

Abstract
Numerous methods have been developed in model systems to deplete or inactivate proteins, to elucidate their functional roles. In C. elegans, a common method for protein depletion is RNA interference (RNAi), in which mRNA is targeted for degradation. Additionally, C. elegans is a powerful genetic organism, amenable to large scale genetic screens and CRISPR-mediated genome editing. However, these approaches largely lead to constitutive inhibition, which can make it difficult to study proteins essential for development or to dissect dynamic cellular processes. Thus, there have been recent efforts to develop methods to rapidly inactivate or deplete proteins, to overcome these barriers. One such method that is proving to be exceptionally powerful is auxin-inducible degradation (AID). In order to administer AID in C. elegans, a 44 amino acid degron tag is added to the protein of interest and an Arabidopsis ubiquitin ligase, TIR1, is expressed in target tissues. Then, the plant hormone auxin is added; auxin mediates an interaction between TIR1 and the degron-tagged protein of interest, which triggers its rapid degradation via the proteasome. Here, we have outlined multiple methods to induce auxin-mediated depletion of target proteins in C. elegans, highlighting the versatility and power of this method.
INTRODUCTION
Understanding the functional roles of proteins is fundamental to biological research. Loss-of-function experiments, in which proteins are inactivated or depleted, are one of the primary ways to understand the contribution of proteins to cellular processes. As a result, many methods have been developed to suppress the activity of proteins (reviewed in (Housden et al., 2017)).
C. elegans is a powerful model organism amenable to a diverse array of genetic approaches for dissecting cellular pathways (reviewed in (Z. Wang & Sherwood, 2011)). In recent years, these approaches have become even easier due to the development of the CRISPR/Cas9 system, wherein one can directly alter the genome by introducing specific mutations or removing entire genes (reviewed in (Kim & Colaiacovo, 2019)). In this approach, the protein Cas9 and a small guide RNA (sgRNA) with a sequence complimentary to the gene of interest are injected directly into the worm germline. The sgRNA and Cas9 form a complex that base pairs with the gene and then Cas9 cleaves the DNA to enable gene editing. Since the genomic edit occurs in the germline, subsequent generations inherit the mutation. Thus, CRISPR provides a robust method for loss-of-function studies.
RNA interference (RNAi) is another common method used to disrupt protein expression. RNAi, originally discovered in nematodes, takes advantage of a mechanism that cells use to regulate the expression of genes by targeting mRNA for destruction (reviewed in (Montgomery, 2004)). The RNase Dicer processes dsRNA into small interfering RNA (siRNA). siRNA is loaded on the RISC complex, which then base pairs with and subsequently degrades target mRNA. In C. elegans, dsRNA can be administered by injection into the germline, feeding, or soaking (methods outlined in (Conte et al., 2015)). Large RNAi libraries are available to enable gene knockdown, which has facilitated many large-scale and targeted RNAi screens in this organism.
Although CRISPR-based mutagenesis and RNAi are extremely powerful, one disadvantage is that they both constitutively suppress protein activity. This can be a problem if a researcher wishes to study a process in an adult worm, but if particular proteins involved in that process also have functions earlier in development. For RNAi, one strategy to get around this problem is to use the injection or soaking methods (injecting or soaking adult worms) or by performing partial feeding RNAi (subjecting adult worms to feeding RNAi, rather than feeding throughout development). However, these methods can sometimes lead to incomplete protein depletion, and even if protein depletion is efficient, it usually occurs over a long time-scale. Thus, in recent years, there has been an effort to develop efficient strategies to deplete or inhibit proteins only at a specific stage. Such methods have the potential to lead to major advances, not only because they allow the study of proteins essential for development, but also because they enable rapid inactivation to study dynamic cellular processes.
Numerous methods have been developed that allow temporal control of protein inhibition in a variety of organisms; some excellent reviews have been recently published that provide an overview of these strategies (R.P. Chen et al., 2019; Natsume & Kanemaki, 2017; Trauth et al., 2019; Verma et al., 2020; Yesbolatova et al., 2019). These approaches use a variety of triggers, including temperature shifts, small molecules, antibodies, and light. Some methods are designed to conditionally alter protein activity, by changing protein localization or interfering with protein-protein interactions. For instance, a protein could be moved to a location where it is unable to perform its normal function (e.g., a transcription factor being forced to leave the nucleus). Alternatively, another strategy is to trigger targeted proteolysis. These methods use a degradation sequence, called a degron, that is attached to the protein of interest. In the presence of an inducer, this sequence enables recognition and destruction of the tagged protein by the proteasome. An advantage of targeted degradation is that it enables rapid and total protein inactivation, since the protein is entirely removed from the cell.
Multiple strategies for degron-mediated protein depletion have recently been developed in C. elegans. One approach co-opts an endogenous protein degradation pathway that normally functions to degrade some germline proteins, including PIE-1, in somatic cells during early development (Armenti et al., 2014; Sallee et al., 2018). PIE-1 contains a 36 amino acid “ZF1” zinc-finger domain that is recognized by the adaptor protein ZIF-1; this adaptor recruits ZF1-containing proteins to an E3 ligase complex, promoting their ubiquitination and degradation by the proteasome. During normal C. elegans development, ZIF-1 is expressed in somatic cells of the early embryo, mediating PIE-1 degradation specifically in those cells. This pathway has now been repurposed by tagging proteins of interest with the ZF1 domain and inducing ZIF-1 expression in other tissues using a heat shock promotor; the ability to control ZIF-1 expression in this manner allows proteins to be degraded with temporal and spatial control (Armenti et al., 2014; Sallee et al., 2018). A related approach co-opts the same degradation pathway using a different strategy. In this modified system, ZIF-1 is fused to a GFP-targeting nanobody and the protein of interest is tagged at the endogenous locus with GFP. Thus, the GFP-tagged protein is degraded in tissues where the nanobody fusion is expressed (S. Wang et al., 2017). These ZIF-1-based strategies offer a robust option for tissue-specific protein depletion. However, one limitation is that these approaches can only be used to degrade proteins in somatic cells; should ZIF-1 expression be induced in the germ line, it would lead to the degradation of PIE-1 and other germline proteins, causing phenotypes unrelated to the protein of interest.
Another strategy recently adapted for C. elegans uses light as an inducer of protein degradation (Hermann et al., 2015). In this method, a photosensitive degron (psd) is fused to a protein of interest. The psd combines a light-switchable LOV2 domain and a sequence containing a Cys-Ala motif that is recognized by the proteasome. Upon illumination with low intensity blue light, the LOV2 domain undergoes a conformational change that unmasks the Cys-Ala motif, triggering proteasome-mediated degradation. This system enables spatial control over illumination and leads to effective protein degradation in the targeted area in approximately an hour.
Finally, a particularly powerful method for protein degradation in C. elegans is the auxin-inducible degron (AID) system. The AID strategy takes advantage of a natural degradation system in plants, whereby the hormone auxin induces proteasome-mediated degradation of proteins containing a 44 amino acid degron sequence. This system was first developed as a means of targeted protein degradation in cell culture (Nishimura et al., 2009) and was subsequently adapted for C. elegans (Zhang et al., 2015). For AID, the protein of interest is endogenously tagged with the degron sequence using CRISPR, and the worm is also engineered to express TIR1, an Arabidopsis E3 ubiquitin ligase. Subsequently, auxin is added, which mediates the interaction between TIR1 and the tagged protein and targets it for degradation via the proteasome (Figure 1).
Figure 1: Auxin-inducible degradation.
