

Abstract
Granular platelet-sized polyphosphate nanoparticles (polyP NPs) were encapsulated in sterically stabilized liposomes, forming a potential, targeted procoagulant nanotherapy resembling human platelet dense granules in both structure and functionality. Dynamic light scattering (DLS) measurements reveal that the Artificial Dense Granules (ADGs) are colloidally stable and that the granular polyP NPs are encapsulated at high efficiencies. High-resolution scanning transmission electron microscopy (HR-STEM) indicates that the ADGs are monodisperse particles with a 150 nm diameter dense core consisting of P, Ca and O surrounded by a corrugated 25 nm thick shell containing P, C and O. Further, the ADGs manifest promising procoagulant activity: Detergent solubilization by Tween 20 or digestion of the lipid envelope by phospholipase C (PLC) allows for ADGs to trigger autoactivation of Factor XII (FXII), the first proteolytic step in the activation of the contact pathway of clotting. Moreover, ADGs’ ability to reduce the clotting time of human plasma in the presence of PLC further demonstrate the feasibility to develop ADGs into a potential procoagulant nanomedicine.
Keywords: Polyphosphate, polyP; Artificial dense granule, ADG; High-resolution scanning transmission electron microscopy, HR-STEM; Electron energy loss spectroscopy, EELS; Factor XII, FXII; Phospholipase C, PLC
Graphical Abstract
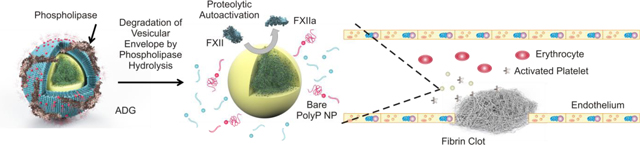
Introduction
Hemorrhagic events arising from trauma in either the civilian or military setting contribute to a significant proportion of avoidable fatality.1–3 In countless combat scenarios proper management of hemorrhage is not immediately feasible without invasive surgical intervention. Topical hemostatic wound dressings and other pharmacological agents, which can be delivered on-demand on the battlefield, are therapeutically inferior in mitigating internal, incompressible bleeding sources.4
Clinical guidelines created to diminish poor outcomes in patients experiencing an uncontrolled post-traumatic bleeding event have focused on restoring normal circulatory perfusion to the wounds after swiftly determining and suppressing the source of bleeding by a trained clinician rather than via administration of a hemostat as a first-line therapy.5, 6 However, these guidelines have largely failed to eradicate trauma-related complications even in developed countries with relatively more medical resources.7 Both private and public entities have invested heavily into developing a targeted, on-demand, broad-spectrum procoagulant agent.4 The Food and Drug Administration (FDA) has approved a myriad of pharmacological therapies in recent years to address external compressible trauma, enhancing survival and reestablishing hemostasis without surgery.8 Hemcon®, for example, is a commercially available bandage composed of a biodegradable polysaccharide, chitosan, which rapidly marginates negatively charged erythrocytes at the trauma site due to the macromolecule’s high positive charge density.9 Nonetheless, Hemcon® is not suitable for the treatment of incompressible wounds. Factor VIIa has also been used to treat internal bleeding.10 However, the efficacy of factor VIIa therapy for treating traumatic injuries is controversial.11 The recombinant clotting factor is expensive and requires special storage and handling conditions.
Nanoscale drug delivery has emerged as a novel therapeutic platform in recent decades with the potential to drastically transform medical treatment,12 promising reduced side effects, enhanced efficacy, and targeted delivery to only effected organs and tissues.13 The conventional strategy to design this next generation of smart drugs entails the encapsulation of therapeutic and/or imaging agents into supramolecularly assembled depots14 such as phospholipid vesicles,15 polymersomes,16, 17 viral capsids,18 polymeric micelles,19 and other self-assembled nanostructures.20 Among these candidate drug architectures, vesicles have garnered the most tangible success, with several liposomal nanoformulations being approved by the FDA to treat a myriad of disorders, especially cancer.21 22 The liposomal envelope of these smart drugs usually contains a small molar percentage of poly(ethylene glycol)-phosphatidylethanolamine (PEG-PE), which is used for steric stabilization and long blood circulation time.23 24
Given both the limitations of the available treatments to improve hemostasis and the promise of nanotherapeutics, numerous candidate nanoparticle hemostats have been developed in recent years. These nanotherapeutics attempt to imitate at least some aspects of platelet morphology and the procoagulant response adjacent to the bleeding site. Anselmo and Mitragotri approach mimicry with their platelet-like nanoparticles (PLNs), synthesized by alternative deposition of the polyelectrolytes bovine serum albumin (BSA) and poly(allylamine) hydrochloride on a polystyrene (PS) core.25, 26 Further functionalization of the PLN with targeting peptide ligands allow PLNs to accumulate at the bleeding site, bind and interact synergistically with the body’s own activated platelets, and rapidly induce coagulation at the site of trauma.26 However, the synthesis of the PLNs involves multiple steps and toxic organic solvents to dissolve the nonbiodegradable polymer.
Inorganic polyphosphate (polyP), on the contrary, is virtually omnipresent in biology and degradable in human blood plasma on therapeutic time scales.27 The polymer is a highly negatively charged macromolecule of orthophosphates, and is often stored intracellularly in a precipitated form as ~250 nm granules28–30 in conjunction with calcium and other divalent and multivalent cations. These organelles, generally termed “acidocalcisomes,” are present in all species including humans.31 These subcellular phosphate-containing bodies are called dense granules in human platelets28 due to their uniformly high electron density. Their contents are secreted upon platelet activation, where they exert potent procoagulant and proinflammatory effects,32 especially on Factor V33 and thrombin-promoted Factor XI activation.34 We have previously shown that polyP self-assembly into granular NPs in the presence of physiological amounts of metal cations is thermodynamically controlled, and that these particles retain their robust procoagulant effects.29
Herein we envision a biomimetic procoagulant nanoparticle composed of a granular polyP NP core encapsulated in a sterically stabilized liposome, which we call an Artificial Dense Granule (ADG). The ADG assembly process is scalable and highly reproducible, and there are no toxic organic solvents or nonbiodegradable materials involved. The size distribution of the ADGs was characterized by dynamic light scattering (DLS) measurements. Electron microscopy techniques demonstrate that the ADGs are virtually indistinguishable from dense granules isolated from human platelets in terms of morphology and structure,28 consisting of a 150 nm uniformly dense core surrounded by a 200 nm shell. Electron energy loss spectroscopy (EELS) confirms the co-localization of P, Ca, and O in the core and P, C and O in the phospholipid lamella. The ADGs are procoagulant as demonstrated by in vitro clotting factor assays: The ADGs can be triggered to initiate FXII autoactivation, the initial step in the contact pathway of blood clotting, via enzymatic hydrolysis by PLC, which modulates platelet degranulation. Furthermore, ADGs manifest contact activity in human plasma, and are roughly equivalent to molecularly dissolved, long-chain polyP at physiologically relevant concentrations.
