

Abstract
Intracranial aneurysm (IA) rupture leads to subarachnoid hemorrhage, a sudden-onset disease that often causes death or severe disability. Although genome-wide association studies have identified common genetic variants that increase IA risk moderately, the contribution of variants with large effect remains poorly defined. Using whole-exome sequencing, we identified significant enrichment of rare, deleterious mutations in PPIL4, encoding peptidyl-prolyl cis-trans isomerase-like 4, in both familial and index IA cases. Ppil4 depletion in vertebrate models causes intracerebral hemorrhage, defects in cerebrovascular morphology and impaired Wnt signaling. Wild-type, but not IA-mutant, PPIL4 potentiates Wnt signaling by binding JMJD6, a known angiogenesis regulator and Wnt activator. These findings identify a novel PPIL4-dependent Wnt signaling mechanism involved in brain-specific angiogenesis and maintenance of cerebrovascular integrity and implicate PPIL4 gene mutations in the pathogenesis of IA.
Saccular IA, defined as an outpouching of a weakened area in the wall of an intracerebral artery, is a considerable health burden. IA has an estimated prevalence of 2–3% in the general population1. Rupture of IAs, which generally occurs without warning signs, leads to aneurysmal subarachnoid hemorrhage, resulting in sudden death before hospital admission in 26% of cases and close to 50% mortality within 1 year, despite recent advances in perioperative management2–4. Identifying genetic factors that contribute to disease pathogenesis might be beneficial for at-risk patients before IA rupture5.
Epidemiological studies and many reports of familial IA suggest that genetic factors play a role in its etiology6–8, but mutations only in a few genes have been implicated9–11. Genome-wide association studies (GWASs) have identified several common risk loci associated with IA12–14; however, by design, GWASs cannot detect rare coding variants with large effect in IA formation. On the contrary, whole-exome sequencing (WES) has the potential to detect such variants and provide unique insights into IA pathophysiology, as well as novel diagnostic and therapeutic entry points.
Results
Identification of a PPIL4 mutation in a family with IA.
To discover candidate coding mutations that cause IA, we performed WES on families with multiple members diagnosed with saccular IA. We applied stringent study inclusion criteria to our cohort of 66 European families, selecting only those with five or more affected individuals; four surpassed this threshold. After WES analysis, co-segregating variants in all affected individuals in each family were identified and used for further variant prioritization. The coding variants were called, annotated and validated as we previously described15–17 (Methods). We searched for variants that were both rare (exome and genome non-Finnish European minor allele frequency (MAF) < 1 × 10−4 in the Genome Aggregation Database (gnomAD)) and deleterious (Combined Annotation Dependent Deletion (CADD) version 1.3 Phred score ≥30) missense (D-Mis; Methods), as well as loss-of-function (LoF) (stop–gain, frameshift and canonical splice site) heterozygous mutations in genes with high arterial expression (above the 75th percentile; Genotype-Tissue Expression (GtEX) database, https://gtexportal.org/home/). In one of the IA families (IA200), we identified 17 variants (with a non-Finnish European MAF < 0.005) that co-segregated in all affected members. Only one of these variants, a single D-Mis mutation (G132S) in PPIL4, encoding peptidyl-prolyl cis-trans isomerase-like 4, met the above criteria (Fig. 1a–d and Supplementary Table 1). This mutation maps to the conserved peptidyl prolyl isomerase-like domain of PPIL4 and is predicted to introduce a damaging steric hindrance of the G132 residue with the neighboring V129 and distant V12 amino acids on the tertiary protein structure (Fig. 1b).
Fig. 1 |. Heterozygous rare and deleterious PPIL4 mutations in familial and index IA cases.
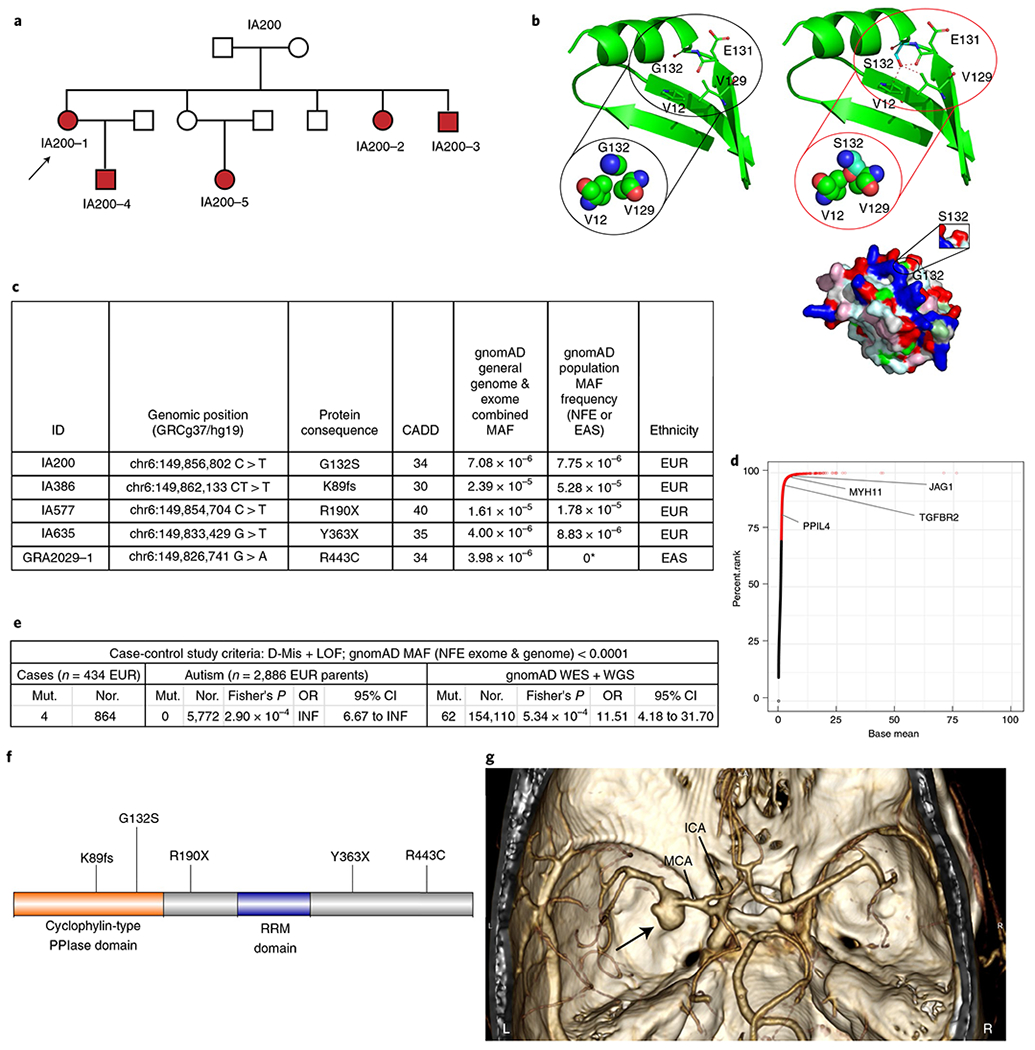
a, Pedigree structure of IA200 (arrow: index case; filled symbols: affected individuals). b, Tertiary structure of PPIL4 (left: wild-type; right: IA-variant) encompassing amino acid 132. Structural clashes are marked with red dashed lines (green: carbon; blue: nitrogen; red: oxygen). Below, molecular surface of the modeled structure (white: hydrophobic; yellow: semi-polar; cyan: polar; blue: positively charged; red: negatively charged; magenta: aromatic; pale green: proline; green: glycine residues). c, Rare and deleterious mutations (Methods) identified in patients with IA. MAFs are reported as general and population MAF. *East Asian (EAS) MAF. d, Scatter plot showing percentile rank of arterial expression enrichment ratio of 18,099 transcripts in the GtEX portal (Methods). Representative genes associated with aneurysm formation with high arterial expression (JAG1, MYH11 and TGFBR2) are indicated. e, Summary of burden analysis (two-sided Fisher’s exact test). f, Schematic of the PPIL4 protein and IA mutations. g, 3D volume-rendered computed tomography angiogram showing a left middle cerebral artery aneurysm (arrow) in a 61-year-old patient (IA577), carrying a stop-gained heterozygous mutation (R190X). n, number of alleles; WGS, whole-genome sequencing; Mut., number of mutant alleles; Nor., number of normal alleles; RRM, RNA recognition motif. EUR, European; INF, infinity; NFE, non-Finnish European.
PPIL4 mutations are enriched in index European IA cases.
We next screened a replication cohort of 430 independent singleton saccular IA probands of European ancestry for rare (non-Finnish European MAF < 1 × 10−4) and deleterious (D-Mis + LoF) mutations in PPIL4 and identified three LoF variants (Fig. 1c,e). No LoF or D-Mis variants were identified in a control cohort of 2,886 Europeans consisting of unaffected parents of children with autism from the Simons Simplex cohort18. We then compared the burden of rare and deleterious variants in all IA index cases to this parental control cohort. Rare and deleterious variants in PPIL4 were significantly enriched in IA cases versus controls (odds ratio (OR) = infinity; Fisher’s P = 2.9 × 10−4). LoF and D-Mis mutations in PPIL4 were also significantly enriched in European IA cases compared to the matched gnomAD dataset19 (OR = 11.51, Fisher’s P = 5.34 × 10−4) (Fig. 1e–g, Extended Data Fig. 1 and Supplementary Tables 2–4). Moreover, we identified an additional three patients with missense mutations and one in-frame deletion of PPIL4 in 430 European and 47 non-European cases, when we relaxed the MAF and deleteriousness thresholds (Extended Data Fig. 1 and Supplementary Tables 2 and 3). However, these variants were not included in the burden analysis.
Hypertension is a risk factor for IA formation20 and rupture21. We thus investigated our cohort of over 600 ethnically diverse individuals diagnosed with early hypertension and identified that approximately 2% indeed carried a diagnosis of IA. We next searched the WES dataset of this cohort and identified a rare D-Mis mutation in PPIL4 (R443C) in a patient of East Asian ancestry who had suffered a ruptured IA (Fig. 1c) and had a family history of intracranial hemorrhage in his now deceased father (Supplementary Table 3).
PPIL4 is essential for vertebrate brain angiogenesis.
PPIL4 belongs to the cyclophilin subgroup of the peptidyl-prolyl cis-trans isomerase (PPIase) protein family (PPIs). It contains an RNA recognition motif and a PPIase-like domain22. PPIs are molecular chaperones that facilitate protein folding by catalyzing the interconversion among geometric proline isomers23,24 and have been intensively studied as targets for the immunosuppressive drugs cyclosporin A, FK506 (tacrolimus) and rapamycin23. To investigate the biological function of PPIL4, which is essentially unknown, we employed Danio rerio (zebrafish) and Xenopus tropicalis (frog), which are well-suited for studying cerebrovascular development and pathology because of their transparent nature and the availability of transgenic reporter lines.
Using the CRISPR–Cas9 system, we first generated ppil4 mutant zebrafish carrying a premature stop codon within exon 5 (Extended Data Fig. 2a). In wild-type zebrafish, Ppil4 expression was restricted to the cranial region (Extended Data Fig. 2b). As expected, ppil4 expression was significantly diminished in heterozygous (ppil4+/−) and homozygous (ppil4−/−) mutant animals (Extended Data Fig. 2b, c), which both lacked gross developmental abnormalities (Extended Data Fig. 2d,e). However, ppil4−/− embryos exhibited necrosis in the dorsal midbrain (optic tectum) at 2 days post-fertilization (dpf) (Extended Data Fig. 2e) and did not survive to adulthood. In contrast, ppil4+/− embryos survived and were fertile.
To assess the role of ppil4 in brain vasculature, we used the tg(kdrl:gfp) line, in which endothelial cells are labeled with green fluorescent protein (GFP). We found that pptl4−/− embryos, as compared to ppil4+/+, exhibited significant reduction in the total number of branches in midbrain central arteries (CtAs) (P = 5.55 × 10−8) (Fig. 2a,b) at 2.5 dpf and several atretic midbrain CtAs sprouting from the parent vessel that were not connected with any other CtAs (Fig. 2a). The total length of midbrain CtAs in ppil4−/− was significantly reduced (P = 1.91 × 10−8) (Fig. 2c). Furthermore, ppil4+/− zebrafish showed significant reduction in number (P = 9.92 × 10−4) and total length of midbrain CtAs, as compared to ppil4+/+ (P = 8.09 × 10−8) (Fig. 2a–c).
Fig. 2 |. Cerebrovascular simplification in ppil4-depleted zebrafish is prevented by overexpression of human PPIL4WT but not by IA-associated PPIL4G132S.
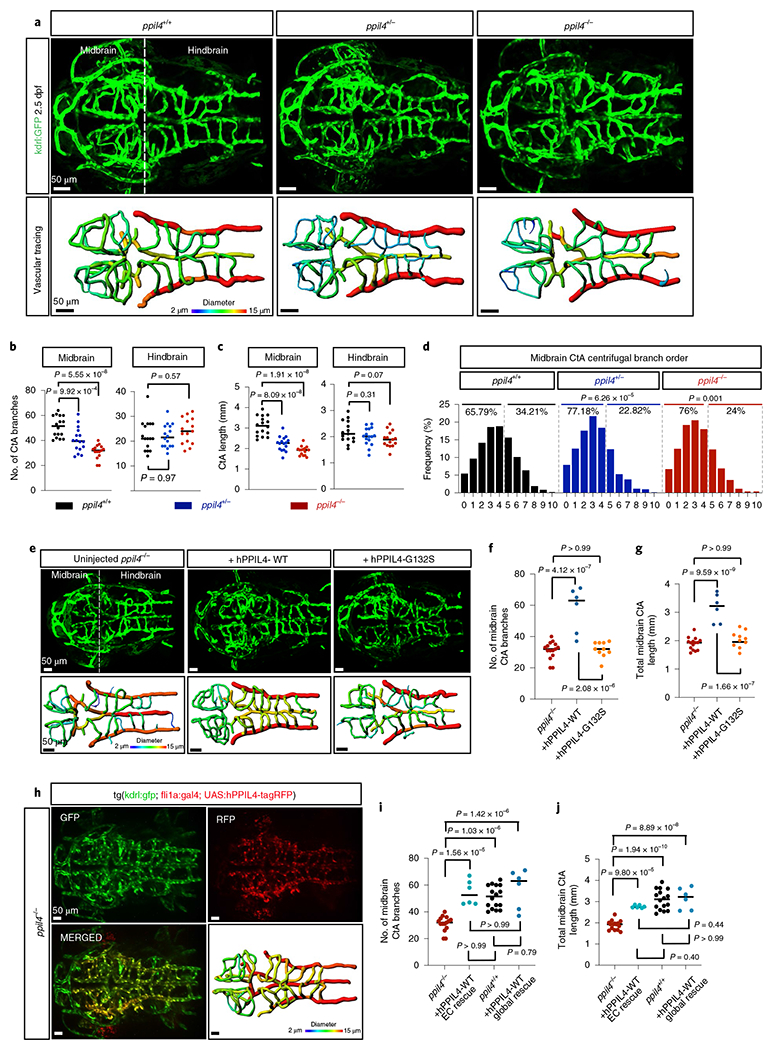
a, MIP of confocal z-stack images and vessel segmentation of ppil4+/+ (n = 16), ppil4+/− (n = 16) and ppil4−/− (n = 15) embryos in tg(kdrl:gfp)zn1 background at 2.5 dpf. b,c, Quantifications of CtA branches (b) and total length (c) in ppil4+/+ (n = 16), ppil4−/− (n = 16) and ppil4−/− (n = 15) embryos. d, Branching level of midbrain CtAs in centrifugal order n = 874, 609 and 521 vessels in ppil4+/+ (n = 16), ppil4+/− (n = 16) and ppil4−/− (n = 14), respectively. e–g, Global rescue experiments. n = 15, 6 and 10 for uninjected, hPPIL4WT- and hPPIL4G132-injected ppil4−/−, respectively. e, MIP of confocal z-stack images and vessel segmentation at 2.5 dpf. f,g, Comparison of midbrain CtA branches (f) and total length (g). h–j, Endothelial-specific rescue experiments. n = 15, 6, 16 and 6 for uninjected ppil4−/−, endothelial-specific rescue of ppil4−/− with hPPIL4-WT, ppil4−/− and global rescue of ppil4−/− with hPPIL4-WT. h, MIP of a representative confocal image of UAS:hPPIL4-WT-tagRFP-injected embryo in ppil4−/−;tg(kdrl:gfp)zn1;tg(fli1a:gal4) background at 2.5 dpf, n = 6. i,j, Comparison of midbrain CtA branches (i) and total length (j). Individual values shown with scatter dot plot and median for all quantifications. Statistical tests: one-way ANOVA followed by Dunnett’s multiple comparison (b,c); pairwise Fisher’s exact test with FDR correction (d). f,g,i,j, One-way ANOVA followed by Bonferroni multiple comparison test. Scale bar, 50 μm. EC, endothelial cell.
To further assess vascular tree geometry, we separated midbrain CtAs into two groups according to branch depth in centrifugal order (h0–5 and h6–10), where vascular networks with higher-level branching indicate higher vascular complexity. Midbrain CtAs with higher branch levels were more abundant in ppil4+/+ than ppil4+/− and ppil4−/− (ppil4+/− versus ppil4+/+ P = 6.26 × 10−5, ppil4−/− versus ppil4+/+ P = 0.001) (Fig. 2d). These results suggest a substantial impairment in cerebrovascular branching in ppil4-depleted zebrafish (Fig. 2a–d).
The cerebrovascular phenotypes observed in ppil4+/− and ppil4−/− mutants persisted at 5.5 dpf (Extended Data Fig. 3a–e), suggesting that they were not the result of a developmental delay. Furthermore, both ppil4+/− and ppil4−/− zebrafish exhibited significant reduction in branch number and total length of hindbrain CtA at 5.5 dpf but not at 2.5 dpf. This is most likely because the hindbrain vasculature is less complex at earlier developmental stages, and the effect of ppil4 depletion becomes more prominent with increased branching and cerebrovascular complexity. Indeed, we found that, in wild-type zebrafish, the CtA branch number increased 4.9-fold (from an average of 22.47–110) between 2.5 and 5.5 dpf in the hindbrain compared to 2.14-fold (51.06–109.3) in the midbrain (Fig. 2b,c and Extended Data Fig. 3d,e). No changes were observed in the trunk vasculature at either stage upon ppil4 depletion (Extended Data Fig. 3f–k), in agreement with the results of in situ hybridization (Extended Data Fig. 2b), suggesting that ppil4 played a role in region-specific angiogenesis.