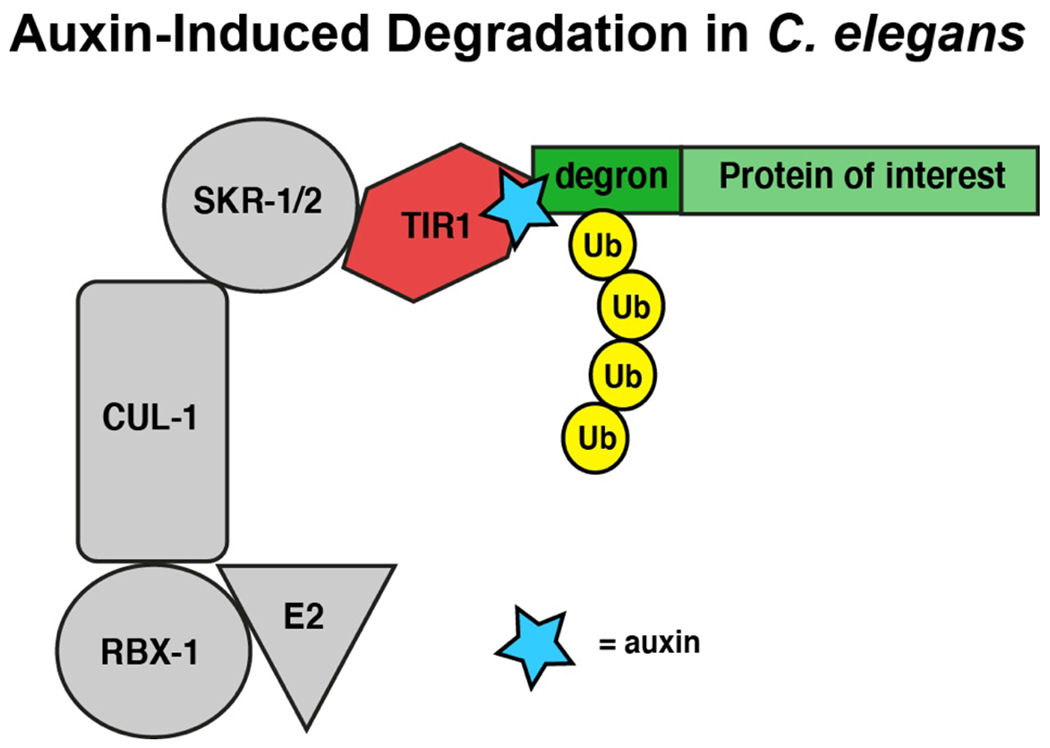
In tissues that express Arabidopsis TIR1 ubiquitin ligase transgene (red) via a tissue specific promoter, addition of auxin (blue) acts like glue to mediate the interaction between TIR1 and the degron-tagged endogenous protein (green). As a result, TIR1 engages machinery in the ubiquitin ligation pathway (grey) and the degron-tagged protein is modified by ubiquitin (yellow). This ubiquitination process targets the degron-tagged protein to the proteasome for degradation and protein function is eliminated. Tissues that do not express TIR1 continue to express the degron-tagged protein at wild-type levels. Eliminating auxin from the system can restore degron-tagged protein expression levels in tissues expressing TIR1 to wild-type levels over time. Therefore, this method provides a rapid and reversible method to eliminate protein expression in specific tissues.
The AID method is highly adaptable and offers a number of advantages. First, like the other conditional depletion methods used in C. elegans, AID provides both spatial and temporal control of protein degradation. Proteins can be depleted in specific tissues by using tissue-specific promotors for TIR1 expression (Figure 2), and also at particular stages of development, given the ability to temporally control auxin treatment. Additionally, in contrast to the ZIF-1-based approaches, AID can be used in the germ line, since it does not rely on an endogenous degradation pathway. The AID method is also convenient, since administering auxin to worms is straightforward and leads to particularly rapid protein depletion. Auxin can be added to the worm plates, which leads to protein depletion on the scale of hours, or worms or isolated cells can be soaked in auxin-containing media, which leads to depletion on the scale of minutes. This timescale is much more rapid than RNAi, and the auxin soaking method is also faster than the other conditional depletion methods currently in use in C. elegans. Finally, the AID system uses a short 44 amino acid degron tag that is unlikely to hinder protein function due to its small size, and proteins of interest can be tagged on either the N- or C-terminus, providing flexibility in experimental design. Given these advantages, the AID method is proving to be extremely valuable in dissecting a variety of processes and pathways.
Figure 2: Tissue specific protein degradation in C. elegans.
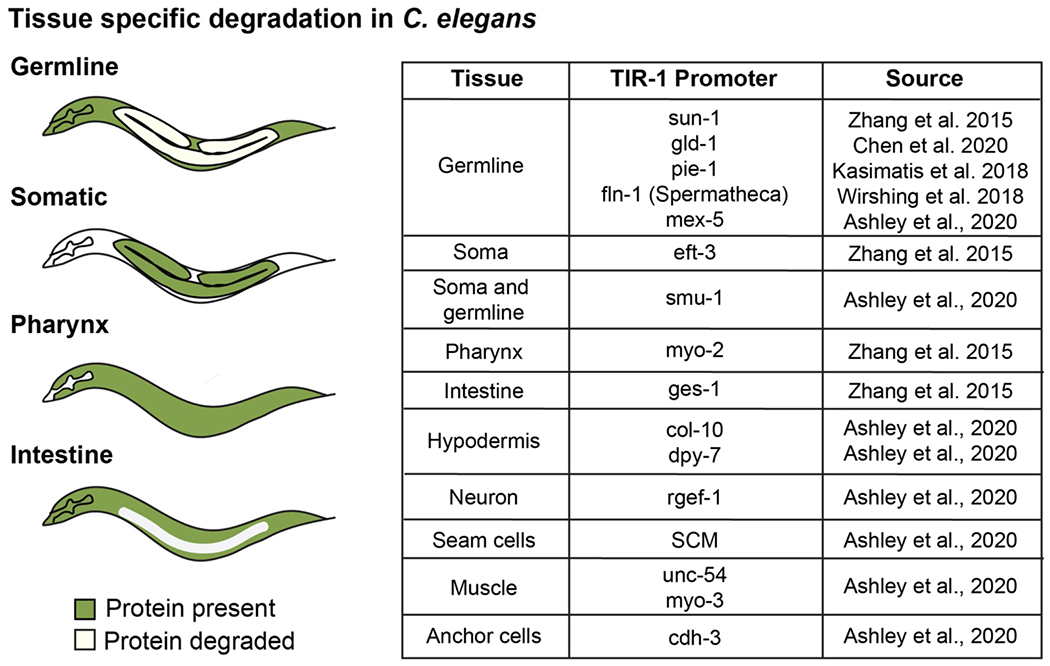
Expression of TIR1 in specific tissues can allow tissue specific degradation of proteins. Some examples of tissue specific degradation upon addition of auxin are diagrammed in C. elegans, wherein green represents regions where the degron-tagged protein continues to be expressed and white represents regions where the degron-tagged protein is degraded due to expression of TIR1 in that tissue. The table highlights some of the tissues in which TIR1 has been successfully expressed, as well as the corresponding tissue-specific promoters that have been used and developed in the field for degron studies thus far (Ashley et al., 2020; J. Chen et al., 2020; Kasimatis et al., 2018; Wirshing & Cram, 2018; Zhang et al., 2015).
In this chapter, we describe multiple strategies for auxin-mediated protein depletion in C. elegans (Figure 3). Our own work has focused on depleting proteins involved in oocyte meiosis (Mullen et al., 2019), so we use this system as an example. In our studies, we use worm strains where the sun-1 promoter is used to drive the expression of TIR1 in the germ line (Figure 2) (Zhang et al., 2015); this expression enables selective depletion of proteins in the gonad, to study meiotic prophase events such as chromosome pairing and recombination, and also in the oocytes, sperm, or embryos that are produced. However, the methods described can be applied to study countless other pathways and processes. For those new to the C. elegans system, we recommend this excellent overview of C. elegans husbandry and the basics of worm growth and maintenance (Stiernagle, 2006) and these descriptions of CRISPR-based genome engineering approaches for generating degron-tagged strains (Dickinson & Goldstein, 2016; Kim & Colaiacovo, 2019).
Figure 3: Schematic of the presented auxin-mediated depletion protocols.
