

Abstract
Estimating cell type composition of blood and tissue samples is a biological challenge relevant in both laboratory studies and clinical care. In recent years, a number of computational tools have been developed to estimate cell type abundance using gene expression data. Although these tools use a variety of approaches, they all leverage expression profiles from purified cell types to evaluate the cell type composition within samples. In this study, we compare 12 cell type quantification tools and evaluate their performance while using each of 10 separate reference profiles. Specifically, we have run each tool on over 4000 samples with known cell type proportions, spanning both immune and stromal cell types. A total of 12 of these represent in vitro synthetic mixtures and 300 represent in silico synthetic mixtures prepared using single-cell data. A final 3728 clinical samples have been collected from the Framingham cohort, for which cell populations have been quantified using electrical impedance cell counting. When tools are applied to the Framingham dataset, the tool Estimating the Proportions of Immune and Cancer cells (EPIC) produces the highest correlation, whereas Gene Expression Deconvolution Interactive Tool (GEDIT) produces the lowest error. The best tool for other datasets is varied, but CIBERSORT and GEDIT most consistently produce accurate results. We find that optimal reference depends on the tool used, and report suggested references to be used with each tool. Most tools return results within minutes, but on large datasets runtimes for CIBERSORT can exceed hours or even days. We conclude that deconvolution methods are capable of returning high-quality results, but that proper reference selection is critical.
Keywords: cell type deconvolution, benchmarking, cell type quantification, gene expression
Introduction
Biological tissues are rarely homogeneous and are instead typically composed of a variety of distinct cell types. The relative abundance of these cell types is fundamental to tissue biology and function, and therefore of frequent interest to the biomedical community. In medical settings, knowledge of cell type populations can provide insight into the nature of a wide range of diseases and, in some cases, inform treatment. In cancer, for instance, the abundance of certain T cells correlates strongly with survival, as well as the efficacy of immunotherapy treatment [1–3]. In laboratory settings, researchers frequently observe gene expression changes that are difficult to interpret without knowledge of cell type composition. Such patterns may be the result of changes of cell type abundance, rather than altered expression in any particular cell type. Cell type deconvolution methods enable researchers to distinguish between these two cases and extract further insights from their experiments.
Existing molecular techniques of cell type quantification can be difficult to apply to large-scale studies or to certain cell types. Fluorescence-activated cell sorting (FACS) is often considered a gold standard method. However, FACS is typically based on a limited number of markers that are selected beforehand, sometimes limiting the cell types that can be quantified. Moreover, standard FACS cannot quantify cell types with unusual morphologies, such as neurons, myocytes and adipocytes. Single-cell RNA-seq methods (scRNA-seq) are becoming increasingly popular, but their cost remains high [4]. Moreover, current scRNA-seq methods capture only a small fraction of cells present in a tissue, and the observed cells may not represent a random sample [5]. Both scRNA-seq and FACS require cells to be dissociated into a single-cell suspension before processing [6]. During this process, some cells may be lysed before they are observed, whereas others remain aggregated and are less likely to be detected. Consequently, subtle alterations during the cell dissociation step can produce dramatic differences in observed cell type fractions [7].
To overcome these limitations, a number of expression-based methods have been developed that aim to serve the biomedical community’s need for accurate estimation of cell type abundances from gene expression data [8–15]. These tools utilize either RNA-seq or microarray expression data to digitally deconstruct tissue samples, a process known as cell type deconvolution. Because it is common for researchers to collect gene expression data for other purposes, utilizing these data to evaluate cell type fractions can extend its utility. Similar deconvolution approaches also exist for DNA methylation data, though the present benchmark focuses on gene expression methods [16]. Due to the large number of expression-based methods currently available, it is often unclear to the user which tool will best suit their needs [17]. Evaluations performed as part of tool publications are often limited in scope, assessing accuracy for only a limited number of datasets, platforms and tissue types [14, 18].
Attempts to benchmark these tools have largely been limited to simulated data, which fails to capture the true complexity of tissue samples in living organisms [17, 19]. Studies that do incorporate clinical data use either imprecise cell quantification methods or very small numbers of samples [20, 21]. A recent benchmarking effort analyzed only nine FACS-sorted samples used for limited validation of simulated results [21]. Conclusions derived from these limited datasets can be misleading and incomplete, and we currently lack a systematic comparison of deconvolution methods evaluated on high-quality data. As such, researchers are left with little guidance as to which deconvolution tool is most suitable for their needs.
In addition, reference data is a requirement for many tools, and existing benchmarking studies do not address the relationship between choice of reference and prediction quality. Here, we explore this relationship by running tools using a variety of reference datasets and reporting performance in each case.
In this study, we compare 12 cell type quantification methods and evaluate their performance using each of 10 separate reference profiles. We evaluate performance on the Framingham dataset, which contains both expression data and cell type composition estimates for 3728 clinical blood samples [22–24]. Cell type composition was evaluated via impedance-based electronic cell counting, a gold standard for high-throughput blood cell type quantification. Expression profiles were measured using an Affymetrix microarray. In addition, we evaluate performance on 300 in silico simulated mixtures (200 peripheral blood mononuclear cell (PBMC) and 100 stromal) and 12 samples of blood cell types mixed in vitro. Our analysis includes a thorough examination of the effect of reference choice on accuracy of deconvolution predictions. Overall, we utilize 3728 clinical samples and 312 simulated mixtures to evaluate the performance of deconvolution methods in the most powerful, complete and unbiased manner to date.
Results
Cell type quantification tools selected for benchmarking
We have selected 12 commonly used cell type quantification tools, which we benchmarked on our datasets in order to evaluate their ability to accurately predict cell type content of tissue samples. The tools included in this study are CIBERSORT (normal and absolute mode) [10, 25], the digital cell quantification (DCQ) algorithm [9], DeconRNASeq [8], dtangle [13], Estimating the Proportions of Immune and Cancer cells (EPIC) [15], Gene Expression Deconvolution Interactive Tool (GEDIT) [18], Microenvironment Cell Populations-Counter (MCP-Counter) [11], nnls [26], quaNTiseq [14], SaVanT [27] and xCell [12] (Table 1).
Table 1.
