

Abstract
Mulberry (Morus spp.) is an economically important plant as the main food plant used for rearing domesticated silkworm and it has multiple uses in traditional Chinese medicine. Two basic chromosome numbers (Morus notabilis, n = 7, and Morus alba, n = 14) have been reported in the genus Morus, but the evolutionary history and relationship between them remain unclear. In the present study, a 335-Mb high-quality chromosome-scale genome was assembled for the wild mulberry species M. notabilis. Comparative genomic analyses indicated high chromosomal synteny between the 14 chromosomes of cultivated M. alba and the six chromosomes of wild M. notabilis. These results were successfully verified by fluorescence in situ hybridization. Chromosomal fission/fusion events played crucial roles in the chromosome restructuring process between M. notabilis and M. alba. The activity of the centromere was another key factor that ensured the stable inheritance of chromosomes. Our results also revealed that long terminal repeat retrotransposons were a major driver of the genome divergence and evolution of the mulberry genomes after they diverged from each other. This study provides important insights and a solid foundation for studying the evolution of mulberry, allowing the accelerated genetic improvement of cultivated mulberry species.
Introduction
Mulberry (Morus spp.), which is recognized as the main food source for silkworm (Bombyx mori), is a deciduous tree of great economic importance. Mulberry originated in the Himalayan foothills and is distributed worldwide, including on the Eurasian, American, and African continents [1]. A draft mulberry genome of the wild species Morus notabilis was published in 2013 [2]. This genome sequence of M. notabilis has provided fundamental resources promoting studies of gene identification, genome evolution, trait development, and genetic improvement in mulberry. Mulberry leaves and fruits have been used as medicines, in beverages, and as functional foods in many countries because they contain abundant biologically active compounds [3], and they have attracted much attention from researchers. Based on published genomic [2], metabolomic [4, 5], and transcriptomic data [2, 6], most of the genes related to the biosynthesis of biologically active compounds in mulberry have been identified in M. notabilis, and the mechanisms regulating secondary metabolite synthesis in mulberry leaves and fruits have been elucidated [5–7]. These findings have contributed to increases in the genetic improvement and utilization of mulberry.
Polyploidy has played an important role in the speciation and evolution of plants. Karyotyping and ploidy-level identification have been employed as tools for parental selection in polyploid plant breeding [8]. Ten different chromosome numbers (14, 28, 35, 42, 49, 56, 84, 112, 126, and 308) have been identified in mulberry, which are related to altered ploidy levels [9, 10], and this information has been invaluable for the polyploid improvement of mulberry. Mulberry is believed to have a basic chromosome number of 14, and this assertion has been widely cited in the literature [11]. The wild mulberry species M. notabilis was first recorded by the German preacher Schneider in 1916 [12]. It has the minimal chromosome number [14] found in Morus and was the first species to be used for mulberry genome sequencing [2]. Karyotyping analyses definitively illustrated that the 14 chromosomes of M. notabilis formed seven pairs, thus indicating a basic chromosome number of seven [2]. Moreover, the mitotic and diakinesis karyotypes of M. notabilis were constructed using fluorescence in situ hybridization (FISH), and the results supported the finding that the basic chromosome number of M. notabilis was seven [9]. Recently, the genome of cultivated mulberry cultivar M. alba ‘Heyebai’ (2n = 28) was published, and the basic chromosome number of M. alba was proposed to be 14 [13]. Phylogenetic analyses of mulberry using nuclear ribosomal internal transcribed spacer (ITS) sequences, chloroplast DNA sequences, and genomic sequences indicated that M. notabilis was a more ancient species than M. alba [1, 10, 14] and that M. alba diverged from M. notabilis ~10 million years ago (MYA) [13, 14]. The karyotypes of M. notabilis and M. alba also showed significant differences [9], even though they were reported to have similar genome sizes [13]. M. notabilis contained 14 chromosomes with clear length differences. In contrast, all of the chromosomes of M. alba were dot or large dot chromosomes, lacking morphological characteristics. To date, the chromosomal evolutionary process in mulberry species remains unclear. A study of mulberry chromosomal evolution should thus contribute to the understanding of the genetic consequences of the evolution and domestication of mulberry [5, 6].
Previous studies have indicated the genome-wide relationships between cultivated plant species and their wild relatives, including citrus [15], soybean [16], tomato [17], and pistachio [18]. These studies have revealed that some genes or regions have experienced artificial selection in cultivated relatives. For example, energy- and reproduction-associated genes have been shown to be under selection in cultivated citrus [15]. Some new quantitative trait loci have also been well characterized in cultivated soybean [16] and tomato [17]. Because long terminal repeat (LTR) retrotransposon elements always account for the largest proportion of a plant genome [19], many studies have demonstrated that LTRs and their associated processes not only shape differences in the architecture of the plant genome within a genus but also contribute to the diversification and speciation of plants [20]. For example, a major burst of different LTRs has contributed to the diversification of apple [20]. LTR insertions lead to changes in the expression of genes related to anthocyanin biosynthesis, which ultimately leads to changes in the fruit phenotypes of grapes and oranges [19, 21, 22]. Similarly, understanding mulberry evolutionary divergence at the genome-wide level is essential for the further improvement of mulberry breeding.
In this paper, we describe the generation of a high-quality M. notabilis genome assembled using Oxford Nanopore Technologies (ONT) ultrahigh-throughput sequencing and high-throughput chromosome conformation capture (Hi-C) technologies. Moreover, we carried out detailed comparative chromosomal analyses between the genomes of M. notabilis and M. alba to examine the genomic evolution process. The mechanisms of genome reorganization and evolution reported in this study will be of value for further genetic research on mulberry species.
Results
Genome sequencing, assembly, and genome annotation
The predicted size of the M. notabilis genome was approximately 401.62 Mb (Supplementary Table 1). To enhance the quality of our genome assembly, approximately 57.29 Gb of raw Nanopore data was generated using the ONT PromethION platform, yielding approximately 142.65-fold coverage (Supplementary Table 2). After filtering out low-quality reads, a total of 48.87 Gb of high-quality reads were used for further assembly. A total of 41.38 Gb of Illumina paired-end reads (103.03-fold coverage) were utilized to correct and polish the assembled genome (Supplementary Table 3). The final 335.39 Mb assembly genome was generated from 650 contigs, with a contig N50 size of 5.72 Mb (Table 1). Then, 40.50 Gb of Hi-C data were used to further improve the integrity of the assembled genome (Supplementary Table 4). All high-quality Hi-C data were mapped to the aforementioned 650 assembled contigs. A total of 327.01 Mb of the 335.39 Mb assembled genome could be anchored to six chromosomes (Fig. 1 and Supplementary Fig. 1). The chromosome-specific probes reported in a previous study [9] could be clearly anchored to the corresponding chromosomes (Supplementary Table 5). Compared with the assembled genome previously generated [2], both the quality and integrity of the genome assembled in the present study were greatly improved (Table 1). In the present study, the longest contig and the contig N50 size of the assembled genome were 15.58 Mb and 5.72 Mb, respectively, whereas the corresponding sizes in previous research were only 0.28 Mb and 0.03 Mb, respectively (Table 1).
Table 1.
Statistics and comparison of the M. notabilis genome assembly obtained in the present study with that previously reported [2].
| M. notabilis v2 | M. notabilis v1 a | M. alba | |
|---|---|---|---|
| Genome size (Mb) | 335.39 | 330.79 | 346.39 |
| Longest contig size (bp) | 15 582 736 | 276 636 | — |
| Contig N50 (bp) | 5 720 535 | 34 476 | 2 710 056 |
| No. of contigs | 650 | 127 741 | 398 |
| Longest scaffold size (bp) | — | 3 477 367 | — |
| Scaffold N50 (bp) | — | 390 115 | — |
| No. of scaffolds | — | 110 759 | — |
| No. of chromosomes | 6 | — | 14 |
| GC content | 35.40% | 35.02% | 34.29% |
| Complete BUSCOsb | 94.03% | 91.06% | 94.30% |
Data collected from [2]. bBenchmarking Universal Single-Copy Orthologs.
Figure 1.
