

Abstract
Covalent inhibitors targeting the main protease (Mpro, or 3CLpro) of SARS-CoV-2 have shown promise in preclinical investigations. Herein, we report the discovery of two new series of molecules that irreversibly bind to SARS-CoV-2 Mpro. These acrylamide containing molecules were discovered using our covalent DNA-encoded library (DEL) screening platform. Following selection against SARS-CoV-2 Mpro, off-DNA compounds were synthesized and investigated to determine their inhibitory effects, the nature of their binding, and to generate preliminary structure-activity relationships. LC-MS analysis indicates a 1:1 (covalent) binding stoichiometry between our hit molecules and SARS-CoV-2 Mpro. Fluorescent staining assay for covalent binding in the presence of cell lysate suggests reasonable selectivity for SARS-CoV-2 Mpro. And lastly, inhibition of enzymatic activity was also observed against a panel of 3CLpro enzymes from different coronavirus strains, with IC50 values ranging from inactive to single digit micromolar. Our results indicate that DEL selection is a useful approach for identifying covalent inhibitors of cysteine proteases.
Keywords: Covalent inhibitor, Covalent DEL selection, SARS-CoV-2 Mpro, COVID-19
Abbreviations: DEL, DNA-encoded library; Mpro, main protease; TCI, tar-geted covalent inhibitors; LC-MS, liquid chromatog-raphy/mass spectrometry; BTK, Bruton's tyrosine kinase; JNK1, c-Jun N-terminal protein kinase 1; Fmoc, 9-fluorenylmethyloxycarbonyl; PCR, polymerase chain reaction; SDS-PAGE, sodium dodecyl sulphate-polyacrylamide gel electrophoresis
Introduction
The pandemic of coronavirus disease (COVID-19) sweeping across the world has caused huge losses to lives and economies since its outbreak in December 2019. [1], [2] Despite the promising efficacy of vaccines in preventing COVID-19 [3], [4], [5], developing effective oral drugs for the treatment of coronavirus infections remains urgent. Various strategies have been applied to find small-molecule drugs for the treatment of coronavirus disease including the repurposing of known anti-viral drugs, high-throughput screening campaigns, virtual screening, and AI-facilitated drug design. [6], [7], [8], [9], [10], [11]
The main protease (Mpro, or 3CLpro) of SARS-CoV-2 virus that causes COVID-19, is one of the potential targets of drug repurposing or de novo design, due to its critical role in coronavirus replication and transcription. [12], [13], [14] In addition, Mpro is a highly conserved cysteine protease among coronaviruses, but with no analogues in the human proteome in terms of recognition sequence and cleavage site [15], which makes it a promising target for designing small-molecule drug candidates. A series of ketone- or aldehyde-based covalent inhibitors of SARS-CoV-2 Mpro that interact with the catalytic Cys145 at the active site of this enzyme have been reported, and crystal structure analysis indicates reversible covalent binding with Cys145 in the main pocket. [16], [17] These inhibitors are generally derived from the previous anti-viral drugs, and possesses a peptidic scaffold and aldehyde functional group for reversible covalent binding. Considering the reported mutations of SARS-CoV-2, it is vital to develop alternative scaffolds and new warheads, to overcome possible resistance mutations of the coronaviruses. [18]
With several covalent inhibitors approved for cancer treatment during the past decade, recent interest in covalent drug discovery has spiked. [19], [20], [21] However, finding targeted covalent inhibitors (TCI) is still challenging. Collections of molecules containing suitable electrophilic warheads are lacking, and a major risk of high-throughput screening of these low chemical diversity collections is the interference by over-reactive electrophiles. [22], [23] An alternative strategy of generating TCIs is to modify known non-covalent ligands or fragments by attaching an electrophilic group that can react with a proximal nucleophilic amino acid, which is usually a cysteine, but can be serine, tyrosine or a few other amino acids as well. [24], [25], [26] However, the pool of known non-covalent ligands to select from is usually not large enough, and the rational design of potent covalent ligands from fragments that bind the target non-covalently can be a daunting task. Therefore, the combination of readily produced DNA-encoded libraries (DEL) of electrophiles coupled with a covalent selection method can provide a promising way to address these limitations and challenges. [27]
DEL is an emerging technology that allows constructing and screening millions to billions of compounds, in a cost-effective and efficient manner. [27], [28], [29], [30], [31], [32], [33], [34], [35] Several laboratories have investigated the use of DELs, employing widely varied formats, to discover covalent hit molecules. Chan et al. reported the discovery of a covalent inhibitor of MAP2K6 with the 50% inhibitory concentration (IC50) 4.5 μM using a template DEL strategy. [36] Zimmermann et al. showed the identification of covalent binders against JNK-1 using an encoded self-assembling chemical library. [37] Zambaldo et al. used a DNA library in a microarray format to discover covalent hits against kinases MEK2 and ERBB2. [38] Zhu et al. demonstrated the use of an on-DNA tool compound with HRV 3C protease. [39] And perhaps most relevant to our work, Guilinger et al. reported a covalent DEL selection to identify Bruton's tyrosine kinase inhibitors. [40] Despite these examples, covalent DEL selection against protein targets remains challenging in a few aspects: 1) the diversity of covalent libraries in terms of electrophiles with different reactivity; 2) the optimal incubation conditions for different protein target and electrophile combinations; and 3) methods of achieving adequate signal-to-noise to identify potential covalent hits from one round of selection, particularly against target classes other than kinases.