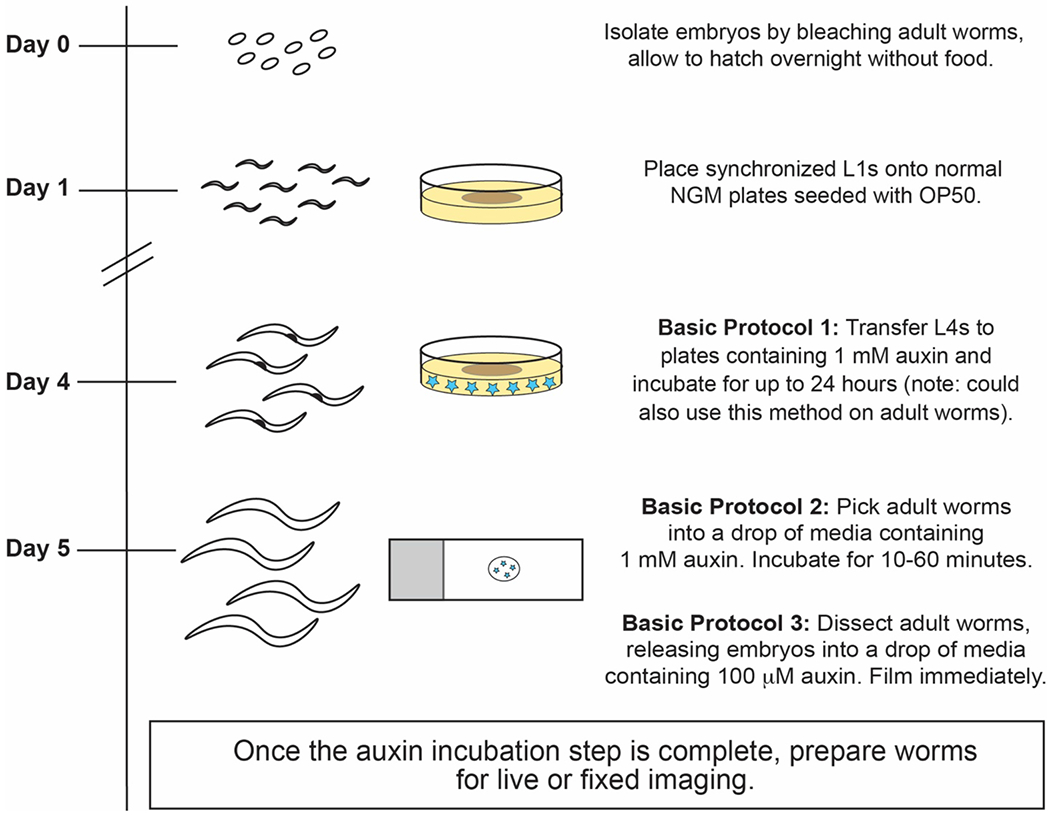
After bleaching adult worms to isolate embryos, synchronized hatched L1s can be placed on regular NGM food plates until they reach the desired stage. For Basic Protocol 1, L4-stage or adult worms (approximately on Day 4-5, depending on growth temperature) can be placed on auxin-NGM plates to initiate depletion; depletion in this method occurs on the scale of hours. For faster protein depletion, worms that are fed on regular NGM plates until adulthood can be soaked in auxin-containing media for up to an hour (Basic Protocol 2) or dissected into auxin-containing media to analyze isolated embryos (Basic Protocol 3) before proceeding with experiments.
STRATEGIC PLANNING
Selection of tags and potential issues with the degron system
One major advantage of the AID system is its versatility. The degron tag can be added to either the N- or C-terminus, or to internal sites in the target protein, making the system viable for nearly any protein of interest. When choosing where to place the degron tag, it is important to consider where essential domains reside on the protein in order to avoid disrupting protein function, and multiple locations may have to be attempted. If the protein of interest has a known loss-of-function phenotype such as embryonic lethality, a screen for that phenotype will indicate whether the location of the degron tag is interfering with its function.
In addition to the degron tag, it is also useful to have a way to visualize the protein of interest to monitor depletion. In the original development of the AID system in C. elegans, degron-tagged proteins were tracked by adding eGFP to the degron-tagged protein (Zhang et al., 2015), and we have also successfully used this strategy. However, we found that some proteins were unable to tolerate both the degron and the GFP tag. When troubleshooting these cases, we found that smaller epitope tags such as HA and 3xFLAG could be successfully substituted for GFP while preserving protein function. We could then use antibodies raised against these epitopes to detect our protein of interest via immunofluorescence. In a few cases, we attempted to add the degron sequence without GFP or an epitope tag, and we were unable to obtain viable strains, suggesting that the proteins were functionally disrupted. In these cases, adding an epitope tag at the protein terminus restored protein function. Considering the degron peptide (KPPAKAQVVGWPPVRSY) has a predicted net charge of +2.9 at physiological pH, we hypothesize that the degron tag may alter the electrostatic behaviors of some proteins when exposed at the terminus. Although we have not tested this hypothesis directly, our experience highlights that multiple tagging strategies may be required to obtain functional strains.
The ability of a protein to tolerate the degron tag can also be enhanced by the use of a flexible linker. In our hands, placing the simple GS(4xG) linker between the endogenous protein and the tags has proved to be sufficient for connecting the degron and epitope tags to a protein of interest without interfering with protein expression or function (epitope::degron::linker::protein for an N-terminal tag or protein::linker::degron::epitope for a C-terminal tag). However, a variety of other linkers have been successfully used for protein tagging in C. elegans (reviewed in (X. Chen et al., 2013)), and we predict that these should be effective for the degron tag as well.
PROTOCOLS
BASIC PROTOCOL 1: Long-term auxin-mediated depletion on plates
The most straightforward method for auxin-mediated depletion in C. elegans is to transfer worms of the desired stage from standard NGM plates to NGM plates containing auxin. In our studies, we work with adult worms that are actively producing oocytes and embryos. Thus, when first assessing the efficiency of auxin-mediated depletion on a new strain, we move L4 (late larval) stage worms from regular plates to auxin-containing plates for 24 hours at 20°C (Figure 3), which allows us to analyze young adults. The L4 stage is easy to identify in C. elegans due to a distinct crescent-shaped clearing made by the maturing vulva, which is located near the center of the worm. Additionally, since the timing of when L4-staged worms start producing oocytes and embryos is well known at a variety of growth temperatures (Altun & Hall, 2009; Wood, 1988), it is straightforward to synchronize worms and then grow the worms on the plates until they reach the desired stage. This 24-hour incubation has led to effective depletion in all of the cases we have tested, mimicking the effects of a completely penetrant RNAi phenotype. However, the original AID study in C. elegans demonstrated that depletion of some proteins can be achieved in less than 4 hours on an auxin plate (Zhang et al., 2015) and we have also found this to be the case for most proteins. Thus, when working with a new strain, we recommend assessing depletion efficiency at multiple timepoints (methods for this are presented in Basic Protocol 4). Moreover, the below protocol can also be optimized to examine depletion during other developmental stages, by moving different-staged worms to the auxin plates.
Materials
400 mM auxin stock solution (See Reagents and Solutions)
M9 media (See Reagents and Solutions)
4 L Flask
ddH2O
NaCl
Bacto-peptone powder
Bacteriological Agar powder
Cholesterol Solution (5 mg/ml in ethanol)
1 M MgSO4 Solution
1 M KH2PO4 Solution
1 mM CaCl2
OP50 E. coli bacteria (Ordered from the Caenorhabditis Genetics Center (CGC), See Reagents and Solutions)
Luria Broth (See Reagents and Solutions)
Disposable glass Pasteur pipettes (Fisherbrand)
6 cm worm plates
Bleach (Clorox)
5 M NaOH
Dissecting microscope (We use Leica M80 microscopes, but a wide variety of dissecting stereo microscopes with a transmitted light source could be used)
Refrigerated Tabletop Centrifuge for spinning 15 ml conical tubes
Preparing auxin plates
In a 4 L flask add 6 g NaCl, 5 g Bacto-Peptone and 34 g Bacteriological Agar.
Add ddH2O to bring total volume to 2 L (which will result in approximately 170 plates).
Swirl to mix ingredients and autoclave.
Remove from autoclave and cool the mixture for 25 minutes on the bench top. If leaving overnight, put in an oven set to 50°C to prevent the media from solidifying.
- After cooling or removal from the 50°C oven, add the following in order:
- 2 ml 1 M CaCl2
- 2 ml Cholesterol solution
- 2 ml 1 M MgSO4
- 50 ml 1 M KH2PO4.
Just before pouring the plates, add the auxin stock to bring the final concentration of auxin to 1 mM.
Swirl the flask to ensure that the auxin is mixed, and then the plates are ready to be poured.
Put the poured plates in a dark and dry place to dry.
While the NGM plates are drying, start overnight OP50 cultures in LB, to be used to spot the plates.
After the plates are dry, spot OP50 bacteria on the plates, spread the spot to cover about half of the plate, and allow it to grow. Note that the bacteria can take longer to grow on auxin-NGM plates than on regular NGM plates, because auxin slows bacterial growth. It is also important to store the plates in a dark and dry place to prevent auxin degradation during this step.
Once the OP50 lawn is grown, the plates are ready for use. Alternatively, the seeded plates can be stored in the dark at 4°C for up to 4 weeks before use.
Auxin-mediated depletion on plates
In order to obtain a substantial number of worms that are synchronized at a specific developmental stage, we use the bleaching protocol described below. Once the worms are synchronized at the desired age, they can be placed on auxin-containing plates to initiate auxin-mediated degradation. If synchronization of a large number of worms is not required, worms can be directly picked onto the auxin plates without performing this bleaching step.