Summary of cell type quantification tools evaluated by this benchmarking study, with details of inputs, algorithm and publication
| Tool | Publication year | Reference format | Tool type | Algorithm | Language |
|---|---|---|---|---|---|
| nnls | 1995 (R package 2012) | Expression matrix | Deconvolution | Constrained linear regression | R |
| DeconRNASeq | 2013 | Expression matrix | Deconvolution | Constrained linear regression | R |
| DCQ | 2014 | Expression matrix | Signature quantification | Elastic net regularized regression | R |
| CIBERSORT | 2015 | Expression matrix | Deconvolution | Support vector regression | R |
| MCP-Counter | 2016 | N/Aa | Signature quantification | Log-sum of marker gene expression | R |
| EPIC | 2017 | Expression matrix | Deconvolution | Constrained linear regression | R |
| SaVaNT | 2017 | Signature gene list | Signature quantification | Log-sum of marker gene expression | Python |
| xCell | 2017 | N/Aa | Signature quantification | Transformed marker gene enrichment scores | R |
| dtangle | 2018 | Expression matrix | Deconvolution | Differential marker gene analysis in log-space | R |
| CIBERSORT (Absolute Mode) | 2018 | Expression matrix | Deconvolution | Support vector regression | R |
| quaNTiseq | 2019 | N/Aa | Deconvolution | Constrained linear regression | R |
| GEDIT | 2021 | Expression matrix | Deconvolution | Constrained linear regression | Python and R |
Note: Included in the table are year of publication, format of required reference data, nature of the algorithm and output values, and the language in which the tool is implemented. Tools are sorted by publication year. Some tools that require expression matrices additionally require lists of signature genes.
aThese tools do not allow for custom references without extensive pre-processing steps or direct modification of the tool’s code.
The tools included in this benchmark fall into two basic categories based on their approach to cell type quantification. The first category is deconvolution tools, which include CIBERSORT, DeconRNASeq, dtangle, EPIC, GEDIT and quaNTiseq [8, 10, 13–15, 18, 26]. These approaches model the observed expression profile of the mixture as a combination of expression profiles of individual cell types, most commonly through application of regression algorithms. As a baseline to compare with other deconvolution tools, we also apply standard, non-regularized linear regression using the nnls package in R [26, 28]. For deconvolution tools, outputs can be interpreted as fractions and will either sum to 1.0 or to <1.0, with the remainder representing the fraction of unknown cell types. These tools differ, however, in both the form of regression (or similar algorithm) and the scale at which they operate. Log-scale algorithms may be capable of producing more efficient estimators, whereas linear methods model biology in a more realistic fashion [13].
Also included in this study are four signature quantification tools, which include DCQ, MCP-Counter, SaVanT and xCell [9, 11, 12, 27]. These tools operate by calculating scores intended to correspond to the relative abundance of each cell type across samples. These scores should not be interpreted as fractions and are, in fact, often unbounded and sum to values >1.0. Although scores for a particular cell type can be compared across samples, comparisons of scores across cell types may not be valid. For example, a sample with a higher score for B cells compared with natural killer (NK) cells does not imply that there are more B cells than NK cells present. The tools MCP-Counter and SaVanT generate signature scores by calculating the log-sum of a set of marker genes [11, 27]. DCQ implements an unbounded elastic net regression to produce enrichment scores, thus inferring relative cell quantities [9]. xCell calculates single sample Gene Set Enrichment Analysis (ssGSEA) enrichment scores for a large number of signatures, then applies a power transformation to the result [12].
Gold standard datasets
In this study, we use both simulated and experimental datasets to evaluate the accuracy of existing deconvolution tools (Table 2). Cell type fractions of these datasets have been evaluated by trusted molecular techniques able to deliver highly accurate cell type proportions. The samples consist of 300 synthetic mixtures prepared in silico using single-cell data, 12 mixtures prepared in vitro and sequenced using microarray, and 3728 clinical samples.
Table 2.
Overview of the gold standard datasets
| Dataset | Sequencing platform | Cell types | Number of samples | Mixture type | Cell quantification method |
|---|---|---|---|---|---|
| Cell mixtures | Illumina H12 Beadchip Microarray | B, mono, NK, neutrophil, CD4 T, CD8 T | 12 | In vitro cell mixtures | Controlled cell mixing in vitro |
| Simulated PBMC | Psuedo-bulk from scRNA-seq | B, mono, NK, CD4 T, CD8 T | 200 | In silico simulated mixtures | Simulated cell mixing in silico |
| Simulated stromal mixtures | Psuedo-bulk from scRNA-seq | B, CD4 T, CD8 T, endothelial, fibroblast, macrophages, mast cells | 100 | In silico simulated mixtures | Simulated cell mixing in silico |
| Framingham Cohort data | Affymetrix Human Exon ST 1.0 Microarray | Neutrophils, lymphocytes, monocytes | 3728 | Human clinical samples | Electrical impedance counting |
Note: Each mixture comes from one of three platforms and consists of 12–3728 samples of varying sets of cell types. Mixtures were either created in silico, mixed in vitro via titration or taken directly from clinical patients. Underlying cell type fractions are either known naturally by nature of mixture generation or evaluated by electrical impedance counting.
The 300 pseudo-bulk synthetic mixtures were prepared using scRNA-seq data. For each mixture, individual cells were randomly selected and their expression profiles summed: 200 of these represent simulated PBMC mixtures containing five common PBMC cell types (B, CD4 T, CD8 T, NK and monocytes). Exactly 100 of these were created using data obtained from a previous study [29] and 100 using data from 10x Genomics (https://www.10xgenomics.com/resources/datasets/). Lastly, a third set of 100 mixtures contained stromal cell types as well (B, CD4 T, CD8 T, macrophage, mast, endothelial and fibroblast cells), also using data from 10x Genomics. Following clustering, PBMC cell type assignment was performed using four to six marker genes for each cell type (Supplementary Figure S1). Cell type assignment of the stromal dataset was performed by 10x Genomics. In each case, 1000 cells were selected in total, their expression values summed and the cell type ratios noted.
In addition, 12 in vitro mixtures were prepared by physical titration of purified cell types in known proportions. Six immune cell types were used to produce these mixtures (B, CD4 T, CD8 T, monocytes, NK, neutrophils), which were combined in varying proportions (Supplementary Figure S2). The mixtures were then profiled using an Illumina HT12 BeadChip microarray. Lastly, the Framingham Cohort data [24] is collected from the blood of healthy individuals and profiled on Affymetrix Human Exon Array ST 1.0 arrays. Gold standard cell type fractions were obtained using electrical impedance.