In this study, we utilized a self-designed and constructed covalent DEL, in combination with an optimized covalent screening method, to discover novel covalent inhibitors of SARS-CoV-2 Mpro. The potential covalent binders were validated through off-DNA compounds synthesis, covalent binding analysis, and functional assay. Our results indicate the feasibility and effectiveness of covalent DEL selection for hit discovery, in particular against viral proteases.
Results and discussion
Covalent DEL Libraries
DNA encoded library technology adopts split-and-pool strategy to generate combinatorial small molecules, each labeled with its unique DNA sequence. Our DNA encoded covalent libraries were designed with three or four cycles of chemistry plus the coupling of electrophiles. After testing the stability of different covalent electrophiles in DNA-compatible reaction conditions, 3 common covalent electrophiles focusing on targeting cysteine were chosen for library construction, to create a complexity of more than 600 million chemical structures. One of library (library 4) is taken as an example (Scheme 1). 191 Fmoc amino acids at cycle-1 (referred to as BB1) were first installed by acylation reaction onto the primary amine on the oligonucleotide headpiece after encoded with 191 unique oligonucleotide tags. After pooling, deprotection of N-Fmoc, the cycle-1 product was split into 28 wells, and encoded with 28 oligonucleotide tags prior to installing 28 Fmoc amino acids at cycle-2 (BB2) by acylation reaction. After pooling, deprotection of N-Fmoc, and splitting again into different wells, 191 unique Fmoc amino acids at cycle-3 (BB3) were reacted though amide bond formation after encoded with 191 unique oligonucleotide tags. Through pooling, N-Fmoc deprotection and encoding with a unique oligonucleotide tag, the electrophiles were then attached to the intermediate libraries by amide bond formation in the last synthetic step. Thus, the same intermediate libraries were reacted with different electrophiles to produce a series of libraries with variable electrophilic warheads. A brief illustration of our covalent DEL libraries are listed in Table S1.
Scheme 1.
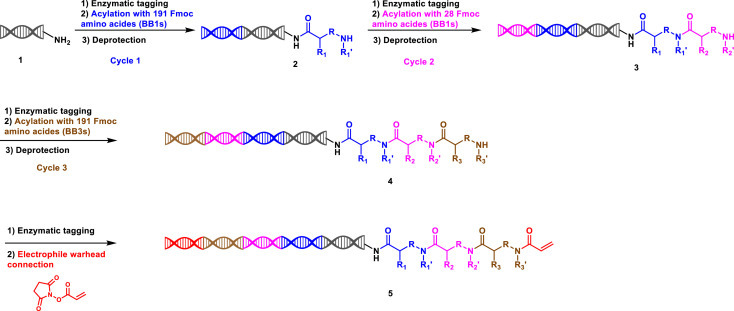
Synthesis of the covalent DNA Encoded Library 4
Covalent DEL Selection
A general procedure of covalent DEL selection includes incubation, immobilization, wash and elution. Note that the incubation time, concentrations of the covalent libraries, and the amount of target protein need to be assessed through condition optimization, as the optimal incubation condition might vary from case to case depending on the accessibility of cysteine among different protein targets and the desired hit profile.
Briefly, 3CLpro was incubated with the 10 pmol covalent DEL pool, comprised with 9 libraries with a total of 1.87 billion diversity in Tris-HCl (pH 7.5) buffer at room temperature for 1 hr. The 3CLpro protein was then immobilized on the Ni-NTA beads matrix through C-terminal 6xHis-tag. After three rounds of washing steps to remove unbound library members, protein: library-member complexes were eluted from the matrix by addition of 250 mM imidazole 30 min, and size exclusion chromatography purification was performed to remove excessive imidazole from the elution. The purified samples were then subjected to PCR amplification and NGS sequencing, similar to our previously published protocols for DEL selection for reversible ligands. [41] It is worth noting that the competitive elution method does not completely eliminate the presence of non-covalent binders.