Day 1:
-
1
Using a glass Pasteur pipette, use 9 ml of M9 to wash worms from three 6 cm plates crowded with gravid adults. Using the glass pipette (instead of a plastic pipette) will ensure that the worms don’t stick to the pipette.
-
2
Place the solution in a standard 15 ml conical tube. If the M9 washes result in a volume less than 9 ml, add extra M9 to bring the final volume to 9 ml.
-
3
Add NaOH to a final concentration of 0.5 M and add bleach to ~20% of the total volume. We use 9 ml M9, 1.5 ml 5 M NaOH, and 3.3 ml bleach for this step.
-
4
Mix the solution by inverting the tube multiple times or gently vortex it to ensure that the solution is well mixed. Allow the worm-bleach solution to sit on the tabletop for approximately 5 minutes. During this time, monitor the worms under a dissecting scope; as soon as the adult worms start to break open and dissolve, proceed to the next step. If the worms are left in bleach for too long, then the embryos may fail to hatch. Therefore, it is helpful to set a timer for this step.
-
5
Using a refrigerated tabletop centrifuge, pellet the embryos for 1 minute at 800xg at 4°C.
-
6
Gently pour off the supernatant without disrupting the embryo pellet.
-
7
Add 15 ml of fresh M9, mix the solution to resuspend the pellet, and again spin for 1 minute at 800xg at 4°C. Repeat this step 2 more times.
-
8
On the last wash, leave a small amount of M9 in the tube and resuspend embryos in the M9 by pipetting up and down with a sterile glass Pasteur pipette. Then use this pipette to drop embryos onto an unseeded (no food) NGM plate.
-
9
Allow embryos to hatch into larval L1s at 15°C overnight.
Day 2:
-
10
Wash hatched L1s off plates using 2-3 ml sterile M9 and collect in a conical tube.
-
11
Use a sterile glass Pasteur pipette to drop L1s onto multiple NGM plates seeded with OP50 (with no auxin). Aim for about 50 worms per plate. It is best to check each drop under a dissecting scope to ensure that you are not putting too many worms on each plate to prevent starvation before the worms reach the L4 stage.
-
12
Grow worms at a temperature between 15°C and 25°C until the desired stage is reached. Worms grow slower at lower temperatures, so worms grown at higher temperatures will reach the L4 or adult stage faster. The developmental timing of C. elegans at various temperatures has been well-documented (Altun & Hall, 2009; Wood, 1988) and this information can be used to modify the growth temperature depending on the desired day you wish to perform your experiments.
-
13
Check worms using a dissecting scope daily to monitor their growth. For 24 hour auxin experiments, proceed to the next step when worms reach the L4 stage; this will ensure that the by the time the incubation is complete the worms will have reached adulthood. Note that if you want to try shorter auxin incubations, you can allow worms to reach adulthood before proceeding.
Day 4:
If your desired stage is L4 or adults, they are typically ready by Day 4 (depending on growth temperature and strain variability).
-
14
Wash worms off NGM plates using 1-2 ml of sterile M9 and collect in a conical tube.
-
15
Use a sterile glass Pasteur pipette to drop worms onto the prepared OP50-auxin plates. Aim for about 30-40 worms per plate and check each plate under a dissecting scope to ensure that you are not putting too many worms on each plate. If there are too many worms on a plate, they may fully consume the OP50 bacteria and starve before the experiment; this could hinder their development and affect their ability to produce oocytes and embryos.
-
16
Allow worms to grow on auxin plates for the desired number of hours. It has been reported that proteins can be depleted in under 4 hours (Zhang et al., 2015), but the depletion timing should be experimentally-determined for each protein of interest. Depletion levels can be analyzed using immunofluorescence or Western Blotting. Alternatively, if the degron-tagged protein was also tagged with GFP or another fluorescent protein, depletion can be assessed by assessing fluorescence intensity. See Basic Protocol 4 for a description of these methods.
-
17
After you have optimized conditions for efficient depletion, use your desired assay to assess the depletion phenotype.
BASIC PROTOCOL 2: Rapid auxin-mediated depletion via soaking
Auxin treatment by soaking can be used to quickly deplete proteins of interest. This procedure depletes proteins on a faster timescale than the plate method described in Basic Protocol 1, sometimes as quickly as 15-20 minutes, but this timing should be verified and optimized for each protein of interest. In our studies, where we wish to analyze phenotypes during oocyte meiosis, we have found that incubations longer than one hour can result in the worms halting ovulation, likely due to lack of food during the soaking procedure. Thus, if we find that we cannot fully deplete a particular protein within this window, we switch to another method. However, we have found that most of the proteins that we have worked with have been successfully depleted in this timeframe. An example of auxin-mediated depletion via soaking is shown in Figure 4; in that experiment the worms were soaked in auxin for 20 minutes before fixing for immunofluorescence.
Figure 4: Combining RNAi with short term auxin-mediated depletion.
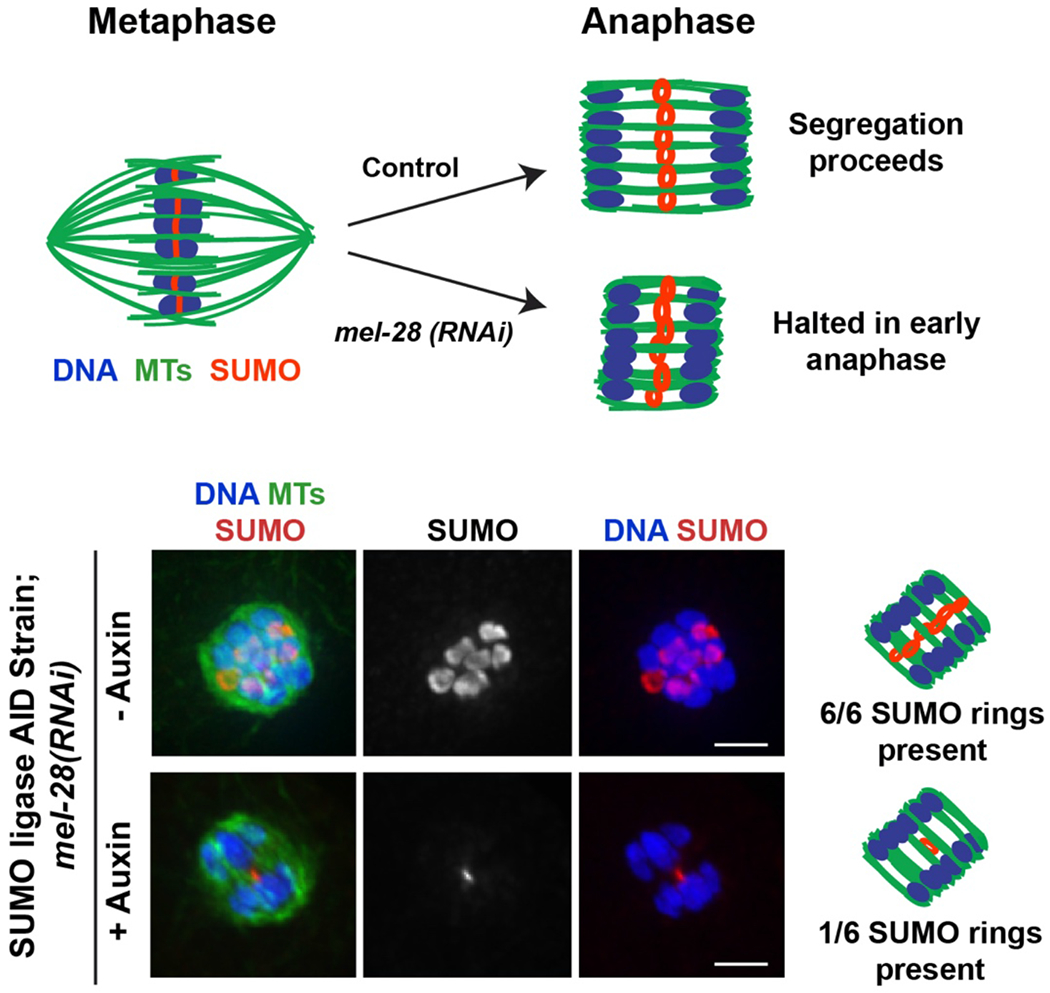
Studying the role of proteins during transient processes such as chromosome segregation is made easier by combining RNAi with the short term degron approach (Basic Protocol 2). As an example, we present depletion of a specific protein during anaphase in oocyte meiosis, modified from a recent study (Davis-Roca et al., 2018). In C. elegans oocytes, a ring complex with a modification known as SUMO (red) forms at the center of each of the six bivalents (blue) (Pelisch et al., 2017; Wignall & Villeneuve, 2009). In anaphase, these rings are left behind in the center of the spindle (green) (Davis-Roca et al., 2018; Davis-Roca et al., 2017; Dumont et al., 2010; Mullen & Wignall, 2017; Muscat et al., 2015). Knocking down the protein MEL-28 arrests spindles at early anaphase (Hattersley et al., 2016), making it possible to study the effects of depleting proteins at this particular stage (diagrams at top). In the example immunofluorescence images (bottom), short term auxin treatment was used to deplete the SUMO conjugating enzyme GEI-17 during this early anaphase arrest, induced by depleting MEL-28 using RNAi (protocol in (Davis-Roca et al., 2018)). MEL-28 depletion alone causes segregation to halt at a stage where all 6 SUMO rings are stabilized (top row of images). In contrast, 20 minutes of auxin treatment via soaking (to deplete GEI-17) results in fewer SUMOylated rings in the anaphase spindle. This suggests that the SUMO conjugating enzyme GEI-17 plays a role in stabilizing the ring complex during anaphase. Scale bar = 2μm.
