

Abstract
Cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors have revolutionized the treatment of hormone-positive metastatic breast cancers (mBCs). They are currently established as standard therapies in combination with endocrine therapy as first- and second-line systemic treatment options for both endocrine-sensitive and endocrine-resistant mBC populations. In the first-line metastatic setting, the median progression-free survival for the three currently approved CDK4/6 inhibitors, palbociclib, ribociclib, and abemaciclib, with aromatase inhibitors is greater than 2 years (palbociclib 27.6 months; ribociclib 25.3 months; and abemaciclib 28.18 months). Although CDK4/6 inhibitors have significant clinical benefits and enable physicians to delay starting chemotherapy, they are expensive and can be associated with drug toxicities. Here, we have performed a systemic review of the reported molecular markers predictive of drug response including intrinsic and acquired resistance for CDK4/6 inhibition in mBC. The rapidly emerging molecular landscape is captured through next-generation sequencing of breast cancers (DNA with or without RNA), liquid biopsies (circulating tumor DNA), and protein analyses. Individual molecular candidates with robust and reliable evidence are discussed in more depth.
INTRODUCTION
Cyclin-dependent kinase 4 and cyclin-dependent kinase 6 (CDK4/6) are members of the serine threonine kinase family, functioning to regulate cell cycle progression during the G1 phase of the cell cycle.1-3 CDK4/6 is activated by D-cyclins to consequently phosphorylate retinoblastoma protein (RB1), triggering the cell cycle cascade. In breast cancers, the recognition that the p16-CDK4/6-cyclin D1 axis is deregulated triggered the development of CDK4/6 inhibitors, now considered an anticancer breakthrough therapeutic strategy for hormone-positive (HR+ve) metastatic breast cancer (mBC).4-7
CONTEXT
Key Objective
To identify robust resistance biomarkers for cyclin-dependent kinase 4 and 6 (CDK4/6) inhibition in metastatic breast cancer through a systematic analysis of the emerging molecular data.
Knowledge Generated
It is recognized that multiple underlying mechanisms cause drug resistance to CDK4/6 inhibitors. These include the loss of retinoblastoma-1 function, aberrant cyclin-E signaling, and CDK6 activity in subpopulations, identifiable from circulating tumor DNA and tumor biopsies. However, there is still a considerable proportion of clinically observed drug resistance, which lacks a molecular explanation.
Clinical Relevance
We can now identify patient populations that will respond poorly to CDK4/6 inhibition through tumor profiling and next-generation sequencing of liquid biopsies before starting treatment and have the opportunity to consider switching to alternative treatment strategies.
RATIONALE FOR SYSTEMATIC REVIEW
The approved CDK4/6 inhibitors have oral bioavailability and are tolerated by most, with signature side effect profiles of neutropenia or diarrhea with growing data recognizing rarer side effects such as prothrombotic effects (1.5% increased risk)8,9 and pulmonary toxicities.10,11 There is a need for a personalized approach to mitigate drug toxicities for patients and to overcome the growing financial burden for health care systems through more effective patient selection. Palbociclib, ribociclib, and abemaciclib are expensive anticancer drugs because they are currently protected by drug patents. Given that patients can derive benefit for 2-3 years and breast cancer is one of the most prevalent solid tumor types,12 the estimated total cost of palbociclib plus letrozole is approximately $256,509 US dollars per treated patient in the United States13 and hence the need for predictive biomarkers of response beyond estrogen receptor positivity (ER+ve).
METHODS
We performed a systematic review of the published literature in accordance with the PRISMA guidance.14 The two key objectives were to (1) identify resistance biomarkers and (2) understand molecular mechanisms underpinning drug resistance for CDK4/6 inhibition in mBC.
The criteria applied for this systemic review were the following:
Inclusion Criteria
mBC
Published literature types: Original research articles and published abstracts. Only systematic reviews or meta-analyses were permissible.
Phase I-III clinical trials, real-world analyses, case reports, and case series
Retrospective or prospective biomarker analyses
Approved CDK4/6 inhibitors
Clinical samples from solid tumors, primary or metastatic biopsy sites, or liquid biopsies
Tissue collection time points: pretreatment, on treatment, or at progression
Types of molecular analytes: DNA aberrations (mutation, copy number aberrations, fusions, deletion events, and promoter methylation), mRNA (gene expression and plasma-derived exosome mRNA), and protein and phosphoproteins.
Selected databases: PubMed, ASCO, European Society for Medical Oncology (ESMO) and ESMO breast, and San Antonio Breast Cancer Conference.
Exclusion Criteria
Nonbreast cancer studies from other solid tumor types
Trials/studies with unclear reported methods on how they identified biomarkers
Early breast cancer studies
Data reported before 2017 **Exception was made for one prospective biomarker RB1 study that was published in 2015 because the authors felt that this work was intellectually ahead of its time15
Overlapping reports from the same research groups presented at multiple conferences; priority was given to the final published articles and published abstracts
General review articles that were not systematic reviews or meta-analyses
Search Criteria and Filters
CDK4/6 biomarker
CDK4/6 biomarker; filter: review
CDK4/6 biomarker; filter: randomized clinical trial
Palbociclib; biomarkers (no filters)
Ribociclib; biomarkers (no filters)
Abemaciclib; biomarkers (no filters)
CDK4/6 resistance; breast
CDK4/6; circulating tumor DNA (ctDNA)
Selection process and data collection process.
The results were screened by one reviewer (U.S.A.), and no automation tools were used. A database was created with study titles, authorships, and abstracts using Microsoft Excel. No relevance was given to funding when reviewing the literature, and both academic and commercially driven studies were included.
The data were prioritized using the following hierarchy:
Clinical data > preclinical data
Phase III clinical studies > phase II > phase I
Prospective research > retrospective research
Biomarker study analyses with control arms for comparators
Significant candidate biomarkers were those with reproducible data, that is, reported by ≥ 3 independent studies
Unbiased analyses sequencing approaches over targeted biased approaches
The planned study outcomes were the identification and prevalence of molecular biomarkers detected from patients treated with CDK4/6 inhibitor combinations/monotherapy. The data were synthesized by checking against the inclusion/exclusion criteria. The results were filtered in three iterations to ensure accuracy and robust representation of the field. No meta-analyses were performed with the data in this study. Studies reporting biomarkers with reported P-values and corrections for false discovery rates in their statistical analyses were preferred.