Experimental Section
Materials and reagents
CaCl2·6H2O, phospholipase C (PLC) from Clostridium perfringens (C. welchii), and secretory phospholipase A2 from honey bee venom (bvsPLA2) were purchased from Sigma-Aldrich (St. Louis, MO). Water was deionized using a Nanopure II filtration system (Barnstead, Dubuque, IA) to 18.2 MΩ-cm. 1,2-dipalmitoly-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (PEG2kDPPE), L-α-phosphatidylcholine (L-α-PC), and Avanti ® Mini Extruder Kit, together with a 200-nm pore-diameter polycarbonate membrane, were purchased from Avanti Polar Lipids (Alabaster, AL). Citrated human pooled normal plasma (PNP) from thirty healthy donors was purchased from George King Biomedical (Overland Park, KS).The polyP preparation used throughout for synthesis of ADGs was Natriumpolyphosphat P70 (BKGP70, approximate range 20–125 phosphate units, mode ~45) from BK Guilini GmbH (Ludwigshafen am Rhein, Germany). PolyP>600 (‘long chain polyP’) used in clotting assays, and polyP>1000, used to synthesize “Artificial Acidocalcisomes,” were size-fractioned as previously described by differential isopropanol precipitation.33 All materials were purchased at standard grades and used as received.
PolyP Nanoprecipitation
Molecularly dissolved polyP was precipitated in aqueous 5 mM CaCl2, pH 5.4 as previously described.29 Briefly, the polyP was micropipetted into the calcium solution and vortexed for 5 s. PolyP precipitated at supersaturation ratios exceeding 100 was diluted with more 5 mM CaCl2, pH 5.4 prior to dynamic light scattering (DLS) characterization (Brookhaven NanoDLS, Brookhaven, NY). Dilution should not alter NP effective diameter as it is hysteretic as demonstrated previously.29
Synthesis of Sterically Stabilized Liposomes
200-nm sterically-stabilized liposomes composed of 95 mol% L-α-PC/ 5 mol% PEG2kDPPE were synthesized by standard protocol via extrusion35 through a 200-nm pore-diameter polycarbonate membrane. Briefly, 14.3 μl of 25 mg/mL PEG2kDPPE and 76.1 μl of 25 mg/mL L-α-PC (both dissolved in chloroform) were micropipetted into a 7 mL glass scintillation vial and dried under a gentle stream of Argon gas. The dried lipid film was then placed under vacuum for an additional 60 min to remove any residual traces of organic solvent. The desiccated lipid cake was subsequently rehydrated with 1 mL of filtered, deionized H2O (DI H2O) and stored for up to two weeks at 4°C. Liposome effective diameter and polydispersity index (PDI) were characterized by DLS after rehydration.
Synthesis of ADGs
5 mM polyP NPs were synthesized as described above. Following precipitation the bare NP effective diameter, polydispersity, and scattering intensity were characterized by DLS before addition of 106.6 μl of 2.6 mM 200-nm L-α-PC/PEG2kDPPE liposomes in DI H2O. The dispersion was bath sonicated for 10 min to encapsulate the granular polyP NPs at high efficiency.
FXII Autoactivation Assay with Detergent
FXII autoactivation was measured in 96-well microplates as described previously36 with some small modifications. In short, ADGs were synthesized at 5 mM [monoP] in 5 mM CaCl2, pH 5.4 as delineated in Synthesis of ADGs above. The ADGs were diluted with more 5 mM CaCl2, pH 5.4 after solubilization with 0.5 % (v:v) Tween 20 or an equivalent volumetric amount of pure DI H2O (as a control) to a final monoP concentration of 500 μM. Final concentrations of FXII zymogen and S-2302 substrate were 50 nM and 0.5 mM, respectively. FXII autoactivation was measured at room temperature.
FXII Autoactivation Assay with PLC from C. welchii
In lieu of solubilization with Tween 20, the 5 mM ADGs were prepared, as above, and diluted into 5 mM CaCl2, 10 μM ZnCl2, 0.1% bovine serum albumin (BSA), 5 mM Tris·HCl, pH 5.5, and preincubated with 30 μg/ml PLC for 20 minutes at 37°C before addition of FXII zymogen and S-2032 chromogenic substrate. BSA was added as a carrier protein for maximal PLC activity. Final concentrations of ADG, FXII zymogen and S-2302 substrate were 500 μM, 50 nM and 0.5 mM, respectively. FXII autoactivation was measured at room temperature.
Determination of PolyP NP Encapsulation Efficiency
A facile assay was devised employing DLS to measure the stability of ADGs after modulation of [Ca2+] with and without the presence of surface-active agents which would disrupt phospholipid vesicular integrity. Bare granular polyP NPs and ADGs at equivalent monoP concentration were monitored for stability by DLS at 5 mM CaCl2 for 30 minutes. Immediately prior to the 30-minute measurement, the calcium concentration was adjusted to 7.5 mM with gentle mixing (to prevent local concentration inhomogeneities) with or without the addition of 0.5% (v:v) Tween 20.
Monitoring Digestion of ADGs by bvsPLA2 and PLC via DLS
The scattering intensity of 5 mM ADGs in 5 mM CaCl2, pH 5.4 at room temperature (RT) was observed in 10-min increments for 30 min to ensure colloidal stability. The ADGs were then diluted 10X in enzyme buffer pre-warmed to 37°C containing either 25 mM Tris·HCl, pH 7.4, 100 mM NaCl, 7.5 mM CaCl2 and 10 μg/ml bvsPLA2 or 50 mM Tris·HCl, pH 5.5, 10 μM ZnCl2, 7.5 mM CaCl2, 0.1% (w:v) BSA, and 1 μg/ml PLC. The scattering intensity continued to be monitored in 10-min intervals at 37°C as phospholipid hydrolysis occurred.