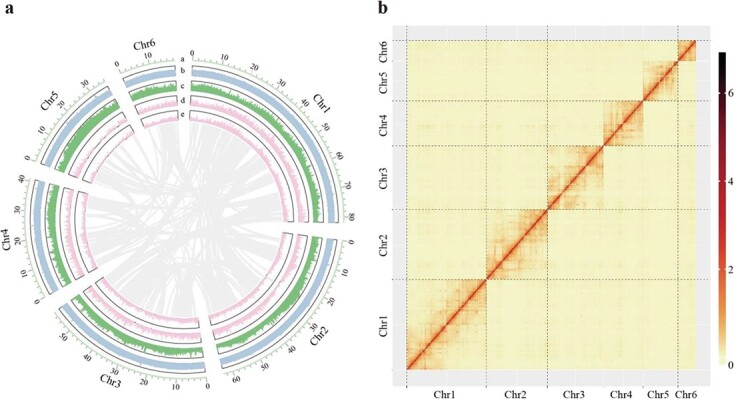
Genome features of M. notabilis. (a) The landscape of the mulberry genome, with a circular representation of the six pseudochromosomes. From inside to outside, gene collinearity, the density of genes on the plus strand, the density of genes on the minus strand, the density of transposable elements, and the density of GC content are shown. The chromosome units of these features are 100 kb. (b) Hi-C-based intrachromosomal interaction heatmap of the M. notabilis genome.
For repetitive sequence identification, a total of 136.68 Mb (accounting for 40.75% of the assembled genome) of transposable element (TE) sequences were identified and annotated by combining de novo and structure-based methods (Supplementary Table 6). Combined with other repetitive sequences, including simple repeats (2.04%), low-complexity sequences (0.37%), and unknown repeat sequences (13.91%), these elements masked 191.43 Mb (57.07%) of the newly assembled M. notabilis genome. Similar methods were used to identify repetitive sequences in the previously assembled M. alba genome, which revealed a total of 123.58 Mb (accounting for 34.64% of the assembled genome) of TEs and masked 188.77 Mb (52.91%) of the assembled M. alba genome (Supplementary Table 6).
For gene prediction, a total of 26 010 protein-coding genes, with an average gene size of 1454 bp, were annotated in the newly assembled M. notabilis genome (Supplementary Table 7). The number of genes in the M. alba genome was 27 012, with an average gene size of 1358 bp (Supplementary Table 7). The present results suggested putative functions of 93.66%, 70.58%, 68.44%, 28.78%, and 54.16% of the M. notabilis genes based on the InterPro, Swiss-Prot, Protein Families (Pfam), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO) databases, respectively (Supplementary Table 7). The corresponding annotated gene proportions in the M. alba genome were 93.97%, 72.26%, 69.43%, 27.99%, and 55.38% based on the InterPro, Swiss-Prot, Pfam, KEGG, and GO databases, respectively (Supplementary Table 7).
Genome and gene collinearity
Phylogenic trees were constructed using 132 single-copy genes, which suggested that M. alba diverged from M. notabilis ~10.99 MYA (Supplementary Fig. 2). We then performed a large-scale genome alignment of the M. notabilis genome sequenced in this study and the genome of M. alba (Fig. 2a), which demonstrated a high level of chromosome synteny between these two genomes (Fig. 2a). The longest chromosome of M. notabilis (chromosome 1) corresponded to four chromosomes (chromosomes 4, 7, 9, and 11) in the M. alba genome. The second-longest chromosome of M. notabilis (chromosome 2) was aligned to three chromosomes (chromosomes 2, 5, and 14) in M. alba. Among the chromosomes in the M. alba genome, a large fragment of chromosome 2 was aligned to chromosome 2 from M. notabilis, but in the reverse direction. We noted that the third-longest chromosome of M. notabilis (chromosome 3) also corresponded to three chromosomes (chromosomes 6, 12, and 13) in the M. alba genome. At the same time, chromosome 4 of M. notabilis could be mapped to two chromosomes (chromosome 3 and chromosome 10) in the M. alba genome. The other two chromosomes, chromosome 5 and chromosome 6, from the M. notabilis genome could be mapped to chromosome 1 and chromosome 8 of M. alba, respectively.
Figure 2.
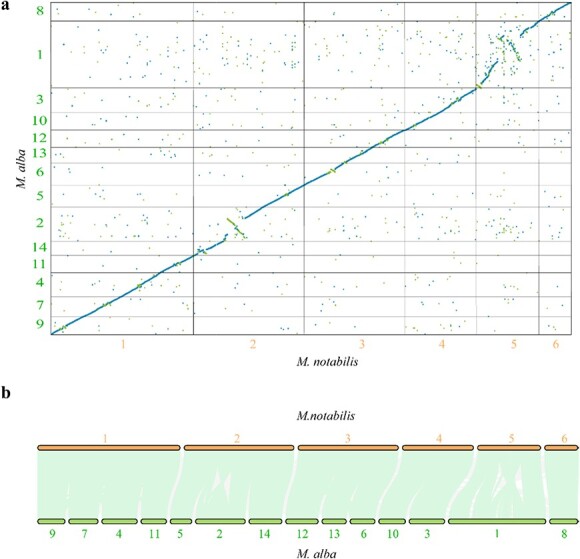
Chromosome rearrangement and gene collinearity analysis between M. notabilis and M. alba. (a) Large-scale genome alignment and comparison of M. notabilis and M. alba ‘Heyebai’. The x-axis denotes the six chromosomes of M. notabilis. The y-axis represents the 14 chromosomes of M. alba ‘Heyebai’. The minimum alignment length displayed was set at 1000 bp. Blue dots denote unique forward alignments. Green dots denote unique reverse alignments. (b) The collinearity of genes between M. notabilis and M. alba. The light green lines represent all collinear genes in the corresponding chromosomes. The gray lines represent other collinear genes. Bold font in orange or dark green denotes the corresponding chromosomes in M. notabilis and M. alba, respectively.
We also analyzed gene collinearity between the two genomes. A total of 36 436 collinear genes (68.72% of a total of 53 022 genes) were identified. If the chromosome location of collinear genes in the two genomes was inconsistent with our genome alignment results, the gene pairs were defined as rearranged genes. A total of 386 rearranged genes were identified in the two genomes based on their positions on the corresponding chromosomes (Fig. 2b, Supplementary Table 8). KEGG analysis suggested that these genes mainly participated in metabolic pathways and the biosynthesis of secondary metabolites (Supplementary Tables 9, 10). Among these genes, 16.06% (62/386) of the genes from M. notabilis could be mapped to 67 KEGG pathways, including metabolic pathways (32/62) and the biosynthesis of secondary metabolites (18/62). A total of 18.13% (70/386) of the genes from the M. alba genome could be assigned to 70 KEGG pathways, including metabolic pathways (38/70) and the biosynthesis of secondary metabolites (22/70).
To investigate chromosomal rearrangements in mulberry without an available genome of the common ancestor of Morus, the distribution of orthologous genes between grapevine (Vitis vinifera) and mulberry was analyzed. In the comparison of the gene order in M. notabilis and M. alba with that in V. vinifera, each region with a succession of dots in mulberry corresponded to three regions in grapevine and vice versa (Supplementary Fig. 3). The results suggested that both M. notabilis and M. alba share the eudicot-common triplication with V. vinifera and that there have been no whole-genome duplication (WGD) events in mulberry since its separation from the Eurosid I clade. The chromosomal structures of M. notabilis and M. alba were also reconstructed according to the composition of the grapevine chromosomes, and the observed correspondence between the reconstructed chromosomes of M. notabilis and M. alba was fairly good (Supplementary Fig. 3).
FISH-based comparative chromosome analyses between M. notabilis and M. alba
FISH was then performed on M. notabilis and M. alba ‘Lunjiao109’ (2n = 28) to test the chromosome collinearity patterns proposed earlier (Fig. 2). Chromosome 1 of M. notabilis corresponded to chromosomes 4, 7, 9, and 11 in M. alba (Fig. 3). In addition, two chromosome constrictions divided the largest diakinesis chromosome (chromosome 1) from M. notabilis into three segments, revealing additional morphological characteristics (Fig. 3e). Chromosome 1 of M. notabilis was chosen as an example here. Next, four single-copy sequence probes (Mn-chr1A, Mn-chr1B, Mn-chr1C, and Mn-chr1D) were designed. The FISH signal patterns of these four single-copy sequence probes were consistent with the physical positions (Fig. 3a–i). Mn-chr1A mapped to the upper distal part of diakinesis chromosome 1 (Fig. 3a). Mn-chr1B and Mn-chr1C mapped to the middle part of diakinesis chromosome 1 (Fig. 3c, e), and Mn-chr1B was located closer to Mn-chr1A and the main chromosome constriction than Mn-chr1C (Supplementary Fig. 4). The tail segment of diakinesis chromosome 1 hybridized to Mn-chr1D. Probes Mn-chr1A, Mn-chr1B, Mn-chr1C, and M-chr1D localized to four different diakinesis chromosomes in ‘Lunjiao109’ (Fig. 3b, d, f, h, and Supplementary Fig. 5). The FISH-based comparative chromosomal analysis results indicated that chromosome 1 of M. notabilis corresponded to chromosomes 4, 7, 9, and 11 in M. alba, as shown in the schematic representation (Fig. 3i).