Study risk of bias assessment.
It was accepted that studies using targeted profiling panels were inherently biased in their result outcomes for molecular biomarkers because these projects were hypothesis-driven and these approaches were in response to the technical challenges of profiling clinical samples, specifically the limitation of good quality tumor tissue available for analyses.
RESULTS
Initially, 1,721 records were identified through searching five online databases (PubMed, ASCO, San Antonio Breast Cancer Conference, ESMO, and ESMO Breast). After three iterations, 111 records were selected, which fulfilled both the inclusion and exclusion criteria (Fig 1). The molecular biomarkers with the strongest body of evidence for their association with drug response are described below.
FIG 1.

PRISMA flow diagram outlining the steps undertaken for systematic review. CDK4/6, cyclin-dependent kinase 4 and 6; ESMO, European Society for Medical Oncology; mBC, metastatic breast cancer; SABC, San Antonio Breast Cancer Conference.
Molecular Biomarkers for Treatment Resistance
Retinoblastoma gene 1.
Retinoblastoma gene 1 (RB1) is a tumor suppressor gene (chromosome 13), and mutations in both alleles cause retinoblastoma cancer. CDK4/6 inhibitors prevent inactivation of RB1 via inhibition of CDK4/6, inducing cell cycle arrest. Hence, RB1 is one of the most studied and reported biomarkers to date. Detecting loss of RB1 function in cancer is challenging because of the multiple molecular mechanisms involved in its inactivation. Consequently, researchers have assessed for loss of RB1 function using ctDNA from plasma, tumor DNA, gene expression (mRNA), and/or protein analyses with immunohistochemistry (IHC). Reported genetic aberrations for RB1 include loss of both alleles or loss of a single copy,16 gene deletions, promoter methylation, or small-range mutations within the RB1 gene or its promotor. Currently, there are at least 10 different RB1 genetic variants reported within the literature, with either monoclonal or polyclonal RB1 mutations detected in ctDNA within patient subpopulations at the time point of developing resistance to CDK4/6 inhibition.
Acquired drug resistance.
Multiple retrospective observational biomarker studies have tested patient samples for RB1 abnormalities from phase II and phase III landmark studies (Table 1 and Figs 2 and 3). The studies consistently report the emergence of new detectable RB1 mutations in 2%-9% of the population at the time when patients develop drug resistance to CDK4/6 inhibition.17,25,26 The development of RB1 mutations in breast cancer is considered a molecular escape mechanism used by tumors in response to treatment selection pressure of G1 cell cycle arrest. For example, a case study with longitudinal ctDNA analyses reports the emergence of RB1 mutation at the time of drug resistance to palbociclib combination, which subsequently could no longer be detected when the patient switched to alternative treatment.20
TABLE 1.
Clinical Data for Molecular RB1 Aberrations in Patients With Metastatic, HR+ve Breast Cancer Treated With CDK4/6 Inhibitor With or Without Endocrine Therapy

FIG 2.

Potential resistance biomarkers from ctDNA analyzed for drug response to CDK4/6 inhibitors in HR+ve mBC (n = 1892; gene alterations associated with drug resistance [ctDNA]). Intrinsic (baseline samples) and acquired resistance biomarkers (end of treatment samples) identified from PALOMA-3 (n = 195 paired samples; palbociclib/placebo plus fulvestrant17; n = 331 baseline28); Pooled analyses for MONALEESA-2, MONALEESA-3, and MONALEESA-7 (n = 1,503 baseline samples; ribociclib/placebo plus endocrine therapy);26 MONARCH-3 (n = 187 paired samples; abemaciclib/placebo plus aromatase inhibitor); and nextMONARCH 1 (n = 79 paired samples; abemaciclib monotherapy arm).25 Genetic aberrations to suggest pathway overactivations are as a result of amplifications in MET, EGFR, and myc and mutations/amplifications in FGFR2 and HER2. Genetic aberrations to suggest loss of pathway signaling: RB1 loss (biallelic or loss of heterozygosity) and loss of CDKN2A(p16)/2B(p15)/2C(p18). AR, gene for androgen receptor; ATM, ataxia telangiectasia mutated; CDKN2A (gene for p16 and p14 proteins), cyclin-dependent kinase inhibitor 2A; CDKN2B (gene for p15); CDKN2C (gene for p18); CHD4, chromodomain helicase DNA binding-4; ctDNA, circulating tumor DNA; EGFR, epidermal growth factor receptor; ER, estrogen receptor; FGFR2, fibroblast growth factor receptor 2; HER2, human epidermal growth factor receptor 2; HR+ve, hormone-positive (ER+ve; HER2–); mBC, metastatic breast cancer; MET, mesenchymal-epithelial transition factor; RB1, Retinoblastoma 1 gene.
FIG 3.