ADG Contact Activity in Human Plasma
Before addition to citrated PNP, 30 μM ADGs were digested with 100 ng/ml PLC from C. perfringens for 20 min in a buffer containing 5 mM CaCl2, 10 μM ZnCl2, 0.1% (w:v) BSA, and 50 mM Tris·HCl, pH 7.4 for 20 min at 37°C. After phospholipase digestion, 50 μl citrated PNP was recalcified with 50 μl of 25 mM CaCl2, 5 mM imidazole, pH 7.4 and 50 μl sample was immediately added (after recalcification). Final concentrations of molecularly dissolved, long-chain polyP (polyP>600) and ADGs were 10 μM. Turbidity was measured at 405 nm every minute for 60 min at room temperature using a Finstriments™ Microplate Reader (MTX Lab Systems, Vienna, Virginia). The resulting sigmoidal absorbance traces were fitted to a Boltzmann sigmoidal function in OriginPro 8.6 (OriginLab Corporation, Northhampton, MA). The time to clot was defined as the inflection point x0±S.E. The percent reduction in the time to clot was normalized against the control (with no activator present).
Conventional Transmission Electron Microscopy
Sample Preparation
Briefly, 10 μl of the undiluted sample was pipetted onto a 300-mesh Holey, copper-on-carbon Formvar grid (Structure Probe Inc., West Chester, PA). After two minutes the liquid that did not evaporate was wicked away by placing the tip of a Kim wipe in close proximity to the liquid droplet. The grid was subsequently allowed to dry at ambient conditions for at least five minutes before examination in a JEM-1220 transmission electron microscope (JEOL Ltd., Tokyo, Japan).
High-Resolution Scanning Transmission Electron Microscopy (HR-STEM)
Methods
The biocompatible graphene sandwich containing the ADG was prepared by: (1) etching the copper layer away from a copper/graphene film to expose the graphene monolayer before transfer to the surface of DI H2O; (2) forming a thin liquid film containing the ADG sample on a standard TEM grid coated with monolayer graphene; and (3) exploiting the evanescent wave generated by total internal reflection to place the grid on top of the graphene monolayer to form the graphene sandwich.37 The EELS spectrum images of the ADGs and the bare particles are background subtracted. In Figure 2B, the P L-edge in the shell EELS spectrum (red) is taken from the shell of the EELS map of the ADG. The P L-edge in the core EELS spectrum (blue) is generated by first extracting the spectrum from the core region of the EELS map, which contains both the core and the shell signal, and then by subtracting the shell spectrum. In Figure 2B, the O, P, and Ca line profiles are normalized to the maximum intensity, while the C line profile is normalized with P at the maximum intensity position.
Figure 2.

A: HAADF images of the ADG (1) and the bare polyP NP (2). B: Elemental and structural maps of the ADG. B1-B4: Individual Ca, P, O and composite (C, Ca, P, O) map extracted from EEL spectrum images with the graphene background subtracted. The color-coded outlines of the Ca, P and O distribution shown in B1-B3 are superimposed in composite map B4. B5: Phosphorous L-edges extracted from the shell and core of the ADG shown in A. B6: Elemental line scan of the C, O, P and Ca map extracted from the graphene background-subtracted spectrum image of the ADG (Figure B1–4) from top left to lower right. C: Elemental maps of the bare particle. C1-C4: Individual Ca, P, and O maps as well as composite (C, Ca, P, O) map extracted from the background subtracted spectrum image.
Sample Preparation
10 mM bare granular polyP NPs and 5 mM ADGs were prepared in biocompatible graphene sandwiches using previously established methodology37 and transferred to JEOL JEM200CF STEM/TEM (JEOL Ltd., Tokyo, Japan) operated at 80 keV for imaging and EELS acquisition. HAADF and ABF images were taken simultaneously.
Results and Discussion
A facile nanoprecipitation process was devised to encapsulate granular platelet-sized polyP into sterically stabilized liposomes rapidly on the benchtop, creating a nanoscale drug delivery platform modeled after the human body’s intrinsic arsenal for controlling hemorrhage. The ADGs consist of a 150-nm granular polyP core surrounded by a 200-nm PEGylated liposome (Figure 1A), mimicking the size and structure of human platelet dense granules.28
Figure 1.

A: Schematic of ADG design and structure. ADGs are 150-nm granular polyP NPs encapsulated in 200-nm sterically stabilized, PEGylated liposomes. B: Illustration of a biocompatible graphene sandwich trapping an ADG for high resolution imaging. To prevent beam-induced morphology change and mass loss in the electron microscopy analysis, the ADG is directely sandwiched between two layers of free-standing graphene.
The nanostructure of the ADGs was subsequently examined by imaging and spectroscopic methods. Conventional transmission electron microscopy (TEM) and high-resolution scanning transmission electron microscopy (HR-STEM) were employed to show that the ADGs possess narrow size distribution and a defined core-shell architecture, with a homogeneously dense polyP granule serving as the core and a ring of lighter electron-density consisting of the phospholipid lamella (Figure 2A, cf. Supporting Information Figure S1). Further, EELS was exploited to map the ADG’s elemental components to prove unambiguously that the synthetic scheme yielded encapsulated polyP particles as intended.
An inherent disadvantage of conventional TEM for structural characterization of biological or biomimetic nanomaterials is radiation damage from the electron beam,38 limiting imaging resolution to length scales of nanometers and spectroscopy to angstroms.39, 40 Standard electron dose and dose rates directed at polyP bodies transform the granular structures into round sponges most likely due to electron beam-induced hydrogen gas bubbling and mass loss, and this therefore necessitates an alternative imaging and structural characterization tool for finer sample elucidation. To overcome these limitations, ADGs suspended in aqueous 5 mM CaCl2 were directly sandwiched between two graphene layers, before transferal to the transmission electron microscope (JEOL JEM-ARM200CF) for high-resolution STEM and EELS analysis37 (Figure 1B). The presence of the graphene monolayers has been shown to substantially abrogate the deleterious electronic effects on the sample, minimizing covalent bond cleavage during electron microscopy visualization of biological materials and biochemical reactions of experimental interest.41–45 As a comparison, bare granular polyP NPs prepared in graphene sandwiches are spherical particles of uniform electron density, with the characteristic white spots, observed by Docampo and Ruiz28 using conventional TEM, being absent (Figure 2A). The polyP granules are approximately 150–200 nm in diameter, in very good agreement with DLS measurements (cf. Supporting Information Figure S3). In contrast to the bare particles, the ADGs exhibit a clear core-shell nanoarchitecture characteristic of a polyP NP encapsulated in a liposome. A high angle annular dark-field (HAADF) image of an ADG reveals a dense, 150-nm core surrounded by an irregularly-shaped 25-nm thick shell of decreased electron density (Figure 2A). These length scales are in good agreement with light scattering data, as the granular polyP NP was measured to have an initial hydrodynamic diameter of ~160 nm and the liposomes were formed via extrusion through a polycarbonate membrane with 200-nm pores.