To elucidate the specificity of the cerebrovascular phenotype and the pathogenicity of the IA-associated PPIL4G132S mutation, we next injected one-cell stage embryos with human wild-type or IA-mutant PPIL4 mRNA (hPPIL4WT and hPPIL4G132S, respectively). Overexpression of hPPIL4WT, but not hPPIL4G132S, prevented the loss (P = 4.12 × 10−7) as well as the reduction in total length of midbrain CtAs (P = 9.59 × 10−9) (Fig. 2e–g). Furthermore, hPPIL4G132S-injected zebrafish exhibited significantly fewer midbrain CtAs than hPPIL4WT-injected fish (P = 2.08 × 10−6), whereas their total length was decreased (P = 1.66 × 10−7) (Fig. 2e–g). These results show that PPIL4 is essential for cerebral angiogenesis, and they support the pathogenicity of this IA-associated PPIL4 mutation.
Endothelial cells represent the primary component of the cerebrovascular network in zebrafish larvae, especially at early embryonic stages before completion of mural cell coverage25. We, therefore, assessed whether the cerebrovascular defects observed in ppil4+/− and ppil4−/− embryos resulted from lack of endothelial cells. Using the endothelial-specific nuclear reporter strain tg(kdrl:NLS-GFP), we discovered that ppil4−/− had significantly fewer endothelial cells in the primordial midbrain channel (PMBC) compared to ppil4+/+ fish at 30 hours post-fertilization (hpf) (P = 0.0384) (Extended Data Fig. 4a,c). The difference became more significant in both ppil4+/− (P = 0.0005) and ppil4−/− (P = 0.0003) mutants when we assessed midbrain vascular sprouts (Extended Data Fig. 4a,c). As expected, endothelial cell abundance in midbrain CtAs was significantly impaired in ppil4+/− (P = 5.17 × 10−8) and ppil4−/− (P = 1.49 × 10−8) zebrafish compared to wild-type at 60 hpf (Extended Data Fig. 4b,d). Endothelial cell number in the PMBC was also significantly lower in ppil4−/− zebrafish compared to wild-type (P = 0.0089) at this stage (Extended Data Fig. 4b,d).
Given the observation of optic tectum apoptosis displayed by the ppil4−/− mutants (Extended Data Fig. 5a,b), we next assessed whether the reduced brain endothelial cell abundance might be due to apoptosis. Caspase-3 immunostaining showed that ppil4+/− and ppil4−/− zebrafish at 2.5 dpf (Extended Data Fig. 5c–j) did not exhibit apoptosis in endothelial cells. In contrast, strong caspase-3 activation was detected in ppil4−/− HU+ neurons and GFAP+ glia, although no gross histologic defects were identified in either cell type (Extended Data Fig. 5k,l).
RNA sequencing (RNA-seq) analysis of brain-enriched wild-type endothelial cells from 2.5-dpf zebrafish (Methods) revealed ppil4 to be among the top 30% of the transcripts (71st percentile); bona fide endothelial markers kdrl and cdh5 were above the 93rd percentile (Extended Data Fig. 6a and Supplementary Table 5). After a brief decrease after 12 hpf, ppil4 expression increased starting at 24 hpf and reached the highest level at 3 months, among the time points explored (Extended Data Fig. 6b). Finally, the expression levels of ppil4 in kdrl:GFP+ cells from brain, liver and heart at 3 months were significantly higher in the brain than liver (P = 5.0 × 10−5) or heart (P = 0.0011) (Extended Data Fig. 6c). Finally, PPIL4 immunostaining of human middle cerebral artery showed prominent endothelial expression (Extended Data Fig. 6d–i). However, PPIL4 is not specific to brain endothelial cells and is expressed in other cell types of the brain parenchyma and vascular tissue, as shown in transcriptome databases26–28.
To elucidate whether PPIL4 has cell autonomous effects in endothelial cells, we performed an in vitro tube formation assay using human umbilical vein endothelial cells (HUVECs). Short haripin RNA (shRNA)-mediated downregulation of PPIL4 resulted in significant reduction in nodes, junctions and branches29 (Extended Data Fig. 6j–n). This suggested that the alterations in cerebrovascular geometry and reduction of vascular complexity observed in the zebrafish model can be attributed to endothelial cell autonomous defects.
We next restored expression of human PPIL4 in ppil4−/− zebrafish endothelial cells using the fli1a promoter. We injected kdrl:GFP ppil4−/− animals in the tg(fli1a:gal4) background with UAS:PPIL4-tagRFP, allowing us to distinguish endothelial cells (GFP+) that also express the injected human WT-PPIL4 (RFP+). Analysis of 2.5-dpf zebrafish revealed that endothelial-specific restoration of PPIL4 prevented the loss of cerebrovascular complexity (Fig. 2h–j and Extended Data Fig. 7a–e). These data suggest that ppil4 expression in endothelial cells is indispensable for a functional vertebrate brain vasculature and that the cerebrovascular phenotype upon ppil4 depletion is associated with its role in endothelial cells.
To gain insight into the molecular alterations underlying the morphologic changes in the cerebral vasculature of ppil4 mutant zebrafish, we investigated the transcriptomic effects of ppil4 loss in brain-enriched endothelial cells at 2.5 dpf. We injected wild-type tg(kdrl:gfp)zn1 zebrafish embryos at the one-cell stage with CRISPR–Cas9 targeting ppil4. We compared RNA-seq results of kdrl-GFP+ endothelial cells isolated from ppil4-CRISPR–Cas9-injected and control F0 zebrafish heads using fluorescence-activated cell sorting (FACS). Differential gene expression analysis revealed significant changes in the expression of 467 genes (Supplementary Table 6). We found that ppil4 depletion resulted in significant downregulation of genes involved in extracellular matrix formation (for example, col1a1a, col1a2, col2a1b, tjp2a, fn1b and gpc1a), suggesting that matrix–endothelial cell reciprocal interactions were perturbed (Extended Data Fig. 7f–h and Supplementary Tables 7 and 8).
Given the substantial changes in cerebrovascular morphology, we evaluated the effect of ppil4 depletion on cerebrovascular hemodynamics. We first calculated the diameter of midbrain CtAs. We found that ppil4−/− zebrafish exhibited significant variability in diameter (Levene’s test P = 1.0 × 10−10) (Fig. 3a) and enrichment for atretic CtAs with a lumen diameter of less than 3 μm (ppil4−/− versus ppil4+/+, Bonferroni-corrected P = 1.33 × 10−6; pairwise Fisher’s exact test). We found an overall reduction of vascular diameter in both ppil4+/− (P = 6.18 × 10−12) and ppil4−/− (P = 3.12 × 10−27) zebrafish compared to wild-type (Fig. 3a). Furthermore, we showed a significant increase in midbrain CtA resistance in ppil4+/− (P = 0.0015) and ppil4−/− (P = 3.98 × 10−10) zebrafish, calculated with the Hagen–Poiseuille equation30 (Fig. 3b). We corroborated these data by microangiography after intracardiac injection with BSA594 at 2.5 dpf, showing increased vascular resistance and lack of flow in midbrain CtAs in ppil4−/− zebrafish (Fig. 3c).
Fig. 3 |. Depletion of ppil4 leads to cerebral hemorrhage.
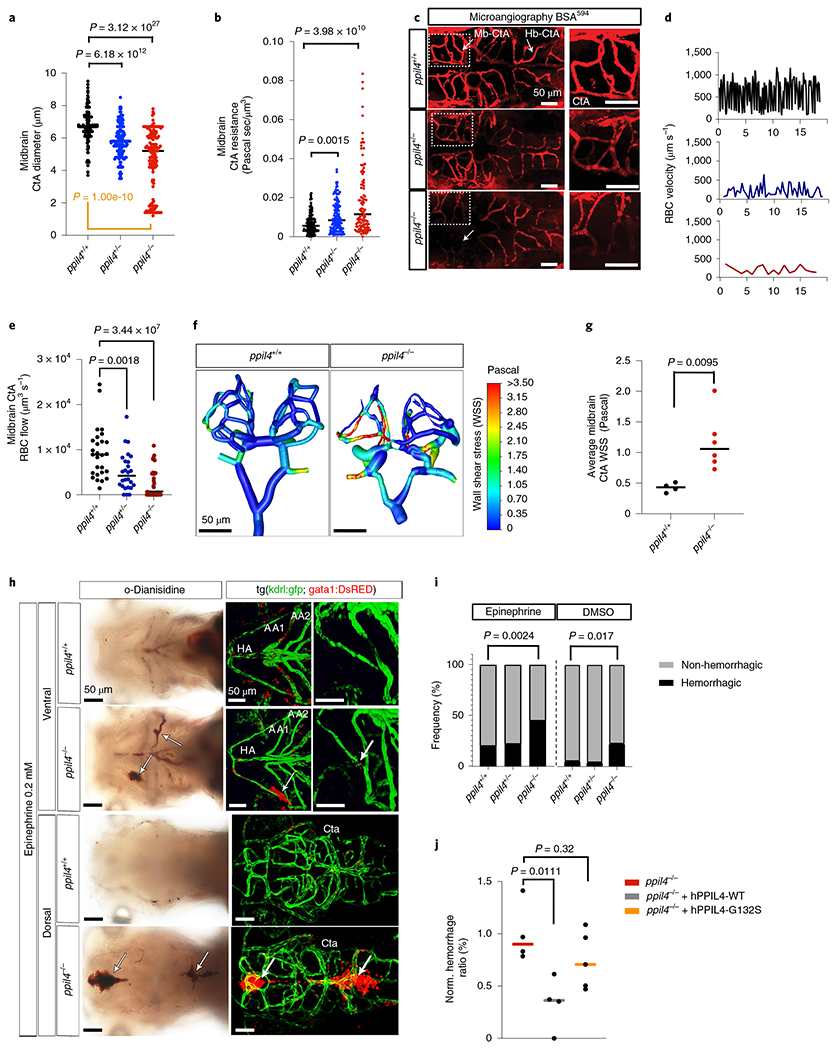
a, Midbrain CtA diameter in ppil4+/+ (n = 153), ppil4+/− (n = 147) and ppil4−/− (n = 140) embryos at 2.5 dpf. Orange bar: Levene’s test. b, Vascular resistance in midbrain CtAs in ppil4+/+ (n = 143), ppil4+/− (n = 129) and ppil4−/− (n = 109) embryos at 2.5 dpf. c, Microangiography by transcardiac injection of BSA594 (2.5 dpf), n = 10 per genotype. d,e, Representative time–velocity plots (d) and comparison of blood flow (e) in midbrain CtAs at 2.5 dpf, n = 27, 26 and 33 vessels in ppil4+/+, ppil4+/− and ppil4−/−, respectively. f,g, Wall shear stress in 3D cerebrovascular models of ppil4+/+ (n = 4) and ppil4−/− (n = 6) zebrafish (2.5 dpf) using ANSYS. h, Bright-field (o-Dianisidine staining) and confocal images of tg(kdrl:GFP;gata1:dsRED) of ppil4+/+ (n = 70) and ppil4−/− zebrafish (n = 78) embryos treated with epinephrine (AA1 and AA2, first and second aortic arch arteries; HA, hypobranchial artery). i, Hemorrhagic events in brain and aortic arch arteries after epinephrine (h) versus DMSO (vehicle) treatment at 72 hpf, nepi = 70, 163 and 78 and nDMSO = 56, 121 and 66 for ppil4+/+, ppil4+/− and ppil4−/−, respectively. j, Normalized frequency of epinephrine-induced hemorrhage in brain and aortic arch arteries in uninjected, hPPIL4WT- or hPPIL4G132S-injected ppil4−/− embryos, n = 4, 4 and 5 sets of biological replicates with 200 zebrafish per set. Individual values are shown with scatter plot and median in a,b,e,g and j. Statistical tests: Kruskal-Wallis test followed by Dunn’s test (a,b); Levene’s test (based on median) (a); one-way ANOVA with Dunnett’s multiple comparison test (e,j); two-tailed Mann-Whitney test (g); pairwise Fisher’s exact test with FDR correction (i). Scale bar, 50 μm. WSS, wall shear stress.
We next calculated red blood cell (RBC) velocity and blood flow in all accessible CtAs and circle of Willis vessels31. We used axial line scanning to analyze ppil4 mutant zebrafish in the tg(kdrl:gfp;gata1:DsRed) background, in which blood vessels and erythroid precursors are labeled with green and red fluorescent protein, respectively (Methods, Fig. 3d and Supplementary Fig. 1a–e). We found that the overall RBC flow in midbrain CtAs was significantly reduced in ppil4+/− (P = 0.0018) and ppil4−/− (P = 3.44 × 10−7) mutants compared to wild-type (Fig. 3e). These data imply that several cerebral vessels, although present, were functionally impaired and, in some cases, completely lacked RBC flow (ppil4−/− versus ppil4+/+, false discovery rate (FDR) = 5.09 × 10−5; ppil4+/− versus ppil4+/+, FDR = 1.6 × 10−2; pairwise Fisher’s exact test). Together, these findings suggest that ppil4 is essential for the formation of a functional cerebrovascular network.
Computational fluid dynamic simulations using ANSYS Fluent software to calculate wall shear stress in midbrain CtAs of ppil4+/+ and ppil4−/− zebrafish predicted increased wall shear stress in the latter (p = 0.0095) (Fig. 3f,g). We observed no significant differences between wild-type and mutant genotypes (ppil4+/− and ppil4−/−) in heart rate, hematocrit or RBC velocity within the circle of Willis (Supplementary Fig. 1b–k), suggesting that impaired circulation in midbrain CtAs resulted from vessel dysmorphology rather than altered systemic hemodynamics.
To elucidate the role of PPIL4 in mediating cerebrovascular integrity, we next subjected zebrafish to hemodynamic stress using epinephrine, which, as expected, induced vasoconstriction in brain vessels of wild-type embryos (Supplementary Fig. 1l,m). ppil4−/− embryos exhibited hemorrhage both in the cerebrum and the aortic arch arteries with greater frequency compared to wild-type, when treated either with epinephrine (P = 0.0024) or DMSO (vehicle control; P = 0.017) (Fig. 3h,i). The cerebral and aortic arch artery hemorrhage observed in epinephrine-treated ppil4−/− zebrafish was prevented by injecting hPPIL4WT (P = 0.0111) but not hPPIL4G132S mRNA at the one-cell stage (Fig. 3j). These results indicate that PPIL4 is essential for cerebrovascular wall integrity, and they provide functional evidence of the pathogenicity of the G132S mutation.
We also assessed the effect of ppil4 depletion in 3-month-old ppil4+/− mutants in the tg(kdrl:gfp;gata1:dsred) background after retro-orbital injection of epinephrine (0.5 mg kg−1). We found that cerebral hemorrhage was more frequently observed in epinephrine-treated ppil4+/− compared to wild-type animals (two-sided Fisher’s exact test, P = 0.0232, OR = 5.48, 95% confidence interval, 1.19–25.56) (Fig.4a–i and Supplementary Fig. 2).
Fig. 4 |. Hemodynamic stress leads to intracranial hemorrhage in adult heterozygous zebrafish.
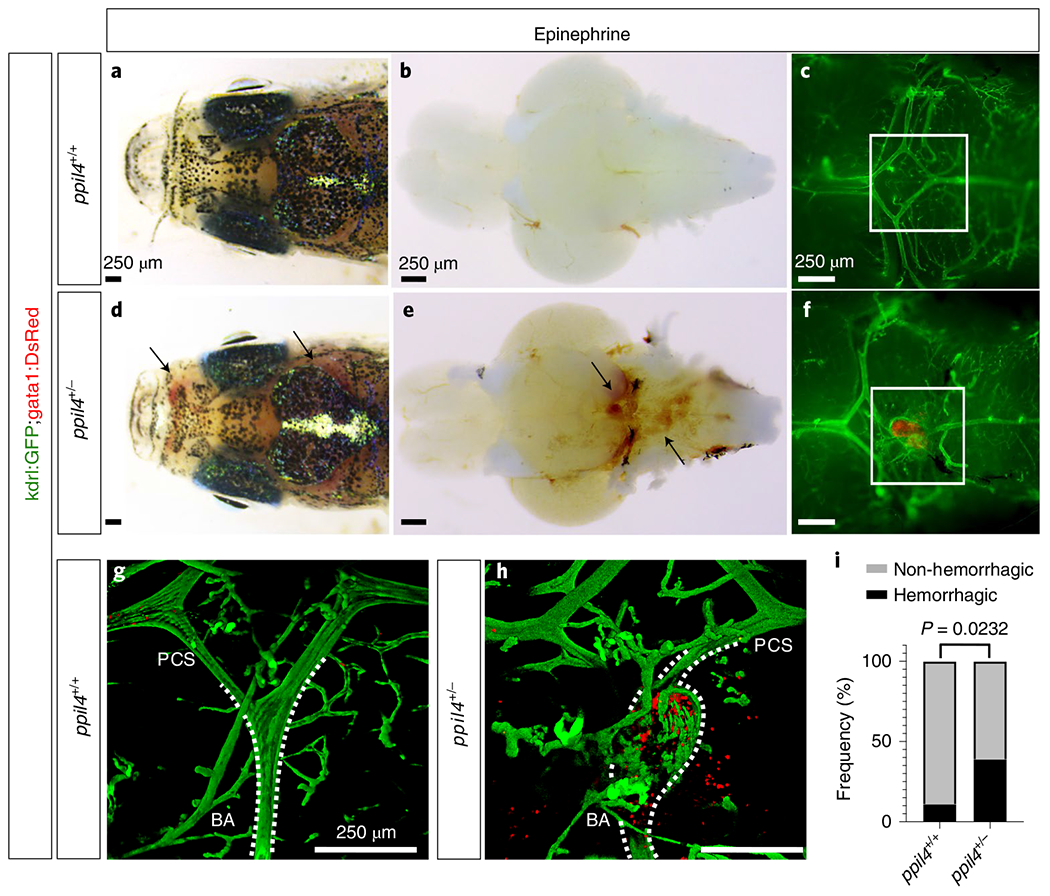
a–h, Administration of epinephrine (0.5 mg kg−1) via retro-orbital injection in 3-month-old wild-type (n = 19) (a-c) and ppil4+/− (n = 51) (d-f) zebrafish in tg(kdrl:gfp;gata1:dsred) background results in intracranial hemorrhage (d-f,h; arrows) or vascular dilation (f,h) in ppil4+/− zebrafish. a,d, Dorsal view of the head. b,e, Ventral view of the brain with bright-field microscopy. c,f, Fluorescence microscopy of ventral vascular structures in the circle of Willis of the brains shown in b and e. g,h, Confocal images of the areas delineated by white squares in c and f. The images are rotated 90° clockwise. BA, basilar artery. i, Quantification of intracranial hemorrhage events in 3-month-old ppil4+/+ (n = 19) and ppil4+/− (n = 51) zebrafish upon epinephrine administration. Statistical test: two-sided Fisher’s exact test.