Materials
Poly-L-Lysine coated microscope slides (Fisher Scientific)
Meiosis Media (See Reagents and Solutions)
Humidity Chamber: Opaque container with a tight lid lined with wet Whatman paper (alternatively, a clear container can be used if covered with aluminum foil to block light).
Worm pick: Can be made by inserting a platinum wire in a glass pipette and melting the glass around the wire to keep it in place (Stiernagle, 2006).
400 mM Auxin solution (See Reagents and Solutions)
Auxin-mediated depletion by soaking whole worms
Dilute the stock auxin solution into M9 or into Meiosis Media to make a solution at the final concentration of 1 mM. Meiosis Media was originally designed for live imaging of the meiotic divisions (Laband et al., 2018), we have found that it can be helpful in keeping worms healthy during prolonged incubations because it contains more nutrients than M9.
Put a 10 μl drop of the auxin-containing media on Poly-L-Lysine slide.
Use a worm pick to collect about 30 worms into the drop. Try to avoid transferring bacteria into the drop in order to prevent further dilution of the solution.
Gently place the slide in a wet humidity chamber with a lid, and let the worms incubate. This timepoint will need to be optimized by attempting various time intervals and may vary by protein.
As with Basic Protocol 2, depletion levels can be analyzed using immunofluorescence or Western Blotting, or by assessing fluorescence intensity if the degron-tagged protein was also tagged with a fluorescent protein (See Basic Protocol 4).
Once conditions that enable efficient depletion are optimized, use your desired assay to assess the resulting phenotype.
BASIC PROTOCOL 3: Acute auxin-mediated depletion of proteins in isolated embryos
One powerful advantage of the auxin system is that it enables tracking of protein depletion and the resulting phenotypes in real time, since the depletion can occur on the order of minutes. Such studies are not possible using traditional depletion methods like RNAi, which require longer incubation times. This advantage is particularly powerful for studying dynamic process such as cell division; proteins can be depleted while filming this process, enabling researchers to determine the effect of depletion on particular cell cycle events. For live imaging of the meiotic and mitotic divisions in C. elegans, embryos can be dissected from worms and imaged ex utero (for a thorough description of these methods, see (Laband et al., 2018)). Thus, combining live imaging with auxin-mediated depletion is straightforward: intact worms are picked into media containing auxin, immediately dissected, and then the isolated embryos are imaged. If the degron-tagged protein also contains a GFP tag, then imaging the GFP enables one to track protein depletion (See Basic Protocol 4), while imaging another fluorescently-tagged protein in different channel enables visualization of the process of interest. We have found that this procedure enables even faster depletion than Basic Protocol 2. Although both methods involve soaking, it is likely that the embryos are exposed to auxin more quickly when they are dissected and isolated, rather than the auxin first having to pass through the worm cuticle, reaching the cells of interest more slowly. In our experience, we have observed depletion of some proteins in oocytes in as little as 10 minutes.
Materials
Meiosis Media (See Reagents and Solutions)
Microscope Slides
Humidity Chamber (See Basic Protocol 2)
Worm pick
400 mM Auxin solution (See Basic Protocol 1)
Dissection Needles: 25G x 5/8 (0.5mm x 16mm)
Auxin-mediated depletion in isolated embryos
Dilute the ethanol auxin stock solution into Meiosis Media to a final auxin concentration of 100 μM. Note that this is a lower concentration than in Basic Protocol 2. Since isolated cells are incubated directly in the auxin solution, less auxin is required for effective depletion compared to soaking whole worms (since auxin does not have to soak into the worm through the cuticle, which may limit the amount that reaches the cells of interest in the short-term auxin incubations).
Pick about 10 worms into a drop of auxin-containing Meiosis Media on a microscope slide.
Use a needle to quickly dissect the worms in half near the vulva and allow oocytes and embryos to spill out into the media drop.
Gently move worm carcasses away from the oocytes and embryos using the needle or worm pick.
At this stage, either mount the isolated embryos for live imaging or use immunofluorescence to analyze depletion and the resulting phenotypes (see the “live ex utero imaging” and “fixed immunofluorescence imaging” sections of Basic Protocol 4 for details).
BASIC PROTOCOL 4: Assessing auxin-mediated depletion
To properly evaluate the results of auxin-mediated depletion experiments, it is important to determine the extent of protein depletion. Multiple methods can be used to assess if the depletion is successful. For each of these methods, it is important to compare auxin-treated worms to control worms treated with vehicle alone. For example, in the soaking method (Basic Protocol 2), control worms should be soaked in media without auxin, for the same amount of time as the auxin-treated worms, and then should be processed in the same way so a comparison can be made between the two conditions. Since the auxin stock solution is made in ethanol, for the vehicle treatment ethanol should be substituted in place of the auxin stock in the relevant protocols. One or more methods outlined below can be used to assess the extent of protein depletion, but in all cases a vehicle-treated control should be set up as a point of comparison.
Methods to analyze protein depletion:
Fixed whole worm imaging
If the degron-tagged protein is also tagged with a fluorescent protein such as GFP, then fixed imaging of whole worms can be used to detect the depletion of the tagged protein. In this method, worms are fixed at the desired timepoint in ethanol, which preserves GFP fluorescence, and then are imaged. Thus, comparing the amount of fluorescence in auxin-treated and untreated worms can reveal the extent of protein depletion.
Materials:
M9 media (See Reagents and Solutions)
Ethanol (room temperature)
Coverslips (20mm x 20mm)
1 mg/ml Hoechst stock (DNA Dye, if DNA staining is desired)
Microscope Slides (commercially available)
Vecta Shield Mounting Media (Vector Laboratories)
Nail polish to seal slides
Whatman Paper
Protocol:
Before starting, prepare the mounting media. If DNA staining is desired, first prepare a Hoechst-M9 solution, diluting the Hoechst stock 1:1000 (1 μl of 1 mg/ml Hoechst in 999 μl M9, for a final concentration of 1 μg/ml). Then mix this solution 1:1 with Vecta Shield mounting media and store in a cool dark location. If DNA staining is not desired, simply dilute the Vecta Shield mounting media 1:1 in M9.
If conducting a long term auxin experiment on plates (Basic Protocol 1), pick about 8-10 worms from the auxin plate into a 5 μl M9 drop on microscope slide before moving to Step 3. For short term experiments where worms are already soaking in auxin-containing media on a microscope slide (Basic Protocol 2), move directly to Step 3.
Once the worms have been picked into media, use the edge of a small piece of Whatman paper to wick away excess liquid in the drop. Since the worms can stick to the Whatman paper, monitor the worms using a dissecting microscope while performing the wicking procedure to make sure that you keep the Whatman paper from touching the worms. If the worms begin to clump and stack on top of each other, carefully move them apart using a pipette tip. Note that you do not need to remove all of the media, but the fixation procedure in step 4 works best if there is only a small amount of liquid left.
Once the amount of liquid is reduced, pipet 5 μl of ethanol onto the worms and allow it to evaporate, which fixes and desiccates the worms. Repeat this step twice.
Once the final ethanol treatment is completely evaporated, add 3-5 μl of the mounting media solution on top of the worms and carefully place a coverslip on top.
Use nail polish to seal the edges of the coverslip to prevent it from moving.
If slides are not imaged immediately, they should be stored at 4°C. It is best to image the worms as soon as possible (and no longer than a week after fixation), as the fluorescence decreases over time. Make sure to image the control and auxin-treated worms on the same day and use the same microscope settings and exposure.
