

PURPOSE
Hematologic toxic effects of peptide receptor radionuclide therapy (PRRT) can be permanent. Patients with underlying clonal hematopoiesis (CH) may be more inclined to develop hematologic toxicity after PRRT. However, this association remains understudied.
MATERIALS AND METHODS
We evaluated pre- and post-PRRT blood samples of patients with neuroendocrine tumors. After initial screening, 13 cases of interest were selected. Serial blood samples were obtained on 4 of 13 patients. Genomic DNA was analyzed using a 100-gene panel. A variant allele frequency cutoff of 1% was used to call CH.
RESULT
Sixty-two percent of patients had CH at baseline. Persistent cytopenias were noted in 64% (7 of 11) of the patients. Serial sample analysis demonstrated that PRRT exposure resulted in clonal expansion of mutant DNA damage response genes (TP53, CHEK2, and PPM1D) and accompanying cytopenias in 75% (3 of 4) of the patients. One patient who had a normal baseline hemogram and developed persistent cytopenias after PRRT exposure showed expansion of mutant PPM1D (variant allele frequency increased to 20% after exposure from < 1% at baseline). In the other two patients, expansion of mutant TP53, CHEK2, and PPM1D clones was also noted along with cytopenia development.
CONCLUSION
The shifts in hematopoietic clonal dynamics in our study were accompanied by emergence and persistence of cytopenias. These cytopenias likely represent premalignant state, as PPM1D-, CHEK2-, and TP53-mutant clones by themselves carry a high risk for transformation to therapy-related myeloid neoplasms. Future studies should consider CH screening and longitudinal monitoring as a key risk mitigation strategy for patients with neuroendocrine tumors receiving PRRT.
INTRODUCTION
Cancer-related mortality is decreasing each year in the United States.1 Therapy-related myeloid neoplasms (t-MNs) are expected to rise in parallel among cancer survivors.2 t-MNs constitute 10%-20% of newly diagnosed acute myeloid leukemia and myelodysplastic syndrome cases and are associated with dismal outcomes.3 Radiation therapy and DNA-damaging chemotherapy are linked to increased risk of t-MNs.2 With the application of sensitive sequencing technologies, the putative origin of t-MN driver mutations such as those involving DNA damage response (DDR) genes like TP53 can be detected even at time of the primary malignant diagnosis. Detection of clonal hematopoiesis (CH) mutations offers the first potential biomarker for risk of subsequent t-MN.4,5 t-MN's biology is a complex interplay of dose; type and duration of cytotoxic therapy; and age, environment, and genetic make-up of the host.2 Comprehensive genomic analysis of at-risk patients undergoing potentially leukemogenic therapies will aid in understanding biology of t-MN and might help to devise a mitigation strategy for t-MN risk.
CONTEXT
Key Objective
Do patients with neuroendocrine tumors (NETs) who experience short-term and long-term hematologic adverse events after peptide receptor radionuclide therapy (PRRT) exposure harbor any underlying clonal predisposition for altered or malignant hematopoiesis?
Knowledge Generated
Blood samples from patients with NETs demonstrated a high prevalence, 62% with clonal hematopoiesis (CH) at the time of diagnosis. On serial sample analysis, CH in DNA damage response genes, ie, TP53, CHEK2, and PPM1D, expanded on longitudinal monitoring after PRRT therapy in three of four patients. The clonal expansions in DNA damage response genes after PRRT were associated with varying degrees of prolonged cytopenias in all the patients with TP53, CHEK2, and PPM1D mutations.
Relevance
Under the stress of potentially leukemogenic therapy (PRRT), TP53, CHEK2, and PPM1D clones preferentially repopulated the hematopoietic compartment and were associated with prolonged cytopenias. Screening and longitudinal monitoring of CH in patients with NET receiving PRRT can be an important risk-modifying strategy for these patients.
t-MN historically has been considered a consequence of DNA damage induced in normal hematopoietic stem or progenitor cells by the effect of genotoxic therapy (Data Supplement). However, this model is falling out of favor as not all patients exposed to genotoxic therapies develop t-MN and in those who develop a t-MN, there is a wide range of latency periods. There is increasing evidence outlining the role of the presence of somatic mutations in hematopoietic stem cells (HSCs) before the exposure to genotoxic therapy as precursors for t-MN.2 As CH and its evolution may serve as a surrogate predictive marker for subsequent t-MN development, we sought to study hematopoietic clonal dynamics and fitness in the context of a modern and potentially leukemogenic therapy.