Optimal reference choice varies across tools
In addition to expression data from heterogeneous tissue samples, most tools require reference data containing expression profiles of pure cell types. These data can be in the form of an expression matrix, a list of signature genes or both. It has been shown that the choice of reference can have a significant impact on the quality of results [30], and as part of this study, we explore the effect of reference choice on tool performance.
As reference data is a requirement for most tools included in this study, we have carefully curated an extensive list of reference datasets (Supplementary Table S1). These references include data from a variety of sources, including scRNA-seq, bulk RNA-seq and multiarray platforms. Each reference contains a different set of cells, with some including stromal cells and others only immune. Each reference also contains a varying number of genes; ImmunoStates [31] and leukocyte matrix containing 22 cell types (LM22) [10] have been curated to contain only a short list of signature genes, whereas other matrices contain the larger set of genes measured by their respective platforms.
Included in this study are 10 cell type quantification tools that accept custom references, and we provide 10 reference datasets to each. The reference profiles used derive from a variety of sources and platforms, including LM22 [10], ImmunoStates [31], 10x Genomics [32], EPIC [15], BLUEPRINT [33] and the Human Primary Cell Atlas [34]. In order to evaluate the effect of reference choice on prediction quality, we systematically evaluate each tool using each reference source. For every set of results, we calculate the Pearson correlation between true cell type fractions and tool output, and treat these correlations as our metric of performance. For tools that predict fractions, we also compare average absolute error (Supplementary Figure S3).
We find that choice of reference has a substantial impact on the quality of results (Figure 1). Some tools produce accurate results when the optimal reference is selected, but high error and even negative correlations when an improper reference is used, particularly when applied to the Framingham dataset. Three of the references tested (10x Immune, EPIC-tumor infiltrating cells (TIC), Human Primary Cell Atlas (HPCA)-Stromal) do not include neutrophils. These naturally perform poorly on the Framingham mixtures, which on average contain 60% neutrophils per mixture. However, many combinations of tools with other references fail to produce high-quality results, often resulting in negative correlations. Although no reference performs best for all tools and datasets, we provide a table of recommended reference choice for each tool based on these results (Table 3). Although we provide recommendations for stromal data, these are based on simulated mixtures with a limited set of cell types. Due to the huge diversity of stromal tissues that are studied, the optimal choice of reference may differ on novel datasets.
Figure 1 .
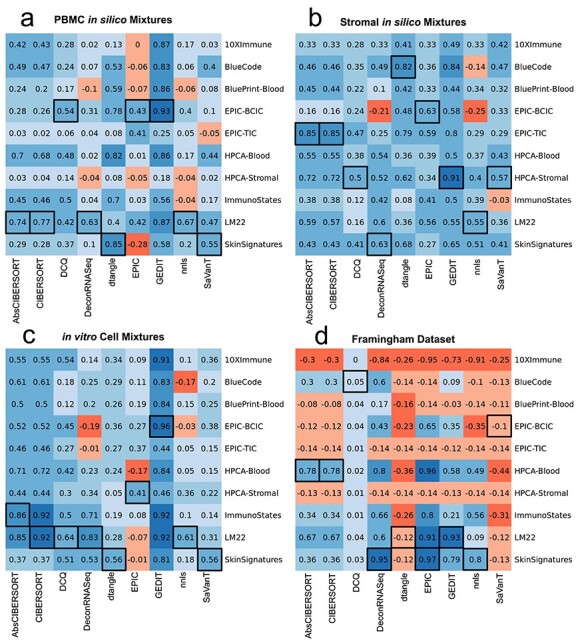
Determining the optimal reference choice for each tool that supports external references. Shown here are Pearson correlations between true cell fractions and tool output for each combination of tool and reference matrix. Results are shown for PBMC and stromal in silico simulated mixtures (A and B), for in vitro mixtures of immune cells (C) and clinical samples from the Framingham Cohort dataset (D). Tools not able to deliver results within 48 h were excluded and are not reported here. Highest correlation for each tool is shown in black boxes. When run on the full Framingham dataset with the Skin Signatures reference, both modes of CIBERSORT failed to produce results after 96 h; results shown here were obtained by segmenting the Framingham dataset into six parts and merging results.
Table 3.
Recommended references for use with each tool in the contexts of blood and stromal samples
| Tool | Recommended reference (blood) | Recommended references (Stromala) |
|---|---|---|
| AbsCIBERSORT | LM22 | EPIC-TIC |
| CIBERSORT | LM22 | EPIC-TIC |
| DCQ | LM22 or EPIC-BCIC | HPCA-Stromal |
| DeconRNASeq | LM22 or Skin Signatures | Skin Signatures |
| dtangle | Skin Signatures | BlueCode or EPIC-TIC |
| EPIC | EPIC-BCIC | EPIC-TIC |
| GEDIT | LM22 or EPIC-BCIC | HPCA-Stromal |
| nnls | LM22 | Skin Signatures |
| SaVaNT | Skin Signatures | HPCA-Stromal |
aStromal recommendations are based on 100 simulated mixtures from scRNA-seq data containing seven cell types (including endothelial, fibroblasts, macrophages and mast cells). The references suggested here may not be ideal for application to other cell types or platforms.
CIBERSORT and GEDIT produce consistently high correlations between estimated and true cell type fractions
Next, we compare the performance of each tool when the optimal reference profile is used. We utilize two metrics to evaluate the accuracy of predicted fractions compared with actual fractions: correlation and absolute error. As performed in the previous section, we compute the Pearson correlation between true cell type fractions and tool output. These outputs can represent either estimated cell type fractions or enrichment scores.
Based on correlation analysis, CIBERSORT and GEDIT produce the most accurate results across all datasets, though both are outperformed by DeconRNASeq and EPIC when applied to the Framingham dataset. For each mixture tested, multiple tools produce highly accurate results (correlation >0.85), though no single tool is able to maintain the highest performance across all datasets (Figure 2).
Figure 2 .