We corroborated the above findings by studying the role of ppil4 in a second vertebrate model, X. tropicalis. Morpholino (MO)-mediated depletion of ppil4 showed a dose-dependent increase in the rate of spontaneous intracerebral hemorrhage at 2 dpf (stages 39 and 40). In addition, co-injection of hPPIL4WT but not hPPIL4G132S mRNA at the one-cell stage rescued the intracerebral hemorrhage phenotype in ppil4 MO-depleted embryos, supporting findings in zebrafish and suggesting that the cerebrovascular function of PPIL4 is conserved in vertebrates (Supplementary Fig. 3a–k).
PPIL4 regulates Wnt signaling activity.
We next elucidated the potential role of Ppil4 in mouse brain vascular endothelial cells by analyzing publicly available single-cell RNA sequencing (scRNA-seq) datasets27,28. We found that Ppil4 displays strong statistical dependency and positive relationship with several genes, including Syt15, Sema3g, Gkn3 and Bmx, expressed at the highest levels (99th percentile) in the artery (Supplementary Table 9) 27,28. In contrast, several genes expressed at the highest levels of brain venous endothelial cells (for example, Tmsb10 (refs. 27,28)) showed negative relationship with Ppil4 (Supplementary Table 9). We next visualized arterio-venous characteristics of Ppil4 expression using pseudotime analysis, which showed stronger expression in arterial compared to venous endothelial cells in the mouse brain (Extended Data Fig. 8a–c and Supplementary Tables 9 and 10). To reveal the cellular pathways most significantly associated with Ppil4 expression, we next performed a hypergeometric test for genes that were positively regulated with Ppil4 in kNN-DREMI32 analysis in mouse cerebrovascular endothelium. Using KEGG pathways33, we found that genes associated with Wnt signaling33, a pathway critical for brain angiogenesis and vascular wall integrity34,35, were significantly enriched within the top-scored genes (95th percentile) (FDR = 3.01 × 10−4) (Extended Data Fig. 8d and Supplementary Table 11).
To gain mechanistic understanding of how PPIL4 regulates cerebral angiogenesis and vessel wall integrity, we next searched for potential interactors. We identified Jumonji domain-containing 6, arginine demethylase and lysine hydroxylase (JMJD6, also known as phosphatidylserine receptor or PTDSR), a known angiogenesis regulator36,37 and Wnt signaling molecule38–40, as a potential binding partner of PPIL4. We observed a probability of interaction of 1.0 in BioPlex, a database of protein–protein interactions generated by affinity purification mass spectroscopy of human proteins41,42. Notably, JMJD6 inactivation in the mouse has been reported to cause cardiovascular abnormalities43,44 and angiogenic sprouting defects36.
Using co-immunoprecipitation in HEK293 cells, we showed that the reciprocal interaction of JMJD6 with PPIL4WT was substantially diminished when PPIL4G132S instead of PPIL4WT was used (Fig. 5a). Subcellular fractionation coupled with co-immunoprecipitation showed that the interaction between JMJD6 and PPIL4G132S was reduced both in cytoplasm and nucleus, as compared to PPIL4WT (Extended Data Fig. 9a). Furthermore, immunostaining of HEK293 cells co-transfected with JMJD6 and either with PPIL4WT or PPIL4G132S showed that the co-localization of the PPIL4G132S with JMJD6 was reduced in the nucleus, as compared to PPIL4WT (Fig. 5b).
Fig. 5 |. PPIL4 potentiates Wnt activity by binding to JMJD6.
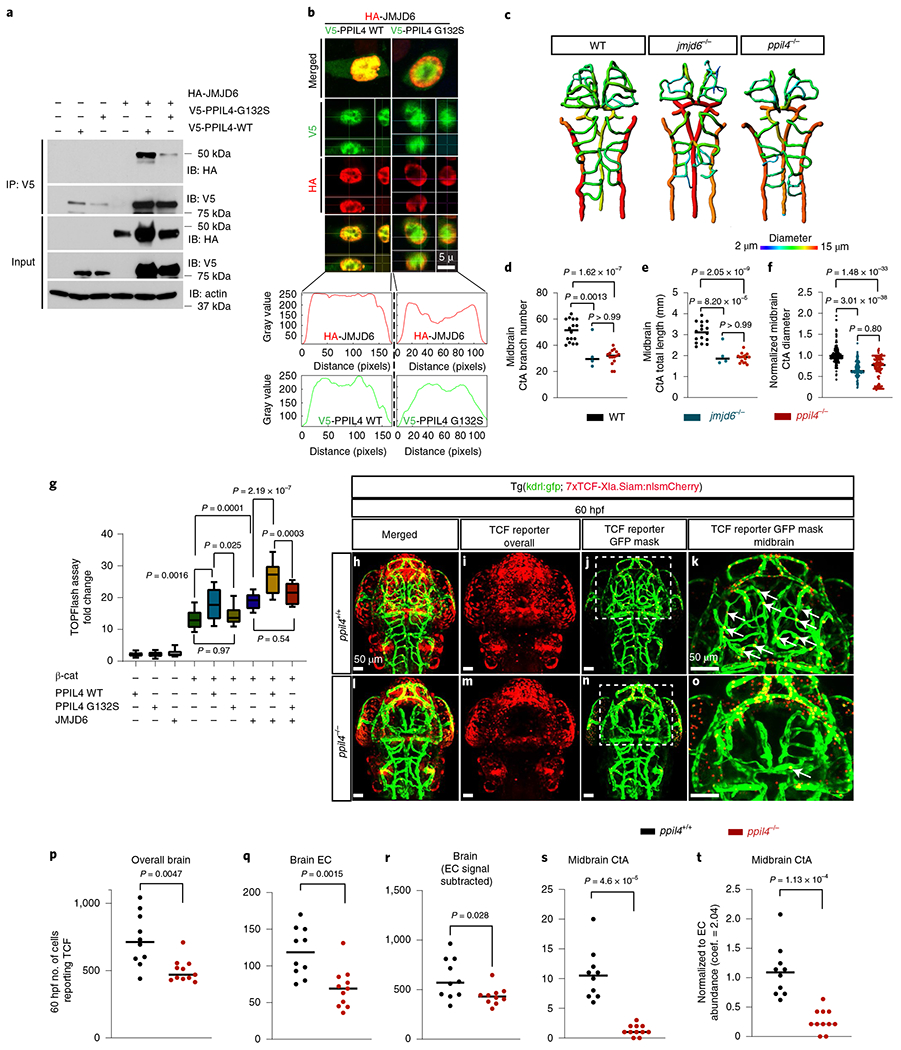
a, Co-immunoprecipitation of JMJD6 with PPIL4WT or PPIL4G132S in HEK293 cells. Input: whole cell lysates, n = 3 biological replicates. b, Orthogonal section from representative confocal images of HEK293 cells expressing HA-JMJD6, V5-PPIL4WT and V5-PPIL4G132S. Lower panels: co-localization of red and green channels within the nucleus, n = 3 biological replicates. c-f, Brain vessel segmentation and phenotypic assessment in wild-type (n = 16), ppil4−/− (n = 15) and jmjd6−/− (n = 4) at 2.5 dpf. g, Effect of transient overexpression of JMJD6, PPIL4WT, PPIL4G132S or β-catenin on Wnt signaling activation (TOPFlash assay), n = 4 biological replicates. h-o, MIP of confocal z-stack images of ppil4+/+ (n = 10) and ppil4−/− (n = 11) embryos in tg(kdrl:gfp; 7xTCF-Xla.Siam:nlsmCherry) background at 60 hpf. j,n, Visualization of endothelial-specific Wnt activity using the Spots Mask application (Imaris). k,o, Higher magnification of the indicated areas in j and n, respectively. p-t, Quantification (Imaris) of TCF reporter signal in ppil4 +/+ (n = 10) and ppil4 −/− (n = 11) embryos at 60 hpf. See Methods for details of image processing and assembly. t, Number of TCF-reporting endothelial cells normalized to endothelial cell number using a coefficient of 2.04 obtained from endothelial cell abundance experiments (Methods). Individual values are shown with scatter dot plot and median in d-f and p-t. In g, the box extends from the 25th to the 75th percentile; the whiskers show the minimum and maximum values; and the line at the center is the median. Statistical tests: one-way ANOVA (d-g), followed by Dunnett’s (d-f) or Sidak’s (g) multiple comparison test. p-t, Two-tailed Student’s t-test. Scale bar, 5 μm in b and 50 μm in h-o. EC, endothelial cell.
To test the effect of JMJD6 depletion on cerebrovascular development, we generated jmjd6 knockout zebrafish carrying a premature stop codon within exon 3 using CRISPR–Cas9 editing. Strikingly, similarly to ppil4−/− mutants, jmjd6−/− zebrafish exhibited necrosis in the optic tectum at 2.5 dpf (Extended Data Fig. 9c) and prominent angiogenesis defects in the midbrain (Fig. 5c and Extended Data Fig. 9e–g). Moreover, midbrain CtA length, diameter and branch number were significantly reduced in the jmjd6−/− zebrafish (Fig. 5d–f), underscoring the phenotypic overlap between the ppil4 and jmjd6 knockout models. This suggested that impaired PPIL4–JMJD6 interaction can explain the cerebrovascular phenotype associated with ppil4 depletion.
JMJD6 was shown previously to activate Wnt signaling by binding to transcription factor 7-like 1 (TCF7L1) and preventing its interaction with the transcriptional co-repressor Groucho39. Furthermore, JMJD6 depletion was reported to cause reduced TCF7L2 protein expression, resulting in suppression of Wnt signaling38. Wnt signaling is indispensable for mediating cerebrovascular integrity34,35, and LoF of several Wnt signaling-related molecules causes cerebrovascular angiogenesis defects remarkably similar to those observed with ppil4 loss45,46. Given the phenotypic similarities, we hypothesized that PPIL4 mediates vertebrate cerebrovascular development and wall integrity through Wnt signaling.
To determine the combined effect of PPIL4 and JMJD6 on Wnt signaling activation and test the effect of IA-associated PPIL4G132S mutation, we next used a TOPFlash luciferase reporter assay in HEK293 cells. We observed a significant increase in Wnt transactivation when we co-transfected PPIL4WT with β-catenin compared to β-catenin alone (P=0.0016). This increase in Wnt signaling transactivation was lost when PPIL4G132S instead of PPIL4WT was used for co-transfection (P=0.025). Furthermore, the combination of PPIL4WT and JMJD6 potentiated β-catenin-induced Wnt transactivation, as compared to JMJD6 alone (P = 2.19 × 10−7), suggesting that the two molecules elicited a synergistic effect on Wnt signaling activation. In contrast, co-transfection of PPIL4G132S with JMJD6 had no such effect (Fig. 5g).
We next tested the effects of PPIL4 depletion on Wnt activity in vivo. We used a Wnt reporter line, tg(7xTCF-Xla.Siam:nlsmCherry)ia5 47, in a ppil4+− kdrl:eGFP background and further in-crossed to generate ppil4−/− Tg(7xTCF-Xla.Siam:nlsmCherry;kdrl:eGFP) zebrafish47. ppil4−/− zebrafish showed significant reduction in Wnt signaling activity in the entire brain, both at 30 hpf (P = 0.0026) (Extended Data Fig. 10a–g,i) and at 60 hpf (P = 0.0047) (Fig. 5h–p,r and Supplementary Fig. 4a–f). The reduction in Wnt signaling activity remained significant when the analysis was restricted to the brain vasculature at 30 hpf (P = 0.0001) (Extended Data Fig. 10a–f,h) and 60 hpf (P = 0.0015) (Fig. 5j,k,n,o,q and Supplementary Fig. 4a–f). Furthermore, Wnt signaling activity (that is, the number of TCF-reporting endothelial cells) in midbrain CtAs at 60 hpf was substantially reduced (P = 4.6 × 10−5) (Fig. 5j,k,n,o,s), and remained significant even after normalizing for endothelial cell number (P = 1.13 × 10−4) (Methods and Fig. 5t). Together, these results suggest that ppil4 is required for Wnt signaling activity in cerebrovascular endothelial cells as well as in the parenchyma. Finally, we tested whether restoring Wnt signaling would prevent the cerebrovascular pathology associated with ppil4 depletion. Treatment of X. tropicalis ppil4 morphants with 6-bromoindirubin-3′-oxime (1 uM), an inhibitor of glycogen synthase kinase 3β and an activator of Wnt signaling48, prevented cerebral hemorrhage (P = 3.17 × 10−14) (Supplementary Fig. 5a,b). Together, these results show that PPIL4 is a novel Wnt signaling molecule and that cerebrovascular defects observed due to ppil4 loss are attributable to attenuated Wnt activity.
Discussion
GWASs have identified several common risk loci associated with IA12–14; however, conclusive identification of specific genes or molecular pathways involved in IA formation and rupture remain largely elusive. Here we provide evidence that rare and deleterious variants in PPIL4 are significantly enriched in patients with IA and are associated with IA risk. Using two well-established vertebrate model systems, we show the role of PPIL4 in vertebrate cerebrovascular wall integrity and hemodynamics. Our findings reveal that PPIL4 depletion causes cerebral hemorrhage in vivo, and this is rescued by human wild-type PPIL4 but not an IA-associated PPIL4 mutation. Furthermore, PPIL4 depletion leads to significant changes in vessel complexity and morphology as well as rupture in midbrain CtAs and aortic arch arteries. Morphologic defects observed in aortic arch arteries, along with abnormal in vitro endothelial tube formation followed by PPIL4 downregulation, suggest that the vascular phenotype associated with PPIL4 depletion is likely due to its primary effect on vascular tissue rather than a defect in brain parenchyma–vascular cell communication. This hypothesis is further supported by endothelial-specific overexpression of human wild-type PPIL4 in ppil4−/− background, which prevents the cerebrovascular defects observed in homozygous mutant zebrafish at 2.5 dpf. Together, these results suggest that endothelial expression of ppil4 plays an important role in establishing a functional cerebrovascular morphology and integrity.
We show that PPIL4 potentiates Wnt signaling, a cellular pathway critical for cerebrovascular development and integrity, by binding to JMJD6, a known angiogenesis regulator and Wnt signaling molecule. IA-associated PPIL4G132S disrupts this interaction, resulting in reduced Wnt signaling activity. The phenotypic overlap between ppil4 and jmjd6 knockout zebrafish further suggests that the impaired PPIL4–JMJD6 interaction explains the cerebrovascular defects caused by PPIL4 depletion.
Together, our results suggest that PPIL4, as an IA susceptibility gene, is a novel and key regulatory molecule in Wnt signaling and vertebrate cerebrovascular development. These results hold potential to improve understanding of IA pathogenesis as well as vertebrate cerebrovascular development, stimulating research into new diagnostic and therapeutic options.
Methods
Human subjects.
The study protocol was approved by the Yale Human Investigation Committee (protocol no. 9406007680). Institutional review board approvals for genetic imaging studies, including written consent forms from all study participants, were obtained by the referring physicians at the participating institutions. There were no charges for the participation.
Discovery, replication and control cohorts.
To identify candidate disease-causing genes, we followed a stringent method and focused and sequenced only the families with five or more affected individuals diagnosed with IA. We identified four families in our cohort meeting these criteria: IA200 (five cases), IA201 (five cases), IA20 (seven cases) and IA112 (six cases). The DNA samples from the IA200 and IA201 families were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research with material transfer agreement number MTI.10482 (family ID: NINDS2126; affected individuals: ND03818 (IA200-1), ND03819 (IA200-2), ND03809 (IA200-3), ND03816 (IA200-4), ND03811 (IA200-5); ND01998 (IA201-1), ND01996 (IA201-2), ND02339 (IA201-3), ND01773 (IA201-4) and ND09131 (IA201-5)). In each family, we whole-exome sequenced at least three individuals, including the index cases. The variants co-segregating in all affected members were identified using Sanger sequencing. Our replication cohort comprises 476 ethnically diverse in-house singleton cases (430 European, 20 African American, nine Mexican, five East Asian and 12 undetermined ethnicity) with a saccular IA diagnosis, which was confirmed by at least one imaging study (computed tomography angiography, magnetic resonance angiography or conventional angiography) in all patients. Genomic DNA was extracted from peripheral blood samples of cases using the Qiagen DNeasy Blood & Tissue Kit (Qiagen, 69504). Following the application of study inclusion criteria below, 430 singleton index European cases remained for the analysis. WES data of the European control cohort were obtained from the Simons Simplex Collection (SSC), which consists of 2,886 European parents of autistic children18. Permission to access the genomic data in the SSC in the National Institute of Mental Health Data Repository was obtained. Written informed consent for all participants was provided by the Simons Foundation Autism Research Initiative.
WES.