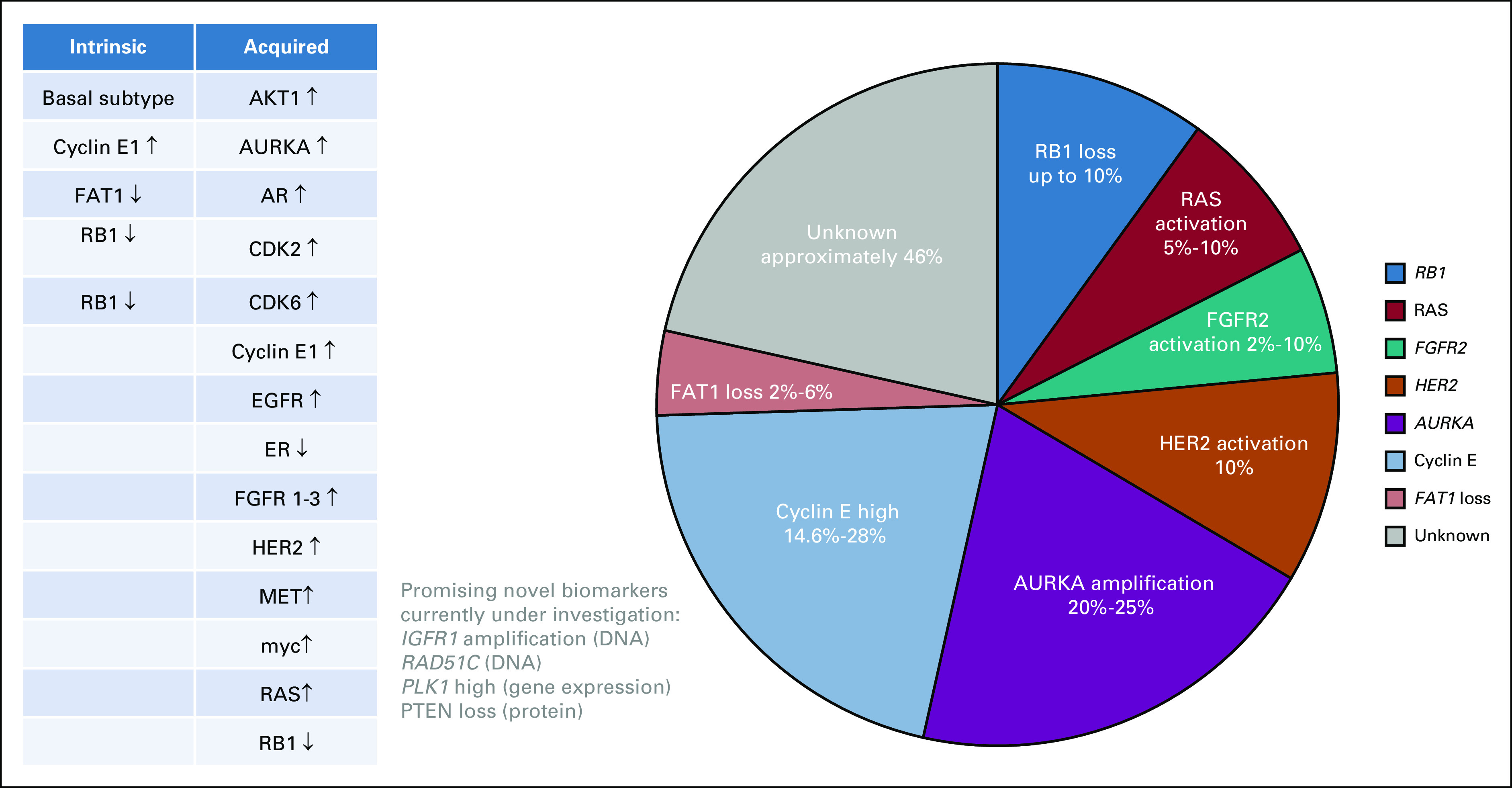
Potential resistance biomarkers from tumor analyses of primary tumor or metastases in patients with HR+ve mBC (n = 1,162; Molecular alterations associated with drug resistance [solid tumors: DNA, RNA, or protein]). Intrinsic and acquired resistance biomarkers from PALOMA2 (n = 454 palbociclib plus letrozole),29 PALOMA3 (gene expression analyses n = 302 paired samples; palbociclib plus fulvestrant),30 the study by Wander et al22 (n = 58; paired samples CDK4/6i plus endocrine therapy), and the study by Li et al21 for FAT1 loss (CDK4/6i plus endocrine therapy; n = 348). AURKA, aurora kinase A; CDK4/6, cyclin-dependent kinase 4 and 6; EGFR, epidermal growth factor receptor; ER, estrogen receptor; FAT1, FAT atypical cadherin 1; FGFR2, fibroblast growth factor receptor 2; HER2, human epidermal growth factor receptor 2; IGFR1, insulin-like growth factor 1 receptor; MET, mesenchymal-epithelial transition factor; PLK, polo-like kinase; RAS, rat sarcoma virus; RB1, Retinoblastoma 1 gene. Left: Biomarkers divided into intrinsic and acquired drug resistance categories, ↓ loss of activity and ↑ increased activity.
In the PALOMA-3 study,17 the developments of new RB1 mutations in ctDNA were the key differences between palbociclib/fulvestrant and the control arm (placebo/fulvestrant). Similar results have been published by independent research groups (see Table 1: Seth Wander et al18; Condorelli et al,19 etc) using the Guardant360 assay for liquid biopsies, and there are published abstracts from MONARCH-3 and nextMONARCH studies (abemaciclib with or without aromatase inhibitors).25 For example, two patients with acquired RB1 mutations in ctDNA on previous CDK4/6 inhibition were intrinsically resistant to subsequent abemaciclib monotherapy and rapidly progressed.18 Exome sequencing of metastatic tumor samples highlighted that those pretreatment biopsies with single-copy RB1 loss evolved by acquiring biallelic RB1 disruptions with point mutation, splice site alteration, or frameshift events in the second allele, detected in 9.8% of tumor samples.22
There is a paucity of prospective interventional clinical trials using biomarkers such as RB1 to select patients for CDK4/6 inhibition treatment. One small interventional phase II, open labeled, single-arm study (n = 37) prospectively selected patients for palbociclib monotherapy using RB1 IHC staining (> 1% on tumor)15 from pretreatment tumor samples. Historically, palbociclib is reported to have limited single-agent activity; however, the response rates were approximately 30% in this RB1+ve, ER+ve cohort that had progressed on two lines of endocrine treatment. All patients with RB1+ve triple-negative breast cancer (TNBC) progressed on palbociclib monotherapy, indicating that RB1 status has low positive predictive value for drug response with stronger evidence for its negative predictive value.
RB1 and PTEN loss.
A translational study collecting serial biopsies and analyzing autopsy samples identified that 2 of 5 patients developed simultaneous RB1 and PTEN loss after developing drug resistance to ribociclib/letrozole combination and 4 of 5 patients acquired either RB1 loss or PTEN loss. IHC analyses were used to ensure that the effects of epigenetic mechanisms were captured for RB1 and PTEN function.24
Intrinsic drug resistance.
The prevalence of RB1 mutations in baseline pretreatment clinical samples is reported between 0% and 5% using ctDNA analyses17,18,21 and in 9% of tumors (IHC for RB1 protein; 51 of 563 patients).23 One study reported that 3% of patients with RB1 loss (n = 9 of 348) treated with CDK4/6 inhibitors demonstrated a significantly shorter median progression-free survival (mPFS) of 3.6 months (95% CI, 2.2 to no response) compared with their RB1 wild-type counterparts.21 The young PEARL study, a randomized phase II clinical trial for premenopausal women, released similar data, linking RB1 loss (4% of arm A) to shorter mPFS intervals (log2 hazard ratio [HR] = 2.26; 95% CI, 0.51 to 4.01; P = .011).27,31
Overactivation of CDK2.