The structure of enriched molecules was decoded through the DNA translation table from the sequencing data set. Further analysis of the affinity selection output was performed using Spotfire and presented in a cube view (Fig. 1 ). Compounds that were over 5 folds enriched than average are shown with the cube, where each axis represented building blocks used in cycle-1, cycle-2, and cycle-3 chemistry, respectively. Each blue dot represents a unique compound, and the size of each dot is proportional to the copy counts of each compound. Through clustering analysis of building blocks in different cycles, unique structural patterns that represent potential hits can be identified. Features sharing one or more building blocks can be easily recognized in the cube view as lines or planes. [41], [42] Analysis of these data revealed several features from two enriched libraries. The first DEL (Panel A) provided two highly enriched (and related) clusters, with the cycle-2 synthon being p-tolyl or 3-(trifluoromethyl)phenyl substituted proline, and an amino acid with a 2-methylallyl substitution at cycle-3. There was no preference for building blocks at cycle-1, and the amino acids varied greatly. There is also a strong feature from the second enriched DEL (Panel B). Enriched building blocks in cycle-2 share a similar structural pattern as piperazine substituted benzoic acids. Building blocks at cycle-1 contain varied amino acids, and building blocks at cycle-3 contain aryl rings attached to the cycle-2 building blocks by Suzuki coupling.
Figure 1.
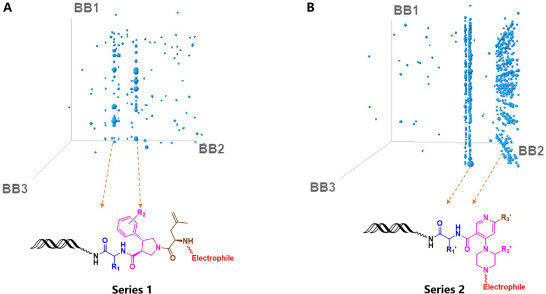
Cubic plot of libraries with preferred scaffolds and the corresponding structural features, where each axis represents one cycle of chemistry, and the size of each dot proportional to the copy counts of a unique compound. (A) For series 1, the BB2 and BB3 showed clear structural features with various BB1. (B) For series 2, the BB2 showed enriched structural features with aromatic substitution of BB3 and various BB1.
We also compared the data performance with that of another reported elution method: heat-on-beads. [39] With the heat-on-beads method, unbound molecules were removed through 5 min heat at 95°C. Molecules bound to target protein immobilized on affinity matrix after the heat elution were directly amplified by PCR on the beads for sequencing. The cubic scatter plot of the heat-on-beads elution method was shown in Fig S1. It was found that in the same covalent libraries, the heat-on-beads elution method did not show clear structural features. It is possible that the on-bead PCR step resulted in higher background signals and lower the signal-to-noise ratio in this case.
On-DNA Compounds Assay
We first re-synthesized the potential hit molecules with their DNA tags attached, employing the same protocols as used in previous library synthesis. [41] These unpurified products were analyzed by LC-MS products and by-products (i.e. truncates) annotated. The product samples were then incubated with SARS-CoV-2 Mpro and a gel shift assay was performed to test for covalent binding. The results of this preliminary assay (Fig S2 and Table S2) showed covalent binding of molecules from both hit series to SARS-CoV-2 Mpro. With these preliminary results in hand, we proceeded with off-DNA synthesis.
Covalent Binding Assay for Off-DNA Compounds
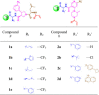
To validate the covalent binding of the potential hits identified based on our data analysis, we synthesized two series of compounds with different substitutions, including target products and potential by-products. The compound synthesis strategy was to keep the main scaffold of each series and explore the varied building blocks as revealed by the data analysis. For series 1 (Fig. 1A), five different amino acids from building block 1 were selected for 3-(trifluoromethyl)phenyl substituted proline scaffold (1a-e) respectively. For series 2 (Fig. 1B), the compounds 2a-b were synthesized to test if the additional building block at cycle-3 is necessary for activity. Compounds 2c-d are the full-length compounds with aromatic substitutions at cycle-3. In all cases, the site of the DNA-linker was replaced with methylamide group. The synthetic route and protocol for each compound are provided in the supporting information. All the compound structures are listed in Table. 1 .