Live ex-utero imaging
If the degron-tagged protein is also engineered with a fluorescent tag such as GFP, then live imaging can be used to detect loss of GFP fluorescence in real time after adding auxin. Moreover, if other proteins in the cell are tagged with a fluorescent tag of a different color, then live imaging can be used to track the behavior or localization of other proteins to visualize phenotypes as the protein of interest is being degraded. The protocol below gives an overview of this type of imaging, but we also recommend a recent methods paper for excellent detailed descriptions of various live imaging methods in C. elegans (Laband et al., 2018).
Materials:
Coverslips (20mm x 20mm)
Vaseline
5 ml syringe
Needle: 25G x 5/8 (0.5mm x 16mm)
Tweezers
Tricaine (optional)
Levamisole (optional)
Protocol:
For the short-term and acute auxin depletion protocols, whole worms or isolated embryos are incubated in auxin-containing media. Thus, it is possible to set up a live imaging experiment and assess depletion of the protein of interest in real time. After picking the worms into media (Basic Protocol 2) or dissecting the embryos (Basic Protocol 3), immediately proceed to step 2. Note that if you want to image live intact worms, the worms must be anesthetized so that they do not move during imaging. To this end, tricaine and levamisole should be added to the media that the worms are picked into in Basic Protocol 2. The final concentrations should be 0.2% tricaine and 0.02% levamisole.
Fill a 5 ml syringe with Vaseline and use the attached needle to draw a circle of Vaseline around the drop with intact or dissected worms. The Vaseline ensures that the coverslip, when placed on top, does not compress and damage the worms and embryos prior to imaging. The Vaseline ring should be approximately 1 to 1.5 cm in diameter, surrounding a 5 μl drop of media in the center.
Gently place the coverslip on top of the Vaseline and drop (use tweezers if necessary); the coverslip will stick to the Vaseline, and the drop will contact the coverslip.
Immediately mount the slide on your desired microscope to begin time lapse imaging.
Fixed Immunofluorescence Imaging
Immunofluorescence imaging using an antibody to the degron tag or using an antibody against the protein of interest itself, if available, can be used to assess depletion efficiency. Alternatively, if the strain was engineered with a secondary tag (e.g. GFP or FLAG), antibodies to these tags can also be used for immunofluorescence. Different immunofluorescence protocols have been optimized for different tissues. We use a protocol that is optimized for oocytes and embryos (modified from (Oegema et al., 2001) and described in (Davis-Roca et al., 2017; Mullen & Wignall, 2017)). However, if you are performing auxin-mediated depletion in other tissues or cell types, follow protocols standard in those fields.
Materials
Anti-GFP antibody (We use Roche, #11814460001 but multiple antibodies are commercially available)
Anti-Degron tag antibody (Anti-mini-AID tag mAb from MBL Life Science M214-3)
Alexa-Fluor secondary antibodies (Invitrogen)
Protocol
Fix worms as desired depending on the tissue being analyzed. We use a methanol fixation method adapted from Oegema, et al., 2001 in order to visualize the oocytes and embryos.
The degron antibody can be used to visualize the protein of interest, in order to monitor depletion. For primary staining of oocytes and embryos, we use this antibody at a 1:1000 dilution. However, it is possible that this concentration may need to be optimized if imaging protein depletion in other tissues. This optimization should be first done using control conditions (without auxin), to ensure that the staining is working efficiently, before launching depletion experiments. If the degron-tagged protein is also tagged with GFP, then a GFP antibody can also be used to monitor depletion. We use the Roche GFP antibody at a 1:500 dilution for staining of oocytes and embryos, but similar to the degron antibody, this may need to be optimized for other tissues.
Follow the immunofluorescence protocol for your tissue of interest, fixing the worms and incubating with the optimized concentration of the primary antibody. We use a protocol designed for oocytes and embryos. In this protocol, primary antibodies are incubated either for 1-2 hours at room temperature, or overnight at 4°C, and this timing is optimized depending on how well the antibodies work. We have found that the degron and GFP antibodies work well using either of these options, so the incubation time can be chosen based on practical considerations, such as the desired experimental timing.
For secondary antibody staining, we use Alexa-Fluor conjugated antibodies from Invitrogen at a 1:500 dilution. This incubation is performed for 2 hours at room temperature.
Upon completion of the immunofluorescence protocol, mount the worms for imaging.
The slides can then be imaged to assess depletion. Be sure to use the same microscope settings and exposures for the control and auxin-treated samples.
Western blotting
Western blotting can be used to detect depletion of the protein of interest, using antibodies to the protein itself or to one of the tags. This technique was successfully used to quantify protein depletion in the original development of the AID system in C. elegans (Zhang et al., 2015). For Western blotting in C. elegans, samples are typically made from whole worms because a large number of worms can be easily obtained and processed (protocol below). However, it is important to note that if your protein of interest is expressed in multiple tissues, then the exact extent of depletion may be difficult to determine using whole-worm samples. This is because the protein will only be depleted in a specific tissue (given that TIR1 is typically expressed using tissue-specific promotors; Figure 2). Alternatively, if your protein is largely expressed in a single tissue, or if gel samples can be made from the isolated tissue of interest, then Western blotting would be a more precise way of assessing protein depletion.
It is also important to note that the dilution of primary antibody used will need to be optimized for each protein of interest as several variables such as antibody specificity and protein abundance will influence what titer is most effective.
Materials:
M9 Media (see Reagents and Solutions)
Unseeded NGM plate
2X SDS Laemmli Sample Buffer (commercially available, we use Bio-Rad)
Polyacrylamide gel
Nitrocellulose membrane
Anti-GFP antibody (See immunofluorescence protocol)
Microcentrifuge
Protocol
After worms have undergone the desired auxin treatment, transfer 100 worms onto an unseeded NGM plate (with no bacterial lawn) and allow them to stay on the plate for five minutes. This serves to minimize bacterial transfer into the sample when you perform the next step.
Wash worms off the plate with 1 ml M9. Pipet the M9 onto the plate, and then use a glass Pasteur pipette to gently swirl the M9 wash around the plate a few times to dislodge the worms. Then suck up the M9 and transfer it to a 1.5 ml tube. Worms can stick to plastic, so it is important to use a glass pipette so that you recover all of the worms from the wash step.
Spin the tube briefly in a microfuge in order to pellet the worms.
Remove all but 15 μl of supernatant, then add an equal volume of 2X SDS Lamelli Sample Buffer. Boil sample for 10 minutes at 95 °C.
Load the worm lysate into a polyacrylamide gel of desired concentration and separate proteins by electrophoresis. The volume of sample you need to load will vary depending on the abundance of your protein of interest. This should be optimized on control worms before initiating the depletion experiments. We recommend loading the entire 30 μl gel sample as an initial test, but this amount can be reduced if the protein is abundant; not every protein requires loading this amount.
Transfer protein onto a nitrocellulose membrane. Protein(s) of interest can then be detected using standard Western blotting procedures with your desired antibody. We have had success with the Roche GFP antibody (described in the immunofluorescence section), used a 1:1000 dilution, and we also have performed successful Westerns using antibodies raised to our protein of interest itself. However, other antibodies could also be optimized and used to determine whether a degron-tagged protein was successfully depleted.
REAGENTS AND SOLUTIONS
Auxin Solution (400 mM)
Weigh out 0.28 g auxin powder (indol-3-Acetic Acid) (Alfa Aesar)
Dilute in 100% ethanol (cooled at 4°C) in a 15 ml conical
Mix well by inverting the tube or gently vortexing to ensure that the auxin powder is completely dissolved in the ethanol.
Since auxin is sensitive to light and temperature, wrap the conical in foil prior to storage
Store at 4°C for up to 3 weeks.
Luria Broth (LB)
5 g yeast extract
10 g tryptone
10 g NaCl
Bring final volume to 1 L with dH2O and autoclave
LB can be store at room temperature for a few months, unless contaminated.