Figure 3.
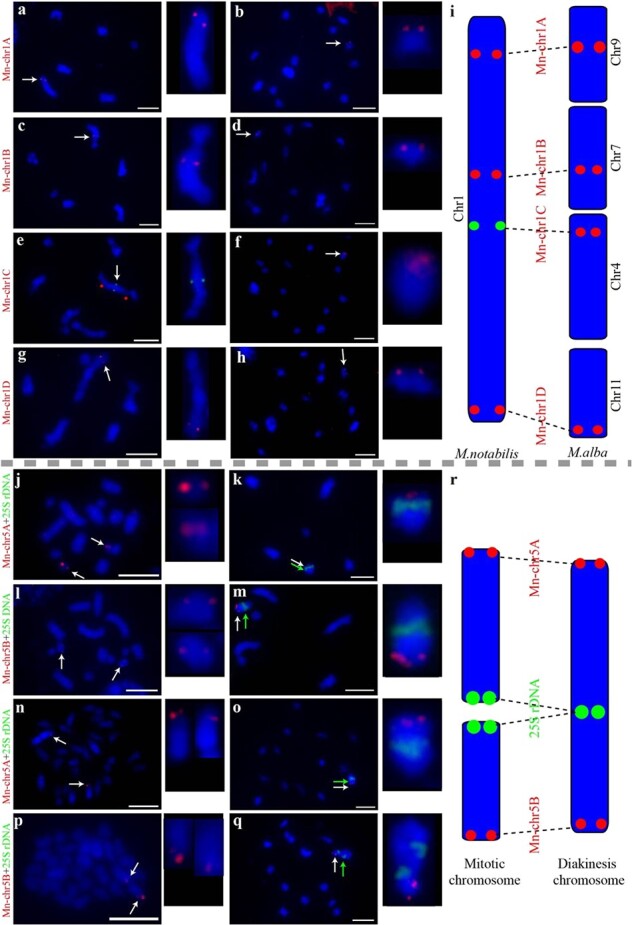
Chromosomal collinearity patterns verified by FISH using single-copy sequence probes and a 25S rDNA probe in M. notabilis and M. alba ‘Lunjiao109’. Probe Mn-chr1A mapped to the upper distal part of diakinesis chromosome 1 of M. notabilis (a) and diakinesis chromosome 9 of ‘Lunjiao109’ (b). Probe Mn-chr1B mapped to the middle part of diakinesis chromosome 1 of M. notabilis (c) and diakinesis chromosome 7 of ‘Lunjiao109’ (d). Probe Mn-chr1C mapped to the middle part of diakinesis chromosome 1 of M. notabilis. Red stars mark two clear chromosome constrictions on chromosome 1 (e). Probe Mn-chr1C mapped to diakinesis chromosome 4 of ‘Lunjiao109’ (f). Probe Mn-chr1D mapped to the tail segment of diakinesis chromosome 1 of M. notabilis (g) and diakinesis chromosome 11 of ‘Lunjiao109’ (h). Chromosomes were stained with DAPI (blue). All probe signals and the chromosomes where they were located are shown to the right in each cell at greater magnification. Arrows indicate the FISH signals of the probes. Scale bars represent 5 μm. Schematic representation of the collinearity pattern between chromosome 1 of M. notabilis and chromosomes 4, 7, 9, and 11 of M. alba. Probe names are shown in red font (i). Probe Mn-chr5A mapped to the smallest mitotic chromosome (chromosome 7) pair of M. notabilis (j) and the terminal region of the short arm of diakinesis chromosome 5 of M. notabilis, which was indicated by the centrally located signal of 25S rDNA (indicated by a green arrow) (k). Probe Mn-chr5B mapped to the mitotic chromosome 5 pair of M. notabilis (l) and the terminal region of the long arm of diakinesis chromosome 5 of M. notabilis, which was indicated by the centrally located signal of 25S rDNA (indicated by a green arrow) (m). Probe Mn-chr5A mapped to the terminal region of one pair of the mitotic chromosomes of ‘Lunjiao109’ (n) and the terminal region of the long arm of diakinesis chromosome 1 of ‘Lunjiao109’, which was indicated by the centrally located signal of 25S rDNA (indicated by a green arrow) (o). Probe Mn-chr5B mapped to the terminal region of one pair of mitotic chromosomes of ‘Lunjiao109’ (p) and the terminal region of the short arm of diakinesis chromosome 1 of ‘Lunjiao109’, which was indicated by the centrally located signal of 25S rDNA (indicated by a green arrow) (q). Chromosomes were stained with DAPI (blue). All probe signals and the located chromosomes are shown to the right in each cell at greater magnification. Arrows indicated the FISH signals of the probes. Scale bars represent 5 μm. Schematic representation of the chromosomal fusion events from mitotic chromosomes to meiotic chromosomes in M. notabilis and M. alba. Probe names are shown in red font (r).
As reported previously [9], mitotic chromosomes 5 and 7 were fused to form diakinesis chromosome 5 of M. notabilis. To illustrate the relationships between the 14 mitotic chromosomes, six diakinesis chromosomes, and six assembled pseudochromosomes, two single-copy sequence probes (Mn-chr5A and Mn-chr5B) from pseudochromosome 5 of M. notabilis were designed, and FISH was performed. As shown in Fig. 3j–m, Mn-chr5A mapped to the smallest dot mitotic chromosome 7 (Fig. 3j) and the distal part of the short arm of diakinesis chromosome 5, which colocalized with the 25S rDNA probe (Fig. 3k). Mn-chr5B mapped to the largest dot mitotic chromosome 5 (Fig. 3l) and another distal portion of diakinesis chromosome 5 (Fig. 3m). The chromosomes of ‘Lunjiao109’ showed similar signal patterns of probes Mn-chr5A, Mn-chr5B, and 25S rDNA. Mn-chr5A mapped to one of three pairs of mitotic chromosomes, which were deeply stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue) (Fig. 3n). Mn-chr5A and 25S rDNA colocalized with the largest diakinesis chromosome (chromosome 1) of ‘Lunjiao109’ (Fig. 3o). In detail, Mn-chr5A hybridized to the distal part of diakinesis chromosome 1, and the centrally located signal pattern was identified as 25S rDNA. Mn-chr5B mapped to another pair of mitotic chromosomes stained deeply by DAPI (Fig. 3p). Mn-chr5B was located in the distal part of the short arm of diakinesis chromosome 1, which was distinguished by the 25S rDNA signal (Fig. 3q). The results of the FISH-based comparative chromosomal analysis indicated that two pairs of mitotic chromosomes from M. notabilis and ‘Lunjiao109’ were fused into one diakinesis chromosome 5 and one diakinesis chromosome 1, respectively (Fig. 3r).
Identification and localization of centromere tandem repeats in M. notabilis and M. alba
The centromere is one of the key elements ensuring the stability of chromosomal inheritance, especially when chromosome fusion and fission occur. The centromere was selected to illustrate the chromosomal evolution process in M. notabilis and M. alba in the present study. Tandem repeats with a repeat monomer length of 175 bp showed high abundance in M. notabilis and M. alba and were treated as prime candidate centromere tandem repeats (Supplementary Figs. 6, 7). The physical location of these tandem repeats indicated that Mn-175 nt was distributed on all chromosomes of M. notabilis in at least two clusters except for chromosome 6, which lacked Mn-175 nt altogether (Fig. 4a). Tandem repeats of Ma-175 nt indicated its physical location on all chromosomes in M. alba in at least one cluster (Fig. 4b). The Mn-175 nt and Ma-175 nt sequences were shown to exhibit 89.14% similarity (Supplementary Fig. 8). Then, Mn-175 nt was labeled with biotin and hybridized to the chromosomes of M. notabilis and ‘Lunjiao109’. The Mn-175 nt probe mapped to all the diakinesis chromosomes of M. notabilis. For example, at least six pairs of signals were observed on diakinesis chromosome 1, whereas diakinesis chromosome 6 showed only one pair of obvious signals (Fig. 4c). Probe Mn-175 nt mapped to all of the diakinesis chromosomes of ‘Lunjiao109’ with seven major signals, and its signal was especially prominent on the two largest diakinesis chromosomes (chromosomes 1 and 2) (Fig. 4d). When homologous chromosomes were pulled to the opposite poles in metaphase I, the Mn-175 nt probe localized to centromere regions (Fig. 4e). The Mn-175 nt probe clearly localized to the centromere regions of nine cruciform bivalents in metaphase II in ‘Lunjiao109’ (Fig. 4f).