A growing body of evidence indicates adaptive activation of CDK2 in response to CDK4/6 inhibition.32,33 Single-cell analysis of CDK2 activity in TNBC cell lines with sensitive (luminal) and resistant phenotypes (basal) unraveled that CDK2 can be activated independent of CDK4/6 and drives cell cycle transition from the G1 to S phase in the absence of CDK4/6. Hence, overactivation of CDK2 activity in cancer cells limits the potency of CDK4/6 inhibitors (palbociclib and ribociclib). In addition, individual cancer cells adapt to CDK4/6 inhibition by increasing CDK2 activity levels earlier in the G1 phase of the cell cycle to bypass CDK4/6 blockade because of higher intracellular cyclin-E1 levels, the binding partner of CDK2.32
cMET—FAK/CDK2 axis.
Preclinical and clinical evidence indicates a role for mesenchymal-epithelial transition factor (c-MET) family receptor tyrosine kinase signaling in the refractory response to CDK4/6 inhibition. CDK2 activation independent of CDK4/6 involves cMET and its downstream effector focal adhesion kinases (FAK).34 In vitro modeling with RB1 breast cancer cell lines and other solid tumor types demonstrated that MET/FAK signaling enabled CDK4/6-independent CDK2 activation and cell cycle transit. MET activation in glioblastoma arose through CDK4/6 inhibitor–induced autocrine production of the cMET ligand hepatocyte growth factor,35 and stromal hepatocyte growth factor is known to activate MET signaling in breast cancer.36 Current clinical data linking cMET aberration with drug resistance are associated with abemaciclib although there are preclinical data with the other CDK4/6 inhibitors. In the phase-II study nextMONARCH, 8% of patients treated with abemaciclib plus tamoxifen demonstrated new genetic changes in MET (mutation, indel, or amplification).25
Aberrant cyclin-E1 signaling.
Several retrospective clinical analyses18,23,30,37 and preclinical studies32,33,38,39 report that either cyclin-E1 gene amplification or elevated gene expression has significant treatment interaction effects for CDK4/6 inhibition, supporting cyclin-E1 as a potential intrinsic resistance biomarker (Fig 3).40,41 In PALOMA-3 (n = 302), palbociclib efficacy was lower in patients with high cyclin-E1 gene expression levels. Tumor profiling was performed using the EdgeSeq Oncology BM Panel for 2,534 cancer-related genes (HTG Molecular Diagnostics, Tucson, AZ) in baseline biopsies before treatment.30 In the palbociclib/fulvestrant treatment arm, the mPFS for the cyclin-E1–high cohort was 50% shorter at 7.4 months versus 14.1 months for the cyclin-E1–low cohort. Cyclin-E1 levels had no significant impact on clinical outcomes for patients treated with placebo/fulvestrant (low Cyclin E1 (CCNE1) = 4.0 v high CCNE1 = 4.8 months). It was also noted that cyclin-E1 mRNA levels were more predictive when measured in metastases compared with archival primary breast tissue.30
There is some conflict within the clinical data for cyclin-E1 as an intrinsic biomarker of resistance. In the PALOMA-2 study, there were no significant treatment interactions for cyclin-E1.23 In the PALOMA-3 study, biomarker analyses identified cyclin-E1 as a resistance biomarker specifically in patients with visceral metastases. These observations generate the hypothesis that utility of cyclin-E1 as an intrinsic resistance biomarker could be context-dependent, for example, relevant in the endocrine-resistant population and those with visceral metastases.29 In the endocrine-sensitive populations recruited within PALOMA-2 and MONALEESA-2 (letrozole v letrozole plus ribociclib) studies, there was a lack of association between cyclin-E1 expression and mPFS.40,42
c-myc.
CDK2 and CDK4/6 can activate c-myc by phosphorylation and gene transcription. In palbociclib-resistant cell lines, there is upregulation of c-myc. Inhibition of CDK2 activity in palbociclib-resistant breast cancer cell lines prevents c-myc upregulation, and the overexpression of c-myc in palbociclib-sensitive cell lines leads to palbociclib resistance.43 In clinical samples, specifically liquid biopsies (Guardant360 assay), 5% of patients who progressed on abemaciclib plus endocrine treatment within the MONARCH-3 study acquired new myc genetic alterations detectable in ctDNA. Nine percent of patients treated within the nextMONARCH-1 study with single-agent abemaciclib25 acquired genetic myc alteration with drug resistance, re-enforcing the hypothesis that myc alteration is an independent biomarker of resistance for CDK4/6 inhibition.
Dysregulation of CDK4.
Currently, there is no strong evidence to support the fact that CDK4 amplification or mutations (DNA) are predictive of drug response. Neither PALOMA-2 nor PALOMA-3 identified significant treatment interactions between genomic events affecting CDK4 gene expression (tumor mRNA) and palbociclib response.28,29 Researchers are currently exploring if CDK4 activity modulates drug response through the quantification of phosphorylation of CDK4 (T172) at a protein level.44
Plasma-derived exosomal CDK4 mRNA.
Systematic analyses of the literature identified one small prospective pharmacogenetic clinical study (N = 40 ECLIPS study), which reported a positive correlation between baseline plasma-derived exosomal CDK4 mRNA copies/ml with response to palbociclib/fulvestrant treatment. There was a correlation between higher pretreatment levels of plasma exosomal CDK4 (mRNA level > 5,050 copies/mL) with better clinical response (partial response/stable disease) and lower baseline CDK4 mRNA levels (≤ 5,050 copies/mL) with shorter mPFS (CDK4-high = mPFS not reached v CDK4-low = 6.45 months, P = .01).45
Overactive CDK6 signaling.
CDK6 amplification.
Cell line models that rendered resistance to palbociclib demonstrated new gene amplification of the CDK6 locus or increased CDK6 mRNA/protein levels. Knockdown of CDK6 restored sensitivity to CDK4/6 inhibition, indicating that drug resistance was attributable to increased levels of CDK6 activity.46,47 In the clinic, higher gene expression levels of CDK6 in patient samples were associated with drug resistance in mBC patients with bone-only disease (PALOMA-3 study).29
FAT1 loss.