Table 1.
Structures of off-DNA resynthesized compounds from two series.
 |
Intact mass spectrometry (MS) of proteins are widely used in the confirmation of covalent binding and has also been developed as a screening method for fragment-based covalent screening. [26] Therefore, the intact MS of SARS-CoV-2 Mpro was analyzed before and after incubation with compounds. Incubations were initially performed with each compound at 100 μM for 1 hr at room temperature, and covalent binding was detected for compounds from both series (Fig. 2 A). We then varied incubation times from 1 hr to 24 hrs and observed a time-dependent increasing of labelling percentage. As shown in Fig. 2B, the maximum labelling percentage was reached after 24 hrs incubation, suggesting the relative slow reactivity of the compound 1e with SARS-CoV-2 Mpro.
Figure 2.
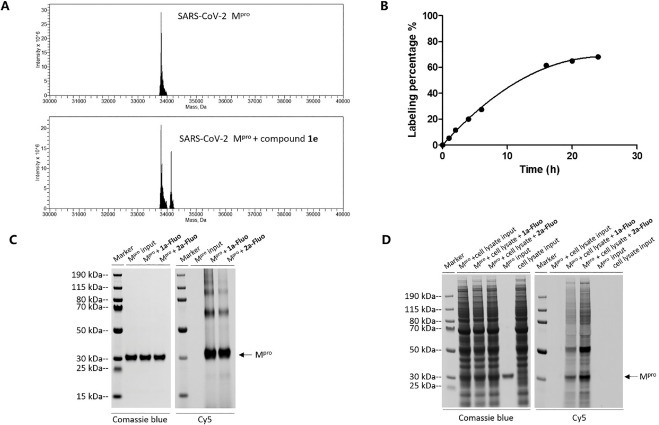
The covalent binding of hit compounds as determined by LC-MS and fluorescent staining assay. (A) Representative protein intact MS of hit compound covalently binding with SARS-CoV-2 Mpro. (B) Time-dependent increasing of the labeling percentage for compound 1e as determined by LC-MS. (C) Fluorescent analogs (Cy5 labeled) of compounds 1a and 2a were synthesized and incubated with SARS-CoV-2 Mpro. SDS-PAGE analysis of the resulting mixtures was then performed and theCy5 signal which overlaps with the protein bands (Coomassie blue) suggested the formation of covalent complex between protein and hit compounds. 1a-Fluo and 2a-Fluo represent compound 1a- and 2a-Fluorescence respectively. (D) Similar fluorescent staining assay as in 2C was performed for covalent binding in the presence of cell lysate. The Cy5 signal which is mainly corresponding to the target protein bands (Coomassie blue) suggested the formation of covalent complex between protein and hit compounds.
As an alternative method to prove the covalent binding of compounds with target proteins, we synthesized fluorescent analogs (Cy5 labeled) of compounds 1a and 2a. These Cy5 labeled compounds were then incubated with SARS-CoV-2 Mpro at 100 μM for 1 hr at room temperature. SDS-PAGE analysis of the resulting mixtures was then performed, and the fluorescent signal of Cy5 was measured and compared with protein bands detected by Coomassie blue staining. The overlap of Cy5 signal and Coomassie blue stained SARS-CoV-2 Mpro indicates covalent binding of compounds 1a-Fluo (compound 1a-Fluorescence) and 2a-Fluo (compound 2a-Fluorescence) (Fig. 2C). To gain a sense of the selectivity of our molecules, we further investigated the reaction of 1a-Fluo and 2a-Fluo with SARS-CoV-2 Mpro in the presence of cell lysate, where the strongest Cy5 signal still overlapped with Coomassie blue stained SARS-CoV-2 Mpro (Fig. 2 D). These results suggest our hit compounds are moderately selective for SARS-CoV-2 Mpro.
Enzymatic activity assay
Next, we investigated the inhibitory effect of our hit molecules against the main protease of different coronaviruses including SARS-2, SARS-1, OC43, MERS, 229E, HKU1 and NL63 CoV. [15] The results are summarized in Table 2 . Compound 1a-b appears to be the most potent overall, and compound 1e possess the lowest IC50 value against SARS-CoV-2 Mpro. Compound 1e also showed the highest covalent binding percentage as judged by LC-MS, suggesting that enzymatic inhibition results from covalent binding. It is not surprising that these compounds show activity against main proteases of other coronaviruses, especially for SARS-CoV-1. The main proteases in different genera possess sequence identity close to 50 %, along with high tertiary and quaternary structural conservation especially in the region around the main pocket. [43] Moreover, we tested the inhibitory effect of our hit compound 1e against the main protease of SARS-CoV-2 at 1 hr and 16 hrs. The results showed that as the incubation time increases, the IC50 decreases (Fig. 3 ), which is consistent with the time-dependent nature of covalent reaction.