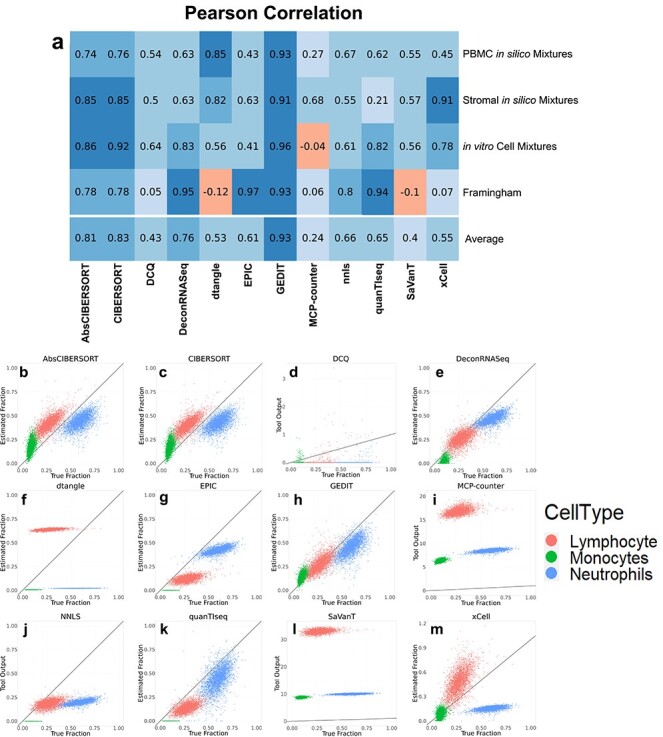
Comparisons between tool outputs and true cell type fractions, as evaluated by gold standard techniques. (A) Pearson correlations between tool output and true fractions for each combination of tool and dataset; here, the optimal reference is used for each dataset (Figure 1). (B–L) Scatter plots visualizing the output of each tool (y-axis) versus cell type fractions as evaluated by automated cell counting (x-axis). Three cell types were evaluated, with lymphocytes, monocytes and neutrophils shown in red, green and blue, respectively. The y = x line is shown in black. For tools that accept custom reference data, the reference data that resulted in the highest correlation is shown here (Figure 1D). Equivalent graphs for the other three datasets are included in supplementary materials (Supplementary Figures S4–S6).
CIBERSORT and GEDIT produce the lowest absolute errors across all datasets
Researchers are often interested in accurately predicting absolute cell type fractions, rather than relative abundances or scores. We, therefore, evaluate the ability of each tool to accomplish this task, including those that normally return non-fractional outputs. Specifically, we normalize each output vector such that the sum across all cell types in the reference sum exactly to 1.0. Absolute error is then calculated as the difference between this adjusted output and true cell type fractions (Figure 3). It is important to note that this approach falls outside of the intended usage of several tools (i.e. DCQ, MCP-Counter, SaVaNT and xCell), since these are not designed for inter-cell type comparisons. These tools are, nonetheless, included in order to comprehensively evaluate all available options for estimating absolute abundances.
Figure 3 .
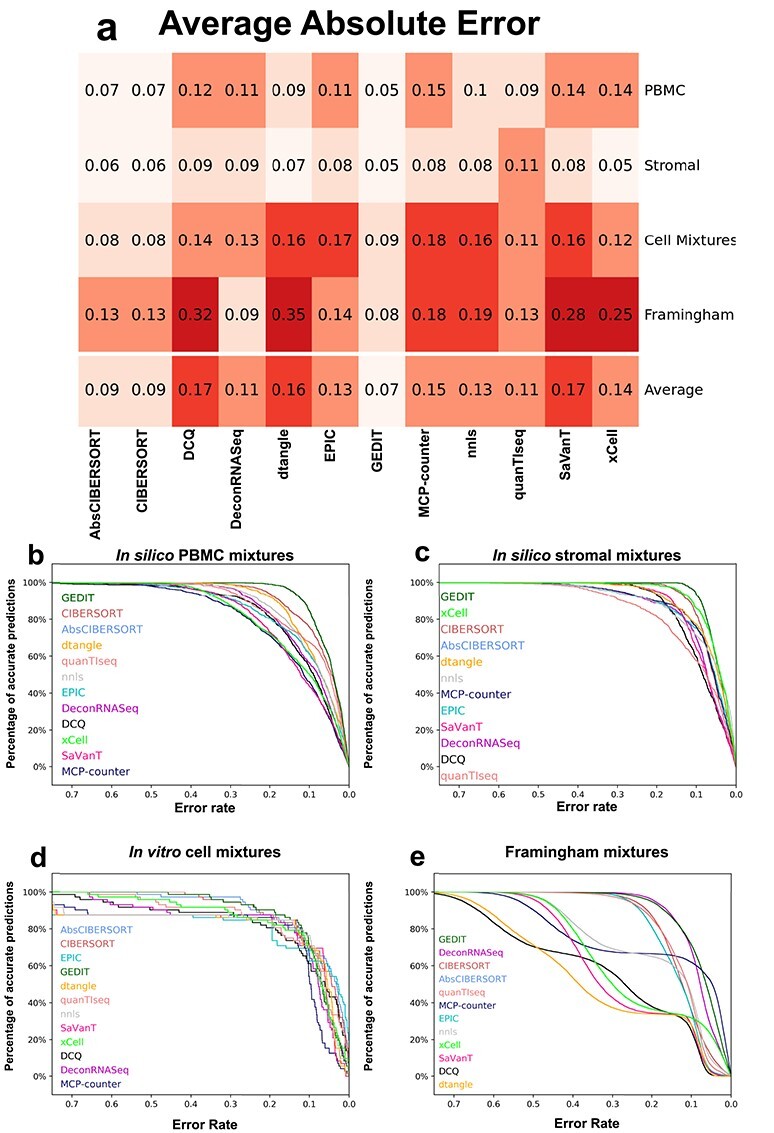
Error values of predictions for each tool and dataset. (A) Absolute error values, averaged across all predictions, for each tool and dataset. (B–E) Distribution of absolute error for all predictions in each dataset. Predictions are considered accurate (y-axis) if error is less than allowed error rate (x-axis). Legend is sorted by decreasing area under the curve (Supplementary Table S2). For each combination of tool and dataset, the reference producing the highest correlation value was used (Figure 1).