Figure 4.
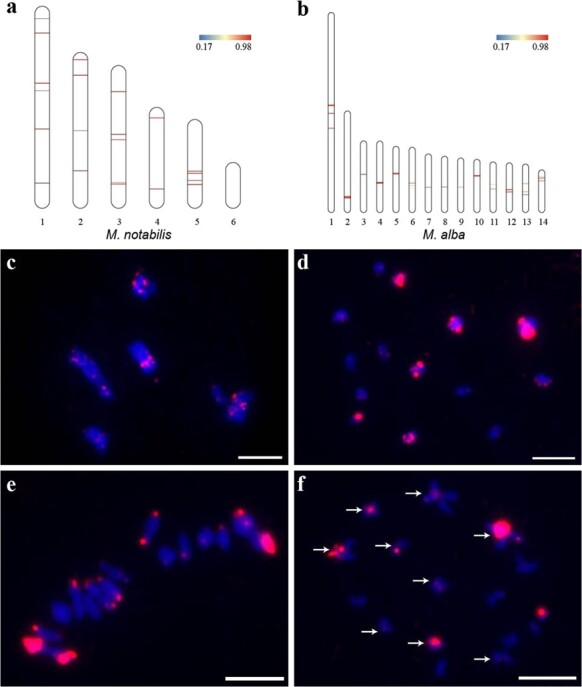
Identification of centromere tandem repeat sequences based on bioinformatic screening and FISH analyses. Schematic representation of the physical locations of Mn-175 nt candidate centromere sequences on the chromosomes of M. notabilis (a) and Ma-175 nt candidate centromere sequences on the chromosomes of M. alba ‘Heyebai’ (b). FISH mapping of Mn-175 nt on the diakinesis chromosomes of M. notabilis (c) and ‘Lunjiao109’ (d), in meiotic metaphase I of ‘Lunjiao109’ (e), and in meiotic metaphase II of ‘Lunjiao109’ (f). Arrows in (f) indicate that Mn-175 nt FISH signals were clearly located in the centromere regions of the cruciform bivalents. Chromosomes were stained with DAPI (blue). Scale bars represent 5 μm.
Comparative genomic analyses of gene families
Comparative analysis of these two genomes indicated that 2138 and 2219 genes were specific to the M. notabilis and M. alba genomes, respectively. When we compared the positional relationships between specific genes and their neighboring collinear genes in the two genomes, a total of 20 and 21 species-specific genes of M. notabilis and M. alba were found to be located in the rearranged regions (Supplementary Table 11). A total of four genes of M. alba were located at the end of the corresponding chromosome (Supplementary Table 11). When we mapped these species-specific genes to KEGG pathways, we found that only 24 specific genes in M. notabilis could be assigned to KEGG pathways and that they were mainly involved in two types of pathways: metabolic pathways (12/24) and the biosynthesis of secondary metabolites (6/24) (Supplementary Table 12). A total of 50 specific genes in the M. alba genome could be mapped to 55 pathways, and these were mainly related to metabolic pathways (19/50) and the biosynthesis of secondary metabolites (12/50) (Supplementary Table 13). To gain insight into gene family evolution, 18 845 groups of orthologous gene families were generated between the two genomes. The results of gene family evolution analysis revealed that a total of 293 and 97 gene families from M. notabilis had undergone expansion or contraction, respectively (Supplementary Tables 14, 15 and Supplementary Figs. 9a, 9c). The numbers of expanded and contracted gene families in M. alba were 372 and 66, respectively (Supplementary Tables 16, 17 and Supplementary Figs. 9b, 9d). KEGG enrichment analysis of the expanded gene families from the two genomes revealed that these genes were predominantly involved in the biosynthesis of secondary metabolites. In contrast, the contracted gene family pathways were mostly related to cyanoamino acid metabolism in M. notabilis and the flavonoid biosynthesis pathway in M. alba.
Furthermore, a total of 33 and 50 genes were shown to have evolved under positive Darwinian selection using codeml from the PAML package with a branch-site model in M. notabilis and M. alba, respectively. Although only six and eight positively selected genes from the M. notabilis and M. alba genomes, respectively, could be mapped to KEGG pathways, the results suggested that these genes were mainly enriched in pathways related to the biosynthesis of secondary metabolites (Supplementary Tables 18, 19).
A significant difference in transposable element content between the two mulberries
A total of 57.07% of the M. notabilis genome could be classified as repetitive sequences, whereas the repetitive sequence proportion in the M. alba genome was 52.91% (Supplementary Table 6). Among these repetitive sequences, the proportions of both total LTRs and the related superfamilies showed a significant difference between these two genomes (Fig. 5a and Supplementary Fig. 10).
Figure 5.
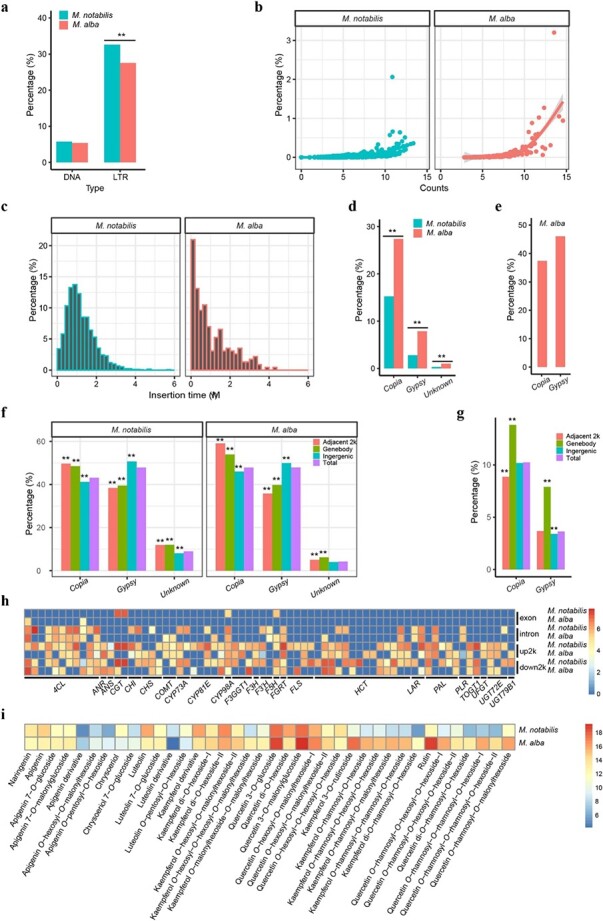
Comparative analysis of long terminal repeats (LTRs) in the genomes of M. notabilis and M. alba. Comparison of DNA and LTR TE proportions between the two genomes. **P < 0.01 (a). Comparison of the copy numbers and proportions of each LTR in the two genomes. The counts of each element indicate the number of element fragments in the genome. The x-axis values were log2 transformed. The y-axis denotes the proportion of each element in the genome. The best-fit regression lines are shown (b). Insertion time distribution and amplification of LTR TEs in the M. notabilis and M. alba genomes. The width of the insertion time bins was 0.2 MYA. The x-axis denotes the insertion times of all full-length LTRs. The values on the y-axis represent the proportion of elements per time interval (c). Proportions and comparison of LTRs belonging to the same families between the two genomes. The proportion of each superfamily was calculated based on the total length of the same family elements of the superfamily/the total length of the elements of the superfamily. **P < 0.01 (d). Proportion of rearranged Copia and Gypsy elements in the M. alba genome. The bars denote the proportion of the corresponding superfamily, which was calculated from the total length of the rearranged elements of the superfamily/total length of the same family elements in the M. alba genome (e). Genome-wide distribution patterns of LTR TEs within and around genes between the two mulberry genomes. The genomes were classified into three regions: gene body regions, adjacent 2 kb upstream and downstream regions, and intergenic regions. The bars denote the proportions of the corresponding superfamily elements in the aforementioned regions. The proportions of superfamily elements within these regions were compared with those in the genome. **P < 0.01 (f). Genome-wide distribution patterns of rearranged LTR TEs in the M. alba genome. The genome was also classified into three regions: gene body regions, adjacent 2 kb upstream and downstream regions, and intergenic regions. The proportions of rearranged superfamily elements within those regions were compared with those in the genome. **P < 0.01 (g). LTRs were located in the gene body or adjacent 2 kb upstream and downstream regions of 71 flavonoid biosynthesis pathway-related genes. Columns in the heatmap represent genes. Rows in the heatmap represent regions of the genes. The number displayed near the color scale indicates the length (log2 transformed) of LTRs in the corresponding area (h). Summary of the differences in flavonoid metabolite content between M. notabilis and M. alba. Columns in the heatmap represent concentrations of flavonoid metabolites. Rows in the heatmap represent the two mulberry species. The color bar denotes the concentrations (log2 transformed) of flavonoid metabolites in the two mulberry species (i).