To the best of our knowledge, this is the first time that EELS has been used to probe the elemental consituents of polyP NPs and their liposomally encapsulated counterparts, ADGs. Figure 2B shows the elemental distribution of Ca, P and O within the ADG particle. The compositional map reveals that the shell consists of C, O, and P, but not Ca, suggesting the presence of a phospholipid envelope. The carbon signal in the shell stems most likely from the PEG brushes and the hydrocarbon tails, glycerol backbone, and headgroup carbons of the phospholipid lamella (cf. Supporting Information Figure S2). The dense core exhibits a homogeneous distribution of Ca, P and O, demonstrating that the granular polyP NPs were successfully encapsulated by the phospholipid envelope. As a control group, Figure 2C shows the elemental distribution of Ca, P, and O of the bare particle. These elemental maps match well with the acquired HAADF image, and show a uniform distribution of Ca, P and O in the bare particle, consistent with the elemental composition of granular polyP NPs. The pixel-size in Figure 2B is larger than in Figure 2C to minimize the effects of electron-dose induced changes to the shell. In order to rule out the possibility that in the ADG, the O and P signals in the core region shown in Figure 2B are only coming from the phospholipid envelope, which also contains O and P, we perform elemental line scans of the C, O, P and Ca to show the distribution of these elements, shown in Figure 2B. Elemental line scans of C and Ca indicates the presence of a Ca core encapsulated by a ~25 nm thick C shell. The shape of the C and Ca signal is consistent with the typical core-shell structure. Unlike the “saddle” shape of C, which indicates the absence of C in the core, the Gaussian shape of the O and P signals indicates that these two elements are present in both the core and the shell. Furthermore, analysis of the P L-edges is used to compare the local P bonding between the core and the shell. Both spectra show a near-edge fine-structure typical for PxOy,46 while the P spectrum from the shell shows an additional pre-peak, indicating a loss in P-O bonding. These EELS data are consistent with our expectation of the P bonding structure in the granular polyP core as well as the liposome shell.
The stability of the ADGs was subsequently investigated for 1 h at room temperature in the same solution in which they were prepared (5 mM CaCl2, pH 5.4) utilizing DLS, measured in 10-min intervals (Figure 3B). The initial effective diameter of the bare polyP granules is approximately 160 nm with a PDI ~0.1 (t=−10 min in Figure 3B), in agreement with our previous report.29 After addition of a stoichiometric excess of sterically stabilized liposomes and 10 min of bath sonication, encapsulated polyP granules are monodisperse with a PDI~0.2, ((Supporting Figure S3), with an effective diameter only marginally bigger than their unencapsulated counterparts (t=0 min in Figure 3B). Further DLS measurements reveal that the diameter remains unchanged ca. 165 nm after 60 min has elapsed, whereas bare polyP NPs (black symbols and line in Figure 3B) in the same solution environment manifest power-law type growth kinetics. At the time of nanoprecipitation the bare polyP granules are approximately 160 nm, ripening to ~220 nm in 1 h. The equivalent power-law growth phenomenon is clearly absent with the ADGs (red symbols and line in Figure 3B). The particle diameter of the ADGs as determined by electron microscopy is comparable qualitatively to the hydrodynamic diameter measured by dynamic light scattering, which strongly suggests that the encapsulated particles observed in HR-STEM are stable in suspension for at least an hour, a time scale sufficient to potentially mitigate bleeding after a severe hemorrhagic event.
Figure 3.

A: Scattering intensity of ADGs after phospholipase digestion. ADGs were prepared in 5 mM CaCl2 at room temperature (RT) and monitored for stability for 30 min before transferal to an enzyme buffer at 37°C containing either 10 μg/mL sPLA2 from bee venom (bvsPLA2) or 1 μg/mL PLC from C. perfringens. The scattering count rate drops significantly for both 10 μg/mL bvsPLA2 and 1 μg/mL PLC ADG digestions, implying that the phospholipases are inducing hydrolysis of the lipid envelope, leading to degradation and agglomeration of ADGs and precipitation of the polyP NP cargo. B: Stability of ADGs in suspension. The average effective diameter was determined for the bare polyP NP (immediately before adding liposome, t=−10 min), immediately after sonication (t=0 min), and every ten minutes thereafter until 1 h had elapsed. The ADGs do not appreciably change in size over the duration of the experiment. The bare polyP NPs, on the other hand, prepared at the same supersaturation ratio, grow in a power-law manner to a mean effective diameter of approximately 220 nm in 1 h. C: Verification of the ADG encapsulation efficiency by measuring ADG diameter shifts after exposure to detergent and changes in calcium concentration. Liposome solubilization by the non-ionic detergent Tween 20, in conjunction with an increase in the calcium concentration, was exploited to judge the ADG encapsulation efficiency semi-quantitatively. In the absence of Tween 20 and an increase in calcium concentration to 7.5 mM at t=30 min, there is no statistically significant perturbation in the ADG effective diameter (solid red dots). However, increasing the calcium concentration after dissolution of the lipid envelope by detergent exposure allows for the polyP NPs to be exposed to the higher amount of calcium, resulting in an increased effective diameter (hollow red dots).
ADG encapsulation efficiency was investigated by modulating the calcium concentration, since the presence of calcium controls the thermodynamic equilibrium of granular polyP NP self-assembly.29 Changes in the calcium concentration would have nearly immediate effects on the polyP NP effective diameter, as the polyP NP size is solely determined by the calcium concentration over an extensive range of polyP supersaturation values.29 5 mM ADGs were synthesized in 5 mM CaCl2, pH 5.4, and monitored by DLS every 10 min. Once 30 min had elapsed the calcium concentration was increased to 7.5 mM and an excess of a non-ionic detergent, Tween 20, (which would not affect polyP nanoprecipitation in a measurable way, cf. Supporting Figure S4) was added to solubilize the vesicular envelope and expose the polyP cargo to the higher calcium environment (Figure 3C). Upon addition of 0.5% (v:v) Tween 20 and increasing the calcium concentration to 7.5 mM, the average particle size rapidly increases above 200 nm (red line with open circles in Figure 3C). 10 min after detergent solubilization, the particle population is no longer unimodal (data not shown), with a Gaussian-averaged effective diameter greater than 300 nm. Visible sedimentation of agglomerated granular polyP occurs 20 min after adding detergent, with the effective diameter bigger than the micron length scale and the scattering count rate tending to 0. In the absence of detergent, an increase in calcium concentration, in contrast, causes only a negligible change in size, with the average effective diameter centered around 165–180 nm (red line with solid circles in Figure 3C), comparable to the instance with no modulation in the calcium concentration (red line with solid circles in Figure 3B). Bare polyP NPs subjected to the same experimental scheme show typical power-law growth before calcium concentration increase and subsequently rapidly precipitate into micron-sized particles after 10 min independent of the presence of detergent. These results suggest that the particle encapsulation efficiency is close to the theoretically maximum value.