Samples were prepared and sequenced at the Yale Center for Genome Analysis. Genomic DNA from blood was isolated using NimbleGen version 2 exome capture reagent (Roche) followed by paired-end sequencing using Illumina HiSeq 2000 or HiSeq 2500. Sequencing reads were mapped to the reference genome GRCh37/hg19 using the GATK Best Practice workflows, which include duplication marking, indel realignment and base quality recalibration, as previously described15–17. Variants, including single-nucleotide changes and small indels, were called using GATK HaplotypeCaller and were further annotated using ANNOVAR (v2015Mar22) for MAF in gnomAD (version 2.1.1)19, and the CADD version 1.3 algorithm49 was used to predict the deleteriousness of missense variants. The ClinVar database50 was used to populate the relationship between identified variants and human disease phenotypes, resulting in removal of 17 individuals with known disease-causing mutations.
Principal component analysis.
EIGENSTRAT software51 was used to analyze single-nucleotide polymorphisms in cases and HapMap samples with known ethnicities. Principal component analysis was performed to cluster the studied samples with HapMap samples using R software (version 3.4.1)52. The ethnicities of the cases were determined based on their best clustered ethnicity group against HapMap populations.
Kinship analysis and related sample removal.
Sample relationships were confirmed using the pairwise identity-by-descent (IBD) calculation in PLINK53. An orthogonal analysis that calculated the overlap of high-confidence rare variants (MAF = 0% in ExAC, 1000 Genomes and EVS) between each pair of individuals was conducted to reassure the results. If the IBD sharing or rare variant sharing between a pair of samples was 20% or more, the sample with greater sequence coverage was kept in the analysis and the other discarded. This analysis removed six related individuals.
Variant filtering.
Variant filtration for the co-segregating genes within the four families—IA20, IA112, IA200 and IA201—was performed according to variant sequencing quality, MAF and deleteriousness of the variant and arterial expression of the gene. Only the variants with high sequencing quality were included in the analysis (that is, VQSR ‘PASS’, read depth ≥8 and genotype quality score ≥20). We filtered the rare dominant variants with an exome and genome non-Finnish European MAF <1 × 10−4 in gnomAD. We further filtered the genes that displayed greater than 75th percentile average arterial expression (GtEX average transcripts per million (TPM) in aorta, coronary artery and tibial artery). Only LoF variants (canonical splice site, frameshift insertion/deletion and stop–gain) and deleterious missense variants (annotated as CADD version 1.3 Phred score ≥30) were used for further analysis. The same variant quality, deleteriousness and MAF thresholds were used for burden analysis.
Case–control burden analysis.
A two-tailed Fisher’s exact test was performed to compare the mutational burden of LoF + D-Mis mutations in 434 European individuals (430 singleton index cases and four familial index cases) versus 2,886 European autism parents18 and European individuals (non-Finnish + Finnish) in the gnomAD19. The same filtering criteria were applied to filter the variant calls from the control sets. The total number of alleles evaluated per gene was taken as the maximum of the allele numbers reported for all positions in a gene. An additional three missense mutations (IA510, IA553 and IA447) and one in-frame mutation (IA242) were identified in the IA replication cohort when MAF and deleteriousness thresholds were relaxed. These mutations are reported in Supplementary Tables 2 and 3. However, these mutations were not included in the case–control burden analysis.
Sanger sequencing.
All co-segregating variants found in WES analysis of IA200, IA201 and IA20 families and the PPIL4 variants identified in the IA cohort were confirmed by Sanger sequencing following standard protocols, and the polymerase chain reaction (PCR) amplification was carried out in an S1000 Thermal Cycler (Bio-Rad). No co-segregating variants were identified in affected family members of IA112.
Modeling of the protein structure of PPIL4.
A modeled structure of the cyclophilin-like domain of PPIL4 was created on the PHYRE2 web server in intensive mode using cyclophilin CeCYP16-like domain of the serologically defined colon cancer antigen 10 from Homo sapiens (Protein Data Bank: 2HQ6). Structural models were rendered using the PyMOL Molecular Graphics System, version 2.0 (Schrödinger).
In vitro experiments with human samples and cell lines.
Culture conditions and transduction of lentiviral particles.
Primary HUVECs were obtained from the Yale University Vascular Biology and Therapeutics Core facility. Cell culture conditions were maintained at 37 °C and 5% CO2. EGM-2 Endothelial Cell Growth Medium-2 BulletKit (Lonza, CC-3162) was used to grow HUVECs. Lentiviral shRNA particles targeting human PPIL4 were purchased from Sigma-Aldrich (pLKO vector TRCN0000229276; TRC2). Control experiments were carried out using non-target shRNA lentiviral particles purchased from Sigma-Aldrich (SHC016V-1EA; TRC2). Instructions were followed as recommended by the manufacturer’s protocol for effective transduction and generating stable cell lines. For luciferase and co-immunoprecipitation experiments, HEK293 cells (American Type Culture Collection, cat. no. CRL-1573) were cultured in DMEM supplemented with 10% FCS and transfected using Lipofectamine 2000 (Life Technologies) according to vendor instructions.
Capillary-like tube formation assay.
The tube formation assay using HUVECs was performed on 24 culture plates coated with Matrigel (BD Biosciences, cat. no. 356237), according to the manufacturer’s instructions. HUVECs were seeded onto the coated plates and incubated at 37.8 °C for 18 h, which was followed by Calcein AM (BD Biosciences, cat. no. 354216) staining. Images were captured using fluorescence microscopy and analyzed by ImageJ, Angiogenesis Analyzer.
RNA isolation and sample preparation for quantitative real-time PCR.
The Split RNA Extraction Kit (Lexogen, 008.48) was used for RNA extraction from an equal number of cells in at least three independent biological replicates for every condition. After a total RNA isolation as described above, cDNA generation was done using iScript cDNA Synthesis Kit (Bio-Rad, 1708890). RT–qPCR experiments were performed for assessment of differential expression of PPIL4 and for confirmation of RNA-seq results. The following primer sets were used: PPIL4: F- CCATTTGATGACCCTCCTGATT, R- TGCTCCTATTCGACCACTATCT; GJA4: F- CCTGGAGAAGTTGCTGGAC, R- GAGGATGCGGAAGATGAAGAG; NOTCH1: F- GTCAACGCCGTAGATGACC, R- TTGTTAGCCCCGTTCTTCAG; GPR124: F- AGAAGGTGGAGATCGTGGT, R- CTGAGGTGAAGGGATACTGC; ELN: F- GCAAACCTCTTAAGCCAGTTC, R- GCCAGCCTTAGCAGCTTTATAG. iTaq Universal SYBR Green Supermix (Bio-Rad, 1708890) was used for qPCR reaction, and Bio-Rad CFX384 Real Time System was used for cDNA amplification and quantification. Samples were run in 384-well plates in triplicate as multiplexed reactions with a normalizing internal control (TATA-binding protein (TBP); GeneCopoeia, cat. no. HQP017928). All PCR assays were performed at least three times in three independent experiments. Relative expression of PPIL4 was calculated using the 2−ΔΔCt method. Expression of the genes in wild-type HUVECs was calculated relative to TBP expression level. Statistical analyses were performed using GraphPad Prism, version 8.1.0.
Cell fractionation and immunoprecipitation.
Whole-cell extracts were prepared using a Tris-based buffer, and lysates were cleared by centrifuging at 12,000g for 15 min. Supernatants were used for immunoprecipitation with Anti-V5 or Anti-HA antibodies and Protein G DynaBeads (Invitrogen, 10003D). For fractionation studies, a NE-PER kit (Thermo Fisher Scientific, 78833) was used, and fractions were diluted with Tris-based lysis buffer before immunoprecipitation. Immunoprecipitates were resolved by SDS-PAGE and subjected to western blot analysis using standard methods and visualized by chemiluminescence.
Antibodies.
Immunoprecipitation.
Goat anti-V5 (Bethyl Laboratories, cat. no. A190-119A) was used at 1–2 μg mg−1 of lysate or at 1:1,000 dilution.
Western blot.
Primary antibodies.
All primary antibodies were used at 1:1,000 dilution for western blot experiments. Mouse anti-V5 (Life Technologies, isotype IgG2a, R960-25), mouse anti-HA (Millipore, 05-904; isotype: IgG3; clone: HA.C5), mouse anti-ACTB (Millipore Sigma-Aldrich, a1978; clone: AC-15), anti-lamin A/C (Cell Signalling Technology, 2032S) and anti-alpha tubulin (T5168, Sigma-Aldrich; clone: B-5-1-2). Secondary antibodies. All secondary antibodies were used at 1:5,000 dilution for western blot experiments: goat anti-mouse IgG light chain specific HRP-conjugated (115-035-174, Jackson ImmunoResearch), goat anti-mouse IgG Fcy fragment specific HRP-conjugated (115-035-071, Jackson ImmunoResearch) and donkey anti-rabbit IgG, HRP-linked F(ab’)2 fragment (Amersham, NA9340).
Immunostaining.
Primary antibodies.
Anti-PPIL4 (mouse, Sigma-Aldrich, WH0085313M1; clone: 1C10), used at 5 μg ml−1; anti-JMJD6 (rabbit, Abcam, ab64575), used at 1 μg ml−1; anti-PECAM (mouse, Thermo Fisher Scientific, MA5-13188; clone JC/70 A), used at 1:100; anti-PPIL4 (rabbit, Thermo Fisher Scientific, PA5-30859), used at 1:100; human VE-cadherin antibody (goat, R&D Systems, AF938), used at 10 μg ml−1; and anti-PPIL4 (rabbit, Sigma-Aldrich, Human Protein Atlas Antibodies, HPA031600), used at 1 μg ml−1. Secondary antibodies. All secondary antibodies were used at a dilution of 1:1,000: Alexa Fluor 488 anti-mouse IgG (goat, Thermor Fisher Scientific, a11001); Alexa Fluor 633 anti-mouse IgG (goat, Thermo Fisher Scientific, a21052), Alexa Fluor 555 anti-mouse IgG (goat, Thermo Fisher Scientific, a21424); Alexa Fluor 488 anti-rabbit IgG (goat, Thermo Fisher Scientific, a11034); Alexa Fluor 555 anti-rabbit IgG (donkey, Thermo Fisher Scientific, a31572); and Alexa Fluor 555 anti-goat IgG (donkey, Thermo Fisher Scientific, a21432)
hLuciferase (reporter) assay.
Cells were plated in 960-well plates 24 h before transfection and harvested for luciferase assay 48 h after transfection. Luciferase assay was performed using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s instructions. Renilla plasmid was used as an internal control to normalize the firefly luciferase activity.
Evaluation of PPIL4 expression using publicly available GtEX data.
Average arterial expressions (average TPM values of tibial, coronary and aorta) for all genes were obtained from the GtEx portal (https://gtexportal.org/home/). Arterial expression of PPIL4 was found above the 75th percentile among the coding genes (HUGO Gene Nomenclature Committee approved). Arterial enrichment ratio of PPIL4 expression was found using the following ratio: (average arterial expression (TPM)/average expression of all tissues (TPM)). PPIL4 expression was found above the 82nd percentile in the rank list of arterial enrichment ratio (Fig. 1d).
Evaluation of endothelial expression of PPIL4 using publicly available specific scRNA-seq data.
Publicly available mouse brain vasculature scRNA-seq was used for the following experiment27,28. Pipeline and pre-processing were done by filtering for mitochondrial genes, library size and cell doublets and then by undergoing library normalization and log-transformation using ‘scprep’54. Data were then imputed using Markov Affinity-based Graph Imputation of Cells32; cluster number was determined using the silhouette score after linear dimensionality reduction, which revealed two major clusters. ‘Mean difference’ was used to identify specifically expressed genes in two clusters. Characteristics of these clusters were assessed using the top 50 genes specific for venous and arterial endothelial cells in mouse brain vasculature27,28. Using gene set enrichment analysis55, we found that venous endothelial cell genes were enriched in Cluster 0, whereas there was a significant enrichment for the arterial endothelial cell genes in Cluster 1 (FDR and family-wise error rate P = 0 for both clusters). Hence, we putatively named Clusters 0 and 1 as ‘Venous’ and ‘Arterial’, respectively. Visualization was done using the non-linear dimensionality reduction method PHATE56. Gene–gene interaction was elucidated using knn-DREMI, as previously described32. Gene enrichment analysis was performed using KEGG pathways obtained from the Molecular Signatures Database33 among the genes that were positively regulated with Ppil4 in mouse brain vascular endothelium identified in knn-DREMI analysis.
Immunostaining of human cerebral arteries.
Samples of circle of Willis vessels were obtained from the Department of Pathology at the Yale School of Medicine. Transverse sections of the middle cerebral artery were obtained via cryosectioning and immunostained with anti-PECAM (mouse, Thermo Fisher Scientific, MA5-13188) and anti-PPIL4 (rabbit, Thermo Fisher Scientific, PA5-30859) and appropriate secondary antibodies. Images were captured with a Zeiss Lightsheet Z.1 Dual Illumination Microscope and assembled using Zeiss LSM Image Browser. Minimum–maximum adjustments were applied to all channels equally.
Zebrafish experiments.
Zebrafish strains and husbandry.
Zebrafish were raised and maintained at 28.5 °C using standard methods (unless otherwise indicated) and according to protocols approved by the Yale University Institutional Animal Care and Use Committee (2017-11473). Wild-type zebrafish from the AB strain were used to generate mutant ppil4 and jmjd6 lines. The following transgenic lines were crossed with ppil4ya337 mutants and used for this study: tg(kdrl:grcfp) zn1 tg(kdrl:NLS-GFP), tg(kdrl:ras-mCherry)s896, tg(gata1a:DsRed)sd2 and tg(7xTCF-Xla.Siam:nlsmCherry)ia5.
Generation of ppil4ya337 zebrafish mutants.
The ppil4ya337 line was generated by CRISPR–Cas9 mutagenesis. Two single guide RNAs (sgRNAs) were designed using the CRISPRscan algorithm57 to target exon 5 of the genomic region encoding for the ppil4 zebrafish protein: gRNA(a): 5′-taatacgactcactataGGGAGAGGTCACGGAGGGTAgttttagagctagaa-3′ and gRNA(b): 5′-taatacgactcactataGGGTTCGGAGAGGTCACGGAgttttagagctagaa-3′. sgRNAs and Cas9 were synthetized as previously described58, and 30 pg of sgRNAs and 100 pg of Cas9 RNA were injected into one-cell stage embryo. Mosaic founders were raised and outcrossed. Genomic DNA from the caudal fin of adults was extracted and processed as previously described59. Founder carrying a mutation of 11-bp deletion (−11Δ) was identified and characterized. LoF was assessed by both in situ hybridization and qPCR. F2 generation was established in several transgenic backgrounds, and homozygote (ppil4−/−), heterozygote (ppil4+/−) and wild-type (ppil4+/+) animals were identified by 6-FAM fluorescent PCR fragment analysis using the following primers: forward: 5′-TTTCTGATCACCACAGGAGAGA-3′ and reverse: 5′-TTCCTACCTGATGTCCTGGAA-3′.
Generating JMJD6 zebrafish mutants.
jmjd6 mutant lines were generated by CRISPR–Cas9 mutagenesis. An sgRNA was designed using the CRISPRscan algorithm57 to target exon 3 of the genomic region encoding for the jmjd6 zebrafish protein: gRNA(a): 5′-taatacgactcactataGGTTCAGGGCCACAAGCGCgttttagagctagaa-;3′ sgRNA and Cas9 were synthetized as previously described58, and 30 pg of sgRNA and 100 pg of Cas9 RNA were injected into one-cell stage embryo. Mosaic founders were raised and outcrossed. Genomic DNA was extracted from the caudal fin of adults. Founder carrying a mutation of 2-bp deletion (−2Δ) was identified and characterized. LoF was assessed by qPCR and Sanger sequencing. F2 generation was established in several transgenic backgrounds, and homozygote (jmjd6−/−), heterozygote (jmjd6+/−) and wild-type (jmjd6+/+) animals were identified by Sanger sequencing using TTGCATTGAAATATCGAATTTAGAG and CTAGGCTAGTTTTTATAGAAGATGG primers.
Rescue experiments.
Targeted mutagenesis was performed to generate PPIL4 G132S mutation carried by affected individuals in IA200. The capped mRNAs were synthesized from the linearized plasmids using the T7 mMessage mMachine Transcription Kit (Thermo Fisher Scientific, AM1344). Both wild-type PPIL4 mRNA and the mRNA sequences harboring human mutation G132S were independently injected at one-cell stage of zebrafish embryos for global rescue experiments. Furthermore, we expressed human wild-type PPIL4 (hPPIL4) specifically in endothelial cells using the fli1a promoter in zebrafish. We injected kdrl:GFP ppil4−/− zebrafish in the Tg(fli1a:gal4) background with UAS:PPIL4-tagRFP. In total, 21 zebrafish were injected and survived to be analyzed. Homogenous overlap of UAS:PPIL4-tagRFP with kdrl:gfp was observed in six zebrafish, which were further used for the phenotypic assessment. For presentation, data were assembled using Imaris (Bitplane) software. Signal intensity of the red channel was individually increased, if needed, to demonstrate homogenous and overlapping expression of tagRFP within the cerebrovascular network.
Live image acquisition and quantitative analysis of zebrafish.
For imaging, zebrafish embryos were treated with 0.003% 1-phenyl-2-thiourea (PTU; Sigma-Aldrich, P7629) at 8 hpf to prevent pigmentation. For live imaging, embryos were anesthetized with 0.5% Tricaine (Sigma-Aldrich, A-5040) in egg water and mounted in 1% low-melting agarose (Bio-Rad, 1613114) and egg water. Images were captured with a Zeiss LSM 710 or a Leica Microsystems SP5 confocal microscope using a ×25 objective. Maximum intensity projections (MIPs) were generated with Imaris software. The 3D structure of brain vasculature was reconstructed using Imaris or Neurolucida software (MBF Bioscience). The dorsal cerebral venous system (mesencephalic, dorsal longitudinal and posterior cerebral veins) was removed either during image acquisition or by using ‘Clipping Plane’ (Imaris) during image processing. The midbrain and hindbrain blood vessels were traced and skeletonized, and the quantitative measures of the branch number, the total CtA length (mm) and the branch depth (centrifugal order) were obtained with Imaris software. These measurements were used to characterize the complexity of the vascular network. Average diameter (μm) was calculated using Neurolucida software. Bright-field images were acquired with a Nikon Eclipse 80i microscope with a ×20 objective.
Quantification of RBC velocity and blood flow.