LTRs showed lower copy numbers and proportions in the M. notabilis genome than in the M. alba genome (Fig. 5b and Supplementary Fig. 11). Insertion time analysis of all full-length LTRs in the two genomes indicated that most of these elements were inserted into the corresponding genomes within the past 4 MYA (Fig. 5c). Peak frequencies of LTR insertions were found at approximately 1 MYA and 0.2 MYA in M. notabilis and M. alba, respectively. Compared with M. notabilis, the distribution pattern of full-length LTRs in M. alba resembled a negative exponential distribution.
To further understand the relationship of LTRs between these two genomes, all LTRs belonging to the same family or species-specific family were identified between the two genomes. As shown in Fig. 5d and Supplementary Fig. 12a, the proportions of LTRs belonging to the same family of the Copia and Gypsy superfamilies in the M. alba genome were significantly higher than those in the M. notabilis genome. MorusRLC0002 and MorusRLG0001 accounted for 25.89% and 42.34% of the total length of the same families of Copia and Gypsy, respectively, making the greatest contribution to the burst of these families in M. alba (Supplementary Table 20). In regard to the species-specific families in the two genomes, the proportion of specific families in the M. notabilis genome was significantly higher than that in the M. alba genome (Supplementary Fig. 12b). The largest specific family, MnotRLC0001, which accounted for 30.09% of the total length of the specific families of Copia, contributed most to the burst of special Copia families in the M. notabilis genome (Supplementary Table 20). If one LTR belonging to the same family was located on other noncorresponding chromosomes in the two genomes, it was regarded as a rearranged LTR. Further analysis suggested that rearranged LTRs occupied a large proportion of the total number of LTRs in M. alba, including 37.43% and 46.07% of Copia and Gypsy elements, respectively (Fig. 5e).
In light of the observations that there were significantly different total LTR contents in the two genomes and that rearranged LTRs accounted for a large fraction of the total LTRs in M. alba, the distribution patterns of the LTRs in the two genomes were analyzed to determine whether any of these superfamilies tended to be distributed within or around genes in each respective genome. LTRs with similar distribution patterns in the two genomes were therefore studied (Fig. 5f). Both Copia and unknown LTRs showed a greater tendency to be distributed within gene bodies and adjacent genes (i.e. within a 2 kb adjacent region upstream or downstream of a gene), whereas Gypsy elements were found within the intergenic region more often than expected (Fig. 5f). In regard to the distribution patterns of rearranged LTRs in M. alba, however, both Copia and Gypsy elements were significantly overrepresented in the gene body regions (Fig. 5g). All LTR-related genes were mapped to KEGG pathways, and our KEGG enrichment analysis suggested that such genes in M. alba were enriched with respect to the biosynthesis of secondary metabolites (Supplementary Fig. 13 and Supplementary Table 21). Although these genes were not enriched in the same pathway in M. notabilis, the largest proportion of KEGG mapped genes (26.33%, 933/3542) participated in the same pathway (Supplementary Table 22). Detailed distribution analysis further suggested that LTRs showed different distribution patterns in many flavonoid biosynthesis pathway-related genes, such as ANS, CHS, and UGT (Fig. 5h). It should be mentioned that more LTRs were found within and around such genes in M. notabilis than in M. alba. The results of metabolomic analysis also revealed that many flavonoid metabolites showed significant differences in concentration between these two mulberry species (Fig. 5i). For example, the fold difference of the quercetin O-rhamnosyl-O-rhamnosyl-O-hexoside-I concentrations between the two species reached 12.07 (Fig. 5i).
Discussion
Chromosome number evolution from M. notabilis to M. alba
In this study, the genome of M. notabilis was sequenced using the techniques of ONT and Hi-C and assembled into six high-quality chromosome sequences. The haplotype chromosome number of M. notabilis was seven [2, 17], which was different from the assembled chromosome number. Mitotic chromosomes 5 and 7 fused to form meiotic chromosome 5, as proposed in our previous study [17]. In the present study, two single-copy sequence probes from assembled chromosome 5 were designed to test this chromosomal fusion event. Our results indicated that Mn-chr5A located on chromosome 7 and Mn-chr5B located on chromosome 5 were fused into diakinesis chromosome 5 in M. notabilis (Fig. 3j–m), demonstrating why the genome sequences of M. notabilis were assembled into six chromosomes. The physical lengths of our assembled chromosomes were consistent with the relative lengths of diakinesis chromosomes in M. notabilis, suggesting that a corrected chromosome order should be proposed [17]. A chromosomal fusion event was also identified in M. alba, in which two mitotic chromosomes were fused into diakinesis chromosome 1 (Fig. 3n–r). Thus, chromosomal fusion events from mitotic chromosomes to meiotic chromosomes were shared by ancient wild M. notabilis and cultivated M. alba.
Previously, a consensus was reached that the basic chromosome number of M. notabilis was seven [2]. Recently, Jiao et al. proposed that the basic chromosome number of cultivated mulberry, M. alba, was 14 at the sequence level [13]. Hence, two basic chromosome numbers (n = 7 and n = 14) have been assumed to coexist in the genus Morus. Multiple basic chromosome numbers have been reported in many genera, such as Melampodium (n = 11, n = 9, and n = 14) [23], Gossypium (n = 12 and n = 13) [24], and Cucumis (n = 12 and n = 7) [25]. High-quality genome sequences provide valuable resources for studying the genome-wide evolution of plant species. Chromosomal fission/fusion and polyploidization are the predominant causes of chromosome number evolution [26–29]. Genomic synteny analyses and FISH-based comparative chromosome analyses of M. notabilis and M. alba were also performed in the current study. Our results indicated strong chromosome-level synteny between the six chromosomes of M. notabilis and the 14 chromosomes of M. alba (Fig. 2 and Fig. 3a–i). In addition, no intermediate chromosome number between 14 and 28 has been identified in mulberry. We propose that large-scale chromosomal fission/fusion events resulted in changes in the basic chromosome number of mulberry based on several lines of evidence. Triplication predated mulberry/grapevine separation, and no WGD events occurred in the mulberry lineage after separation (Supplementary Fig. 3). Correspondence was observed between the chromosomal structures of the two mulberries reconstructed from the three ancestral genomes in grapevine (Supplementary Fig. 3), and only 386 rearranged genes were identified in the two mulberry genomes (Fig. 2b, and Supplementary Table 8). Similar findings were observed in monkeyflower (Mimulus), in which at least eight fission events and two fusion events led the basic chromosome number to evolve from 8 to 14 [30]. These results suggest that large-scale chromosomal fission/fusion events drove the speciation of M. notabilis and M. alba.
Centromere evolution in mulberry
As the key element required for faithful chromosome segregation during mitotic and meiotic cell division, centromeres have played important roles in the evolutionof mulberry, especially during the occurrence of large-scalechromosome fission/fusion events. The multiple occurrences of Mn-175 nt on the chromosomes of M. notabilis suggested that inactivated centromeres existed in this genome. This phenomenon has also been observed in maize and wheat [31, 32]. The inactivated centromere sites in M. notabilis provided candidate centromere sites to ensure the accurate inheritance of the broken chromosomes of M. alba, thus avoiding the loss of genetic material. The reactivation of inactivated centromeres has been reported in maize [33]. In addition, chromosome fusion from mitotic chromosomes to meiotic chromosomes was identified in M. notabilis and M. alba in the present study. The fused chromosomes both divided and segregated together normally in the meiotic phases, as previously reported [9], suggesting that a core centromere had formed. The fusion of multiple centromeres into a single centromere has also been reported previously [32], a finding that was consistent with our results. Thus, the inactivation and reactivation of centromeres ensured the stable inheritance of the broken and fused chromosomes of mulberry, making it a good model for further studies of centromere evolution.