Not only do the ADGs structurally resemble human platelet dense granules, they also manifest potentially promising procoagulant effects in vitro like their biological counterparts when triggering agents are used to disassemble the phospholipid envelope and expose the procoagulant polyP cargo. Inspired by the platelet degranulation process that occurs in vivo, we present a paradigm through which hydrolytic enzymes present in human platelets or secreted into the circulation after hemorrhage can be exploited to induce clotting factor activity on a time scale sufficient to cease bleeding.
The targeted delivery of polyP relies on the presence of phospholipases (e.g. PLC and sPLA2), which occur at much higher concentrations at local bleeding sites (at the scale of μg/ml) compared to global baseline physiological levels (at the scale of ng/ml) to quickly degrade phospholipid vesicles. Immediately after traumatic hemmorhage, a concatenation of carefully orchestrated biochemical events must occur near the wound site, with platelets serving as the principal actors in establishing primary hemostasis.47 Platelets adjacent to the trauma site stick to the damaged endothelium creating a temporary “plug,” recruit other platelets via secreted small molecule messengers, and initiate the coagulation cascade by supporting activation of circulating clotting factors.48–50 PLC plays a major role in platelet activation, and is central to the platelet degranulation process51, 52— the exocytosis of dense and α granules into the vasculature, ensuring that high local concentrations of polyP are established in platelet-rich thrombi. PLC is an intracellular lipase which hydrolyzes phospholipids at the 3 position,51 leading to mobilization of intracellular calcium stores and activation of G protein-coupled secondary messenger systems with diverse physiological consequences extending beyond hemostasis and inflammation.53 Although PLC in human platelets exclusively hydrolyzes phosphoinositides,54 numerous other PLC isoforms exist in nature that do not discriminate between phospholipids. Many bacterial toxins referred to as “lecithinases” possess robust PLC activity for phosphatidylcholines and phosphatidylethanolamines.55, 56
An additional strategy employs secreted phospholipases at sites of inflammation, e.g. around tumors and sites of vascular injury, to promote hydrolysis of the vesicular envelope. It has been clinically documented that in human patients with a constellation of inflammatory diseases, such as rheumatism and atherosclerosis, that the serum concentration of secretory phospholipase A2 (sPLA2) can reach 10–30 ng/mL.57 Local plasma concentrations of sPLA2 in the boundary layer directly adjacent to sites of trauma can be 100- to 1000-fold higher, approaching 10 μg/mL. Moreover, several studies have shown that degradation of phospholipid vesicles by phospholipase hydrolysis could be an order faster when conjugated to PEG chains.58 The likely mechanism is that PEG chains cause extrusion of the phospholipid to the exterior of the bilayer, promoting more rapid and facile phospholipase adsorption at the interface.59 Therefore, PEGylated phospholid vesicles, as with our design of encapsulating polyP NPs, not only offer steric stabilization and immunoprotection, but also a means for controlling and targeting drug release upon phospholipid degredation at the sites where phospholipases are overexpressed.
The phenomenon of ADG liposomal envelope digestion by model phospholipases was explored by DLS to estimate time scales of hydrolysis and protein concentrations necessary for polyP cargo release to suitably modulate clotting factor activity. The colloidal stability of ADGs was surveyed in 5 mM CaCl2, pH 5.4 at RT for 30 min before dilution into an enzymatic assay buffer at 37°C. Upon transfer to the enzymatic buffers tailored for optimal PLC and sPLA2 activity, respectively, the scattering intensity marginally drops, likely due to changes in liposome phase behavior and fluidity due to the low melting temperature of L-α-phosphatidylcholine, the major constituent in the sterically stabilized liposomes (Figure 3A). Moreover, the calcium concentration for both buffers is 7.5 mM in order to gauge polyP release after hydrolysis. In the absence of enzyme, the size of the ADGs does not change significantly, suggesting that the majority of the granular polyP NPs are completely encalpsulated. After addition of 1 μg/mL PLC or 10 μg/mL sPLA2 at 30 minutes, however, the scattering intensities drop siginificantly. This suggests that the phospholipases have begun to hydrolyze the liposomal barrier surrounding the polyP NP, leading to polyP NP precipitation in the more calcium-enriched environment, decreasing the total number of colloidal particles due to sedimentation. Further investigation reveals that 30 μg/ml PLC is an ideal concentration for in vitro autoactivation of FXII (Please see Supporting Figure S6).
The ability of ADGs to autoactivate isolated FXII zymogen after detergent-induced solubilzation was first assessed as a proof-of-principle demonstrating that the polyP NP cargo retains its procoagulant functionality. Corroborating Engel et al.’s results that platelet-sized polyP robustly supports zymogen conversion,36 we show under similar experimental conditions that 500 μM ADGs are able to promote autoactivation of FXII after detergent solubilization by 0.5% (v:v) Tween 20, whereas ADGs not treated with detergent are largely inert, exhibiting minimal FXII autoactivity (Figure 4B). Empty sterically stabilized liposomes are also unable to initiate conversion of FXII to FXIIa, suggesting that promotion of autoactivation stems from release of the granular polyP NPs. FXII autoactivity of the bare granular polyP NPs is concentration-dependent, and manifests saturating kinetic behavior with maximal autoactivation occurring at 500 μM monoP for BKGP70, the same polyP used in this study (see Supporting Figure S5 for concentration dependence). Figure 4C shows that ADGs preincubated in a 0.1% (w:v) BSA suspension with PLC for optimal phospholipase activity for 20 min before addition of zymogen initiate conversion of FXII in a manner consistent with ADGs subjected to detergent treatment with Tween 20. ADGs preincubated in the same buffer without PLC are largely “contact inactive”. Likewise, empty sterically stabilized liposomes are unable to support autoactivation of FXII zymogen. The absorbance intensity of empty sterically stabilized liposomes preincubated with the equivalent concentration of PLC does increase slightly during the duration of the experiment; however, this rise is likely due to vesicle aggregation or zymogen activation stemming from interaction with negatively charged degradation products.