Neuroendocrine tumors (NETs) are frequently diagnosed in younger patients, have a relatively indolent clinical course, and are associated with germline predisposition in some cases.6 Several studies have demonstrated varying degrees of t-MN risk after radionuclide treatment including peptide receptor radionuclide therapy (PRRT).7-11 PRRT is a tumor-targeted treatment that uses radiation to induce tumor cell death via β particle–emitting radionuclide.9 The lower rates of progression of primary tumors treated with this agent12 led to US Food and Drug Administration approval of PRRT for somatostatin receptor–positive gastroenteropancreatic NETs in 2018. Hematologic toxicity was the major adverse event with the nadir of cytopenias occurring about a month after therapy.13 Permanent hematologic dysfunction has been reported after therapy, indicating significant injury to hematopoietic precursors and potentially an increased risk for development of t-MNs.11,14,15 To date, the precise stress induced by PRRT on the HSC compartment is largely elusive because of the novelty of this drug.16,17
The aim of our study is to better elucidate the role of PRRT in exerting selective pressure on specific mutant HSCs and their clonal progeny and assess the overall clonal fitness of hematopoietic cells under PRRT-induced stress.
MATERIALS AND METHODS
Patients who received PRRT therapy between March 2018 and August 2019 were identified through our NET Clinic database and the biorepository at our institution, Roswell Park Comprehensive Cancer Center (RPCCC). After initial screening, 20 individuals were identified. Thirteen cases were selected on the basis of availability of at least one whole blood or DNA sample preceding PRRT treatment. Of the 13 cases, serial peripheral blood samples could be obtained for four patients after an informed consent. Clinical and laboratory variables were collected through retrospective chart review and included CBC counts obtained at time of initial sample collection (±30 days). Associations with prior chemotherapy or radiotherapy exposure were based on receipt of any NET-directed systemic therapy (excluding hormone therapy with somatostatin analog) or radiation therapy before the date of initial blood DNA sampling. None of the patients in our analysis had another active hematologic malignancy or a precursor state such as monoclonal gammopathy of undetermined significance or monoclonal B-cell lymphocytosis at the time of peripheral blood next generation sequencing. Patients were followed for the development of subsequent hematologic adverse events with the last follow-up date of January 1, 2021. The study was approved by the Institutional Review Boards of both participating sites (RPCCC and Weill Cornell Medical College). Chi-square tests and Wilcoxon rank-sum tests were used to compare patient and clinical characteristics among patients with and without CH. Patients were stratified on the basis of age, sex, and smoking status. Logistic regression models were used to study the association of covariates with mutational events. All analyses were conducted at a significance level of 0.05.
Mutational Analysis
Method details.
Genomic DNA from mononuclear cells or buffy coats was extracted. Targeted enrichment using a custom pool of biotinylated baits directed to 100 genes involved in CH, cancer, and CVD was performed. After filtering out potential germline and artifacts, variants with a variant allele frequency (VAF) ≥ 1% were reported. A detailed technical description is provided in the Data Supplement.
Quantification and statistical analysis.
Analysis was conducted in R environment v4.0.2 (R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria).18 Figures were produced using the package ggplot2 v3.3.319 and ComplexHeatmap v2.4.3.20 Data summarization and reshaping were performed using plyr v1.8.6, dplyr v1.0.1, and reshape2 v1.4.4 packages. Statistical significance was defined as P < .05. Statistical tests were performed using R software.
The list of genes in the CH panel is provided in the Data Supplement.
RESULTS
Patient Characteristics
Table 1 shows the baseline characteristics of the 13 patients before PRRT exposure. As noted, this cohort had low exposure to prior chemotherapy and radiotherapy (7.7% and 23.1%, respectively).
TABLE 1.