Long terminal repeats and associated processes play major roles in shaping mulberry genomes.
It is worth paying attention to the dynamics of LTRs in these two genomes. Our results indicated that the two mulberry species diverged from each other approximately 10 MYA (Supplementary Fig. 2). Significant differences were found both within a given gene family and in species-specific gene family proportions between the two genomes (Fig. 5d and Supplementary Fig. 12). Combined with the results described earlier, we suggest that after being derived from M. notabilis, the amplification and proliferation of species-specific LTRs were not as high in the M. alba genome as in the M. notabilis genome. In other words, the LTRs of M. notabilis showed more potent activity than those of M. alba. For instance, MnotRLC0001, the largest species-specific family, belonged to the Copia superfamily, contributing the most to the burst of specific Copia in the M. notabilis genome. A possible explanation for this phenomenon may be the different environments in which the wild and cultivated mulberry species grew and evolved. As reported previously, retrotransposons become activated and are quickly amplified under strong natural selection forces, such as unexpected climate change, abiotic stresses, and biotic stresses [21, 34].
In regard to the rearrangement of LTRs in the two genomes following their divergence from one another, an interesting phenomenon was observed. The distribution patterns of the rearranged Copia and Gypsy elements were not completely random, showing a greater tendency to be distributed in gene body regions (Fig. 5g) than to exhibit an overall random distribution pattern (Fig. 5f). Previous studies carried out in different plant species have suggested that numerous unique structural chromosomal rearrangements are largely attributable to transposition and proliferation events of LTRs [35, 36]. Our results were not only consistent with those previously reported in other plants but also indicated that rearranged LTRs exhibited dramatic dynamic changes relative to other species-specific LTRs in mulberry genomes and may play important roles in the diversity of the two mulberry genomes.
Due to the significant difference in the abundance of LTRs in the genomes of the wild and cultivated mulberry species, it was not surprising to find that LTRs may affect gene functions when they are located in the direct vicinity of such genes. In fact, the important roles of these elements as regulators of gene function in many plant genomes have been well demonstrated [21, 22, 37, 38]. Some examples of LTRs regulating gene expression should be mentioned here. When the Gret LTR was integrated upstream of the VvmbyA1 gene, which regulates anthocyanin biosynthesis in the black-skinned grape cultivar Kyoho, grape skin color changed in grape cultivars such as ‘Ruby Okuyama’ and ‘Chardonnay’ containing the integrated Gret LTR [19, 22]. Another LTR, Rider, provides a novel promoter that can drive the expression of the Ruby gene in the flesh of orange fruit, resulting in the development of blood oranges [19, 21]. When we focused on the metabolite differences between the wild and cultivated mulberry species, we found significant differences in flavonoid metabolite contents in these two mulberry species (Fig. 5i). Such a difference was also revealed at the genomic level. As illustrated in the present study, we assigned the members of five different gene sets (rearranged, species-specific, contracted, expanded, and positively selected genes) to KEGG pathways and found that a total of 18, 6, 7, 52, and 2 genes, respectively, were involved in the biosynthesis of secondary metabolites, compared with 22, 12, 6, 55, and 2 genes, respectively, mapped to the biosynthesis of secondary metabolites in M. alba, but none of these genes exhibited a degree of altered gene function comparable to those associated with LTRs. The numbers of LTR-associated genes involved in the biosynthesis of secondary metabolites were as high as 933 and 793 in M. notabilis and M. alba, respectively. For example, a total of 73 genes associated with flavonoid biosynthesis were flanked by LTRs or contained LTRs in the two genomes (Fig. 5h). Considering these results in conjunction with the roles LTRs have been shown to play in previous reports, we assert that LTRs are a major driver in genome divergence and the evolution of mulberry species.
In conclusion, we assembled a high-quality chromosome-level genome sequence of M. notabilis. Our results illustrated that chromosome fissions/fusions were the key mechanism underlying the evolution of differences in the basic chromosome number between M. notabilis and M. alba. Subsequently, LTRs played critical roles in determining differences in the architecture of these two mulberry genomes. Our high-quality genome sequence of M. notabilis provides a valuable resource for the study of the origin, genome evolution, domestication, and subsequent genetic improvement of mulberry.
Materials and methods
Plant materials, genome sequencing
Fresh young leaves were collected from the wild mulberry species M. notabilis growing wild in Ya’an, Sichuan Province, China. The leaves were immediately frozen in liquid nitrogen and stored. Total DNA was prepared using a standard cetyltrimethylammonium bromide method. After extraction, the concentration, integrity, and purity of the DNA were determined using a NanoDrop spectrophotometer (A260/A280 = 1.8, A260/A230 = 2.0–2.2) and Qubit. Then, genomic DNA was repaired using the NEBNext FFPE DNA Repair Mix Kit (M6630, USA) and was subsequently processed using the Ligation Sequencing Kit (SQK-LSK109, UK) according to the manufacturer’s instructions. The high-quality large segment library was premixed with loading beads and sequenced on the ONT PromethION platform with the ONT sequencing reagents kit (EXP-FLP001.PRO.6, UK) and a corresponding R9 cell, according to the manufacturer’s instructions.
For Hi-C sequencing, the chromosomal structure was crosslinked with formaldehyde, and the genomic DNA was digested using HindIII, as previously described [39]. A Hi-C library with a 300 bp–700 bp insert size was constructed and subsequently sequenced on the Illumina NovaSeq 6000 platform.
De novo genome assembly
The genome size of M. notabilis was estimated with GenomeScope2 (v2.0) [40] using high-quality Illumina reads. All raw reads generated on the ONT platform were first corrected with Canu (v1.8) software [41]. Thereafter, the error correction and de novo assembly tool NECAT (v0.01, https://github.com/xiaochuanle/NECAT) was used to assemble the original contigs. After initial assembly, the contigs generated were immediately corrected by aligning ONT reads from three rounds using Racon (v1.3.3) [42]. Then, Pilon (v1.23) [43] was used to polish the contigs for three rounds with the Illumina paired-end (PE) reads to generate the final contigs, which were assessed by evaluating the mapping ratio to Illumina reads and completeness and integrity by using Benchmarking Universal Single-Copy Orthologs (BUSCO) (v3) [44].
For chromosome-level assembly, all raw Hi-C data reads were cleaned to remove adapter sequences and low-quality PE reads to generate high-quality clean data. Then, the high-quality Hi-C data were truncated at putative Hi-C junctions and mapped to contigs using the Burrows-Wheeler Aligner (v0.7.7) software package. HiC-Pro (v2.8.1) [45] was used to filter valid reads. Only uniquely mapped interaction paired reads were selected and used to cluster and order the genome sequences onto chromosomes using LACHESIS [46] and Juicebox (v1.11.08) [47].
Repetitive element and genome annotation
Repetitive elements were identified through two strategies: de novo and structural signature-based identification. The de novo identification of TEs was performed using RepeatModeler (v2.0.1, http://www.repeatmasker.org/RepeatModeler/) based on our assembled genome to generate a de novo repeat library, which was named denovoLib. LTR_finder (v1.07) [48] and LTR_harvest, which was distributed with GenomeTools (v1.5.10) [49], were used to identify LTR retrotransposons. Subsequently, LTR_retriever (v2.8.7) [50] was used to improve the accuracy of LTR identification from the outputs of RepeatModeler and LTR_harvest. MGEScan-nonLTR (v2) [51] was used to identify non-LTR retrotransposons. The results generated from our structural signature-based strategy were combined to form another repeat library, named signatureLib. The two repeat libraries (denovoLib and signatureLib) were combined to generate a custom library as the input library for RepeatMasker (v4.1.0, http://www.repeatmasker.org/RepeatMasker/) to annotate and mask repetitive elements in our assembled genome. LTR retrotransposons were classified into the corresponding superfamilies using LTR_retriever (v2.8.7) [50]. Thereafter, these LTR retrotransposons were classified into families using CD-hit (v4.8.1, http://weizhong-lab.ucsd.edu/cd-hit/) based on the 5′ LTR/3′ LTR sequences, according to the 80–80–80 rule [52]. To estimate the evolutionary insertion time of LTR retrotransposons, the two LTRs of an intact LTR retrotransposon were retrieved using an in-house Perl script and aligned using MUSCLE (v3.8.31). Then, their nucleotide divergence was calculated using the baseml module, which was deployed in PAML (v4.9) [53]. Finally, the insertion time was calculated using the Formula T = K/2r as reported previously [51], where K represents the divergence between the two LTRs, and r indicates the number of substitutions per synonymous mutation site per year (5.62 × 10−9) [54].