FAT1 is a tumor suppressor gene and a member of the cadherin superfamily, which has been found to interact with the beta-catenin and Hippo signaling pathways. FAT1 mutations are present in approximately 2% of primary tumors and 6% of metastatic biopsies from HR+ve breast cancers. Loss of FAT1 increases CDK6 expression via downregulation of Hippo-signaling pathway in cell line models.21 Protein expression levels of FAT1 were lower in CDK4/6-resistant versus CDK4/6-sensitive breast cancer patient-derived xenograft models. Retrospective analyses of patient samples (HR+ve n = 348) confirmed that mBC patients with deleterious FAT1 mutations in their pretreatment biopsies (MSK-IMPACT) had poorer clinical outcomes with CDK4/6 inhibition (mPFS = 2.4 months) compared with the FAT wild-type population. The degree of FAT1 inactivation influenced the degree of CDK4/6 resistance, which was most pronounced in patients showing biallelic FAT1 inactivation versus missense mutations21 (Fig 3).
c-myc/miR-29B-3p/CDK6 axis.
MicroRNAs are noncoding RNAs that control both the stability and translation of protein-coding mRNAs and are estimated to regulate 30%-60% of the human genome. MicroRNAs regulate the cell cycle by targeting the cyclin-dependent kinases and cyclins.48 Silencing microRNAs that negatively regulate CDK6 results in CDK6 activation. Preclinical modeling has demonstrated that c-myc mediated downregulation of the miR-29b-3p and stimulated CDK6 activity, and this was associated with palbociclib insensitivity.49
Tyrosine kinase receptor signaling.
Overactivation of upstream signaling pathways, such as PI3-AKT kinase, rat sarcoma virus (RAS), or fibroblast growth factor receptor (FGFR) signaling, stimulates downstream signaling pathways in cancer cells through cyclin-D, the binding partner for CDK4. The first study to publish whole-exome sequencing analyses of metastatic tumor biopsies from HR+ve patients (n = 58 patients; n = 59 biopsies) treated with CDK4/6 inhibitors plus endocrine treatment identified eight different molecular resistance mechanisms. 80% of the resistant tumors carried genomic alterations in at least one of these eight potential resistance mechanisms22:
Biallelic RB1 disruption (9.8%) → RB1 downregulation
AKT1 mutations/amplification (12.2%) → AKT1 activation
KRAS/HRAS/NRAS mutations/amplification (9.8%) → RAS activation
FGFR2 mutations/amplification (7.3%) → FGFR activation
Human epidermal growth factor receptor 2 (HER2) mutations (12.2%) → HER2 activation
Cyclin-E2 amplification (14.6%) → cyclin-E activation
Aurora kinase A amplification (26.8%) → aurora kinase A amplification
Loss of ER expression on IHC (7.3%) → loss of estrogen signaling.22
Loss of ER expression is reported in cell lines with acquired resistance to CDK4/6 inhibition driven by CDK6 amplification46 although loss of ER expression could be attributed to endocrine resistance.
Retrospective clinical analyses from two independent clinical studies, MONALEESA-22 and MONARCH-3,3 also characterized the heterogenous drug resistance landscape driven via receptor tyrosine kinase signaling pathways (Figs 2 and 3). In the MONARCH-3 study, genetic aberrations in epidermal growth factor receptor (8%) and FGFR1 (7%) were detected in the abemaciclib-resistant population.25 Biomarker analyses from the MONALEESA-2 study (ribociclib/letrozole v placebo/letrozole; n = 391; NanoString 230-gene nCounter GX Human Cancer Reference panel) discovered that patients with low receptor tyrosine kinase gene expression levels derived more clinical benefit from ribociclib drug combinations (HR = 0.41; 0.27 to 0.61).42
Thymidine kinase 1 activity.
Thymidine kinases (TKs) are phosphotransferases involved in DNA replication, and TK1 activity is a surrogate marker for tumor proliferation. TKs are downstream of CDK4/6 and controlled via the RB1-E2F transcription regulatory axis. Two published studies have reported associations between plasma thymidine kinase 1 (TK1) activity and response to palbociclib. In the first study,50 gene expression was higher in cell lines with acquired palbociclib resistance versus sensitive cell lines. These data were validated using clinical samples from the Phase II TRend study, which demonstrated that plasma TK1 activity (DiviTum assay) fell in response to palbociclib treatment in the majority of mBC patients with a subsequent rise in TK1 activity levels at the time of developing drug resistance. The minority of patients (n = 8) with an initial rise in TK1 activity after commencing CDK4/6 inhibition experienced worse clinical outcomes compared with their counterparts (mPFS = 3.0 months; 95% CI, 2.7 to not available v 9 months 95% CI, 5.8 to 12.0; P = .002).50
A second study reported similar trends, with an initial fall in TK1 mRNA copies/ml detected from plasma-derived exosomal mRNA levels quantified using digital droplet polymerase chain reaction. A significant increase in TK1 mRNA copies/ml was observed in patients with progressive disease in the ECLIPS study (P = .01).45 Preliminary data from recent conference proceeding with larger patient cohorts suggest that plasma TK1 enzyme activity could be a prognostic biomarker for clinical response to palbociclib treatment, with TK1 activity levels potentially providing a dynamic marker for drug response at earlier time points to recognize the emergence of treatment resistance.51,52
Novel Upcoming Molecular Biomarker Candidates
CDHD4, ATM, and CDKN2 family CDK inhibitors.
Pooled meta-analyses of the MONALEESA studies (–2, –3, and –7) with ribociclib and endocrine therapy highlighted previously unrecognized resistance biomarkers from baseline patient ctDNA (n = 1,503). Novel candidates identified include the following: (1) chromodomain-helicase DNA binding protein 4 (CDHD4), an epigenetic transcriptional repressor (2%); (2) ataxia-telangiectasia gene (ATM) involved in DNA repair (3%); (3) CHK2, a downstream effector of ATM53,54; and (4) members of the CDKN2/INK family cyclin-dependent kinase inhibitors (CDK2A(p16)/CDK2B(p15)/CDN2C(p18) (2%), which negatively regulate CDK4 and CDK6 (Fig 2).26
Polo-like kinase 1.