Figure 4.

A: Illustration of phospholipase-induced degradation of the lipid envelope. Secretory phospholipases such as human sPLA2 encountered at locally high concentrations near sites of trauma and inflammation, as well as intracellular isoforms like PLC, involved in platelet degranulation, are envisioned to degrade the liposome vesicle, exposing the granular polyP NP to induce FXII autoactivation. B: Autoactivation of FXII zymogen via detergent-triggered solubilization of ADG vesicular envelope. ADGs initiate autoactivation of FXII after addition of 0.5% (v:v) Tween 20, similar to the bare polyP NPs (as a positive control). In contrast, ADGs without detergent treatment yield no significant conversion of FXII to FXIIa. As a negative control empty liposomes at the same concentration elicit no FXII autoactivation. C: FXII autoactivation after ADG preincubation with PLC. ADGs were incubated with 30 μg/ml PLC (from C. perfringens) at 37°C for 20 min before initiation of FXII autoactivation in a buffer containing BSA to ensure optimal phospholipase digestion. Bare polyP NPs without BSA were included as a positive control. ADGs preincubated with PLC manifest a similar rate of absorbance increase to both bare polyP NPs and ADGs digested with Tween 20. ADGs without PLC do not measureably promote FXII autoactivation at the concentration tested. Empty liposomes digested with PLC show some absorbance increase, which is likely a result of vesicle aggregation after phospholipid hydrolysis.
Interestingly, 500 μM bare polyP NPs in the presence of 0.1% (w:v) BSA evince no contact activity over the 120 min assay, whereas bare polyP NPs without BSA elicit robust FXII autoactivation within 1 h (Figure 4C). BSA is an established divalent metal chelator, with low dissociation constants for Ca2+, Zn2+ and Cu2+.60, 61 Moreover, zinc cations greatly accelerate FXII autoactivation, and hence signficantly reduce the autoactivation doubling time.62, 63 The presence of millimolar amounts of divalent metals like Ca2+ and Zn2+ ensures that the predominant polyP species is either an NP or polyP-metal complex (see Supporting Information Figure S7-8 for zinc’s nanoprecipitative effects). It is well established that foreign contact activators manifest as either negatively charged colloids or granules like kaolin64 or as polymer-metal complexes, e.g. ellagic acid coordinated with copper.65 The fact that BSA effectively lowers the free ionic concentration of these precipitative divalent metal cations and completely abrogates polyP NP contact activity in this simplified in vitro model of the contact pathway of blood clotting suggests that divalent metal ion complexation to the polymer may be required for polyP to elicit FXII zyomgen autoactivation. Additionally, recent work by Ursula Jakob and colleagues at the University of Michigan provides compelling evidence that polyP is a nonspecific protein binder, facilitating protein folding in a fashion comparable to conventional protein chaperones.66 Surface plasmon resonance measurements have identified numerous blood proteins that bind to polyP, including thrombin.67 We have also observed by DLS that bare polyP NPs may complex with BSA via a calcium-mediated process.29 However, enzymatically triggered hydrolysis of ADGs by PLC even in the presence of BSA still yields siginificant proteolytic autoactivation (Figure 4C). Although there is a high concentration of serum albumin in human plasma, polyP still manages to function as a very potent procoagulant agent, implying that the inhibitory effect of serum albumin cannot be too strong. Nonetheless, the precise mechanism by which BSA and polyP interact demands further investigation. Taken together, these data suggest that PLC hydrolysis of the lipid envelope is occurring, quickly exposing a negatively charged surface to autoactivate the FXII zymogen, and, further, that the sterically stabilized liposome shields the granular polyP NP from interacting with FXII and other proteins like BSA over a duration of at least two hours.
In addition to assessing the ability of ADGs to catalyze the conversion of FXII zyomgen to FXIIa in isolation, the particles’ contact activity was determined in pooled normal plasma (PNP) by measuring changes in turbidity upon coagulation. ADGs were preincubated for 20 min with 100 ng/mL PLC from C. perfringens before initiation of clotting. Figure 5 shows that 10 μM ADGs + 100 ng/ml PLC reduce the time to clot equivalently compared to 10 μM molecularly dissolved, long-chain polyP, with 21% and 19% diminutions compared to control, respectively. 10 μM ADGs and 100 ng/ml PLC alone both clot slightly more rapidly than control (with no activator present). The fact that ADGs minimally reduce the clotting time suggests that either a small fraction of the polyP remains unencapsulated, or that some polyP can be transported across the relatively fluid ADG bilayer, of which 95 mol% is the low Tm L-α-PC, when it is diluted into PNP. Given that PLC also reduces the time to coagulation compared to control implies that the phospholipase possesses marginal intrinsic contact activity at the concentration tested. It is also noteworthy that molecularly dissolved, long-chain polyP is not as robust a contact activator as previous reports in the literature demonstrate29, 33; however, the assay protocol was suitably modified to gauge the ADG procoagulant activity, accounting for the divergent behavior. Altogether, PLC is able to digest the ADG lipid envelope, allowing the polyP granule inside to initiate the contact pathway of blood coagulation.
Figure 5. Contact activity of ADGs in human plasma.

A: Absolute clotting time in human plasma. 10 μM ADGs + 100 ng/ml PLC are comparably procoagulant to 10 μM molecularly dissolved, long-chain polyP. 10 μM ADGs without trigger and 100 ng/ml PLC alone marginally reduce the time to clot compared to the control (with no activator present). Absolute clotting time is given by the fitted inflection point of a Boltzmann sigmoidal growth function ± S.E. of the averaged absorbance traces of each sample group (n=11). * represents p<0.05 for a two-sided t test compared to control (without activator). B: Clotting times given as percent reduction from the control.
Conclusion
We present the facile, rapid, and inexpensive synthesis of a polyP NP encapsulated in a liposome as a potential procoagulant nanotherapy imitating human platelet dense granules. All the materials are biocompatible and biodegradable, and the process could conceivably be scaled in an economical manner. High-resolution TEM and EEL spectrum imaging of ADGs confirm the nanostructure of a 200-nm sterically stabilized liposome shell encapsulating a 150-nm diameter granular polyP NP core. A label-free direct graphene sandwich approach was used in electron microscopy experiments to prevent beam-induced mass loss and morphology change. Light scattering measurements of ADGs in suspension indicate that the procoagulant nanoparticle is colloidally stable. Triggered initiation of FXII autoactivation by ADGs via detergent solubilization or phospholipase hydrolysis of the liposomal envelope in an in vitro model lends direction in the future for an in vivo investigation into ADGs’ efficacy in activating the contact system of clotting with the objective to limit blood loss following a severe hemorrhagic event.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are indebted to L.J. at the UIC RRC EMS for her assistance with imaging ADGs, as well as her experience and judgment with evaluating biological materials. The authors would like to acknowledge Dr. Q.Q. from BNL for her help in writing the EELS background subtraction DigitalMicrograph script.