The axial line scan option in a Zeiss 710 confocal microscope was used to determine RBC velocity. A line was drawn inside the lumen and parallel to the longitudinal axis of the vessel. The average length of the line was 9.451 ± 0.067 μm. The scan speed was chosen as maximum per second (Hz), which was 1,058.2 Hz for 20,000 cycles (945.45-μs scan time per cycle). Total scan duration was 18.9 s for every measurement. RBC velocity was measured in 555 brain vessels of 25 zebrafish (7 ppil4+/+, 11 ppil4+/− and 7 ppil4−/−). A snapshot of the analyzed area was taken to determine the diameter of the scanned vessel for flow quantifications. A kymograph was generated in TIFF format after every measurement by using the Zeiss LSM Image Browser. The following pre-processing steps were followed. Erythrocyte recognition and segmentation were performed using Fiji (ImageJ). First, the image was processed using ‘noise despeckle’ and ‘enhance contrast’ steps. Additionally, brightness and contrast were appropriately adjusted if needed. Then, the erythrocytes were segmented, thresholding was adjusted and the slope of the erythrocytes was calculated automatically using the ‘Analyze particle’ option in the ‘Analyze’ panel. Next, the velocity of each erythrocyte was calculated using the following equation: velocity = (1/tan(a)) × (length of the y axis of the kymograph / duration of the total scan).
The time–velocity plot generally displayed a clean sinusoidal rhythm, indicating systolic and diastolic cycles. We averaged systolic peak velocities and end diastolic velocities separately to measure the mean velocity. We next calculated the flow in midbrain CtAs using the following equation: flow = velocity × π × radius2.
Evaluation of the cardiac function.
Cardiac function was assessed by quantifying heart rate and RBC velocity in large brain arteries (circle of Willis vessels). The comparison was only performed between the vessels, which did not show any significant difference in terms of RBC velocity, such as posterior communicating segment (PCS), internal carotid artery (ICA) segment and metencephalic artery (MtA).
Vascular resistance calculations.
The Hagen–Poiseuilles equation—R = 8Ln/π × r4)—was used for calculating the resistance, where R represents resistance, L represents length, n represents viscosity, π is the pi number and r is the radius. Diameter and length were calculated using Imaris software.
Blood flow simulations and evaluation of the wall shear stress using ANSYS Fluent 19.2 computational fluid dynamic software.
Computational simulations of the blood flow and wall shear stress calculations were performed in 3D vascular models of zebrafish cerebrovascular network using ANSYS Fluent 19.2 software. Midbrain and hindbrain central arteries as well as circle of Willis vessel segmentation were obtained in light sheet z-stack images followed by Imaris Filament Application (Bitplane 9.6). After the segmentation steps, 3D models were saved as WRL files and pre-processed in SpaceClaim (ANSYS SpaceClaim 19.2) software. The models were then meshed in ANSYS using computational fluid dynamics application settings. Meshing orthogonal quality was calculated, and the objects that were less than 0.01 were excluded. Inlets and outlets were determined in the 3D model. Mass flow rate was determined as 5.66 × 10−10 (kg s−1), which was calculated based on the axial line scanning flow calculations. Flow rate weight for each inlet and outlet were determined according to diameter. Blood density and viscosity were considered as 1,060 kg m−3 and 0.00278 kg ms−1, respectively. For each analysis, 300 iterations were performed, and convergence occurred between 200 and 270. The flow characteristics of the blood were considered as incompressible and laminar.
Quantification and comparison of hematocrit levels.
Hematocrit is a major modifier of the viscosity, and the flow is dependent on the amount of RBCs. As previously mentioned, the erythrocyte number analyzed during 18.9 s of scan time was significantly correlated with the mean velocity. Thus, to compare the hematocrit in zebrafish, we first assessed the number of the RBCs in the kymographs of the vessels, such as PCS, ICA and MtA, which did not show any significant difference in RBC velocity. An additional measurement was performed to calculate the hematocrit by taking the fraction of RBC pixels over the total number of pixels, as previously reported31.
Whole-mount in situ hybridization and immunofluorescence imaging.
Embryos were raised to the desired developmental stage in 0.003% PTU to prevent pigmentation and fixed with 4% formaldehyde overnight. The next day, embryos were washed four times with PBS-Tween and stored at −20 °C in 100% methanol for at least 2 h. To generate the ppil4 sense and antisense riboprobes, a fragment of ppil4 coding sequence was amplified using the following primers: forward: 5′-ACTTCCTGAAACTGTGCAAAATC-3′ and reverse: 5′-AGTCTTGCTTGCTTTTCACTTTG-3′. The 1,002-bp fragment was then cloned into a pGEM-T Easy Vector (Promega, A1360). ppil4 riboprobe was synthesized and labeled with digoxigenin using the DIG RNA Labeling Kit (SP6/T7, Roche Applied Bioscience, 11175025910). Fluorescence in situ hybridization (FISH) was performed as previously described60. Riboprobes were hybridized overnight at 60 °C. The FISH signal was detected using a TSA Plus Fluorescence Kit (PerkinElmer, NEL741E001KT). The same immunohistochemistry protocol was applied for both gastrula stage and 2.5-dpf embryos, with some modifications. For 2.5-dpf embryos, fixed samples were rehydrated with a graded series of methanol/PBS, with final wash in PBS-Tween. Samples were then digested in 0.125% trypsin for 5 min and incubated for at least 2 h in blocking solution (0.8% Triton-X, 10% normal goat serum, 1% BSA, 0.01% sodium azide in PBS-Tween) at room temperature before incubation with primary antibody at 4 °C. For gastrula stage samples, digestion with trypsin was not performed.
The following primary antibodies were used for imaging. (1) Chicken anti-GFP (AbCam, ab13970), used at 1:250 and 1:300. (2) Rabbit anti-caspase-3 (BD Pharmingen, 559565; clone: C92-605). (3) Rabbit anti-caspase-3 (BD Pharmingen, 559565; clone: C92-605), used at 1:250. (4) Mouse anti-GFAP (Abcam, ab4648; clone: 2A5), used at 1:300. (5) Mouse anti-HU (Molecular Probes, A-21271; clone: 16A11), used at 1:250. (6) For in-situ hybridization, anti-DIG POD primary antibody (Fab fragment from sheep; Roche, 11207733910), used at 1:1,000. Goat polyclonal antibodies coupled to Alexa Fluor 488, 546 and 647 dyes (Thermo Fisher Scientific) were used at 1:300 as secondary antibodies: goat Alexa Fluor 488 anti-chicken IgG, A-11039; goat Alexa Fluor 488 anti-mouse IgG, A-11001; goat Alexa Fluor 488 anti-rabbit IgG, A-11008; goat Alexa Fluor 546 anti-mouse IgG, A-11003; goat Alexa Fluor 546 anti-rabbit IgG, A-11035; goat Alexa Fluor 647 anti-mouse IgG, A-21235; and goat Alexa Fluor 647 anti-rabbit IgG, A-21244. When immunohistochemistry was combined with FISH, 1:300 chicken anti-GFP (Abcam, ab13970) was incubated with anti-DIG POD primary antibody, and Alexa Fluor 488 goat anti-chicken IgG (Thermo Fisher Scientific) secondary antibody was applied as the last step after TSA reaction. Processed samples were mounted in 1% low-melting-point agarose in 1× PBS. Pictures were taken with a Leica Microsystems SP5 confocal microscope using a ×25 immersion objective.
o-Dianisidine staining.
Staining RBCs was performed on at least three separate clutches. Non-fixed embryos were stained by o-Dianisidine (Sigma-Aldrich, D9143) for 45 min in the dark. Stained embryos were then fixed, and embryos showing head hemorrhages were counted and genotyped. Bright-field pictures were acquired with a Nikon Eclipse 80i microscope with a ×20 objective.
Epinephrine treatment.
To induce hemodynamic stress in zebrafish embryos, epinephrine (Sigma-Aldrich, E4642-5G) was added to egg water with 0.003% PTU to a final concentration of 0.2 mM. Embryos in the double transgenic background Tg(gata1a:DsRed; kdrl:grcfp)sd2;zn1 were treated from 2.5 dpf to 3 dpf. For rescue experiments, embryos were first injected with the indicated mRNA at one-cell stage. Embryos were then processed for o-Dianisidine staining or used for live imaging. The effect of ppil4 depletion in adult heterozygous zebrafish was assessed by administrating 0.5 mg kg−1 of epinephrine in Ringer’s solution via retro-orbital injection in 3-month-old kdrl:GFP/gata1:dsred zebrafish derived from ppil4+/− in-crosses. Zebrafish were injected twice a day for 4 d and then killed; then, brains were dissected, fixed, imaged and genotyped. Hemorrhage was assessed under bright-field microscopy blinded to genotype.
TUNEL assay.
Apoptotic cells were detected by TUNEL assay. Whole-month embryos at 2.5 dpf were fixed in 4% paraformaldehyde overnight and stored in 100% methanol at −20 °C. The TUNEL assay was performed using the ApopTag Red In Situ Apoptosis Detection Kit (Millipore, S7165). Stained samples were genotyped and imaged.
Quantitative RT–PCR in zebrafish embryos.
Total RNA from adult fin clips or pooled embryo heads or whole bodies was isolated using TRIzol reagent (Life Technologies, 15596026), according to the manufacturer’s protocol. Next, 0.5–0.1 μg of total RNA was used to synthesize cDNA with the iScript cDNA Synthesis Kit (Bio-Rad, 1708890), following the manufacturer’s standard protocol. RT–qPCR primers were designed with an amplicon size range of 110–160 bp with CG clamp at the 3′ end using Primer3 software and tested for specificity by blasting the sequences against the zebrafish genome on Ensembl BLAST. Zebrafish β-actin was used as a reference gene (sequences are available upon request). Amplification efficiency of each primer was determined from analysis of a serial dilution of control cDNA. The efficiency of all the primers was greater than 90% (slope = −3.22 to −3.41 and R2 = 99%). qPCR was performed in a total of 10 μl containing 2 μl of cDNA, 5 μl of Roche FastStart Universal SYBR Green Master Mix (Rox) (Sigma-Aldrich, 4913850001) and 300 nM of the forward and reverse primers. The reaction was carried out on a Bio-Rad CFX-384 real-time PCR system under the following conditions: activation of FastStart Taq DNA Polymerase at 95 °C for 10 min followed by 40 cycles of 15-s denaturation at 95 °C and 1-min annealing at 60 °C. After this run, melting curve analysis was performed to identify primer dimers. Genomic DNA contamination and cross-contamination was checked by including no RT control for each sample and no template control for every different gene analyzed during the preparation of samples. Each sample was run in triplicate for each gene to be assayed. Data analyses were conducted using CFX Manager software, version 3.1 (Bio-Rad). Relative expression of each gene was calculated using the 2-ΔΔCt method. Statistical analyses were performed using GraphPad Prism, version 8.1.0.
FACS total RNA extraction.
To assess ppil4 relative expression levels, both endothelial (Kdrl:GFP+) and non-endothelial (Kdrl:GFP−) cells were isolated by FACS from Tg(kdrl:grcfp)zn1 whole-mount embryos at different developmental stages (0.5 dpf, 1 dpf, 3 dpf and 90 dpf). For RT–qPCR experiments, at least three biological replicates were used. For RNA-seq sample preparation, Tg(kdrl:gfp)zn1 embryos were injected with 30 pg of ppil4 sgRNA and 100 pg of Cas9 RNA and raised to 54 hpf. Approximately 100 embryos were collected per biological replicate. Embryos were then dissected, and the head portion was cell dissociated. Endothelial cells (kdrl:GFP+) were isolated by FACS exclusively from the heads of embryos in three independent biological replicate sets for the ‘ppil4 sgRNA + 100 pg of Cas9 RNA’-injected group and two independent biological replicate sets for the control group and prepared for RNA-seq. Four biological replicate sets of wild-type embryos were used to determine transcriptome-wide gene expression levels in brain-enriched endothelial cells. Total RNA was extracted using TRIzol reagent (Life Technologies, 15596026), according to the manufacturer’s protocol.
RNA-seq and data analysis in brain-enriched endothelial cells.
RNA integrity number (RIN) was assessed using an Agilent Bioanalyzer 2100, and samples with an RIN less than 7.5 were excluded. Ribozero depletion was performed, and samples were loaded four per HiSeq 2500 lane (paired-end, 75-bp sequencing). We first trimmed the low-quality bases and removed adapter sequences using cutadapt61. RNA-seq reads were aligned to the reference zebrafish (danRer11) transcriptome using STAR62. HTSeq was conducted for quantification of the transcript counts63. Differential gene expression analysis was performed using DESeq2 (ref. 64). Ensembl and gene symbol IDs mapping in zebrafish were downloaded from the BioMart website65. For differentially expressed genes, enrichment analysis was performed using significantly (FDR < 0.05) differentially expressed genes within the ‘GO-Cellular Components’ obtained from the Metascape database66. Average gene expression (fragments per kilobase of transcript per million mapped reads) in brain-enriched zebrafish endothelial cells from four wild-type biological replicates is presented as Supplementary Table 5.
Assessment of in vivo Wnt signaling activity with Tg(7xTCF-Xla.Siam:nlsmCherry)ia5.
Tg(kdrl:gfp)zn1 ppil4+/− zebrafish were crossed with Tg(7xTCF-Xla.Siam:nlsmCherry)ia547 to generate the ppil4+/− Tg(kdrl:grcfp; 7xTCF-Xla.Siam:nlsmCherry) line. Images were captured using a Leica Microsystems SP5 confocal microscope. The dorsal cerebral venous system (mesencephalic, dorsal longitudinal and posterior cerebral veins) was removed in all images of 60-hpf zebrafish blinded to the genotype either during image acquisition or by using Clipping Plane during image processing. The dorsal cerebrovascular venous network was not removed in 30-hpf zebrafish. Imaris software was used to quantify the overall and endothelial-specific TCF reporter activity by quantifying the number of cells with TCF reporter activity. Although an overall reduction in red channel intensity was observed in ppil4−/− zebrafish (especially in otic vesicle and optic tectum area), the intensity was not used to quantify TCF reporter activity, given that channel intensity could be affected by several factors. TCF reporter activity in overall brain and specific to GFP-expressing endothelial cells was identified and quantified using the ‘Mask’ option within the ‘Spots’ application by counting the number of cells with TCF signal (that is, mCherry+). Overlapped regions/cells (GFP+, nlsmCherry+) were detected automatically in Imaris using user-defined thresholds and pseudo-colored red to visualize TCF-reporting endothelial cells. After the quantification step, images were assembled by overlaying the automatically detected vascular TCF signal and the overall red channel signal from the nlsmCherry+ cells in ‘TCF-reported-overall’ and the ‘MERGED’ panels in Fig. 5, Extended Data Fig. 10 and Supplementary Fig. 4. All analyses were performed blinded, before genotyping. The embryos were grouped according to genotype after measurement of Wnt reporter activity individually. Statistical analysis was performed as described above. The number of TCF-reporting endothelial cells was normalized according to the ratio of midbrain CtA endothelial cell number between ppil4−/− and ppil4+/+ zebrafish using a coefficient of 2.04, which was obtained from the quantification of endothelial cell numbers in 60-hpf zebrafish from both genotypes in the tg(kdrl:NLS-GFP; kdrl:ras-mCherry) background.
Microangiography.
Embryos at 2.5 dpf were anesthetized using 1× tricaine in fish water and injected into the heart with 67-kDa BSA Alexa Fluor 594 conjugate (Thermo Fisher Scientific, A13101). The success of injection was monitored under a stereo-microscope equipped with a dsRed filter set (Leica). Injected embryos were then mounted in 1% low-melting-point agarose (Bio-Rad), and live images were captured 10–30 min after injection with a Leica SP5 confocal microscope using a ×25 objective. Embryos were fixed 30 min after injection. Embryos were imaged using a Leica Microsystems SP5 confocal microscope using a ×25 objective.
X. tropicalis experiments.
Frog husbandry and microinjection of MO and mRNA in X. tropicalis embryos. Xenopus were housed and cared for according to established protocols approved by the Yale University Institutional Animal Care and Use Committee (2015-11035). Embryos were raised to appropriate stages in 1/9MR+ gentamycin. Using standard protocols, we injected antisense oligonucleotide MOs, fluorescent tracers and/or mRNAs into one- or two-cell Xenopus embryos and assayed gene expression by western blot and cranial bleeding by visual inspection and o-Dianisidine staining. The following MO oligonucleotides were used for this study: PPIL4 translational MO (6–12 ng per embryo; 5′-AAGCACCGCCATTCTACTTCGTCCA-3′), standard control MO (6–9 ng per embryo; 5′-CCTCTTACCTCAGTTACAATTTATA-3′) or mCherry tagged mRNA (200 pg per embryo). Mini-Ruby (Invitrogen, D3312) was used for tracer in MO injections. We generated human PPIL4 mRNA as described in the in vitro experiment section. To test the specificity of the phenotype observed upon MO injection, we rescued PPIL4 morphants by co-injecting 9 ng per embryo of the PPIL4 MO (5′-AAGCACCGCCATTCTACTTCGTCCA-3′) with 200 pg per embryo of human wild-type PPIL4 mRNA at one-cell stage. The same rescue experiment was repeated with 200 pg per embryo of human PPIL4 mRNA carrying the missense mutation G132S to test the pathogenic variant’s functionality found in the IA200 family.
Assessment of cerebral hemorrhage and vascular dysmorphology in X. tropicalis embryos.
To assess cranial hemorrhage, frog embryos were collected at stage 39 (dpf 3) and anesthetized with benzocaine. Cranial hemorrhage was scored by visual inspection using a Zeiss SteREO Discovery V20 microscope. Whole-embryo o-Dianisidine (Sigma-Aldrich, D9143) staining was performed to confirm cranial hemorrhage phenotype at stage 39 and vascular dysmorphology at stage 46 (dpf 4). In brief, embryos were collected at corresponding stages and stained for 15 min in the dark in a solution consisting of 0.6 mg ml−1 of o-Dianisidine, 0.01 M sodium acetate (pH 4.5), 0.65% H2O2 and 40% (v/v) ethanol. Embryos were examined by a Zeiss SteREO Discovery V20 microscope. Images were opened with Fiji (ImageJ) Color Mode: ‘Default’ and ‘Autoscale’ using either CZI files or TIFF files generated by Zeiss ZEN lite blue edition software. Contrast enhancement was performed using the ‘Enhance Contrast’ application in the ‘Process’ tab by performing 0.3% pixel saturation in whole image if necessary. Both the ‘autoscale’ and ‘enhanced contrast’ options were applied to all channels and the whole image.
