkes J. S.; Zaworotko M. J. Air and Water Stable 1-Ethyl-3-Methylimidazolium Based Ionic Liquids. J. Chem. Soc., Chem. Commun. 1992, 13, 965–967. 10.1039/C39920000965. [DOI] [Google Scholar]
- Alcalde E.; Dinarès I.; Ibáñez A.; Mesquida N. A Simple Halide-to-Anion Exchange Method for Heteroaromatic Salts and Ionic Liquids. Molecules 2012, 17, 4007–4027. 10.3390/molecules17044007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil P.; Schwiertz M.; König A. Metathesis of Ionic Liquids: Continuous Ion Exchange by Donnan Dialysis. Chem. Eng. Technol. 2014, 37, 919–926. 10.1002/ceat.201200322. [DOI] [Google Scholar]
- Vu P. D.; Boydston A. J.; Bielawski C. W. Ionic Liquids via Efficient, Solvent-Free Anion Metathesis. Green Chem. 2007, 9, 1158–1159. 10.1039/B705745H. [DOI] [Google Scholar]
- Srour H.; Rouault H.; Santini C. C.; Chauvin Y. A Silver and Water Free Metathesis Reaction: A Route to Ionic Liquids. Green Chem. 2013, 15, 1341–1347. 10.1039/C3GC37034H. [DOI] [Google Scholar]
- Dinarès I.; Garcia de Miguel C.; Ibáñez A.; Mesquida N.; Alcalde E. Imidazolium Ionic Liquids: A Simple Anion Exchange Protocol. Green Chem. 2009, 11, 1507–1510. 10.1039/B915743N. [DOI] [Google Scholar]
- Hofmeister F. Zur Lehre von der Wirkung der Salze. Arch. Für Exp. Pathol. Pharmakol. 1888, 24, 247–260. 10.1007/BF01918191. [DOI] [Google Scholar]
- Dupont D.; Depuydt D.; Binnemans K. Overview of the Effect of Salts on Biphasic Ionic Liquid/Water Solvent Extraction Systems: Anion Exchange, Mutual Solubility, and Thermomorphic Properties. J. Phys. Chem. B 2015, 119, 6747–6757. 10.1021/acs.jpcb.5b02980. [DOI] [PubMed] [Google Scholar]
- Naert P.; Rabaey K.; Stevens C. V. Ionic Liquid Ion Exchange: Exclusion from Strong Interactions Condemns Cations to the Most Weakly Interacting Anions and Dictates Reaction Equilibrium. Green Chem. 2018, 20, 4277–4286. 10.1039/C8GC01869C. [DOI] [Google Scholar]
- Onghena B.; Valgaeren S.; Vander Hoogerstraete T.; Binnemans K. Cobalt (II)/Nickel (II) Separation from Sulfate Media by Solvent Extraction with an Undiluted Quaternary Phosphonium Ionic Liquid. RSC Adv. 2017, 7, 35992–35999. 10.1039/C7RA04753C. [DOI] [Google Scholar]
- Vereycken W.; Riaño S.; Van Gerven T.; Binnemans K. Extraction Behavior and Separation of Precious and Base Metals from Chloride, Bromide, and Iodide Media Using Undiluted Halide Ionic Liquids. ACS Sustainable Chem. Eng. 2020, 8, 8223–8234. 10.1021/acssuschemeng.0c01181. [DOI] [Google Scholar]
- Van Roosendael S.; Regadío M.; Roosen J.; Binnemans K. Selective Recovery of Indium from Iron-Rich Solutions Using an Aliquat 336 Iodide Supported Ionic Liquid Phase (SILP). Sep. Purif. Technol. 2019, 212, 843–853. 10.1016/j.seppur.2018.11.092. [DOI] [Google Scholar]
- Rydberg J.Solvent Extraction Principles and Practice, Revised and Expanded; CRC Press: New York, 2004, pp. 335–348. [Google Scholar]
- Gaudernack B.; Hannestad G.; Hundere I.. Process for Separation of Yttrium from the Lanthanides. US Patent US3,821, 352, 1974.
- Lu D.; Horng J. S.; Hoh Y. C. The Separation of Neodymium by Quaternary Amine from Didymium Nitrate Solution. J. Common Met. 1989, 149, 219–224. 10.1016/0022-5088(89)90489-X. [DOI] [Google Scholar]
- Riaño S.; Sobekova Foltova S.; Binnemans K. Separation of Neodymium and Dysprosium by Solvent Extraction Using Ionic Liquids Combined with Neutral Extractants: Batch and Mixer-Settler Experiments. RSC Adv. 2020, 10, 307–316. 10.1039/C9RA08996A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellens S.; Goovaerts R.; Möller C.; Luyten J.; Thijs B.; Binnemans K. A Continuous Ionic Liquid Extraction Process for the Separation of Cobalt from Nickel. Green Chem. 2013, 15, 3160–3164. 10.1039/C3GC41519H. [DOI] [Google Scholar]
- Parmentier D.; Paradis S.; Metz S.; Wiedmer S.; Kroon M. Continuous Process for Selective Metal Extraction with an Ionic Liquid. Chem. Eng. Res. Des. 2016, 109, 553–560. 10.1016/j.cherd.2016.02.034. [DOI] [Google Scholar]
- Mattison P. L.; McCurry P. M. J.. Preparation of Quaternary Ammonium Compounds. US Patent US6,586, 632, 2003.
- Vereycken W.; Riaño S.; Van Gerven T.; Binnemans K. Determination of Chlorides in Ionic Liquids by Wavelength Dispersive X-Ray Fluorescence Spectrometry. ACS Omega 2021, 6, 13620–13625. 10.1021/acsomega.1c00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argiropoulos G.; Cattrall R. W.; Hamilton I. C.; Paimin R. Determination of the Quaternary Ammonium Ion Components of Aliquat 336 by Electrospray Mass Spectrometry. Anal. Chim. Acta 1998, 360, 167–169. 10.1016/S0003-2670(97)00674-0. [DOI] [Google Scholar]
- Lee G. L.; Cattrall R. W.; Daud H.; Smith J. F.; Hamilton I. C. The Analysis of Aliquat-336 by Gas Chromatography. Anal. Chim. Acta 1981, 123, 213–220. 10.1016/S0003-2670(01)83173-1. [DOI] [Google Scholar]
- Larsson K.; Binnemans K. Separation of Rare Earths by Split-Anion Extraction. Hydrometallurgy 2015, 156, 206–214. 10.1016/j.hydromet.2015.04.020. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.