Funding Sources
The characterization of the ADG clotting activity was sponsored by the US Army Medical Research and Materiel Command (WQ81XWH-11-2-0021). The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702-5014 is the awarding and administering acquisition office. This work made use of the JEOL JEM-ARM200CF in the Electron Microscopy Service (Research Resources Center, UIC). The acquisition of the UIC JEOL JEM-ARM200CF was supported by a MRI-R2 grant from the National Science Foundation [DMR-0959470]. The contents of this article do not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred.
Footnotes
ASSOCIATED CONTENT
Supporting Information Available
Calculation of PolyP Aggregation Number in a Representative Granular PolyP NP; Calculation of Stoichiometric Amount of Lipids for Theoretically Complete Encapsulation of the Granular PolyP NPs; Conventional TEM of ADGs; HR-STEM of ADGs and Bare PolyP NPs; Colloidal Stability & Particle Size Distribution of ADGs Prepared for HR-STEM Imaging; Nanoprecipitation of PolyP in Aqueous Media Containing Tween 20; FXII Autoactivity of Bare PolyP NPs; ADG-Induced FXII Autoactivity: Dependence on PLC Concentration; PolyP Nanoprecipitation in Aqueous Media Containing Ca2+/Zn2+; Colloidal stability of ADGs and Empty PEGylated Vesicles in BSA/Ca2+ Suspensions. This material is available free of charge via the Internet at http://pubs.acs.org.
Conflict of Interest
Y.L. and A.J.D. are co-inventors on a pending patent application related to this work. S.A.S. and J.H.M. hold several patents and are co-inventors on pending patent applications on the therapeutic usage of polyP.
References
- 1.Sauaia A; Moore FA; Moore EE; Moser KS; Brennan R; Read RA; Pons PT J. Trauma Acute Care Surg 1995, 38, 185–193. [DOI] [PubMed] [Google Scholar]
- 2.Holcomb JB Crit. Care 2004, 8, S57–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kauvar DS; Wade CE Crit. Care 2005, 9, S1–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alam HB; Burris D; DaCorta JA; Rhee P. Mil. Med 2005, 170, 63–69. [DOI] [PubMed] [Google Scholar]
- 5.Spahn DR; Cerny V; Coats TJ; Duranteau J; Fernández-Mondéjar E; Gordini G; Stahel PF; Hunt BJ; Komadina R; Neugebauer E. Crit. Care 2007, 11, R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossaint R; Bouillon B; Cerny V; Coats TJ; Duranteau J; Fernández-Mondéjar E; Hunt BJ; Komadina R; Nardi G; Neugebauer E. Crit. Care 2010, 14, R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kauvar DS; Lefering R; Wade CE J. Trauma Acute Care Surg 2006, 60, S3–S11. [DOI] [PubMed] [Google Scholar]
- 8.Cox ED; Schreiber MA; McManus J; Wade CE; Holcomb JB Transfusion 2009, 49 Suppl 5, 248S–255S. [DOI] [PubMed] [Google Scholar]
- 9.Brown MA; Daya MR; Worley JA J. Emerg. Med 2009, 37, 1–7. [DOI] [PubMed] [Google Scholar]
- 10.Beekley AC Crit. Care Med 2008, 36, S267–S274. [DOI] [PubMed] [Google Scholar]
- 11.Nishijima DK; Zehtabchi S. Ann. Emerg. Med 2009, 54, 737–744. [DOI] [PubMed] [Google Scholar]
- 12.Farokhzad OC; Langer R. ACS Nano 2009, 3, 16–20. [DOI] [PubMed] [Google Scholar]
- 13.Murday JS; Siegel RW; Stein J; Wright JF Nanomedicine 2009, 5, 251–273. [DOI] [PubMed] [Google Scholar]
- 14.Peer D; Karp JM; Hong S; Farokhzad OC; Margalit R; Langer R. Nat. Nanotechnol 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- 15.Torchilin VP Nat. Rev. Drug Discov 2005, 4, 145–160. [DOI] [PubMed] [Google Scholar]
- 16.Discher DE; Ahmed F. Annu. Rev. Biomed. Eng 2006, 8, 323–341. [DOI] [PubMed] [Google Scholar]
- 17.Meng F; Zhong Z; Feijen J. Biomacromolecules 2009, 10, 197–209. [DOI] [PubMed] [Google Scholar]
- 18.Kovacs EW; Hooker JM; Romanini DW; Holder PG; Berry KE; Francis MB Bioconjugate Chem. 2007, 18, 1140–1147. [DOI] [PubMed] [Google Scholar]
- 19.Huh KM; Lee SC; Cho YW; Lee JW; Jeong JH; Park KJ Control. Release 2005, 101, 59–68. [DOI] [PubMed] [Google Scholar]
- 20.Gref R; Domb A; Quellec P; Blunk T; Muller RH; Verbavatz JM; Langer R. Adv. Drug Deliv. Rev 2012, 64, 316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain RK; Stylianopoulos T. Nat. Rev. Clin. Oncol 2010, 7, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barenholz YJ Control. Release 2012, 160, 117–134. [DOI] [PubMed] [Google Scholar]
- 23.Papahadjopoulos D; Allen TM; Gabizon A; Mayhew E; Matthay K; Huang SK; Lee KD; Woodle MC; Lasic DD; Redemann C; Martin FJ Proc. Natl. Acad. Sci. U.S.A 1991, 88, 11460–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen TM Adv. Drug Deliv. Rev 1994, 13, 285–309. [Google Scholar]
- 25.Zhou Z; Anselmo AC; Mitragotri S. Adv. Mater 2013, 25, 2723–7. [DOI] [PubMed] [Google Scholar]
- 26.Anselmo AC; Modery-Pawlowski CL; Menegatti S; Kumar S; Vogus DR; Tian LL; Chen M; Squires TM; Sen Gupta A; Mitragotri S. ACS Nano 2014, 8, 11243–11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith SA; Mutch NJ; Baskar D; Rohloff P; Docampo R; Morrissey JH Proc. Natl. Acad. Sci. U.S.A 2006, 103, 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruiz FA; Lea CR; Oldfield E; Docampo RJ Biol. Chem 2004, 279, 44250–44257. [DOI] [PubMed] [Google Scholar]