Baseline Patients' Characteristics
Commonly used therapies after baseline sample collection included temozolomide (23%), everolimus (38%), capecitabine (8%), and nintedanib (8%). Patients received a median of four PRRT cycles (range 2-4 cycles). Seventy-five percent (9 of 12) of the cohort developed cytopenias after PRRT exposure. Persistent cytopenias (defined as cytopenia that persisted at least 3 months beyond last PRRT exposure, with no chemotherapy or targeted therapy exposure during those 3 months) were observed in 54% of patients (Table 2). The median laboratory follow-up time after last PRRT dose was 7 months (range 0-23 months). Three patients had dose interruptions or cancelations because of various reasons. One patient's last dose of PRRT was canceled because of persistent thrombocytopenia (patient 3). Cycle 3 was delayed for patient 8 because of gastroenterologic side effects, and the patient never received the remaining two cycles because of sudden death. Patient 11 received only two cycles because of lack of clinical benefit. The median time for development of cytopenias from first exposure to PRRT was 3 months. Table 2 demonstrates the development of neutropenia, thrombocytopenia, and anemia determined from the CBC obtained at baseline (pre-PRRT exposure), 6 months after PRRT exposure, at the last follow-up after PRRT exposure (graded using the NCI CTCAE v5.0 hematologic toxicity scale) and other details regarding PRRT exposure.
TABLE 2.
Development of Neutropenia, Thrombocytopenia, and Anemia Determined From the CBC Obtained at Baseline (pre-PRRT exposure), 6 Months After PRRT Exposure, and at the Last Follow-Up After PRRT Exposure (graded using the NCI CTCAE v5.0 hematologic toxicity scale)

CH Prevalence Before PRRT Exposure
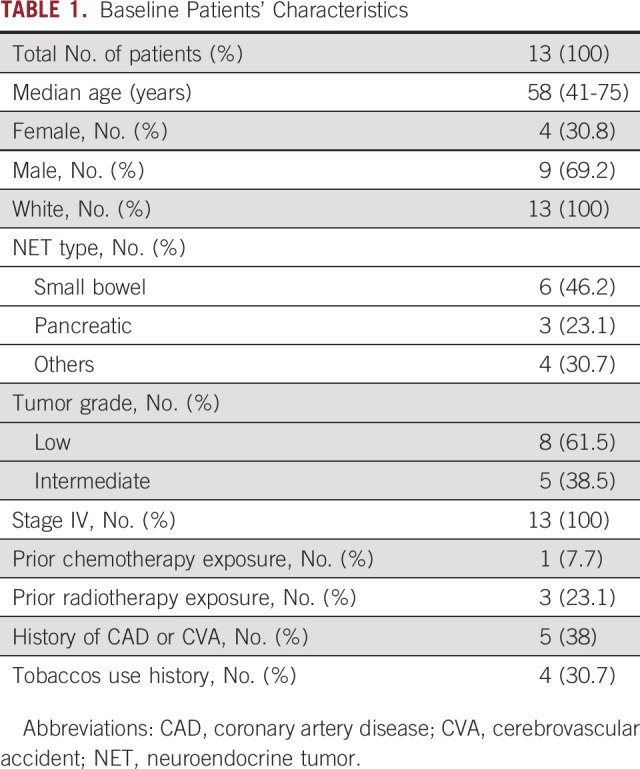
The overall prevalence of CH in this cohort with minimal prior chemotherapy and radiotherapy exposure was 62% (8 of 13). TET2 was the most commonly mutated gene, followed by mutations in ASXL1, CHEK2, PPM1D, and TP53. DNMT3A mutations, often considered an advanced age-related clonal event,21 were not common in this cohort, perhaps because of the younger age of this group. Figure 1 shows the mutational landscape of myeloid neoplasm–associated genes in the pre-PRRT exposure cohort, along with clinical and laboratory parameters. Table 3 shows gene mutations identified, types of mutations, VAFs, and associated nucleotide and protein changes as a result of these mutations (the detailed call set report is provided in the Data Supplement). Analysis of age and cytopenias in the CH+ group versus CH– group (Data Supplement) showed significant association of age with CH (P = .047) and a trend toward significant association between absolute neutrophil count and CH, P = .093.
FIG 1.
Oncoprint for patients with neuroendocrine tumors along with clinical and laboratory parameters. VAF, variant allele frequency.
TABLE 3.