Table 2.
Summary of the enzymatic activity and inhibitory activity against SARS-CoV-2 infection.
| Compound # | SARS-2a IC50 (μM) | SARS-1a IC50 (μM) | OC43a IC50 (μM) | MERSa IC50 (μM) | 229Ea IC50 (μM) | HKU1a IC50 (μM) | NL63a IC50 (μM) |
|---|---|---|---|---|---|---|---|
| 1a | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 1b | 22.8 | 47.8 | >100 | >100 | >100 | >100 | >100 |
| 1c | 61.6 | 61.7 | >100 | >100 | >100 | >100 | >100 |
| 1d | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 1e | 2.0 | 3.5 | 37.4 | >100 | 1.8 | 19.5 | >100 |
| 2a | 36.5 | 74.2 | 45.9 | >100 | >100 | 52.2 | >100 |
| 2b | 51.0 | 93.1 | 66.6 | >100 | >100 | 80.1 | >100 |
| 2c | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 2d | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| GC376 | 0.013 | 0.026 | 0.014 | 0.153 | 0.026 | 0.012 | 0.052 |
The IC50 value as determined by the enzymatic assays of the corresponding main protease. The values were calculated from ten data points with two independent determinations.
Figure 3.
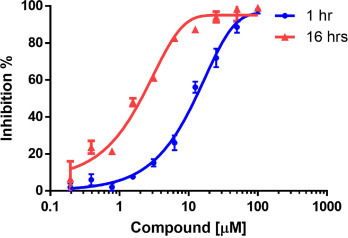
Time-dependent enzymatic assay of compound 1e for SARS-CoV-2 main protease. The IC50 value with 1 hr and 16 hrs incubation time were 15.9 μM and 1.9 μM respectively. The values were calculated from ten data points with three independent determinations.
SARS-CoV-2 Antiviral Assay
We wonder if these compounds are functionally active in the cellular assay of SARS-CoV-2 infection. With reference to the reported strategy on antiviral assays in co-dosing P-glycoprotein transport inhibitor to reduce cellular efflux, the cell was treated with compound 1e ten-point dose from 100 μM in the presence of 2 μM CP-100356, which is a known P-glycoprotein transport inhibitor to suppress the high efflux potential of the assay cell line. [44] As shown in Fig. 4 , compound 1e showed inhibitory effect against SARS-CoV-2 infection by the cellular assay. The 50% effective concentration (EC50) was around 33 μM while it was found with moderate cytotoxic activity up from 100 μM. The cytotoxicity and relative higher EC50 in cell-based assay might be arise from the off-target effects previously identified from gel shift assay in the presence of cell lysate. Further optimization work is needed to address this problem to achieve higher potency and selectivity.
Figure 4.
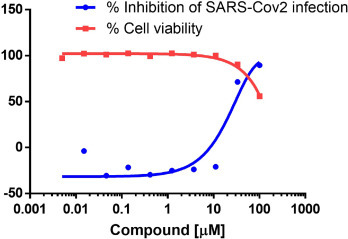
The inhibition of SARS-CoV-2 infection and the effect on cell viability of compound 1e. The antiviral EC50 value was 33 μM, CC50 value was more than 100 μM. The values were calculated from ten data points with two independent determinations, using 2 uM CP-100356 only group as a control for 0% inhibition.
Molecular Modelling
In an effort to understand the key interactions between SARS-CoV-2 Mpro and the covalent binders, we performed covalent docking on 1e and 2a. [45] Our docking results showed that both 1e and 2a formed hydrogen bonds with Gly143 in the active site (Fig. 5 ). However, their binding modes were significantly different. In compound 1e, the 3-(trifluoromethyl)phenyl group was accommodated in the S4 subpocket consisting of Met165, Leu167, Phe185, Gln189, and Gln192. [46, 47] Meanwhile, the R1 moiety at cycle one, a benzyl group in 1e, was fit into another subsite S1 comprising of Phe140, Asn142, His163, Glu166, and His172, which might explain the weaker binding affinities of 1a and 1d in this series, as the proper size and the aromaticity of the R1 group seem to be important (Fig. 5 A). A hydrogen bond was also formed between the terminal methylamide group and Glu166. For compound 2a, a plausible binding mode was observed and shown in Fig. 5 B. The R1’ group of 2a was near the S4 pocket, but did not extend into it. Furthermore, the significantly reduced binding affinity of 2c and 2d may be caused by the steric hindrance between the non-hydrogen moieties of R3’group and Thr25 in the vicinity. The methylamide group at cycle-1 for both 1e and 2a was found to at the edge of the binding pocket, which allows for the exit of the DNA tag. These results shed insight on the further hit-to-lead optimization to develop compounds with better activity.