Patients with high gene expression levels of polo-like kinase 1 (PLK-1), treated with palbociclib in the phase III study PEARL, had a worse mPFS in a multivariate model (5.7 months) versus patients with low PLK-1 pretreatment tumors (9.3 months; HR = 1.64; 95% CI, 1.25 to 2.34; P = .0008; adjusted model for confounders: age, site of disease, sites of metastasis, prior chemotherapy, and Ki67). In the capecitabine cohort, there were no differences in clinical outcomes for patients with high or low gene expression of PLK-1 (9.9 months v 9.4 months, PLK1-high v PLK1-low; HR = 0.82; 95% CI, 0.56 to 1.21; P = .3189).37
Insulin-like growth factor 1 receptor.
Early data suggest that insulin-like growth factor 1 receptor amplification may be associated with resistance to CDK4/6 inhibition. This is currently being explored in cell line models and patient samples.55
Breast cancer subtypes and drug resistance.
Luminal breast cancers are sensitive to CDK4/6 inhibition in preclinical and clinical scenarios. This observation is supported by preclinical data sets from ER+ve breast cancers,33 TNBC,32 and HER2+ve cancers.56 Patients with luminal A and luminal B subtypes benefit from palbociclib/endocrine treatment23,29 irrespective of whether the primary or metastatic biopsy site is profiled for gene expression analyses. A retrospective correlative biomarker analysis of intrinsic subtypes and efficacy across the MONALEESA phase III studies (MONALEESA-2, MONALEESA-3, and MONALEESA-7 trials) with 1,160 patients (ribociclib: n = 672; placebo: n = 488)) was conducted using NanoString technologies. The authors highlighted that they did not use the standardized 50-gene PAM50 test, and only 36 of the 50 PAM50 genes were available.57
The key outcome from this study was that all breast cancer subtypes gained a significant clinical benefit with ribociclib/endocrine treatment except the basal-like subtype (mPFS: ribociclib 3.71 months v placebo 3.58 months; HR, 1.15; 95% CI, 0.46 to 2.83; P = .77). This was reflected by the HRs for the different subtypes: HER2E-enriched (HR, 0.39; 95% CI, 0.25 to 0.60; P < .001), luminal B (HR, 0.52; 95% CI, 0.38 to 0.72; P < .001), luminal A (HR, 0.63; 95% CI, 0.49 to 0.83; P < .001), and normal-like (HR, 0.47; 95% CI, 0.30 to 0.72; P < .001).57 Interestingly, the HR+ve basal tumors (n = 30; 2.6%) in this MONALEESA data set demonstrated high expression levels of cyclin-E1, epidermal growth factor receptor, and p16/cyclin-dependent kinase inhibitor 2A, with low expression of the luminal-related genes, similar to the TNBC phenotype. This translation study provides a strong rationale to use intrinsic subtyping of breast cancers for prospective allocation of CDK4/6 inhibitor combination treatment in future clinical trials.
In conclusion, CDK4/6 inhibition with endocrine treatment has become the gold standard treatment choice for HR+ve mBC. Multiple biomarker studies have highlighted that there are diverse resistance mechanisms underpinning the CDK4/6-resistant phenotype. CDK4/6 is a downstream target within a cancer cell, and therefore, there are multiple upstream interconnecting signaling networks, such as receptor tyrosine signaling pathways, which a cancer cell can activate to circumvent cell cycle arrest in the G1 phase of the cell cycle. One of the greatest challenges is distinguishing between mechanisms causing resistance to CDK4/6 inhibition and endocrine resistance. Therefore, we have given preferences to translational studies incorporating an endocrine control arm in their analyses and single-agent palbociclib or abemaciclib monotherapy studies.
Molecular mechanisms selectively affecting CDK4/6 inhibitor response include loss of RB1 function, which is less common in patients with CDK4/6 inhibitor–naïve breast cancer. Up to 9% of patients with acquired drug resistance can be attributed to genetic aberrations in RB1 gene or downstream molecular events causing loss of RB1 function. Aberrant cyclin E1/E2 signaling and overactive CDK2 activity are an alternative resistance mechanism, which may be more relevant in people with prior endocrine resistance. Novel resistance mechanisms such as FAT1 loss resulting in higher CDK6 activity are now recognized because of convincing data associating drug response with clinical outcomes.
Currently, the molecular biomarker landscapes for liquid biopsies and solid tumors are distinct (Figs 2 and 3). This reflects the differences in detectability of genetic aberrations and the different sequencing approaches used by researchers such as hypothesis-driven targeted sequencing panels vs. unbiased whole-exome sequencing approaches. Overall, there is now a greater appreciation of the molecular complexities associated with drug resistance, and generally, a resistance biomarker acquired at the time of developing drug resistance appears to also be predictive of de novo drug resistance before patients start a CDK4/6 inhibitor drug combination.
In conclusion, multiple pharmacologically tractable biologic mechanisms associated with drug resistance are emerging in small patient subsets and these all converge to generate the resistant phenotype. The resistance biomarkers can be used in future clinical studies to select out patient subpopulations with less favorable molecular profiles for CDK4/6 inhibitors. In addition, these mechanisms could be harnessed to combat drug resistance development or to re-establish treatment response through the identification of novel drug combinations.
Finally, although systematic assessments of patient-derived materials have identified a sizable network of recurrent resistance markers, a considerable portion of clinical observed resistance currently lacks a molecular explanation, indicating a clear need for continuous discovery-based preclinical and clinical approaches.
Rebecca Roylance
Leadership: Google health
Honoraria: Daiichi Sankyo Europe GmbH, Daiichi Sankyo/Astra Zeneca, Daiichi Sankyo/Astra Zeneca
Consulting or Advisory Role: Lilly, Daiichi Sankyo Europe GmbH, IQVIA, Daiichi Sankyo/Astra Zeneca
Travel, Accommodations, Expenses: Roche/Genentech, Daiichi Sankyo Europe GmbH, Daiichi Sankyo Europe GmbH, BMS
No other potential conflicts of interest were reported.