- 29.Donovan AJ; Kalkowski J; Smith SA; Morrissey JH; Liu Y. Biomacromolecules 2014, 15, 3976–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Docampo R; Moreno SN Parasitol. Today 1999, 15, 443–448. [DOI] [PubMed] [Google Scholar]
- 31.Docampo R; de Souza W; Miranda K; Rohloff P; Moreno SN Nat. Rev. Microbiol 2005, 3, 251–261. [DOI] [PubMed] [Google Scholar]
- 32.Muller F; Mutch NJ; Schenk WA; Smith SA; Esterl L; Spronk HM; Schmidbauer S; Gahl WA; Morrissey JH; Renne T. Cell 2009, 139, 1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith SA; Choi SH; Davis-Harrison R; Huyck J; Boettcher J; Rienstra CM; Morrissey JH Blood 2010, 116, 4353–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi SH; Smith SA; Morrissey JH Blood 2011, 118, 6963–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mui B; Chow L; Hope MJ Methods Enzymol. 2003, 367, 3–14. [DOI] [PubMed] [Google Scholar]
- 36.Engel R; Brain CM; Paget J; Lionikiene AS; Mutch NJ J. Thromb. Haemost 2014, 12, 1513–1522. [DOI] [PubMed] [Google Scholar]
- 37.Wang CH; Qiao Q; Shokuhfar T; Klie RF Adv. Mater 2014, 26, 3410–3414. [DOI] [PubMed] [Google Scholar]
- 38.Egerton RF; Li P; Malac M. Micron 2004, 35, 399–409. [DOI] [PubMed] [Google Scholar]
- 39.Egerton RF, Electron Energy-Loss Spectroscopy in the Electron Microscope. Springer: 2011. [Google Scholar]
- 40.Wu JS; Kim AM; Bleher R; Myers BD; Marvin RG; Inada H; Nakamura K; Zhang XF; Roth E; Li SY; Woodruff TK; O’Halloran TV; Dravid VP Ultramicroscopy 2013, 128, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park J; Park H; Ercius P; Pegoraro AF; Xu C; Kim JW; Han SH; Weitz DA Nano Lett. 2015, 15, 4737–4744. [DOI] [PubMed] [Google Scholar]
- 42.Wojcik M; Hauser M; Li W; Moon S; Xu K. Nat. Commun 2015, 6, 7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Q; Smith JM; Park J; Kim K; Ho D; Rasool HI; Zettl A; Alivisatos AP Nano Lett. 2013, 13, 4556–4561. [DOI] [PubMed] [Google Scholar]
- 44.Chen Q; Smith JM; Rasool HI; Zettl A; Alivisatos AP Farad. Discuss 2014, 175, 203–214. [DOI] [PubMed] [Google Scholar]
- 45.Kashyap S; Woehl TJ; Liu X; Mallapragada SK; Prozorov T. ACS Nano 2014, 8, 9097–9106. [DOI] [PubMed] [Google Scholar]
- 46.Favron A; Gaufrès E; Fossard F; Lévesque P; Phaneuf-L’Heureux A; Tang N; Loiseau A; Leonelli R; Francoeur S; Martel R. arXiv preprint arXiv:1408.0345 2014. [DOI] [PubMed]
- 47.Broos K; Feys HB; De Meyer SF; Vanhoorelbeke K; Deckmyn H. Blood Rev. 2011, 25, 155–167. [DOI] [PubMed] [Google Scholar]
- 48.Coughlin SR Arterioscler. Thromb. Vasc. Biol 1998, 18, 514–518. [DOI] [PubMed] [Google Scholar]
- 49.Coughlin SR Nature 2000, 407, 258–264. [DOI] [PubMed] [Google Scholar]
- 50.Furie B; Furie BC N. Eng. J. Med 2008, 359, 938–949. [DOI] [PubMed] [Google Scholar]
- 51.Brass LF; Shaller CC; Belmonte EJ J. Clin. Invest 1987, 79, 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flaumenhaft R. Arterioscler. Thromb. Vasc. Biol 2003, 23, 1152–1160. [DOI] [PubMed] [Google Scholar]
- 53.Taylor SJ; Chae HZ; Rhee SG; Exton JH Nature 1991, 350, 516–518. [DOI] [PubMed] [Google Scholar]
- 54.Rittenhouse-Simmons SJ Clin. Invest 1979, 63, 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Titball RW J. Appl. Microbiol 1998, 84, 127s–137s. [PubMed] [Google Scholar]
- 56.Roberts MF; Otnaess AB; Kensil CA; Dennis EA J. Biol. Chem 1978, 253, 1252–1257. [PubMed] [Google Scholar]
- 57.Nevalainen TJ; Haapamaki MM; Gronroos JM Biochim. Biophys. Acta 2000, 1488, 83–90. [DOI] [PubMed] [Google Scholar]
- 58.Jorgensen K; Davidsen J; Mouritsen OG FEBS Lett. 2002, 531, 23–27. [DOI] [PubMed] [Google Scholar]
- 59.Majewski J; Kuhl T; Gerstenberg M; Israelachvili J; Smith GJ Phys. Chem. B 1997, 101, 3122–3129. [Google Scholar]
- 60.Foghandersen N. Clin. Chem 1977, 23, 2122–2126.21044 [Google Scholar]
- 61.Masuoka J; Saltman PJ Biol. Chem 1994, 269, 25557–25561. [PubMed] [Google Scholar]
- 62.Bernardo MM; Day DE; Olson ST; Shore JD J. Biol. Chem 1993, 268, 12468–12476. [PubMed] [Google Scholar]
- 63.Bernardo MM; Day DE; Halvorson HR; Olson ST; Shore JD J. Biol. Chem 1993, 268, 12477–12483. [PubMed] [Google Scholar]
- 64.Renne T; Schmaier AH; Nickel KF; Blomback M; Maas C. Blood 2012, 120, 4296–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bock PE; Srinivasan KR; Shore JD Biochemistry 1981, 20, 7258–7266. [DOI] [PubMed] [Google Scholar]
- 66.Gray MJ; Wholey WY; Wagner NO; Cremers CM; Mueller-Schickert A; Hock NT; Krieger AG; Smith EM; Bender RA; Bardwell JCA; Jakob U. Mol. Cell 2014, 53, 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Choi SH; Collins JN; Smith SA; Davis-Harrison RL; Rienstra CM; Morrissey JH Biochemistry 2010, 49, 9935–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.