Western blot.
Control and morphant embryos were collected at stage 39 and crushed using a pestle and centrifuged at 12,000g for 10 min at 4 °C to separate protein from lipids and debris. For protein isolation, Xenopus embryos were placed in 1× RIPA buffer (10 μl per embryo) (Cell Signaling Technology, 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg ml−1 of leupeptin) supplemented with protease inhibitors (Complete, Roche, 04693116001). We quantified protein concentration in supernatants using DC Protein Assay (Bio-Rad, 5000111). Western blotting was performed following standard protocols, using anti-PPIL4 (Thermo Fisher Scientific, WH0085313M1, used at 5 μg ml−1) primary antibody and anti-mouse HRP-conjugated secondary antibody (Jackson ImmunoResearch, 715–035–150, 1:15,000 dilution).
Anti-GAPDH (Ambion, AM4300, 1:5,000 dilution) was used as a loading control. Quantifications of protein level changes were calculated using Fiji/ImageJ software by normalizing to GAPDH levels.
Statistics.
All ordinal data obtained from zebrafish phenotypic assessment, and which showed normal distribution, were analyzed using one-way ANOVA followed by Bonferroni, Dunnett or Sidak multiple comparison tests. The Kruskal–Wallis with Dunn’s multiple comparison test was used for the data that do not display normal distribution. For nominal data, Fisher’s exact test, pairwise Fisher’s exact test or the chi-square test was used accordingly after taking sample size and number of variables tested into consideration. Levene’s test was used to test the homogeneity of variances within the datasets. The hypergeometric test was used to determine the significance of overlap between two datasets. Statistical tests used specifically in each experiment are detailed in the text or the figure legends. All statistical tests were performed using RStudio version 1.4.1106 or GraphPad Prism version 8.1.0. SPSS (version 26) was used when GraphPad Prism did not calculate exact P values of the multiple correction tests. P values less than 0.05 were considered statistically significant.
Extended Data
Extended Data Fig. 1 |. Sanger sequencing confirmation of PPIL4 variants identified by whole-exome sequencing (WES).
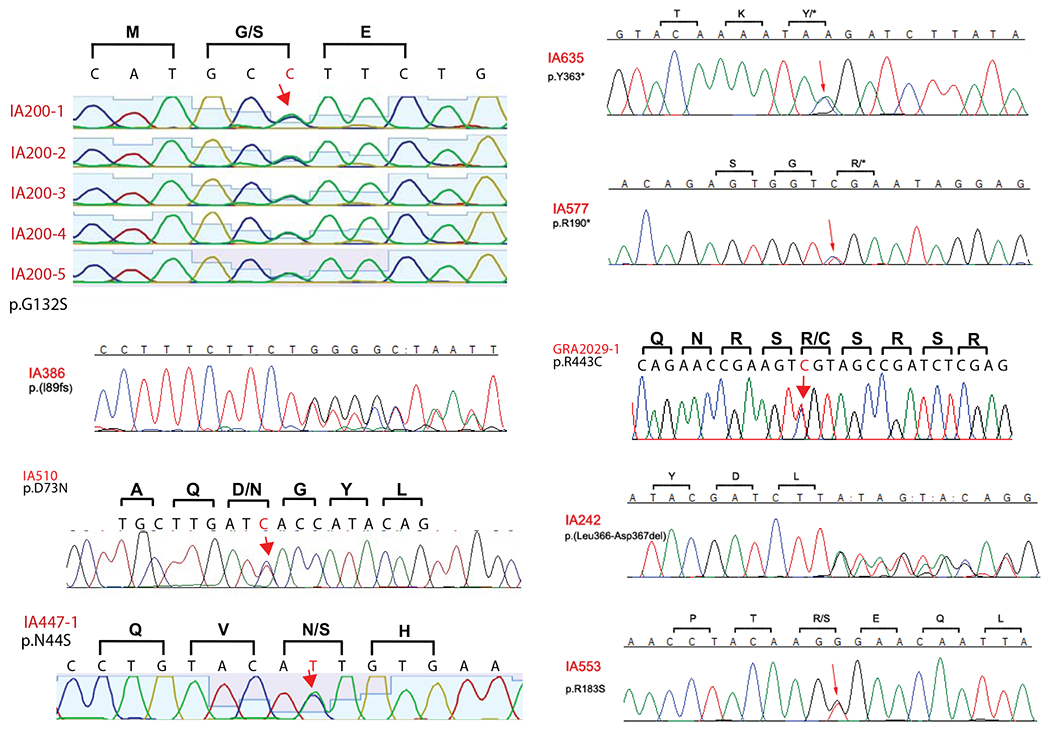
Sanger sequencing confirming heterozygous PPIL4 variants identified in IA patients. Locations of single nucleotide variations (red arrows), deletions (”:”) and the consecutive overlapping sequence. * represents stop gain mutation.
Extended Data Fig. 2 |. Gross morphologic assessment of ppil4 mutant zebrafish.
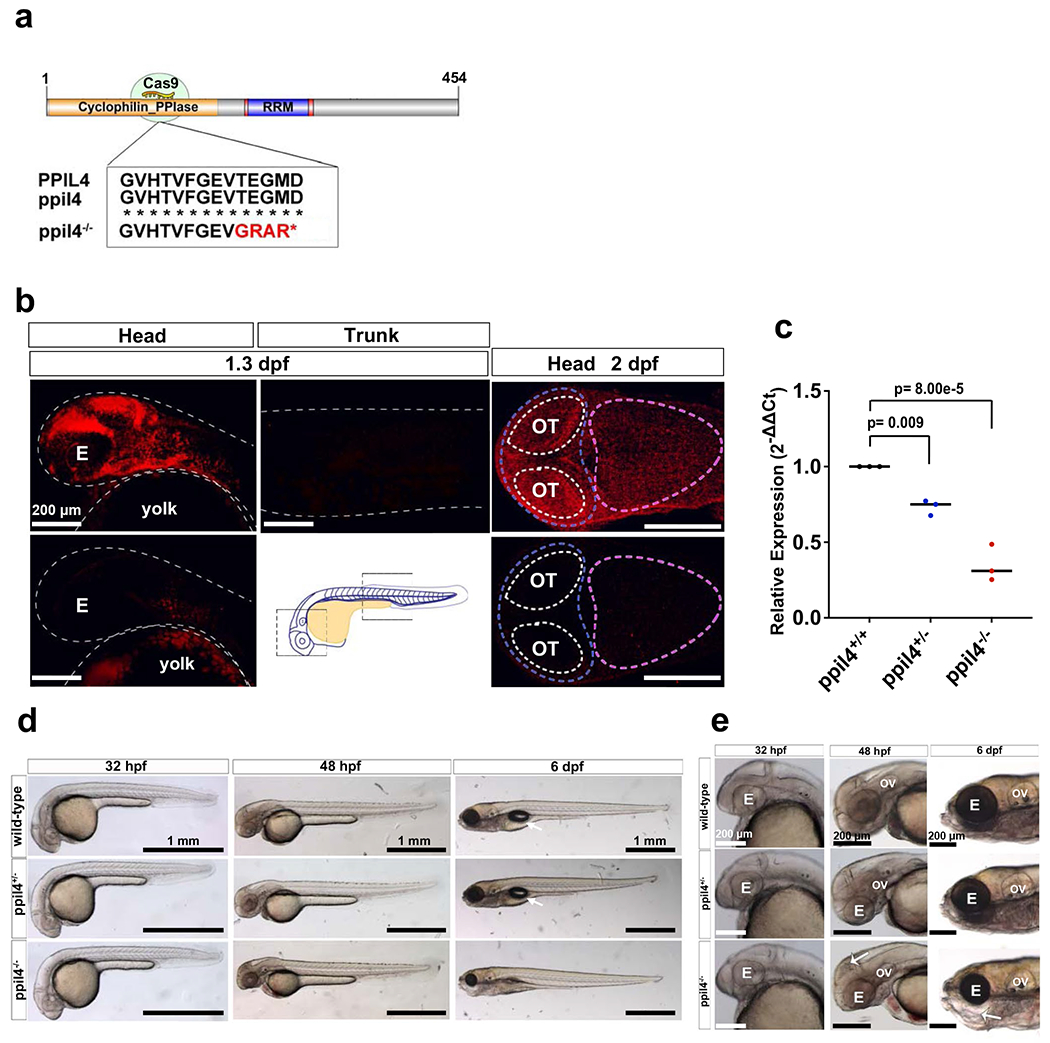
a, Schematic of the zebrafish ppil4 protein showing the location of the stop codon generated in the PPIase domain by Crispr-CAS9. A deletion of 11 bp in exon 5 causes a frameshift and a premature stop codon. b, Fluorescent in situ hybridization for ppil4 in ~1.3 and ~2 dpf wild type embryos (upper row). ppil4 is expressed in the head region but not in the trunk. ppil4 expression is lost in ppil4−/− mutants (lower row). The midbrain (blue-dashed line), hindbrain (pink-dashed), and optic tectum (white-dashed) boundaries are indicated, n= 3 sets of biological replicates with 30 zebrafish (each set) for both timepoints. c, Reduction in ppil4 expression in heterozygous and homozygous mutant zebrafish validated by qPCR. Values shown as fold change in different genotypes (X-axis) relative to wild type (Y-axis), beta actin was used for normalization, n= 3 sets of biological replicates. d, Bright-field images of wild-type, ppil4+/− and ppil4+/− embryos at different stages of development. No gross morphological defects were observed in heterozygous mutants. Arrows (6 dpf): swim bladder. e, ppil4−/− zebrafish manifest necrosis in the head at 48 hpf (white arrow), not evident at 32 hpf and lower jaw abnormality at 6 dpf (white arrow), n=3 sets of biological replicates. Data presented as individual scatter plot with median. Statistical test: One-way ANOVA with Dunnett’s multiple comparison test. E= eye, OV= otic vesicle, OT= Optic tectum. Scale bar: 200 μm in b and e; 1 mm in d.
Extended Data Fig. 3 |. The impact of ppil4 depletion on cerebrovascular network is persistent at 5.5 dpf.
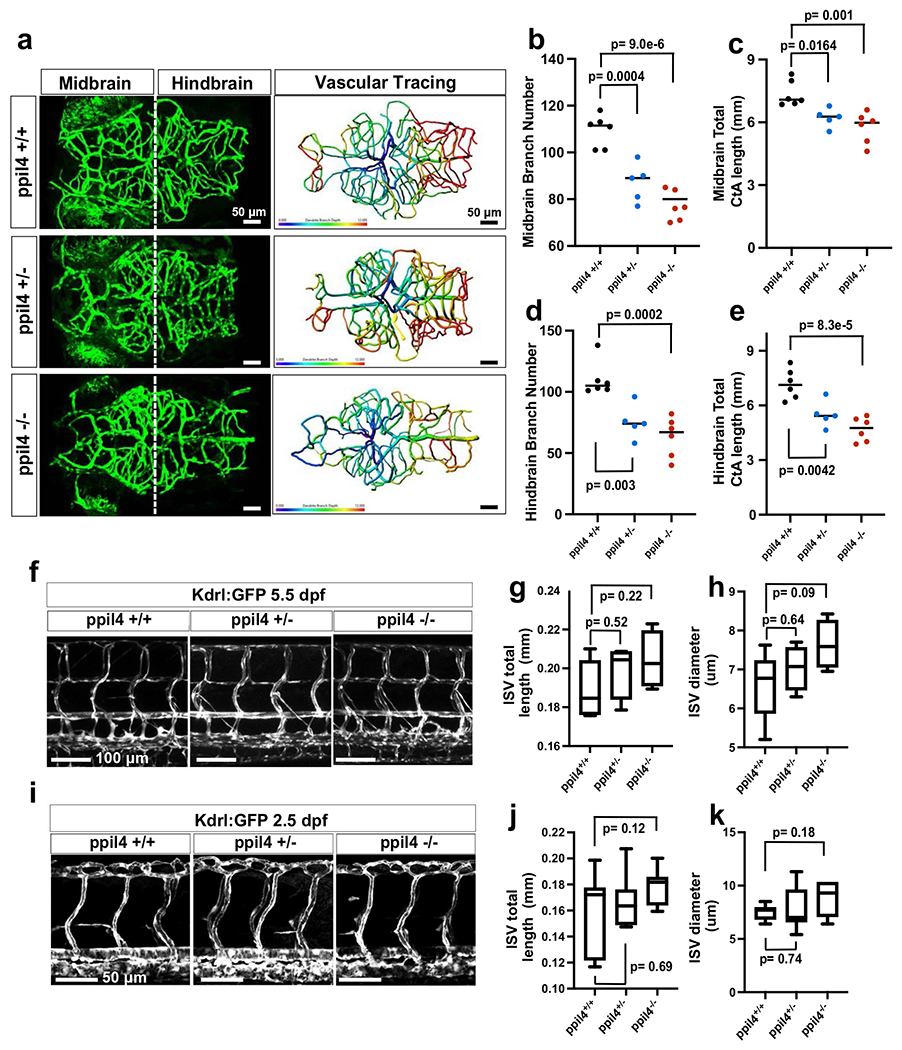
a, Left panels: Maximum intensity projection (MIP) of representative confocal z-stack images in 5.5 dpf old ppil4+/+ (n=6), ppil4+/− (n=5) and ppil4−/− (n=6) embryos in the tg(kdrl:gfp)zn1 background (dorsal-view and caudal facing left). Right panels: Brain vessel segmentation in the larvae shown at left (Imaris). Colors represent branch depth ranging from 0 to 12 (higher and lower branch depth shown in red and blue, respectively). b-e, Quantification of midbrain CtA branch number (b) and length (c); Quantification of hindbrain CtA branch number (d) and length (e) in ppil4+/+ (n=6), ppil4+/− (n=5) and ppil4−/− (n=6) embryos, f-h, Confocal images (f) and comparative assessment (g,h) of trunk vasculature in 5.5 dpf old ppil4+/+ (n=7), ppil4+/− (n=4) or ppil4−/− (n=4) zebrafish. i-k, Confocal images (i) and comparative assessment (j,k) of trunk vasculature in 2.5 dpf old, n=9 per genotype. Individual values shown with scatter dot plot and median in b-e. The box extends from the 25th to 75th percentile. The whiskers show the minimum and the maximum values, while the line in the middle of the box is median in g,h,j and k. Statistical tests: One-way ANOVA followed by Dunnett’s multiple correction for all comparisons. Scale bar: 50 μm in a and i, 100 μm in f.
Extended Data Fig. 4 |. Loss of ppil4 results in reduction in endothelial cell number.
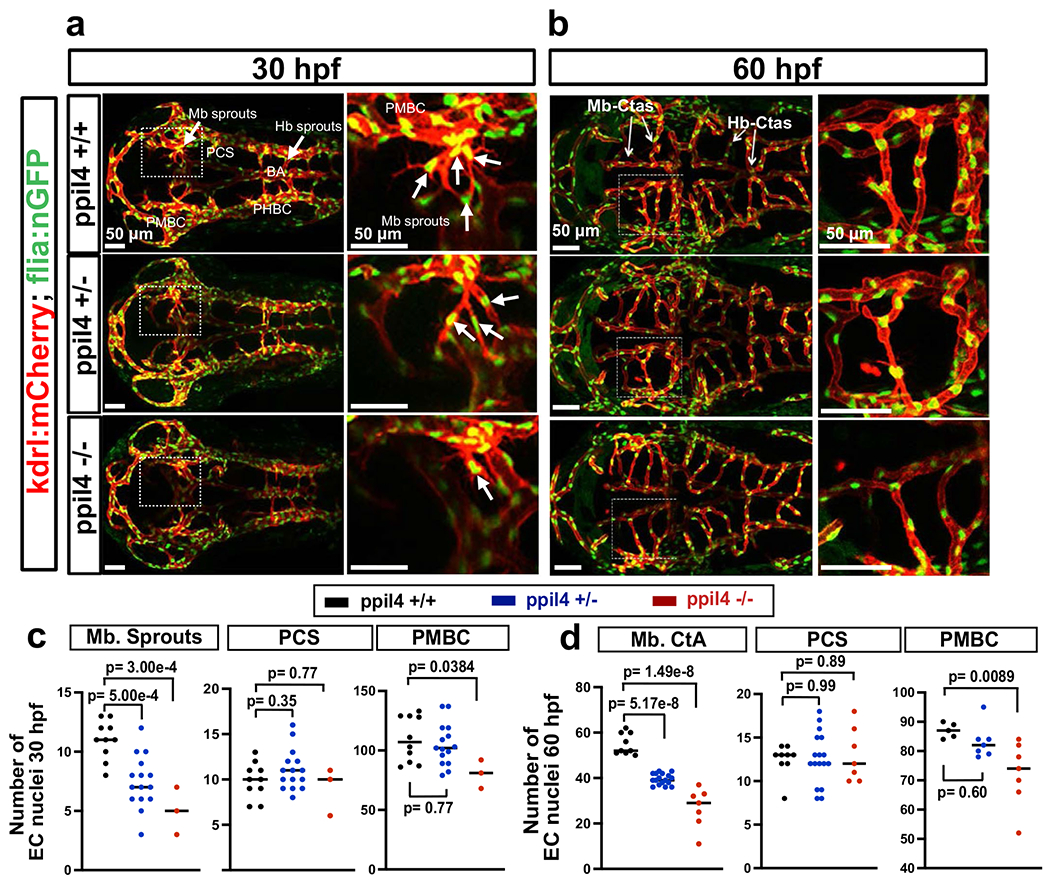
a,b, Confocal images of the cerebral vasculature of (a) 30 and (b) 60 hpf zebrafish in the tg(kdrl:mCherry; fli1:nGFP) background, expressing mCherry and GFP respectively in the cell membrane and nuclei of endothelial cells (ECs) (dorsal view). Arrows indicate angiogenic sprouting in midbrain (a), and hindbrain CtAs (b). c, Comparison of EC number at 30 hpf ppil4+/+ (n=10), ppil4+/− (n=15), and ppil4−/− (n=3) embryos. d, Comparison of EC number in the cerebral arteries of 60 hpf ppil4+/+ (n=9), ppil4+/− (n=17), and ppil4−/− (n=7) embryos. Individual values presented with scatter dot plot and median for all quantifications. Statistical tests: One-way ANOVA followed by Dunnett’s multiple comparison test for all comparisons. Abbreviations: Mb= Midbrain, Hb= Hindbrain, BA= Basilar Artery, PCS= Posterior communicating segment, PMBC= Primordial midbrain channel, PHBC= Primordial hindbrain channel, CtA= Central Artery. Scale bar: 50 μm in a and b.