M9 media
1.5 g of KH2PO4
3 g of Na2HPO4
2.5 g of NaCl
Bring final volume to 500 ml with dH2O and autoclave
After autoclaving, add 100 μl of 1 M MgSO4
M9 can be stored at room temperature for a few months
Meiosis Media
0.5 mg/ml Inulin from dahlia tubers (CAS Number 9005-80-5)
25 mM HEPES, pH 7.5
60% Leibovitz’s L-15 Media (Gibco 11415-049)
20% Heat-inactivated Fetal Bovine Serum
Sterile filter the mixture in a cell culture hood before use
Meiosis Media can be store at 4°C for up to 3 weeks
OP50 Bacteria for spotting plates
Streak a plate of OP50 bacteria from freezer stock and allow to grow overnight in a 37°C incubator
Pick a single colony from the plate and grow in LB at 37°C in a shaker
OP50 bacteria solution can be stored at 4°C for 3 days
COMMENTARY
Background Information
The auxin-inducible degron (AID) method is powerful because it can be used for acute depletion of proteins of interest. The outlined methods enable depletion of proteins on the scale of hours (if worms are incubated on auxin-containing plates; Basic Protocol 1) or minutes (if worms or isolated cells are soaked in auxin-containing media; Basic Protocols 2 and 3). These timescales are much faster than constitutive depletion methods such as RNAi, enabling studies of highly dynamic processes.
Additionally, the AID strategy can be easily combined with conventional RNAi methods. RNAi can be administered in C. elegans using feeding, soaking, or injection methods (detailed in (Conte et al., 2015)). Using the degron system, it is then possible to sequentially knockdown another protein in that particular RNAi condition. As an example, we have used this approach, combining RNAi with short-term auxin-mediated depletion (Basic Protocol 2) to induce cell cycle delays, in order to study the roles of proteins in particular processes. In a recent study, we aimed to deplete a SUMO-conjugating enzyme specifically during anaphase, to determine if SUMOylation played a role at that stage (Davis-Roca et al., 2018). However, this was difficult in unperturbed worms, since chromosome segregation occurs over a very short period of time. We therefore took advantage of a discovery by others that knocking down the protein MEL-28 halts oocytes in early anaphase (Hattersley et al., 2016). Thus, by growing worms expressing degron-tagged GEI-17 (a SUMO-conjugating enzyme in C. elegans (Pelisch et al., 2014; Pelisch et al., 2017)) on mel-28 RNAi plates, we were able to enrich for anaphase spindles and then deplete GEI-17 by soaking worms in auxin for 20 minutes (Basic Protocol 2). As shown in Figure 4, this treatment led to a reduction in SUMO staining during anaphase, demonstrating that we were able to visualize effects of auxin-mediated depletion at that specific cell cycle stage.
Another reason one might want to combine RNAi with the AID method is that RNAi-mediated depletion of two or more proteins in a worm can be difficult due to reduced RNAi efficiency when multiple proteins are depleted. In such cases, RNAi can be combined with Basic Protocol 1. Worms can be grown on RNAi plates that were poured with IPTG (for an overview of RNAi methods in C. elegans see (Conte et al., 2015)) and 1mM auxin (Basic Protocol 1), and then the resulting phenotypes can be analyzed.
Critical Parameters and Troubleshooting
For Basic Protocol 1, it is essential to keep the auxin solution away from light and in a cool and dry location because auxin is a plant hormone that is photosensitive. Similar precautions must be used for auxin plates when they are being stored, after being seeded with OP50 bacteria. It is also important to make fresh auxin solution every 2-3 weeks or when dark residue begins accumulating in the auxin stock solution; this ensures efficacy of the stock solution. Similarly, fresh plates must be poured every month.
As an alternative to auxin (indol-3-acetic acid), a recent study has demonstrated that synthetic auxin substitutes such as 1- naphthaleneacetic acid (NAA) can also be effectively used to conduct auxin experiments in C. elegans (Martinez et al., 2020). NAA is photostable and water-soluble, which would eliminate the need for dissolving in ethanol and for keeping the stock solution away from light. For detailed information about using NAA for depletion experiments in C. elegans, we recommend this recent article (Martinez & Matus, 2020).
For Basic Protocol 2, auxin can be diluted into M9 or into Meiosis Media (Laband et al., 2018)). The Meiosis Media is richer than M9, and we have found that it can be helpful in keeping worms healthy in the soaking protocol, especially for prolonged incubations. For each protein of interest, we optimize the time point for depletion, and the timing can vary from protein to protein. Some proteins can be efficiently depleted in as little as 15-20 minutes, and in most cases, complete depletion occurs within 60 minutes.
For Basic Protocol 4, sometimes the GFP or degron tags are inaccessible and the antibody staining does not work in the immunofluorescence procedure. Therefore, if one method for accessing the extent of auxin-inducible depletion fails, other methods should be tried. Additionally, the concentration of antibodies can be varied to ensure appropriate staining in the immunofluorescence protocol. Additionally, for the live ex utero imaging procedure, ensure that you don’t put pressure on the coverslip during the mounting process as it could damage the oocytes/embryos. Instead, gently place the coverslip on the Vaseline while monitoring the embryos under the microscope.
Understanding Results
We have outlined multiple protocols that enable depletion of degron-tagged proteins in C. elegans. Auxin treatment can be administered through different modes (plates and soaking) and over variable periods of time (long-term, on the scale of hours, and short-term, on the scale of minutes). Additionally, Basic Protocol 4 lists methods that can be used to assess the success of the depletion protocol.
Direct readouts of successful depletion are live imaging of GFP-tagged proteins of interest as well as immunofluorescence against the degron-tagged protein. If using an antibody-based immunostaining approach to assess depletion, absence of antibody staining after auxin treatment can be used to ensure that the depletion is successful; when comparing the extent of staining to control worms that were incubated with vehicle only, it is important to keep all of the imaging parameters consistent (exposure times, etc). An indirect method to assess auxin-inducible depletion can include the absence or change in localization of other cellular proteins that are dependent on the degron-tagged protein of interest. Downstream phenotypes such as embryonic lethality or developmental arrests can also provide indirect readouts of successful depletion, provided such phenotypes have been established by other methods. The type of readouts will be dependent on the designed experiment, the protein of interest, the expected phenotypes of the experiment, as well as the length of depletion. Examples of a successful auxin experiment and data interpretation are presented in Figure 4.
Time Considerations
The timeline of each AID experiment is dependent on the depletion kinetics of the protein of interest. When first performing AID experiments, it is important to determine the amount of time required for the protein of interest to be effectively depleted by the chosen method. This can be done by treating worms with auxin and assessing depletion at multiple time points. For Basic Protocol 1, we start by analyzing depletion at 1 hour increments, and for the more rapid soaking method (Basic Protocol 2) we initially use 10 minute increments; this timing can then be further refined to find the most optimal timepoint. If combining plate-based AID with feeding RNAi, the auxin plates used for depletion must also be seeded with the appropriate RNAi bacteria so that the relevant siRNA remains present during auxin treatment. Thus, RNAi plates containing auxin must be prepared ahead of time (protocols for RNAi detailed in (Conte et al., 2015)).
Once auxin treatment is complete, worms must be prepared for live or fixed imaging. Preparation for live imaging or for fixed whole worm imaging is quick and can typically be completed in minutes. For fixed immunofluorescence imaging of embryos, worm dissection takes 5-10 minutes, and then cells are fixed. This fixation step takes approximately 45 minutes in the methanol fixation protocol we use (but varies depending on the fixation method), antibody staining varies from 2 hours to overnight depending on the antibodies used, washes after antibody incubations are rapid, and mounting takes less than ten minutes (protocol modified from (Oegema et al., 2001)).
ACKNOWLEGMENTS
We would like to thank Drs. Liangyu Zhang and Abby Dernburg, who pioneered the AID system in C. elegans, for their invaluable help and advice as we began working with the degron system in our lab. We also thank Wignall lab members past and present for their contributions in optimizing these protocols, especially Gabriel Cavin-Meza and Dr. Tim Mullen. This work was supported by Cellular and Molecular Basis of Disease (CMBD) Training Grant NIH T32 GM00806 (to HEH) and NIH R01GM124354 (to SMW). Microscopy was performed at the Biological Imaging Facility at Northwestern University, supported by the Chemistry for Life Processes Institute, the NU Office for Research, and the Department of Molecular Biosciences.