The error of predicted fractions varies greatly depending on the exact combination of tool and mixture. CIBERSORT and GEDIT are able to maintain high accuracy across the majority of datasets and for many choices of reference matrix. For all datasets, the ‘absolute’ mode of CIBERSORT produces nearly identical results to the default mode, with slightly higher error in some cases. xCell performs best on the in silico simulated mixtures, but produces high error and low correlation for some cell types in the in vitro mixture (e.g. B Cells).
For all tools, we observe some of the highest errors when applied to the Framingham data. This is likely due to complexities of biological samples that are not adequately modeled by either form of simulation. One possibility is that the simulated mixtures are composed of purified cell types and do not include the full range cell substates that are normally present in living biological tissue. Expression profiles for these non-canonical cells may be effectively missing from available reference data, particularly when using tools like xCell and MCP-Counter that rely on a single built-in reference source.
Runtime varies substantially depending on tools and references
We evaluate the scalability of deconvolution by varying the size of inputs and recording the central processing unit (CPU) time required by each tool. Specifically, we randomly subsample the Framingham Cohort data into batches varying in size from 10 to 5052 samples. We then record CPU time required to run each tool as a function of input size. We exclude xCell from this analysis, since it does not support an easily accessible command line interface.
Furthermore, we explore the effect of reference choice on resource requirements (for the tools that support custom references). Specifically, two separate references are applied and runtime recorded in each case; these references are the larger HPCA reference containing 19 715 genes, and the smaller LM22 reference containing only 547 genes. We find that the size of the reference matrix has a substantial effect on the running time of certain tools, in particular CIBERSORT (both absolute and default modes). When the smaller reference is used, all tasks complete relatively quickly, with the slowest run taking 1.1 h. However, when the larger HPCA reference is used, runtimes for some tools (specifically, both modes of CIBERSORT) can reach over 24 h for large numbers of samples (Figure 4).
Figure 4 .
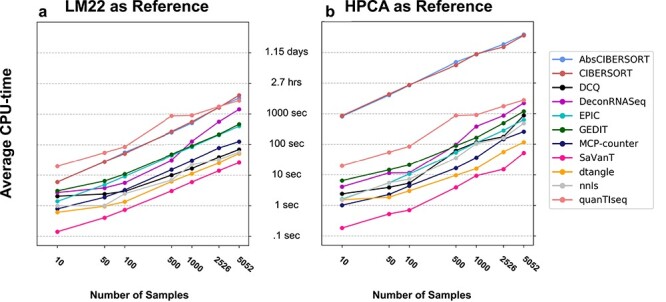
Time requirements for each tool for varying numbers of samples. Subsamples of varying size were randomly selected from the Framingham dataset. Each tool was run twice using LM22 (A) and the Human Primary Cell Atlas (B) as reference data; the larger HPCA produces longer runtimes in most cases. Each point represents the average of 20, 10, 10, 5, 5, 2, 1 runs for input sizes of 10, 50, 100, 500, 1000, 2526, 5052, respectively. The default settings for MCP-Counter and quanTIseq were used, as they do not support external references.
Discussion
Transcriptomics-based cell type deconvolution is an increasingly popular approach for estimating the cell type composition of heterogeneous samples. However, current tools and reference profiles are numerous, and it is important that researchers have a clear way of determining the best choices for their needs. Here, we perform a comprehensive benchmarking study to systematically evaluate the performance of various computational deconvolution tools across 4040 transcriptomics samples using accurate molecularly defined gold standard.
Although no single tool produces the best results across all types of datasets, CIBERSORT (both modes), DeconRNASeq and GEDIT are able to produce reliable results (average error <0.15, correlation >0.6) across all four datasets. Relative to deconvolution tools, signature quantification approaches like DCQ, MCP-Counter, SaVaNT and xCell tend to perform poorly by both metrics (correlation and error), particularly when applied to the Framingham dataset. Even a standard linear regression (nnls) outperforms all four of these methods in many cases.
It may be possible to obtain improved results for some tools by adjusting particular inputs or parameter settings. For example, carefully selecting a set of genes that distinguishes important cell types may improve the quality of results. However, users that desire accurate results across many contexts, and without substantial parameter tuning, will likely be best served by the tools CIBERSORT, DeconRNASeq and GEDIT.
Tools are much more likely to fail (producing high error and low correlation) when applied to the Framingham dataset. Some references are not suitable for application to the Framingham data, due to lack of neutrophil entries. However, the tools DCQ, dtangle and SaVaNT appear unable to produce quality estimates for this dataset regardless of the reference selected. Clearly, performing deconvolution on clinical samples taken directly from living organisms presents challenges not fully captured by either in silico or in vitro simulations. One contributing factor may be natural heterogeneity within cell types. For example, not all neutrophils are identical, but vary across developmental and inflammatory trajectories. Immature or otherwise unusual neutrophils are more likely to be excluded when isolating purified cell types. As a result, simulated mixtures (whether in silico or in vitro) may be primarily composed of canonical cells that more closely resemble those present in reference matrices.
Although deconvolution methods are capable of returning high-quality results, this accuracy is often contingent upon the selection of a proper, high-quality reference dataset, presenting a fundamental limitation to expression-based cell type quantification as a whole. Moreover, a viable reference in one circumstance may fail when combined with a different tool and mixture. Utilizing GEDIT combined with the EPIC ‘Blood Circulating Immune Cells’ reference produces the most accurate results for the in vitro mixtures dataset. However, this same reference produces highly inaccurate predictions on the same mixture when DeconRNASeq is used.
In order to provide clearer direction in the application of deconvolution tools, we provide a table of recommended references. Although the best choice depends on the tool being used, the reference LM22 appears the most reliable for application to blood data. Results for stromal data are more mixed, but matrices EPIC-TIC, the Human Primary Cell Atlas and Skin Signatures appear the most likely to produce accurate results. In addition to providing some of the best overall results, CIBERSORT and GEDIT are also robust relative to other tools. These tools fail less often and are likely good choices for application to novel data, particularly when using novel reference sources.
With the increasing availability of single-cell data, researchers will likely have greater access to high-quality reference data in the future. This is especially relevant for scientists studying highly specific cell types or cell subtypes, a research direction that single-cell technology has enabled to an extent not previously possible. Public sources may lack expression profiles for highly specific cell types, but researchers that are able to perform even a small number of single-cell experiments can utilize the results as a reference source and apply deconvolution to larger numbers of samples.