Gene prediction in the genome was carried out by two strategies: de novo and homolog-based prediction. The MAKER (v2.31.10) genome annotation pipeline was utilized for the whole process. Gene evidence was produced by assembling RNA-Seq reads from male flowers, winter buds, bark, root, fruit, and leaves using Trinity (v2.9.0) [55]. The custom repeat library generated as described earlier was used as the input repeat library. Protein homology evidence was obtained from M. notabilis. Two ab initio gene predictors, Augustus (v3.3.3) [56] and GeneMark-ES (v3.60) [57], were used. The keep_preds parameter was set to 1 to ensure that no gene models were lost from the legacy annotations. Thereafter, amino acid sequences were searched against the InterProScan database [58]. The quality_filter.pl script, distributed with MAKER, was used to filter gene models based on domain content and evidence-based support (Annotation Edit Distance, AED < 1 and/or Pfam domain present). The retained amino acid sequences were then searched against other databases, including the Swiss-Prot, Pfam, KEGG, and GO databases. To compare the two mulberry species, we also performed genome annotation using the M. alba ‘Heyebai’ genome using the same methods described earlier.
Genome and gene collinearity
MUMmer (v3.1) [59] was used to carry out genome alignment between M. notabilis and M. alba. The DotPrep.py (https://github.com/dnanexus/dot) script was used to visualize the genome–genome alignment results. TBtools (v1.055) [60] was used to generate and visualize gene synteny between the two genomes. OrthoFinder (v2.4.0) [61] was used to identify orthologous gene pairs between grapevine and mulberry. The V. vinifera genome, which is by far the closest to the ancestral genome, was used to reconstruct mulberry chromosomal structures.
Gene family annotation and comparative genomic analyses
Homologous genes and species-specific genes from different plant species (M. notabilis, M. alba, Pyunus persica, and Cannabis sativa) were identified with OrthoFinder (v2.4.0) [61]. To construct a phylogenetic tree between these species, the integrated platform PhyloSuite (v1.2.2) [62] was used. All single-copy genes were combined and aligned using MAFFT (v7.471) [63] in codon alignment mode. Then, the alignments were refined using the codon-aware program MACSE (v2.03) [64]. This was followed by the application of Gblocks (v0.91b) [65] to remove ambiguously aligned fragments. All sequences from one species were concatenated into one sequence, and ModelFinder [66] was utilized to select the best-fit partition model using the Bayesian information criterion. Finally, IQ-TREE (v1.6.12) [67] was used to construct a phylogenetic tree according to the best-fit model generated earlier. After this, MCMCtree, implemented in PAML (v4.9) [53], was used to calculate the divergence time between these species, and the time was calibrated with the reported time [2, 13]. CAFÉ (v4.2.1) [68] was used to analyze the expansion and contraction of gene families in the two genomes. Enrichment analyses were performed using clusterProfiler (v3.16.1).
Analyses of positively selected genes
A species tree was built as mentioned earlier. All one-to-one orthologs identified by OrthoFinder (v2.4.0) [61] were combined and aligned using MAFFT (v7.471) [63]. PAL2NAL (v14) [69] was used to convert the protein alignment to a codon-based nucleotide alignment. After removing ambiguously aligned regions using Gblocks (v0.91b) [65], codeml from the PAML (v4.9) package [53] was used to estimate ω values (Ka/Ks) with a branch-site model to identify genes that had undergone Darwinian positive selection [53]. Genes with a false discovery rate value of ≤0.05 were considered to be positively selected genes.
Chromosome preparation
The mitotic and diakinetic meiotic chromosomes of M. notabilis and M. alba ‘Lunjiao109’ were prepared as described previously [9]. For mitotic chromosome preparation, young leaves were pretreated with 2 mM 8-hydroxyquinoline at room temperature for 3 h, fixed in 9:1 ethanol/glacial acetic acid at 4°C for 4 h, and then stored in 70% ethanol at −20°C. The leaves were then washed three times in distilled water and digested in an enzyme solution composed of 2% (w/v) cellulase Onozuka R-10 (YaKult, Japan) and 1% (w/v) pectolyase Y-23 (YaKult, Japan) (pH 5.5) at 37°C for 2.5 h. Young inflorescences were directly fixed in 9:1 ethanol/glacial acetic acid at 4°C for 4 h and stored in 70% ethanol at −20°C. Anthers were digested in an enzyme solution of 2% (w/v) cellulase Onozuka R-10 (YaKult, Japan) and 1% (w/v) pectolyase Y-23 (YaKult, Japan) (pH 5.5) at 37°C for 4 h. Digested leaves and anthers were rinsed with 70% ethanol and macerated into fine suspensions. The cells were resuspended in glacial acetic acid, and a drop of the suspension was added to a glass slide. Then, the slides were screened for well-spread chromosome preparations under an Olympus IX73 microscope (Olympus, Tokyo, Japan).
Probe development and FISH
Single-copy sequences with a length >10 000 bp were screened using BLASTn against the genome sequence of M. notabilis. Repeat sequences were removed through primer design using Primer3. Six out of the 11 sequences (Mn-chr1A, Mn-chr1B, Mn-chr1C, and Mn-chr1D; Mn-chr5A and Mn-chr5B) are shown in Supplementary Table 23. Candidate centromere tandem repeats from M. notabilis and M. alba were screened using Tandem Repeats Finder (TRF) with parameters = [2, 7] (weights for matches, mismatches, and indels, respectively) [70]. A minimum alignment score of 80 and a maximum period size of 1000 were used in the identification of tandem repeats. A plasmid vector containing two copies of Mn-175 nt, was synthesized by TsingKe Biological Technology (Beijing, China). Single-copy, Mn-175 nt, and 25S rDNA sequence probes were labeled, and FISH was performed as described previously [9]. The primers used in this study are listed in Supplementary Table 23. Images were captured with a DP80 charge-coupled device camera attached to an Olympus IX73 microscope (Olympus, Tokyo, Japan). The images were processed with Adobe Photoshop CS6 and Adobe Illustrator CS6.
Acknowledgments
This project was supported by the National Key Research and Development Program (Grant No. 2018YFD1000602) and the Chongqing Research Program of Basic Research and Frontier Technology (Grant No. cstc2018jcyjAX0407).
Author contributions
N.H. conceived and designed the study, Q.Z. contributed to sample preparation, B.M. performed genome assembly and genome annotation, B.M., Y.X., and N.H. carried out comparative genomics analyses, Y.X. and Y.T. performed FISH, B.M. and D.L. carried out metabolomics analysis, B.M. and Y.X. prepared the figures and tables and wrote the draft manuscript, and N.H. revised the manuscript. All authors read and approved the final manuscript.
Data availability
All the data generated during the present study have been deposited in the Genome Sequence Archive (GSA) and the Genome Warehouse (GWH) database (PRJCA004004).
Conflicts of interest statement
The authors declare that there are no conflicts of interest.
Supplementary data
Supplementary data is available at Horticulture Research Journal online.