Extended Data Fig. 5 |. ppil4−/− zebrafish exhibit apoptosis in neurons and radial glia, but not in endothelial cells.
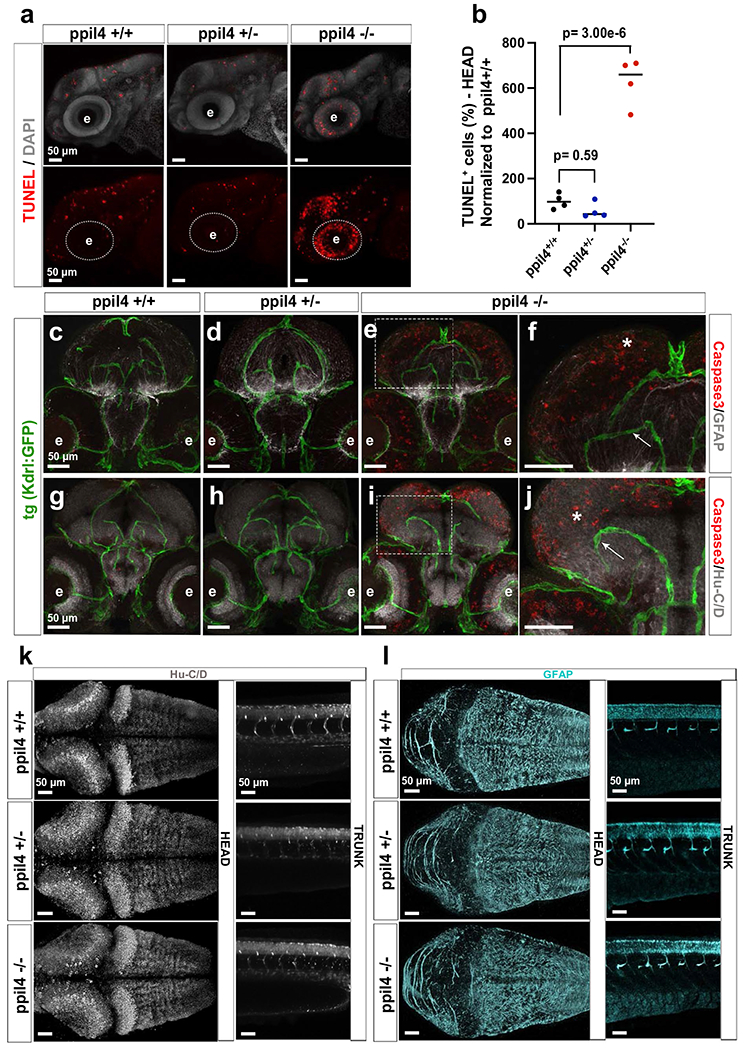
a,b, Representative confocal images of 2.5 dpf embryos, where ppil4−/− mutants exhibit an increase in TUNEL-positive cells in the head region, n=4 sets biological replicates with 30 zebrafish per set. c-j, Cross-sections of the head. Embryos at 2.5 dpf in tg(kdrl:gfp)zn1 background were stained for Caspase-3 and HU (neurons) or GFAP (radial glia). Apoptosis was detected in neurons and radial glia (white asterisks), but not in endothelial cells (white box; arrows). Confocal images, n= 3 sets biological replicates with 30 zebrafish per set. k,l, Whole mount confocal images of 2.5 dpf embryos showing no difference in (k) neuronal (HU+) and (l) radial glial (GFAP+) population in ppil4 mutant genotypes, n= 3 sets biological replicates with 30 zebrafish per set. Dorsal view of the head region (left panel), lateral view of the trunk (right panel). Individual values shown with scatter dot plot and median. Statistical test: (b) One-way ANOVA followed by Dunnett’s multiple comparison test. Abbreviations: TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling; Fb: forebrain; Mb: midbrain; Hb: hindbrain; e=eye. Scale bar: 50 μm.
Extended Data Fig. 6 |. PPIL4 expression in endothelial cells.
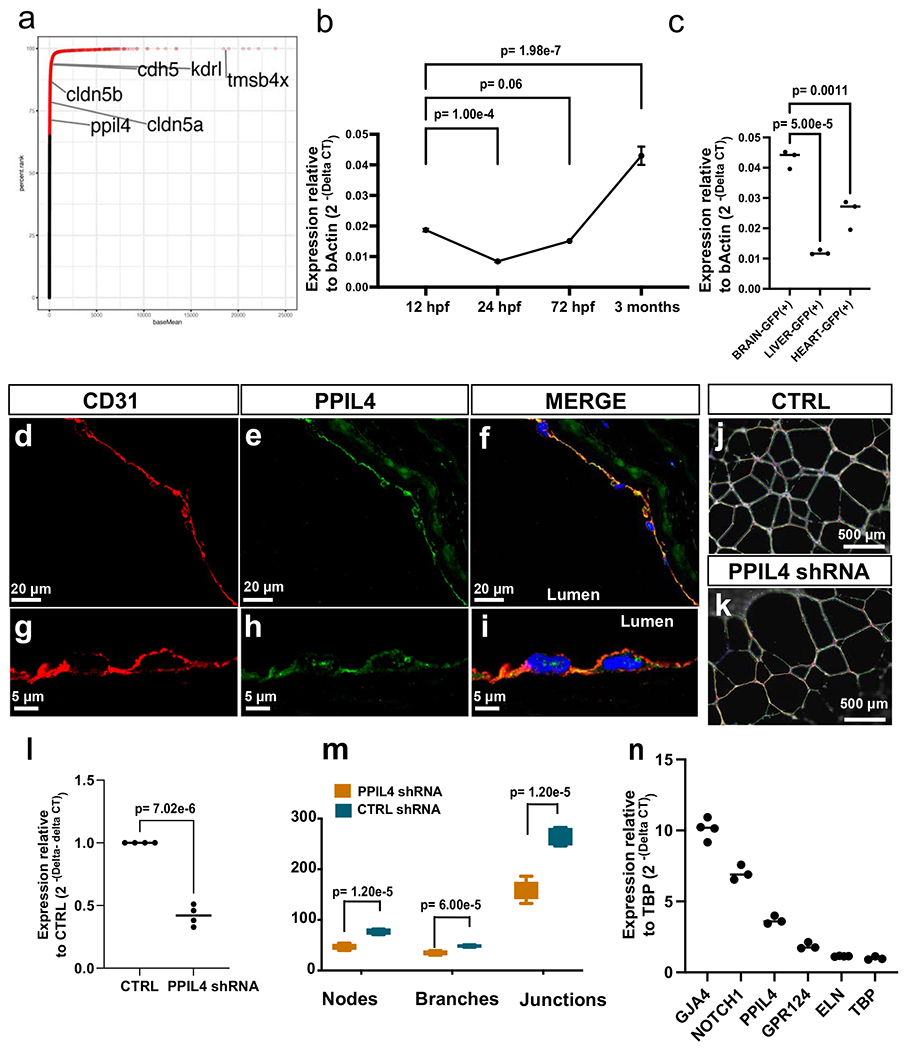
a, Percentile rank of 15,521 transcripts expressed (FPKM) in brain-enriched endothelial cells (EC) in wildtype zebrafish (2.5 dpf), n=4 sets of biological replicates. b, ppil4 expression in zebrafish brain-enriched EC’s at 12, 24, 72 hpf and 3 months (relative to beta actin levels), n=3 sets of biological replicates. c, ppil4 expression levels in kdrl-GFP+ endothelial cells in the brain, liver, and the heart in 3 months old tg(kdrl:gfp) zebrafish (relative to beta actin levels), n=3 sets of biological replicates. d-i, Double immunostaining with pan-endothelial marker CD31 and PPIL4 demonstrating overlap in endothelial cell layer of human middle cerebral artery, n=4 biological replicates. j,k,m, In vitro tube formation assay showing significant impairment in branch formation in shRNA-treated compared with non-target control shRNA treated HUVECs, n=5 biological replicates. l, Reduction in PPIL4 expression in shRNA treated HUVEC validated by qPCR. Values shown as fold change relative to control (relative to TATA-binding protein [TBP]); n=4 biological replicates. m, Reduced number of nodes, branches, and junctions upon PPIL4 downregulation; n=5 biological replicates. n, Expression of PPIL4 and a list of angiogenesis-associated genes in wild type HUVECs (relative to TBP); n= 3 biological replicates. Individual values shown with scatter plot as mean with standard deviation in b; and median in c,l,n. In m, the box extends between 25th-75th percentile. The whiskers show the minimum and the maximum values. Central line is median. Statistical tests: (b,c) One-way ANOVA followed by Dunnett’s multiple comparison. (l,m) Two-tailed Student t-test.
Extended Data Fig. 7 |. Endothelial cell-specific overexpression of PPIL4 in ppil4−/− embryos restores cerebrovascular network simplification.
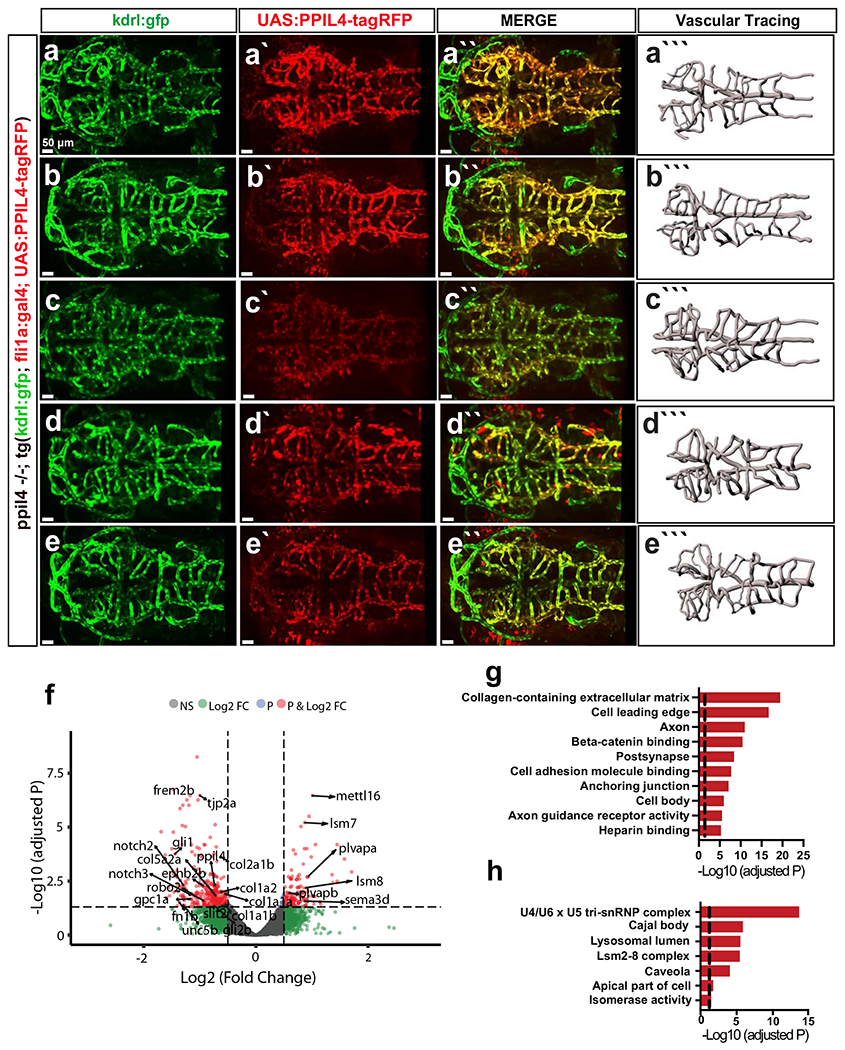
a-e Maximum intensity projection (MIP) of five representative confocal z-stack images of UAS:hPPIL4-WT-tagRFP injected embryos in (ppil4−/−; Tg(kdrl:gfp) zn1; tg(fli1a:gal4)) background at 2.5 dpf (dorsal view; caudal facing left), n=6. GFP+/RFP+ brain vessels, where GFP marks native endothelial cells and RFP endothelial cells overexpressing human WT-PPIL4. a-e, showing GFP-tagged kdrl expressing embryonic endothelial cells and cerebral vessels. a′-e′, PPIL4 overexpression is restricted to endothelial cells. a″-e″ Overlay of red and green channels. a‴-e‴ Vascular tracing performed using Imaris Filament application. f, Volcano plot representing differentially expressed genes in RNA-sequencing results of brain enriched endothelial cells upon abrogation of ppil4 (2.5 hpf). Dots represent genes and colors indicate Log2 fold change (FC) and FDR thresholds with genes meeting both (red), those over Log2FC (blue), those meeting FDR (green), or neither (grey). Horizontal and vertical dashed lines indicate, respectively, thresholds for significance (FDR <0.05) and Log2 FC>0.5. See methods for details of the experiment. g,h, Significant terms from the GO-Cellular Component for significantly differentially downregulated (g) and upregulated (h) genes. Dashed lines showing threshold for significance (FDR=0.05). Scale bar: 50 μm.
Extended Data Fig. 8 |. Genes with strong positive statistical co-dependency with PPIL4 expression are enriched in brain arterial endothelial cells and Wnt signaling pathway.
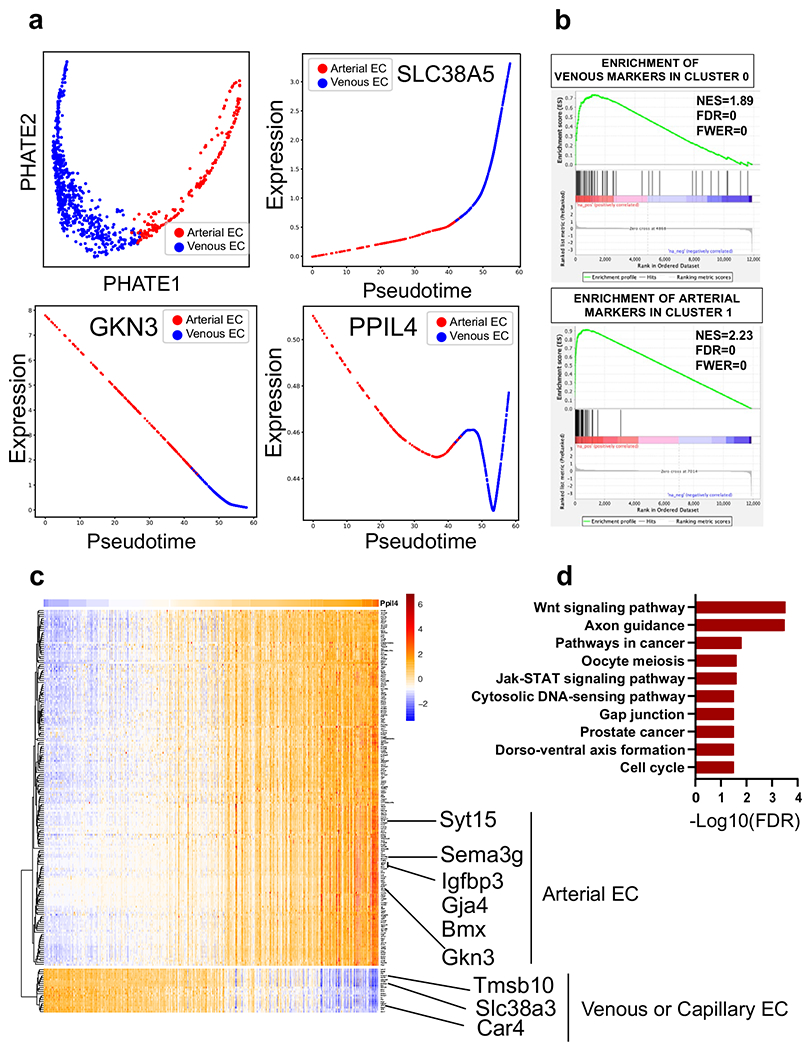
Analysis of publicly available scRNAseq mouse brain endothelial cell datasets obtained from Vanlandewijck et al. & He, L. et al. (PMID: 29443965). a, PHATE plot identifies two major groups in brain endothelial cells, arterial and venous (top left panel). Pseudotime analysis demonstrating PPIL4 expression predominantly in arterial endothelial cells when compared to venous (SLC38A5) and arterial (GKN3) markers. b, GSEA revealing that the top 50 genes associated with venous endothelial cells are significantly enriched among the top ranked genes specifically expressed in Cluster 0. Similarly, GSEA showing significant enrichment of the top 50 arterial endothelial genes among the top ranked genes in Cluster 1. c, Expression of 200 genes with top knn-DREMI score (y axis) ordered by DREVI-based clustering and by peak expression along PPIL4 (x axis). d, Bar plots showing significantly enriched KEGG pathways for genes above 95th percentile knn-DREMI score and positive relationship with PPIL4.
Extended Data Fig. 9 |. PPIL4-WT binding to JMJD6 in both nucleus and cytoplasm and overlapping phenotypes in ppil4−/− and jmjd6−/− zebrafish embryos.