Most tools return results within 30 min, even on datasets larger than 5000 samples. However, CIBERSORT (both modes) can sometimes require extremely long runtimes, potentially taking hours or even days to return results, and limiting its application to large datasets. This is most likely due to the support vector regression used by this tool, which is computationally intensive compared with simpler regressions or signature-based approaches used by other methods. It is possible to reduce runtimes by selecting reference matrices with fewer genes, such as LM22 or ImmunoStates, though this may limit the range of cell types that can be predicted.
Several regression-based tools (CIBERSORT, DeconRNASeq, GEDIT) and reference data sources (LM22, EPIC, HPCA) produce reliable, accurate results for all datasets. For many tools, however, prediction quality varies dramatically depending on the provided dataset and reference source. As such, researchers applying deconvolution to novel datasets (and especially novel cell types) may wish to run deconvolution using multiple tools and/or references. By examining the consistency of results across multiple conditions, one can differentiate between real biological patterns and technical artifacts.
Methods
PBMC and stromal single-cell mixtures
We obtained two human datasets from 10x Genomics (https://www.10xgenomics.com/resources/datasets/) and one human dataset from gene expression omnibus (GEO) (accession GSE103322) [28]. The PBMC mixtures were created in two batches, each producing 100 mixtures. For the first set, we used 1000 cells for each sorted cell type (monocytes, CD8 and CD4 T cells, B cells, NK cells). For each cell type, we randomly selected 1–1000 cells, and then we sum the expression of all the selected cells to create a synthetic mixture. The process was repeated 100 times, thus 100 mixtures were created. For PBMC2, we first clustered the cells and identified the cell types for the dataset. Then, we used the same five cell types in PBMC2 and created the 100 mixtures the same way as we did from PBMC1. For stromal cells, we created 100 mixtures the same way as we did for PBMC2, except for that we included two additional cell types not typically found in blood (fibroblasts and mast cells) and excluded monocytes and NK cells. For each mixture, the true fraction for each cell type is calculated as the number of cells of that cell type selected, divided by the total number of cells across all cell types.
Framingham data
The Framingham Heart Study (FHS) is a population-based study, predominantly of European ancestry, consisting of an ongoing series of primarily family-based cohorts first developed in 1948 and based in Framingham, MA, USA; it comprises the Original [22], Offspring [23] and Third Generation [24] cohorts. FHS gene expression, blood cell counts, subject and sample metadata was obtained from dbGap (phs000007) and represents multiple batches processed by the same laboratory. Gene expression data was measured using an Affymetrix Human Exon 1.0 ST microarray and was processed (filtered and normalized) as in [35], resulting in 5058 available FHS samples, of which 3728 had available blood cell counts. Blood cell counts were obtained through a complete blood count using the Coulter HmX Hematology Analyzer (Beckman Coulter, Inc.) [36, 37].
The ‘gold standard’ used in this benchmark is cell percent as reported by counting on a Beckman Coulter HmX Hematology Analyzer. The following metrics from whole blood were obtained—glycated hemoglobin A (HbA1c), basophil count and percent, eosinophil count and percent, hematocrit, hemoglobin, lymphocyte count and percent, mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), mean corpuscular volume (MCV), monocyte count and percent, mean platelet volume (MPV), neutrophil count and percent, platelet count, red blood cell (RBC), red-blood-cell distribution width (RDW), White Blood Cell (WBC). Further information is available at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd004086.1.
Selected tools
We have selected available cell type quantification tools able to infer the abundances of immune cell types based on the gene expression profiles. In total, we have identified 12 tools, which either predict cell type fractions or produce signature or enrichment scores. xCell was run using the online interface at https://xcell.ucsf.edu/, but all other tools were installed on the Hoffman2 Cluster at University of California, Los Angeles (UCLA). Each task was provided 16 GB of random access memory (RAM) and 48 h of runtime. When applied to the Framingham dataset, five combinations of tool and reference failed to complete within 48 h and were run again with a time limit of 96 h. Two combinations (CIBERSORT + Skin Signatures and AbsCibersort + Skin Signatures) still failed to complete within this time frame. In these cases, the Framingham dataset was split into six batches, and the results for each batch merged after running the tools. In the case of AbsCIBERSORT, the reference matrix was additionally rounded to two decimal places, which allowed the task to complete within the time limit.
As part of this project, we sent correspondence to the authors of each tool describing the settings used and inquiring if adjustments are appropriate. Most tools were run under default settings, with some modifications as described below. Comprehensive parameter tuning was not performed due to concerns regarding overfitting and how to apply equal effort to the tuning of each tool.
When evaluating absolute error for each tool, outputs were converted into fractions. Specifically, scores for each sample were normalized such that the sum of scores across all cell types (including ‘other’, when provided) equals 1.0.
CIBERSORT
We requested the CIBERSORT code from the authors’ website https://cibersort.stanford.edu/download.php and have installed CIBERSORT version 1.04 on the UCLA Hoffman2 cluster (R version 3.6.0). We ran all deconvolution tasks using both the default CIBERSORT mode and the absolute mode and reported both results. As the statistical outputs of CIBERSORT (e.g. P-values) are not considered in our analysis, we ran with zero permutations to reduce resource usage. Quantile normalization was used for the cell mixtures and Framingham datasets, but disabled for the PBMC and stromal datasets; this follows the author recommendations regarding application to microarray and RNA-seq data, respectively.
DCQ
DCQ was installed as part of the ComICS package in R. For each reference matrix used, we designated all genes present in that matrix as marker genes.
DeconRNASeq
The DeconRNASeq R package was installed from Bioconductor and run using default settings.
dtangle
The dtangle R package (version 2.0.9) was obtained from the comprehensive R archive network (CRAN) and installed on the Hoffman2. As part of the wrapper to run the function, genes not shared between the mixture and reference matrices were excluded (otherwise this caused a crash). All observed values x in both matrices were then transformed to log2(1 + x), as dtangle takes log-transformed data as input. Alternate choices for the parameter n_markers were tested, but did not consistently return better results compared with the default setting of 0.1 (Supplementary Figure S8). The dtangle() function was therefore used with default settings.