Supplementary Material
References
- 1. Nepal MP, Ferguson CJ. Phylogenetics of Morus (Moraceae) inferred from ITS and trnL-trnF sequence data. Syst Bot. 2012;37:442–50. [Google Scholar]
- 2. He N, Zhang N, Qi Wet al. Draft genome sequence of the mulberry tree Morus notabilis. Nat Commun. 2013;4:2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu YF, Li H, Zhang Bet al. Nutritional and functional components of mulberry leaves from different varieties: evaluation of their potential as food materials. Int J Food Prop. 2018;21:1495–507. [Google Scholar]
- 4. Li D, Ma B, Xu Xet al. MMHub, a database for the mulberry metabolome. Database (Oxford). 2020;2020. 10.1093/database/baaa011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li D, Chen G, Ma Bet al. Metabolic profiling and transcriptome analysis of mulberry leaves provide insights into flavonoid biosynthesis. J Agr Food Chem. 2020;68:1494–504. [DOI] [PubMed] [Google Scholar]
- 6. Li H, Yang Z, Zeng Qet al. Abnormal expression of bHLH3 disrupts a flavonoid homeostasis network, causing differences in pigment composition among mulberry fruits. Hortic Res. 2020;7:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu J, Wan J, Wang Det al. Comparative transcriptome analysis of key reductase genes involved in the 1-Deoxynojirimycin biosynthetic pathway in mulberry leaves and cloning, prokaryotic expression, and functional analysis of MaSDR1 and MaSDR2. J Agric Food Chem. 2020;68:12345–57. [DOI] [PubMed] [Google Scholar]
- 8. Sattler MC, Carvalho CR, Clarindo WR. The polyploidy and its key role in plant breeding. Planta. 2016;243:281–96. [DOI] [PubMed] [Google Scholar]
- 9. Xuan Y, Li C, Wu Yet al. FISH-based mitotic and meiotic diakinesis karyotypes of Morus notabilis reveal a chromosomal fusion-fission cycle between mitotic and meiotic phases. Sci Rep. 2017;7:9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xuan YH, Wu Y, Li Pet al. Molecular phylogeny of mulberries reconstructed from ITS and two cpDNA sequences. Peerj. 2019;7:e8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Venkatesh KH, Nijagunaiah R, Munirajappa . Cytogenetical studies in some diploid mulberry varieties (Moraceae). Cytologia. 2013;78:69–72. [Google Scholar]
- 12. Schneider, CK. In: Plantae Wilsonianae: An Enumeration of the Woody Plants Collected in Western China for the Arnold Arboretum of Harvard University during the Years 1907, 1908, and 1910 (ed. by Charles Sprague Sargent). Cambridge: The University Press, 1916, 293–4. [Google Scholar]
- 13. Jiao F, Luo R, Dai Xet al. Chromosome-level reference genome and population genomic analysis provide insights into the evolution and improvement of domesticated mulberry (Morus alba). Mol Plant. 2020;13:1001–12. [DOI] [PubMed] [Google Scholar]
- 14. Zeng Q, Chen H, Zhang Cet al. Definition of eight mulberry species in the genus Morus by internal transcribed spacer-based phylogeny. PLoS One. 2015;10:e0135411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X, Xu Y, Zhang Set al. Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat Genet. 2017;49:765–72. [DOI] [PubMed] [Google Scholar]
- 16. Zhou Z, Jiang Y, Wang Zet al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol. 2015;33:408–14. [DOI] [PubMed] [Google Scholar]
- 17. Lin T, Zhu G, Zhang Jet al. Genomic analyses provide insights into the history of tomato breeding. Nat Genet. 2014;46:1220–6. [DOI] [PubMed] [Google Scholar]
- 18. Zeng L, Tu X-L, Dai Het al. Whole genomes and transcriptomes reveal adaptation and domestication of pistachio. Genome Biol. 2019;20:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lisch D. How important are transposons for plant evolution? Nat Rev Genet. 2013;14:49–61. [DOI] [PubMed] [Google Scholar]
- 20. Daccord N, Celton J-M, Linsmith Get al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat Genet. 2017;49:1099–106. [DOI] [PubMed] [Google Scholar]
- 21. Butelli E, Licciardello C, Zhang Yet al. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell. 2012;24:1242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kobayashi S, Goto-Yamamoto N, Hirochika H. Retrotransposon-induced mutations in grape skin color. Science. 2004;304:982–2. [DOI] [PubMed] [Google Scholar]
- 23. McCann J, Schneeweiss GM, Stuessy TFet al. The impact of reconstruction methods, phylogenetic uncertainty and branch lengths on inference of chromosome number evolution in American daisies (Melampodium, Asteraceae). PLoS One. 2016;11:e0162299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Udall JA, Long E, Ramaraj Tet al. The genome sequence of Gossypioides kirkii illustrates a descending dysploidy in plants. Front Plant Sci. 2019;10:1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang L, Koo D-H, Li Det al. Next-generation sequencing, FISH mapping and synteny-based modeling reveal mechanisms of decreasing dysploidy in Cucumis. Plant J. 2014;77:16–30. [DOI] [PubMed] [Google Scholar]
- 26. Cheng YB, Shang D, Lup Met al. Whole genome-wide chromosome fusion and new gene birth in the Monopterus albus genome. Cell Biosci. 2020;10:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mayrose I, Lysak MA. The evolution of chromosome numbers: mechanistic models and experimental approaches. Genome Biol Evol. 2021;13:evaa220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang ZY, Wang XY. Evolutionary genomics model of chromosome number reduction and B chromosome production (in Chinese). Sci Sin Vitae. 2020;50:524–37. [Google Scholar]
- 29. Wang XY, Jin D, Wang Zet al. Telomere-centric genome repatterning determines recurring chromosome number reductions during the evolution of eukaryotes. New Phytol. 2015;205:378–89. [DOI] [PubMed] [Google Scholar]
- 30. Fishman L, Willis JH, Wu CAet al. Comparative linkage maps suggest that fission, not polyploidy, underlies near-doubling of chromosome number within monkeyflowers (Mimulus; Phrymaceae). Heredity. 2014;112:562–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao Z, Fu S, Dong Qet al. Inactivation of a centromere during the formation of a translocation in maize. Chromosome Res. 2011;19:755–61. [DOI] [PubMed] [Google Scholar]
- 32. Zhang W, Friebe B, Gill BSet al. Centromere inactivation and epigenetic modifications of a plant chromosome with three functional centromeres. Chromosoma. 2010;119:553–63. [DOI] [PubMed] [Google Scholar]
- 33. Han F, Gao Z, Birchler JA. Reactivation of an inactive centromere reveals epigenetic and structural components for centromere specification in maize. Plant Cell. 2009;21:1929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Casacuberta E, Gonzalez J. The impact of transposable elements in environmental adaptation. Mol Ecol. 2013;22:1503–17. [DOI] [PubMed] [Google Scholar]
- 35. Ungerer MC, Strakosh SC, Zhen Y. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr Biol. 2006;16:R872–3. [DOI] [PubMed] [Google Scholar]
- 36. Parisod C, Badaeva ED. Chromosome restructuring among hybridizing wild wheats. New Phytol. 2020;226:1263–73. [DOI] [PubMed] [Google Scholar]
- 37. Bariah I, Keidar-Friedman D, Kashkush K. Where the wild things are: transposable elements as drivers of structural and functional variations in the wheat genome. Front Plant Sci. 2020;11:585515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma B, Xin Y, Kuang Let al. Distribution and characteristics of transposable elements in the mulberry genome. Plant Genome. 2019;12:180094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rao SSP, Huntley MH, Durand NCet al. A 3D map of the human genome at Kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ranallo-Benavidez TR, Jaron KSet al. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11:1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koren S, Walenz BP, Berlin Ket al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vaser R, Sovic I, Nagarajan Net al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walker BJ, Abeel T, Shea Tet al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simao FA, Waterhouse RM, Ioannidis Pet al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2. [DOI] [PubMed] [Google Scholar]
- 45. Servant N, Varoquaux N, Lajoie BRet al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burton JN, Adey A, Patwardhan RPet al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31:1119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Durand NC, Robinson JT, Shamim MSet al. Juicebox provides a visualization system for Hi-C contact maps with unlimited Zoom. Cell Syst. 2016;3:99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gremme G, Steinbiss S, Kurtz S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013;10:645–56. [DOI] [PubMed] [Google Scholar]
- 50. Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176:1410–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rho M, Tang H. MGEScan-non-LTR: computational identification and classification of autonomous non-LTR retrotransposons in eukaryotic genomes. Nucleic Acids Res. 2009;37:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wicker T, Sabot F, Hua-Van Aet al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–82. [DOI] [PubMed] [Google Scholar]
- 53. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. [DOI] [PubMed] [Google Scholar]
- 54. Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101:12404–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grabherr MG, Haas BJ, Yassour Met al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–U130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stanke M, Diekhans M, Baertsch Ret al. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–44. [DOI] [PubMed] [Google Scholar]
- 57. Ter-Hovhannisyan V, Lomsadze A, Chernoff YOet al. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jones P, Binns D, Chang H-Yet al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kurtz S, Phillippy A, Delcher ALet al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen C, Chen H, Thomas HRet al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202. [DOI] [PubMed] [Google Scholar]
- 61. Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang D, Gao F, Jakovlić Iet al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 2020;20:348–55. [DOI] [PubMed] [Google Scholar]
- 63. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ranwez V, Douzery EJP, Cambon Cet al. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol Biol Evol. 2018;35:2582–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77. [DOI] [PubMed] [Google Scholar]
- 66. Kalyaanamoorthy S, Minh BQ, Wong TKFet al. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nguyen LT, Schmidt HA, Haeseler Aet al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Han MV, Thomas GWC, Lugo-Martinez Jet al. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013;30:1987–97. [DOI] [PubMed] [Google Scholar]
- 69. Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data generated during the present study have been deposited in the Genome Sequence Archive (GSA) and the Genome Warehouse (GWH) database (PRJCA004004).