AUTHOR CONTRIBUTIONS
Conception and design: Uzma S. Asghar, Sibylle Mittnacht
Collection and assembly of data: Uzma S. Asghar, Ruhi Kanani
Data analysis and interpretation: Uzma S. Asghar, Rebecca Roylance, Sibylle Mittnacht
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Rebecca Roylance
Leadership: Google health
Honoraria: Daiichi Sankyo Europe GmbH, Daiichi Sankyo/Astra Zeneca, Daiichi Sankyo/Astra Zeneca
Consulting or Advisory Role: Lilly, Daiichi Sankyo Europe GmbH, IQVIA, Daiichi Sankyo/Astra Zeneca
Travel, Accommodations, Expenses: Roche/Genentech, Daiichi Sankyo Europe GmbH, Daiichi Sankyo Europe GmbH, BMS
No other potential conflicts of interest were reported.
REFERENCES
- 1.Rugo HS, Finn RS, Dieras V, et al. : Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res Treat 174:719-729, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hortobagyi GN, Stemmer SM, Burris HA, et al. : Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol 29:1541-1547, 2018 [DOI] [PubMed] [Google Scholar]
- 3.Johnston S, Martin M, Di Leo A, et al. : MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 5:5, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asghar U, Witkiewicz AK, Turner NC, et al. : The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 14:130-146, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Leary B, Finn RS, Turner NC: Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13:417-430, 2016 [DOI] [PubMed] [Google Scholar]
- 6.Turner NC, Ro J, Andre F, et al. : Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med 373:209-219, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Finn RS, Martin M, Rugo HS, et al. : Palbociclib and letrozole in advanced breast cancer. N Engl J Med 375:1925-1936, 2016 [DOI] [PubMed] [Google Scholar]
- 8.Thein KZ, Htut TW, Ball S, et al. : Venous thromboembolism risk in patients with hormone receptor-positive HER2-negative metastatic breast cancer treated with combined CDK 4/6 inhibitors plus endocrine therapy versus endocrine therapy alone: A systematic review and meta-analysis of randomized controlled trials. Breast Cancer Res Treat 183:479-487, 2020 [DOI] [PubMed] [Google Scholar]
- 9.Gervaso L, Montero AJ, Jia X, et al. : Venous thromboembolism in breast cancer patients receiving cyclin-dependent kinase inhibitors. J Thromb Haemost 18:162-168, 2020 [DOI] [PubMed] [Google Scholar]
- 10.Jazieh KA, Budd GT, Dalpiaz N, et al. : Can CDK4/6 inhibitors cause fatal lung injury? Expert Rev Anticancer Ther 19:917-919, 2019 [DOI] [PubMed] [Google Scholar]
- 11.Sarkisian S, Markosian C, Ali Z, et al. : Palbociclib-induced pneumonitis: A case report and review of the literature. Cureus 12:e8929, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torre LA, Bray F, Siegel RL, et al. : Global cancer statistics, 2012. CA Cancer J Clin 65:87-108, 2015 [DOI] [PubMed] [Google Scholar]
- 13.Mistry R, May JR, Suri G, et al. : Cost-effectiveness of ribociclib plus letrozole versus palbociclib plus letrozole and letrozole monotherapy in the first-line treatment of postmenopausal women with HR+/HER2– advanced or metastatic breast cancer: A U.S. payer perspective. J Manag Care Spec Pharm 24:514-523, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.PRISMA : PRISMA Transparent Reporting of Systemic Reviews and Meta-Analyses, 2020 [Google Scholar]
- 15.DeMichele A, Clark AS, Tan KS, et al. : CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res 21:995-1001, 2015 [DOI] [PubMed] [Google Scholar]
- 16.Knudsen ES, Nambiar R, Rosario SR, et al. : Pan-cancer molecular analysis of the RB tumor suppressor pathway. Commun Biol 3:158, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Leary B, Cutts RJ, Liu Y, et al. : The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov 8:1390-1403, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wander SA, Han HS, Zangardi ML, et al. : Clinical outcomes with abemaciclib after prior CDK4/6 inhibitor progression in breast cancer: A multicenter experience. J Natl Compr Canc Netw 1-8, 2021. [DOI] [PubMed] [Google Scholar]
- 19.Condorelli R, Spring L, O'Shaughnessy J, et al. : Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 29:640-645, 2018 [DOI] [PubMed] [Google Scholar]
- 20.Xu B, Krie A, De P, et al. : Utilizing tumor and plasma liquid biopsy in treatment decision making for an estrogen receptor-positive advanced breast cancer patient. Cureus 9:e1408, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Razavi P, Li Q, et al. : Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the Hippo pathway. Cancer Cell 34:893-905.e8, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wander SA, Cohen O, Gong X, et al. : The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor-positive metastatic breast cancer. Cancer Discov 10:1174-1193, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finn RS, Liu Y, Zhu Z, et al. : Biomarker analyses of response to cyclin-dependent kinase 4/6 inhibition and endocrine therapy in women with treatment-naive metastatic breast cancer. Clin Cancer Res 26:110-121, 2020 [DOI] [PubMed] [Google Scholar]
- 24.Costa C, Wang Y, Ly A, et al. : PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kalpha inhibitors in breast cancer. Cancer Discov 10:72-85, 2020 [DOI] [PubMed] [Google Scholar]
- 25.Goetz MP, Hamilton EP, Campone M, et al. : Acquired genomic alterations in circulating tumor DNA from patients receiving abemaciclib alone or in combination with endocrine therapy. J Clin Oncol 38, 2020. (suppl; abstr 3519) [Google Scholar]
- 26.Andre F, Su F, Solovieff N, et al. : Pooled ctDNA analysis of the MONALEESA (ML) phase III advanced breast cancer (ABC) trials. J Clin Oncol 38, 2020(suppl; abstr 1009) [DOI] [PubMed] [Google Scholar]
- 27.Park K, Kim GM, Jung KH, et al. : Exploratory biomarker analysis of Young-PEARL [palbociclib plus exemestane with GnRH agonist versus capecitabine in premenopausal women with HR (hormone receptor)-positive, HER2-negative metastatic breast cancer (MBC)] study. Cancer Res 81, 2020. (suppl 4; abstr PS5-19) [Google Scholar]