EPIC
We downloaded EPIC from the author’s GitHub repository (https://github.com/GfellerLab/EPIC) and installed it on the Hoffman2 Cluster. The tool provides two built-in reference datasets (tumor-infiltrating cells and blood circulating immune cells). When running the tool using these datasets, we use the default mode that utilizes additional data regarding reference profile variability and amount of messenger RNA per cell. For the other eight reference datasets used in this study, these additional data are not available and thus were not included as inputs. The cell fractions outputs are taken as cell type estimates.
GEDIT
GEDIT version 1.6 was obtained from the GitHub repository https://github.com/BNadel/GEDIT. Necessary packages were installed, specifically random, numpy, glmnet, RColorBrewer and gplots. It was run using default settings.
MCP-Counter
The MCP-Counter code was obtained from the github (https://github.com/ebecht/MCPcounter) and installed on Hoffman2 along with its dependencies devtools and curl. The MCPcounter.estimate() function was used to produce predictions. ‘HUGO_symbols’ was designated, and otherwise default settings were used. We contacted the authors regarding use of external references, but this appears to require direct modification of the MCP-Counter code and was therefore not performed in this study.
nnls
The Lawson–Hanson algorithm for least squares was implemented in the nnls package in R. A simple wrapper was written, which selected genes shared between the reference and mixtures matrices and ran the regression on each sample. The outputs were then normalized, such that the predictions for each sample summed to 1.0.
quanTIseq
The quanTIseq code was obtained from the GitHub https://github.com/icbi-lab/quanTIseq and run on the Hoffman2 Cluster; installation via docker or singularity both failed on the cluster. Specifically, the quantiseq_pipeline.sh script was called using the command of the form ‘./quanTIseq_pipeline.sh --inputfile=$MIXTUREFILE --outputdir=$OUTPUTFILE --pipelinestart=decon’.
SaVanT
The code for SaVanT was obtained from its authors and run using 50 signature genes per cell type.
xCell
xCell was run using the online tool found at http://xcell.ucsf.edu/. The bulk expression data is submitted under ‘upload gene expression data’ and the default gene signatures were used (xCell, n = 64). The RNA-seq option was selected for PBMC and stromal datasets but not for CellMixtures or Framingham data.
Reference data
Reference data was obtained from a variety of sources, as described in Supplementary Table S1. The HPCA reference matrix contained a wide variety of cells that were not present in any of our mixtures. As such, we subsetted this reference matrix in order to produce two versions more appropriate for the data used in the present study. These two versions contain seven blood cell types (B, CD14+ monocytes, CD16+ monocytes, NK, neutrophils, CD4+ T and CD8+ T) and six blood and stromal cell types (B, CD4+ T, CD8+ T, endothelial, fibroblast, macrophage), respectively.
Cell type matching
Depending on the exact tool and reference matrix used, prediction outputs could be labeled as any one of 219 cell types and subtypes (Supplementary Table S3). For each output, we either match the output with a cell type quantified in the mixture, or note that it does not match any. In some cases, this matching is trivial (e.g. B_cells, B-Cells, and BCells are all noted as ‘B Cells’), and in other cases, the outputs represent cell subtypes (e.g. ‘naive B-Cells’ and ‘memory B Cells’ were also noted as ‘B Cells’). The final output for each cell type in the mixtures was calculated as the sum of outputs matched with that cell type; thus, predictions for a general cell type are computed as the sum of the subtypes for that cell. The table of output interpretation is included as a supplementary file.
Supplementary Material
Brian B. Nadel is a postdoctoral researcher at the University of California, Los Angeles. His research focuses on cell type deconvolution and differing patterns of gene expression across cell types.
Meritxell Oliva is a postdoctoral scholar in the Department of Public Health Sciences at the University of Chicago. Her work focuses on the investigation of sex differences in the human transcriptome and in the identification of contexts in which genetic variants manifest in molecular changes that causally impact phenotypes (GxE).
Benjamin L. Shou is a medical student at the Johns Hopkins School of Medicine. His research focuses on the analysis of genomic, imaging, and clinical datasets related to cardiovascular disease.
Keith Mitchell is a graduate student working in the University of California, Davis Bioinformatics Core. His work includes data analysis and construction of content management systems.
Feiyang Ma is a postdoctoral researcher at the University of California, Los Angeles. His research focuses on single cell data analysis and applying bioinformatics tools in immunology research.
Dennis J. Montoya is an assistant researcher at the University of California, Davis School of Medicine. His research includes the development and application of computational methods to relate high-throughput sequencing data to clinical phenotypes.
Alice Mouton is currently a Funds for Scientific Research ( postdoctoral researcher in the Conservation Genetics lab at the University of Liege (Belgium). She is working at the interface between genomic and epigenomic in the context of conservation and evolutionary biology in mammals.
Sarah Kim-Hellmuth is a principal investigator at the University Hospital Ludwig-Maximilians University Munich. Her research focuses on the context-specificity of genetic gene regulation in humans.
Barbara E. Stranger is an Associate Professor at Northwestern University Feinberg School of Medicine. Her research focuses on genetics of gene expression and context-specific genetic effects on complex traits.
Matteo Pellegrini is a Professor of Molecular Cell and Developmental Biology at the University of California, Los Angeles. His lab focuses on the development of bioinformatics tools to analyze epigenomics data.
Serghei Mangul is an Assistant Professor at the University of Southern California. His research focuses on developing and applying novel and robust data-driven computational approaches to accelerate the diffusion of genomics and biomedical data into translational research and education.
Availability of data and materials
Code and results of the current study are available in the GitHub repository, https://github.com/Mangul-Lab-USC/benchmarking-transcriptomics-deconvolution. Data from the Framingham Heart Study-Cohort can be requested here: https://biolincc.nhlbi.nih.gov/studies/framcohort/.
Authors’ contributions
B.B.N. performed much of the analysis and installed some tools. M.O. provided access to the Framingham dataset and assisted in manuscript preparation. B.L.S. installed and ran several deconvolution tools. K.M. assisted in data analysis and manuscript preparation. F.M. created the simulated pseudo-bulk datasets from scRNA-seq data. D.J.M. provided the in vitro cell mixtures. A.M. assisted with tool installation and execution. S.K.M. assisted in manuscript preparation. B.E.S., M.P. and S.M. provided oversight and assisted in manuscript preparation. S.M. conceived of the presented idea.