Figure 5.
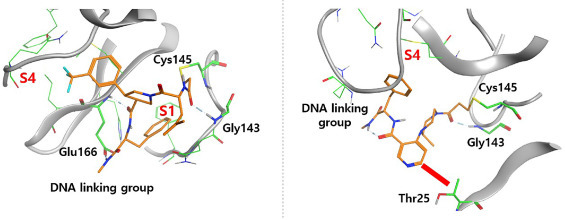
The covalent docking poses of compounds (A) 1e and (B) 2a. Orange and green: carbon; Blue: nitrogen; Red: oxygen; Yellow: sulfur; Cyan: fluorine. The red bar in (B) indicates the potential steric hindrance when R3’ is a heavy atom or a functional group.
To date, the main challenge in developing novel covalent inhibitors is a lack of chemically diverse compound collections available for high-throughput screening. To circumvent this problem, drug hunters often focus on the addition of electrophiles to known non-covalent binders using rational design techniques. However, this strategy has disadvantages including long time-lines and a lack of chemical starting points. [48] Herein we demonstrate the use of DNA-encoded libraries to directly discover covalent hit molecules against the protease SARS-CoV-2 Mpro, complementing recently published work targeting a kinase using similar techniques for library production and selection. [40] Two novel covalent hit series against SARS-CoV-2 Mpro were discovered. These hit molecules bind SARS-CoV-2 Mpro covalently in a 1:1 stoichiometry. Enzymatic activity against a panel of main proteases was determined, and the most potent molecule (1e) shows activity against 6 of the 8 main protease homologs. The most potent compound also showed inhibitory effect against SARS-CoV-2 in cellular assay. Lastly, the binding model of our hit molecules with the main pocket of SARS-CoV-2 Mpro was analyzed by the molecular modelling, and further optimization appears possible.
Supplemental material
Supplemental_Material_for_Discovery_of_SARS-CoV-2_Main_Protease_Covalent_Inhibitors_from_a_DNA-Encoded_Library_Screening_by_Wenji,_et_al.pdf
Supplemental material is available online with this article. Including experimental details of the preparation and characterization of all target compounds, experimental procedure and supporting figures.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Author contributions
Rui Ge: covalent DEL selection, experiment design, and manuscript preparation. Zuyuan Shen: project tracking, off-DNA compounds preparation, figure preparation and manuscript preparation. Jian Yin: molecular modelling, figure preparation, and manuscript preparation. Wenhua Chen: DEL selection data analysis. Qi Zhang and Yulong An: covalent DEL construction. Dewei Tang and Alexander L. Satz: manuscript proofreading and editing. Wenji Su: overall study design, manuscript proofreading and editing. Letian Kuai: overall study design, manuscript proofreading and editing.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors were employed by WuXi AppTec, and their research and authorship of this article was completed within the scope of their employment with WuXi AppTec.
Acknowledgments
We are grateful to Dr. Yue Li, Shunjie Pan, Jing He and Youlang Yuan for expert opinion and support of this work. We would like to thank the CSU team in WuXi AppTec for assistance in synthesis of off-DNA compounds. We also thank coronavirus platform in WuXi biology for assistance in enzymatic activity assay.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.slasd.2022.01.001.