- 28.O'Leary B, Cutts RJ, Huang X, et al. : Circulating tumor DNA markers for early progression on fulvestrant with or without palbociclib in ER+ advanced breast cancer. J Natl Cancer Inst 113:309-317, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finn RS, Cristofanilli M, Ettl J, et al. : Treatment effect of palbociclib plus endocrine therapy by prognostic and intrinsic subtype and biomarker analysis in patients with bone-only disease: a joint analysis of PALOMA-2 and PALOMA-3 clinical trials. Breast Cancer Res Treat 184:23-35, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner NC, Liu Y, Zhu Z, et al. : Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J Clin Oncol 37:1169-1178, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park YH, Kim TY, Kim GM, et al. : Palbociclib plus exemestane with gonadotropin-releasing hormone agonist versus capecitabine in premenopausal women with hormone receptor-positive, HER2-negative metastatic breast cancer (KCSG-BR15-10): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 20:1750-1759, 2019 [DOI] [PubMed] [Google Scholar]
- 32.Asghar US, Barr AR, Cutts R, et al. : Single-cell dynamics determines response to CDK4/6 inhibition in triple-negative breast cancer. Clin Cancer Res 23:5561-5572, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herrera-Abreu MT, Palafox M, Asghar U, et al. : Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res 76:2301-2313, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang C, Stockwell SR, Elbanna M, et al. : Signalling involving MET and FAK supports cell division independent of the activity of the cell cycle-regulating CDK4/6 kinases. Oncogene 38:5905-5920, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olmez I, Zhang Y, Manigat L, et al. : Combined c-Met/Trk inhibition overcomes resistance to CDK4/6 inhibitors in glioblastoma. Cancer Res 78:4360-4369, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho-Yen CM, Jones JL, Kermorgant S: The clinical and functional significance of c-Met in breast cancer: A review. Breast Cancer Res 17:52, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guerrero-Zotano A, Zielinski C, Gil-Gil M, et al. : Plk1 expression & efficacy of palbociclib in advanced hormonal receptor-positive breast cancer patients from PEARL study (GEICAM 2012-03). Cancer Res 81, 2021. (4 suppl; abstr PS2-01) [Google Scholar]
- 38.Guarducci C, Bonechi M, Benelli M, et al. : Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer 4:38, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vijayaraghavan S, Karakas C, Doostan I, et al. : CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun 8:15916, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCartney A, Migliaccio I, Bonechi M, et al. : Mechanisms of resistance to CDK4/6 inhibitors: Potential implications and biomarkers for clinical practice. Front Oncol 9:666, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knudsen ES, Shapiro GI, Keyomarsi K: Selective CDK4/6 inhibitors: Biologic outcomes, determinants of sensitivity, mechanisms of resistance, combinatorial approaches, and pharmacodynamic biomarkers. Am Soc Clin Oncol Educ Book 40:115-126, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hortobagyi GN, Paluch-Shimon S, Petrakova K, et al. : First-line ribociclib (RIB) + letrozole (LET) in hormone receptor-positive (HR+), HER2-negative (HER2–) advanced breast cancer (ABC): MONALEESA-2 biomarker analyses. J Clin Oncol 36, 2018. (suppl; abstr 1022) [Google Scholar]
- 43.Pandey K, Park N, Park KS, et al. : Combined CDK2 and CDK4/6 inhibition overcomes palbociclib resistance in breast cancer by enhancing senescence. Cancers (Basel) 12:3566, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raspe E, Coulonval K, Pita JM, et al. : CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol Med 9:1052-1066, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Del Re M, Bertolini I, Crucitta S, et al. : Overexpression of TK1 and CDK9 in plasma-derived exosomes is associated with clinical resistance to CDK4/6 inhibitors in metastatic breast cancer patients. Breast Cancer Res Treat 178:57-62, 2019 [DOI] [PubMed] [Google Scholar]
- 46.Yang C, Li Z, Bhatt T, et al. : Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36:2255-2264, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schoninger SF, Blain SW: The ongoing search for biomarkers of CDK4/6 inhibitor responsiveness in breast cancer. Mol Cancer Ther 19:3-12, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bueno MJ, Malumbres M: MicroRNAs and the cell cycle. Biochim Biophys Acta 1812:592-601, 2011 [DOI] [PubMed] [Google Scholar]
- 49.Ji W, Zhang W, Wang X, et al. : c-myc regulates the sensitivity of breast cancer cells to palbociclib via c-myc/miR-29b-3p/CDK6 axis. Cell Death Dis 11:760, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCartney A, Bonechi M, De Luca F, et al. : Plasma thymidine kinase activity as a biomarker in patients with luminal metastatic breast cancer treated with palbociclib within the TREnd trial. Clin Cancer Res 26:2131-2139, 2020 [DOI] [PubMed] [Google Scholar]
- 51.Cabel L, Tanguy ML, Lerebours F, et al. : Plasma thymidine kinase 1 activity and outcome of ER+ HER2– metastatic breast cancer patients treated with palbociclib and endocrine therapy. Cancer Res 80, 2020. (suppl 4; abstr P5-01-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luca L, Tyekucheva S, Hilbers FS, et al. : Serum thymidine kinase activity in patients with luminal metastatic breast cancer treated with palbociclib and fulvestrant within the PYTHIA trial. Cancer Res 81, 2021. (suppl 4; abstr PS5-05) [Google Scholar]
- 53.Anurag M, Haricharan S, Ellis MJ: CDK4/6 inhibitor biomarker research: Are we barking up the wrong tree? Clin Cancer Res 26:3-5, 2020 [DOI] [PubMed] [Google Scholar]
- 54.Haricharan S, Punturi N, Singh P, et al. : Loss of MutL disrupts CHK2-dependent cell-cycle control through CDK4/6 to promote intrinsic endocrine therapy resistance in primary breast cancer. Cancer Discov 7:1168-1183, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wander SA, Mao P, Lloyd MR, et al. : Igf1r mediates cdk4/6 inhibitor (cdk4/6i) resistance in tumor samples and in cellular models. Cancer Res 81, 2021. (suppl 4; abstr PD7-08) [Google Scholar]
- 56.Finn RS, Dering J, Conklin D, et al. : PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 11:R77, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prat A, Chaudhury A, Solovieff N, et al. : Correlative biomarker analysis of intrinsic subtypes and efficacy across the MONALEESA phase III studies. J Clin Oncol 39:1458-1467, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]