Funding
BN was supported by the Biomedical Big Data Training Grant 5T32LM012424–03. AM was supported by an NSF grant (award number: 1457106) and the Quantitative and Computational Biology Collaboratory Postdoctoral Fellowship (UCLA). SKH is supported by the Marie-Skłodowska Curie fellowship H2020 grant 706636, the Reinhard-Frank Stiftung and the Helmholtz Young Investigator grant VH-NG-1620. BES was supported by National Cancer Institute grant R01CA229618 and National Institute of Health grant U01HG007598. SM was partially supported by the National Science Foundation grant 2041984.
References
- 1. Gentles AJ, Bratman SV, Newman AM, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015;21:938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fridman WH, Pagès F, Sautès-Fridman C, et al. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12(4):298–306. [DOI] [PubMed] [Google Scholar]
- 3. Li B, Zhao H, Severson E, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 2016;17(1):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ziegenhain C, Reinius B, Vieth B, et al. Comparative analysis of single-cell RNA sequencing methods. Mol Cell 2017;65(4):631–43.e4. [DOI] [PubMed] [Google Scholar]
- 5. Ren X, Kang B, Zhang Z. Understanding tumor ecosystems by single-cell sequencing: promises and limitations. Genome Biol 2018;19(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen G, Ning B, Shi T. Single-cell RNA-Seq technologies and related computational data analysis. Front Genet 2019;10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hines WC, Su Y, Kuhn I, et al. Sorting out the FACS: a devil in the details. Cell Rep 2014;6(5):779–81. [DOI] [PubMed] [Google Scholar]
- 8. Gong T, Szustakowski JD. DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics 2013;29(8):1083–5. [DOI] [PubMed] [Google Scholar]
- 9. Altboum Z, Barnett-Itzhaki Z, Steuerman Y, et al. Digital cell quantification identifies global immune cell dynamics during influenza infection. Mol Syst Biol 2014;10(2):720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Newman AM, Gentles AJ, Liu CL, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Becht E, Buttard B, Giraldo NA, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17(1):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hunt GJ, Freytag S, Bahlo M, et al. dtangle: accurate and robust cell type deconvolution. Bioinformatics 2019;35(12):2093–2099. doi: 10.1093/bioinformatics/bty926. [DOI] [PubMed] [Google Scholar]
- 14. Finotello F, Mayer C, Plattner C, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 2019. doi: 10.1101/223180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Racle J, de Jonge K, Baumgaertner P, et al. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017;6:e26476. doi: 10.7554/eLife.26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Decamps C, Prive F, Bacher R, et al. Guidelines for cell-type heterogeneity quantification based on a comparative analysis of reference-free DNA methylation deconvolution software. BMC Bioinformatics 2020;21:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mangul S, Lam AKM, Martin LS, et al. Systematic benchmarking of omics computational tools. Nat Commun 2019;10(1):1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nadel BB, Ma F, Lopez D, et al. The Gene Expression Deconvolution Interactive Tool (GEDIT): accurate cell type quantification from gene expression data. Giga Science 2021;10(2):giab002. 10.1093/gigascience/giab002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sturm G, Zhang JD, Finotello F, et al. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019;35(14):i436–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jimenez-Sanchez A, Cast O, Miller M. Comprehensive benchmarking and integration of tumour microenvironment cell estimation methods. Cancer Res 2019;79:6238–6246. doi: 10.1101/437533. [DOI] [PubMed] [Google Scholar]
- 21. Cobos FA, Alquicira-Hernandez J, Powell JE, et al. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat Commun 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dawber TR, Meadors GF, Moore FE. Epidemiological approaches to heart disease: the Framingham study. Am J Public Health 1951;41:279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feinleib M, Kannel WB, Garrison RJ, et al. The Framingham offspring study design and preliminary data. Prev Med 1975;4(4):518–25. [DOI] [PubMed] [Google Scholar]
- 24. Splansky GL, Atwood LD, Corey D, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol 2007;165(11):1328–35. [DOI] [PubMed] [Google Scholar]
- 25. AbsCIBERSORT, Newman AM, et al. CIBERSORT website, 2018. https://cibersort.stanford.edu/ (20 October 2018, date last accessed).
- 26.Lawson CL, Hanson RJ. 1995. Solving Least Squares Problems. Philadelphia, Pa: Society for Industrial and Applied Mathematics. http://epubs.siam.org/ebooks/siam/classics_in_applied_mathematics/cl15. [Google Scholar]
- 27. Lopez D, Montoya D, Ambrose M, et al. SaVanT: a web-based tool for the sample-level visualization of molecular signatures in gene expression profiles. BMC Genomics 2017;18(1):824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mullen KM, van Stokkum IHM. nnls: the Lawson-Hanson algorithm for non-negative least squares (NNLS). R package version 1.4. https://CRAN.R-project.org/package=nnls. [Google Scholar]
- 29. Puram SV, Tirosh I, Parikh AS, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171(7):1611–24.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frishberg A, Brodt A, Steuerman Y, et al. ImmQuant: a user-friendly tool for inferring immune cell-type composition from gene-expression data. Bioinformatics 2016;32(24):3842–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vallania F, Tam A, Lofgren S, et al. Leveraging heterogeneity across multiple datasets increases cell-mixture deconvolution accuracy and reduces biological and technical biases. Nat Commun 2018;9(1):4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017;8(1):14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martens JHA, Stunnenberg HG. BLUEPRINT: mapping human blood cell epigenomes. Haematologica 2013;98(10):1487–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mabbott NA, Baillie JK, Brown H, et al. An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics 2013;14(1):632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wheeler HE, Shah KP, Brenner J, et al. Survey of the heritability and sparse architecture of gene expression traits across human tissues. PLoS Genet 2016;12(11):e1006423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kannel WB, Feinleib M, McNamara PM, et al. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol 1979;110(3):281–290. doi: 10.1093/oxfordjournals.aje.a112813. [DOI] [PubMed] [Google Scholar]
- 37. Splansky GL, Corey D, Yang Q, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute's Framingham Heart Study: Design, Recruitment, and Initial Examination. Am J Epidemiol 2007;165(11):1328–1335. 10.1093/aje/kwm021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code and results of the current study are available in the GitHub repository, https://github.com/Mangul-Lab-USC/benchmarking-transcriptomics-deconvolution. Data from the Framingham Heart Study-Cohort can be requested here: https://biolincc.nhlbi.nih.gov/studies/framcohort/.