Appendix. Supplementary materials
References
- 1.Paules C.I., Marston H.D., Fauci A.S. Coronavirus Infections-More than Just the Common Cold. JAMA. 2020;323:707–708. doi: 10.1001/jama.2020.0757. [DOI] [PubMed] [Google Scholar]
- 2.Dhama K., Khan S., Tiwari R., et al. Coronavirus disease 2019-COVID-19. Clin Microbiol Rev. 2020;33 doi: 10.1128/CMR.00028-20. e00028-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao J., Zhao S., Ou J., et al. COVID-19: Coronavirus vaccine development updates. Frontiers in immunology. 2020;11:602256–602257. doi: 10.3389/fimmu.2020.602256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabaan A.A., Al-Ahmed S.H., Sah R., et al. SARS-CoV-2/COVID-19 and advances in developing potential therapeutics and vaccines to counter this emerging pandemic. Ann Clin Microbiol Antimicrob. 2020;19:40. doi: 10.1186/s12941-020-00384-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y.D., Chi W.Y., Su J.H., et al. Coronavirus vaccine development: from SARS and MERS to COVID-19. J Biomed Sci. 2020;27:104. doi: 10.1186/s12929-020-00695-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrison C. Coronavirus puts drug repurposing on the fast track. Nat Biotechnol. 2020;38:379–381. doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- 7.Asselah T., Durantel D., Pasmant E., et al. COVID-19: Discovery, diagnostics and drug development. J Hepatol. 2021;74:168–184. doi: 10.1016/j.jhep.2020.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh A.K., Brindisi M., Shahabi D., et al. Drug development and medicinal chemistry efforts toward SARS-coronavirus and covid-19 therapeutics. ChemMedChem. 2020;15:907–932. doi: 10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khare P., Sahu U., Pandey S.C., et al. Current approaches for target-specific drug discovery using natural compounds against SARS-CoV-2 infection. Virus Res. 2020;290 doi: 10.1016/j.virusres.2020.198169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riva L., Yuan S., Yin X., et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature. 2020;586:113–119. doi: 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y., Wang F., Tang J., et al. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit Health. 2020;2:e667–e676. doi: 10.1016/S2589-7500(20)30192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y.C., Yang W.H., Yang C.S., et al. Structural basis of SARS-CoV-2 main protease inhibition by a broad-spectrum anti-coronaviral drug. Am J Cancer Res. 2020;10:2535–2545. [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M.Y., Zhao R., Gao L.J., et al. SARS-CoV-2: Structure, biology, and structure-based therapeutics development. Front Cell Infect Microbiol. 2020;10 doi: 10.3389/fcimb.2020.587269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin Z., Du X., Xu Y., et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 16.Dai W., Zhang B., Jiang X.M., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee R., Perera L., Tillekeratne L.M.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov Today. 2021;26:804–816. doi: 10.1016/j.drudis.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badua C., Baldo K.A.T., Medina P.M.B. Genomic and proteomic mutation landscapes of SARS-CoV-2. J Med Virol. 2021;93:1702–1721. doi: 10.1002/jmv.26548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sutanto F., Konstantinidou M., Domling A. Covalent inhibitors: a rational approach to drug discovery. RSC Med Chem. 2020;11:876–884. doi: 10.1039/d0md00154f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh A.K., Samanta I., Mondal A., et al. Covalent inhibition in drug discovery. ChemMedChem. 2019;14:889–906. doi: 10.1002/cmdc.201900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Cesco S., Kurian J., Dufresne C., et al. Covalent inhibitors design and discovery. Eur J Med Chem. 2017;138:96–114. doi: 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- 22.Dalton S.E., Campos S. Covalent small molecules as enabling platforms for drug discovery. Chembiochem. 2020;21:1080–1100. doi: 10.1002/cbic.201900674. [DOI] [PubMed] [Google Scholar]
- 23.Gehringer M., Laufer S.A. Emerging and re-emerging warheads for targeted covalent inhibitors: Applications in medicinal chemistry and chemical biology. J Med Chem. 2019;62:5673–5724. doi: 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- 24.Lonsdale R., Ward R.A. Structure-based design of targeted covalent inhibitors. Chem Soc Rev. 2018;47:3816–3830. doi: 10.1039/c7cs00220c. [DOI] [PubMed] [Google Scholar]
- 25.Keeley A., Petri L., Abranyi-Balogh P., et al. Covalent fragment libraries in drug discovery. Drug Discov Today. 2020;25:983–996. doi: 10.1016/j.drudis.2020.03.016. [DOI] [PubMed] [Google Scholar]