REFERENCES
- Altun ZF, & Hall DH (2009). Introduction. WormAtlas, doi: 10.3908/wormatlas.1.1. [DOI] [Google Scholar]
- Armenti ST, Lohmer LL, Sherwood DR, & Nance J (2014). Repurposing an endogenous degradation system for rapid and targeted depletion of C. elegans proteins. Development, 141(23), 4640–4647. doi: 10.1242/dev.115048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley G, Duong T, Levenson MT, Martinez MAQ, Hibshman JD, Saeger HN, … Ward JD (2020). Expanding the Caenorhabditis elegans auxin-inducible degron system toolkit with internal expression and degradation controls and improved modular constructs for CRISPR/Cas9-mediated genome editing. BioRxiv, 10.1101/2020.05.12.090217. [DOI] [Google Scholar]
- Chen J, Mohammad A, Pazdernik N, Huang H, Bowman B, Tycksen E, & Schedl T (2020). GLP-1 Notch-LAG-1 CSL control of the germline stem cell fate is mediated by transcriptional targets lst-1 and sygl-1. PLoS genetics, 16(3), e1008650. doi: 10.1371/journal.pgen.1008650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RP, Gaynor AS, & Chen W (2019). Synthetic biology approaches for targeted protein degradation. Biotechnol Adv, 37(8), 107446. doi: 10.1016/j.biotechadv.2019.107446 [DOI] [PubMed] [Google Scholar]
- Chen X, Zaro JL, & Shen WC (2013). Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev, 65(10), 1357–1369. doi: 10.1016/j.addr.2012.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte D Jr., MacNeil LT, Walhout AJM, & Mello CC (2015). RNA Interference in Caenorhabditis elegans. Curr Protoc Mol Biol, 109, 26 23 21–26 23 30. doi: 10.1002/0471142727.mb2603s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Roca AC, Divekar NS, Ng RK, & Wignall SM (2018). Dynamic SUMO remodeling drives a series of critical events during the meiotic divisions in Caenorhabditis elegans. PLoS genetics, 14(9), e1007626. doi: 10.1371/journal.pgen.1007626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Roca AC, Muscat CC, & Wignall SM (2017). Caenorhabditis elegans oocytes detect meiotic errors in the absence of canonical end-on kinetochore attachments. J Cell Biol, 216(5), 1243–1253. doi: 10.1083/jcb.201608042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, & Goldstein B (2016). CRISPR-Based Methods for Caenorhabditis elegans Genome Engineering. Genetics, 202(3), 885–901. doi: 10.1534/genetics.115.182162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont J, Oegema K, & Desai A (2010). A kinetochore-independent mechanism drives anaphase chromosome separation during acentrosomal meiosis. Nature cell biology, 12(9), 894–901. doi: 10.1038/ncb2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattersley N, Cheerambathur DK, Moyle MW, Stefanutti M, Richardson A, Lee K, … Desai A (2016). A Nucleoporin Docks Protein Phosphatase 1 to Direct Meiotic Chromosome Segregation and Nuclear Assembly. Developmental cell, 38, 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann A, Liewald JF, & Gottschalk A (2015). A photosensitive degron enables acute light-induced protein degradation in the nervous system. Current biology : CB, 25(17), R749–750. doi: 10.1016/j.cub.2015.07.040 [DOI] [PubMed] [Google Scholar]
- Housden BE, Muhar M, Gemberling M, Gersbach CA, Stainier DY, Seydoux G, …. Perrimon N (2017). Loss-of-function genetic tools for animal models: cross-species and cross-platform differences. Nature reviews. Genetics, 18(1), 24–40. doi: 10.1038/nrg.2016.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasimatis KR, Moerdyk-Schauwecker MJ, & Phillips PC (2018). Auxin-Mediated Sterility Induction System for Longevity and Mating Studies in Caenorhabditis elegans. G3 (Bethesda), 8(8), 2655–2662. doi: 10.1534/g3.118.200278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, & Colaiacovo MP (2019). CRISPR-Cas9-Guided Genome Engineering in Caenorhabditis elegans. Curr Protoc Mol Biol, 129(1), e106. doi: 10.1002/cpmb.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laband K, Lacroix B, Edwards F, Canman JC, & Dumont J (2018). Live imaging of C. elegans oocytes and early embryos. Methods in cell biology, 145, 217–236. doi: 10.1016/bs.mcb.2018.03.025 [DOI] [PubMed] [Google Scholar]
- Martinez MAQ, Kinney BA, Medwig-Kinney TN, Ashley G, Ragle JM, Johnson L, … Matus DQ (2020). Rapid Degradation of Caenorhabditis elegans Proteins at Single-Cell Resolution with a Synthetic Auxin. G3 (Bethesda), 10(1), 267–280. doi: 10.1534/g3.119.400781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MAQ, & Matus DQ (2020). Auxin-mediated Protein Degradation in Caenorhabditis elegans. Bio Protoc, 10(8). doi: 10.21769/BioProtoc.3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery MK (2004). RNA interference: historical overview and significance. Methods in molecular biology, 265, 3–21. doi: 10.1385/1-59259-775-0:003 [DOI] [PubMed] [Google Scholar]
- Mullen TJ, Davis-Roca AC, & Wignall SM (2019). Spindle assembly and chromosome dynamics during oocyte meiosis. Current opinion in cell biology, 60, 53–59. doi: 10.1016/j.ceb.2019.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen TJ, & Wignall SM (2017). Interplay between microtubule bundling and sorting factors ensures acentriolar spindle stability during C. elegans oocyte meiosis. PLoS genetics, 13(9), e1006986. doi: 10.1371/journal.pgen.1006986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat CC, Torre-Santiago KM, Tran MV, Powers JA, & Wignall SM (2015). Kinetochore-independent chromosome segregation driven by lateral microtubule bundles. Elife, 4, e06462. doi: 10.7554/eLife.06462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, & Kanemaki MT (2017). Conditional Degrons for Controlling Protein Expression at the Protein Level. Annu Rev Genet, 51, 83–102. doi: 10.1146/annurev-genet-120116-024656 [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, & Kanemaki M (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nature methods, 6(12), 917–922. doi: 10.1038/nmeth.1401 [DOI] [PubMed] [Google Scholar]
- Oegema K, Desai A, Rybina S, Kirkham M, & Hyman AA (2001). Functional analysis of kinetochore assembly in Caenorhabditis elegans. J Cell Biol, 153(6), 1209–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch F, Sonneville R, Pourkarimi E, Agostinho A, Blow JJ, Gartner A, & Hay RT (2014). Dynamic SUMO modification regulates mitotic chromosome assembly and cell cycle progression in Caenorhabditis elegans. Nat Commun, 5, 5485. doi: 10.1038/ncomms6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch F, Tammsalu T, Wang B, Jaffray EG, Gartner A, & Hay RT (2017). A SUMO-Dependent Protein Network Regulates Chromosome Congression during Oocyte Meiosis. Mol Cell, 65(1), 66–77. doi: 10.1016/j.molcel.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallee MD, Zonka JC, Skokan TD, Raftrey BC, & Feldman JL (2018). Tissue-specific degradation of essential centrosome components reveals distinct microtubule populations at microtubule organizing centers. PLoS Biol, 16(8), e2005189. doi: 10.1371/journal.pbio.2005189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T (2006). Maintenance of C. elegans. WormBook, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauth J, Scheffer J, Hasenjager S, & Taxis C (2019). Synthetic Control of Protein Degradation during Cell Proliferation and Developmental Processes. ACS Omega, 4, 2766–2778. [Google Scholar]
- Verma R, Mohl D, & Deshaies RJ (2020). Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol Cell, 77(3), 446–460. doi: 10.1016/j.molcel.2020.01.010 [DOI] [PubMed] [Google Scholar]
- Wang S, Tang NH, Lara-Gonzalez P, Zhao Z, Cheerambathur DK, Prevo B, …. Oegema K (2017). A toolkit for GFP-mediated tissue-specific protein degradation in C. elegans. Development, 144(14), 2694–2701. doi: 10.1242/dev.150094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, & Sherwood DR (2011). Dissection of genetic pathways in C. elegans. Methods in cell biology, 106, 113–157. doi: 10.1016/B978-0-12-544172-8.00005-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wignall SM, & Villeneuve AM (2009). Lateral microtubule bundles promote chromosome alignment during acentrosomal oocyte meiosis. Nature cell biology, 11(7), 839–844. doi: 10.1038/ncb1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirshing ACE, & Cram EJ (2018). Spectrin regulates cell contractility through production and maintenance of actin bundles in the Caenorhabditis elegans spermatheca. Mol Biol Cell, 29(20), 2433–2449. doi: 10.1091/mbc.E18-06-0347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood WB (1988). The Nematode Caenorhabditis elegans. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory. [Google Scholar]
- Yesbolatova A, Tominari Y, & Kanemaki MT (2019). Ligand-induced genetic degradation as a tool for target validation. Drug Discov Today Technol, 31, 91–98. doi: 10.1016/j.ddtec.2018.11.001 [DOI] [PubMed] [Google Scholar]
- Zhang L, Ward JD, Cheng Z, & Dernburg AF (2015). The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development. doi: 10.1242/dev.129635 [DOI] [PMC free article] [PubMed] [Google Scholar]