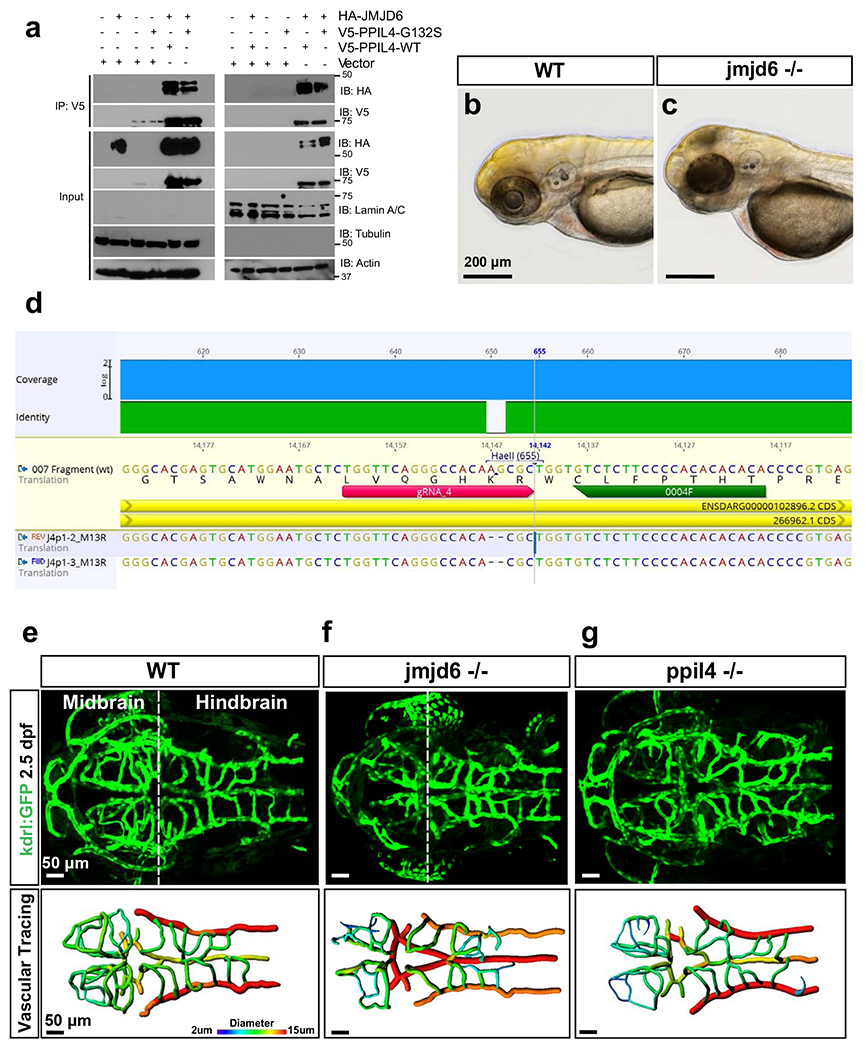
a, Lysates of HEK293 cells expressing HA-JMJD6, V5-PPIL4WT and V5-PPIL4G132S were separated into cytoplasmic and nuclear fractions. Cell lysate (input) and V5-IP of cytoplasmic and nuclear fractions were subjected to immunoblotting with anti-HA, anti-V5, lamin A/C (nuclear fraction [right]), tubulin (cytoplasmic fraction [left]) and actin antibodies. The blots shown are representative of three biological replicates. b,c Bright-field images showing necrosis in the head of jmjd6−/− 2.5 dpf embryos. d, Location of sgRNA designed to target jmjd6 leading to a 2bp deletion in exon 3 and a premature stop codon. e-g, Maximum intensity projection (MIP) of representative confocal z-stack images of 2.5 dpf wild type (n=16), jmjd6−/− (n=4), and ppil4−/− (n=15) embryos in the tg(kdrl:gfp)zn1 background (top panels). Brain vessel segmentation in same larvae obtained by Imaris software; colors represent vessel diameter (bottom panels). Homozygous deletion of jmjd6 or ppil4 individually results in dramatic reduction in midbrain CtA complexity and impairment in vascular morphology. Scale bar: 200 μm in b and c; 50 μm in e-g.
Extended Data Fig. 10 |. ppil4 depletion leads to impaired activation of Wnt signaling in brain parenchyma and brain ECs of 30 hpf zebrafish.
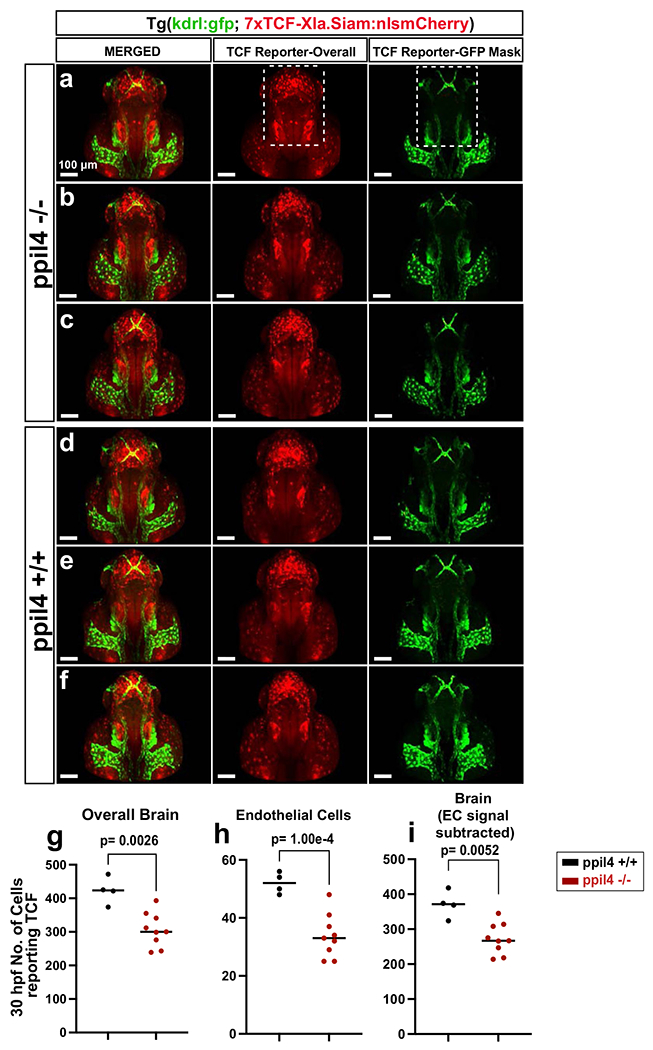
a-c, Maximum intensity projection (MIP) of confocal z-stack images of three representative 30 hpf ppil4−/− and d-f, ppil4++/+ embryos in double transgenic tg(kdrl:gfp; 7xTCF-Xla.Siam:nlsmCherry) background to visualize Wnt signaling activity (red) and endothelial cells (green) (dorsal view and caudal facing up). TCF reporter signal is quantified using the Spots application in Imaris demonstrating loss of TCF reporting cells in brain parenchyma as well as in midbrain CtAs of ppil4−/− embryos. Endothelial specific Wnt-activity is calculated using Spots-Mask for GFP channel in the designated area in a. See methods for details of image processing and presentation. g-i, Quantification of number of TCF reporting cells using the Spots application in Imaris software showing significant decrease in Wnt-activity in ppil4−/− embryos, in (g) overall brain and (h) brain endothelial cells compared with wild type. (i) Wnt signaling activity in overall brain after subtracting the Wnt activity in endothelial cells. n= 4, and 9 embryos for ppil4+/+ and ppil4−/− respectively. Individual values presented with scatter dot plot and median for all quantifications. Statistical tests performed: Two-tailed Student t-test. Scale bar: 100 μm.
Supplementary Material
Acknowledgements
We are indebted to the patients and families who have contributed to this study.
We would like to thank M. Cavanaugh for the zebrafish husbandry. Funding: This work was supported by Yale University funds (to M. Günel) and National Institutes of Health grants 4R01NS057756-10 (to M. Günel.), 1R01NS111935-01 (to M. Günel, A.L. and K.M.-G.) and R01 NS109160-04 and R01 HL130246-05 (to S.N.).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-021-01572-7.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
In-house codes are available at https://doi.org/10.5281/zenodo.5539900.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41591-021-01572-7.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-021-01572-7.
Peer review information Nature Medicine thanks John Kolega, R. Loch Macdonald, Alan Shuldiner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Anna Maria Ranzoni was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Data availability
All summary statistics for case–control burden analysis are fully detailed in Supplementary Tables 1–4. Each variant in Supplementary Tables 1 and 2 is annotated with information regarding genomic location, variant effect, amino acid change, CADD (version 3) score for deleteriousness and general and subpopulation MAF in gnomAD and ExAC. In addition, individual-level phenotypic data from patients with PPIL4 mutations are provided in Supplementary Table 3. Case–control burden analysis (two-tailed Fisher’s exact test) result for 17 genes that are co-segregating in all affected individuals with general gnomAD MAF < 0.005 is shown in Supplementary Table 3. Additionally, sequencing data of all patients in the IA cohort with PPIL4 mutations have been deposited in the European Genome-phenome Archive under accession number EGAS00001005518. Zebrafish expression data are reported in Supplementary Tables 5 and 6. Additionally, public expression and genomics datasets were obtained from the GtEX portal (https://gtexportal.org/home/), the database of gene expression in adult mouse brain and lung vascular and perivascular cell (https://betsholtzlab.org/VascularSingleCells/database.html) and the Genome Aggregation Database (https://gnomad.broadinstitute.org/). Gene enrichment analysis was performed using the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/) and Metascape (https://metascape.org/gp/index.html#/main/step1). CRISPRscan was used for sgRNA design (https://www.crisprscan.org/). The BioPlex (version 3.0)(https://bioplex.hms.harvard.edu/explorer/home) database was used to explore potential protein interactors of PPIL4. VarCards67 (http://159.226.67.237/sun/varcards/) was used for variant annotation of the data individually downloaded from the gnomAD website. Source data are provided with this paper.
References
- 1.Vlak MH, Algra A, Brandenburg R & Rinkel GJ Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: a systematic review and meta-analysis. Lancet Neurol. 10, 626–636 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Korja M, Lehto H, Juvela S & Kaprio J Incidence of subarachnoid hemorrhage is decreasing together with decreasing smoking rates. Neurology 87, 1118–1123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindbohm JV, Kaprio J, Jousilahti P, Salomaa V & Korja M Risk factors of sudden death from subarachnoid hemorrhage. Stroke 48, 2399–2404 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Korja M et al. Cause-specific mortality of 1-year survivors of subarachnoid hemorrhage. Neurology 80, 481–486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kissela BM et al. Subarachnoid hemorrhage: a preventable disease with a heritable component. Stroke 33, 1321–1326 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Graf CJ Familial intracranial aneurysms: report of four cases. J. Neurosurg 25, 304–308 (1966). [DOI] [PubMed] [Google Scholar]
- 7.Schievink WI, Schaid DJ, Michels VV & Piepgras DG Familial aneurysmal subarachnoid hemorrhage: a community-based study. J. Neurosurg 83, 426–429 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Bor ASE, Rinkel GJ, van Norden J & Wermer MJ Long-term, serial screening for intracranial aneurysms in individuals with a family history of aneurysmal subarachnoid haemorrhage: a cohort study. Lancet Neurol. 13, 385–392 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Santiago-Sim T et al. THSD1 (thrombospondin type 1 domain containing protein 1) mutation in the pathogenesis of intracranial aneurysm and subarachnoid hemorrhage. Stroke 47, 3005–3013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bourcier R et al. Rare coding variants in ANGPTL6 are associated with familial forms of intracranial aneurysm. Am. J. Hum. Genet 102, 133–141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou S et al. RNF213 is associated with intracranial aneurysms in the French-Canadian population. Am. J. Hum. Genet 99, 1072–1085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasuno K et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat. Genet 42, 420 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasuno K et al. Common variant near the endothelin receptor type A (EDNRA) gene is associated with intracranial aneurysm risk. Proc. Natl Acad. Sci. USA 108, 19707–19712 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bilguvar K et al. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat. Genet 40, 1472 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van der Auwera GA et al. From FastQ data to high-confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10. 11–11.10. 33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krumm N et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet 47, 582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cras TY et al. Determinants of the presence and size of intracranial aneurysms in the general population: the Rotterdam Study. Stroke 51, 2103–2110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor CL, Yuan Z, Selman WR, Ratcheson RA & Rimm AA Cerebral arterial aneurysm formation and rupture in 20,767 elderly patients: hypertension and other risk factors. J. Neurosurg 83, 812–819 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Zeng L et al. Molecular cloning, structure and expression of a novel nuclear RNA-binding cyclophilin-like gene (PPIL4) from human fetal brain. Cytogenetic Genome Res. 95, 43–47 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Hanes SD Prolyl isomerases in gene transcription. Biochim. Biophys. Acta 1850, 2017–2034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang K, Schmid FX & Fischer G Catalysis of protein folding by prolyl isomerase. Nature 329, 268–270 (1987). [DOI] [PubMed] [Google Scholar]
- 25.Ando K et al. Clarification of mural cell coverage of vascular endothelial cells by live imaging of zebrafish. Development 143, 1328–1339 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanlandewijck M et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480 (2018). [DOI] [PubMed] [Google Scholar]
- 28.He L et al. Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci. Data 5, 180160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeCicco-Skinner KL et al. Endothelial cell tube formation assay for the in vitro study of angiogenesis. J. Vis. Exp e51312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hillen B, Drinkenburg BA, Hoogstraten HW & Post L Analysis of flow and vascular resistance in a model of the cricle of Willis. J. Biomech 21, 807–814 (1988). [DOI] [PubMed] [Google Scholar]
- 31.Kamoun WS et al. Simultaneous measurement of RBC velocity, flux, hematocrit and shear rate in vascular networks. Nat. Methods 7, 655 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Dijk D et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liberzon A In: Stem Cell Transcriptional Networks 153–160 (Springer, 2014). [Google Scholar]
- 34.Daneman R et al. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl Acad. Sci. USA 106, 641–646 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stenman JM et al. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247–1250 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Boeckel J-N et al. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl Acad. Sci. USA 108, 3276–3281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider JE et al. Identification of cardiac malformations in mice lacking Ptdsr using a novel high-throughput magnetic resonance imaging technique. BMC Dev. Biol 4, 16 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou D et al. Inhibition of JMJD6 expression reduces the proliferation, migration and invasion of neuroglioma stem cells. Neoplasma 64, 700–708 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Zhang X et al. JmjC domain-containing protein 6 (Jmjd6) derepresses the transcriptional repressor transcription factor 7-like 1 (Tcf7l1) and is required for body axis patterning during Xenopus embryogenesis. J. Biol. Chem 290, 20273–20283 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Yang Y & Zhang X MiR-770 inhibits tumorigenesis and EMT by targeting JMJD6 and regulating WNT/β-catenin pathway in non-small cell lung cancer. Life Sci. 188, 163–171 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Huttlin EL et al. Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huttlin EL et al. The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Böse J et al. The phosphatidylserine receptor has essential functions during embryogenesis but not in apoptotic cell removal. J. Biol 3, 15 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akhtar S, Gremse F, Kiessling F, Weber C & Schober A CXCL12 promotes the stabilization of atherosclerotic lesions mediated by smooth muscle progenitor cells in Apoe-deficient mice. Arterioscler. Thromb. Vasc. Biol 33, 679–686 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Vanhollebeke B et al. Tip cell-specific requirement for an atypical Gpr124-and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. eLife 4, e06489 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho C, Smallwood PM & Nathans J Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b-specific signaling in mammalian CNS angiogenesis and blood-brain barrier regulation. Neuron 95, 1056–1073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moro E et al. In vivo Wnt signaling tracing through a transgenic biosensor fish reveals novel activity domains. Dev. Biol 366, 327–340 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Sato N, Meijer L, Skaltsounis L, Greengard P & Brivanlou AH Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med 10, 55–63 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Landrum MJ et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet 38, 904 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Wang C et al. Ancestry estimation and control of population stratification for sequence-based association studies. Nat. Genet 46, 409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen X et al. MLL-AF9 initiates transformation from fast-proliferating myeloid progenitors. Nat. Commun 10, 1–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mootha VK et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Moon KR et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol 37, 1482–1492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moreno-Mateos MA et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR–Cas9 targeting in vivo. Nat. Methods 12, 982 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Narayanan A et al. In vivo mutagenesis of miRNA gene families using a scalable multiplexed CRISPR/Cas9 nuclease system. Sci. Rep 6, 32386 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kasper DM et al. MicroRNAs establish uniform traits during the architecture of vertebrate embryos. Dev. Cell 40, 552–565. e555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brend T & Holley SA Zebrafish whole mount high-resolution double fluorescent in situ hybridization. J. Vis. Exp 1229 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 62.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anders S, Pyl PT & Huber W HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smedley D et al. BioMart—biological queries made easy. BMC Genomics 10, 1–12 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li J et al. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 46, D1039–D1048 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All summary statistics for case–control burden analysis are fully detailed in Supplementary Tables 1–4. Each variant in Supplementary Tables 1 and 2 is annotated with information regarding genomic location, variant effect, amino acid change, CADD (version 3) score for deleteriousness and general and subpopulation MAF in gnomAD and ExAC. In addition, individual-level phenotypic data from patients with PPIL4 mutations are provided in Supplementary Table 3. Case–control burden analysis (two-tailed Fisher’s exact test) result for 17 genes that are co-segregating in all affected individuals with general gnomAD MAF < 0.005 is shown in Supplementary Table 3. Additionally, sequencing data of all patients in the IA cohort with PPIL4 mutations have been deposited in the European Genome-phenome Archive under accession number EGAS00001005518. Zebrafish expression data are reported in Supplementary Tables 5 and 6. Additionally, public expression and genomics datasets were obtained from the GtEX portal (https://gtexportal.org/home/), the database of gene expression in adult mouse brain and lung vascular and perivascular cell (https://betsholtzlab.org/VascularSingleCells/database.html) and the Genome Aggregation Database (https://gnomad.broadinstitute.org/). Gene enrichment analysis was performed using the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/) and Metascape (https://metascape.org/gp/index.html#/main/step1). CRISPRscan was used for sgRNA design (https://www.crisprscan.org/). The BioPlex (version 3.0)(https://bioplex.hms.harvard.edu/explorer/home) database was used to explore potential protein interactors of PPIL4. VarCards67 (http://159.226.67.237/sun/varcards/) was used for variant annotation of the data individually downloaded from the gnomAD website. Source data are provided with this paper.