- 26.Resnick E., Bradley A., Gan J., et al. Rapid covalent-probe discovery by electrophile-fragment screening. J Am Chem Soc. 2019;141:8951–8968. doi: 10.1021/jacs.9b02822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodnow R.A., Jr, Dumelin C.E., Keefe A.D. DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat Rev Drug Discov. 2017;16:131–147. doi: 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- 28.Satz A.L., Kuai L., Peng X. Selections and screenings of DNA-encoded chemical libraries against enzyme and cellular targets. Bioorg Med Chem Lett. 2021;39 doi: 10.1016/j.bmcl.2021.127851. [DOI] [PubMed] [Google Scholar]
- 29.Favalli N., Bassi G., Scheuermann J., et al. DNA-encoded chemical libraries - achievements and remaining challenges. FEBS Lett. 2018;592:2168–2180. doi: 10.1002/1873-3468.13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song M., Hwang G.T. DNA-encoded library screening as core platform technology in drug discovery: Its synthetic method development and applications in DEL synthesis. J Med Chem. 2020;63:6578–6599. doi: 10.1021/acs.jmedchem.9b01782. [DOI] [PubMed] [Google Scholar]
- 31.Madsen D., Azevedo C., Micco I., et al. An overview of DNA-encoded libraries: A versatile tool for drug discovery. Prog Med Chem. 2020;59:181–249. doi: 10.1016/bs.pmch.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 32.Ottl J., Leder L., Schaefer J.V., et al. Encoded library technologies as integrated lead finding platforms for drug discovery. Molecules. 2019;24:1629. doi: 10.3390/molecules24081629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao G., Huang Y., Zhou Y., et al. Future challenges with DNA-encoded chemical libraries in the drug discovery domain. Expert Opin Drug Discov. 2019;14:735–753. doi: 10.1080/17460441.2019.1614559. [DOI] [PubMed] [Google Scholar]
- 34.Cochrane W.G., Malone M.L., Dang V.Q., et al. Activity-based DNA-encoded library screening. ACS Comb Sci. 2019;21:425–435. doi: 10.1021/acscombsci.9b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kodadek T., Paciaroni N.G., Balzarini M., et al. Beyond protein binding: recent advances in screening DNA-encoded libraries. Chem Commun (Camb) 2019;55:13330–13341. doi: 10.1039/c9cc06256d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan A.I., McGregor L.M., Jain T., et al. Discovery of a covalent kinase inhibitor from a DNA-encoded small-molecule library x protein library selection. J Am Chem Soc. 2017;139:10192–10195. doi: 10.1021/jacs.7b04880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmermann G., Rieder U., Bajic D., et al. A specific and covalent JNK-1 ligand selected from an encoded self-assembling chemical library. Chemistry. 2017;23:8152–8155. doi: 10.1002/chem.201701644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zambaldo C., Daguer J.P., Saarbach J., et al. Screening for covalent inhibitors using DNA-display of small molecule libraries functionalized with cysteine reactive moieties. Med. Chem. Commun. 2016;7:1340–1351. [Google Scholar]
- 39.Zhu Z., Grady L.C., Ding Y., et al. Development of a selection method for discovering irreversible (Covalent) binders from a DNA-encoded library. SLAS Discov. 2019;24:169–174. doi: 10.1177/2472555218808454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.John P.G., Archna A., Martin A., et al. Novel irreversible covalent BTK inhibitors discovered using DNA-encoded chemistry. Bioorg. Med. Chem. 2021;42 doi: 10.1016/j.bmc.2021.116223. [DOI] [PubMed] [Google Scholar]
- 41.Kuai L., O'Keeffe T., Arico-Muendel C. Randomness in DNA encoded library selection data can be modeled for more reliable enrichment calculation. SLAS Discov. 2018;23:405–416. doi: 10.1177/2472555218757718. [DOI] [PubMed] [Google Scholar]
- 42.Su W., Ge R., Ding D., et al. Triaging of DNA-encoded library selection results by high-throughput resynthesis of DNA-conjugate and affinity selection mass spectrometry. Bioconjug Chem. 2021;32:1001–1007. doi: 10.1021/acs.bioconjchem.1c00170. [DOI] [PubMed] [Google Scholar]
- 43.Roe M.K., Junod N.A., Young A.R., et al. Targeting novel structural and functional features of coronavirus protease nsp5 (3CL pro, M pro) in the age of COVID-19. J Gen Virol. 2020;102 doi: 10.1099/jgv.0.001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffman R.L., Kania R.S., Brothers M.A., et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J Med Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molecular Operating Environment (MOE) 1010 Sherbooke St. West, Suite #910; Montreal, QC, Canada: 2021. Chemical Computing Group ULC. 2019.01. H3A 2R7. [Google Scholar]
- 46.doi: 10.1126/science.abb3405. [DOI]
- 47.10.3389/fmolb.2020.599079. [DOI]
- 48.Hansen R., Peters U., Babbar A., et al. The reactivity-driven biochemical mechanism of covalent KRAS(G12C) inhibitors. Nat Struct Mol Biol. 2018;25:454–462. doi: 10.1038/s41594-018-0061-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.