Patients With Mutational Events in Specific Genes, Associated Nucleotide and Protein Change, and Baseline and Serial Sample VAFs %

Serial Sampling Samples After PRRT Exposure
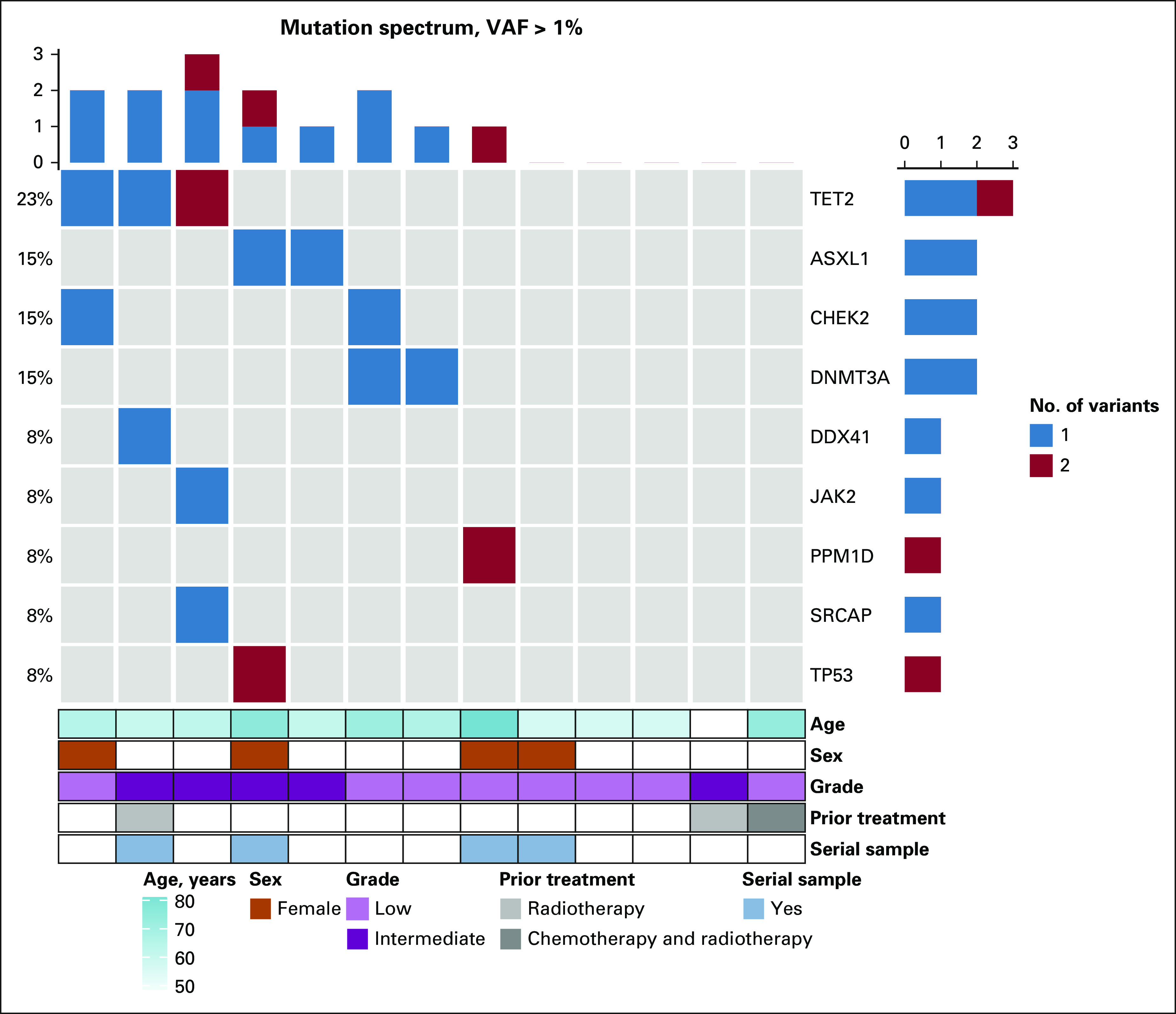
Serial sampling samples after PRRT exposure were obtained for four patients in the baseline cohort (Fig 2). Clinical features and clinical course of these four patients along with clonal trajectory of CH genes during therapy are described in detail below.
FIG 2.
The clonal trajectories of mutation events. Serial analysis of patients exposed to PRRT. DNA specimens from four patient cases (patients 10, 2, 4, and 5) exposed to PRRT and serial samples after PRRT exposure were analyzed at serial time points. (A) Line graphs indicate change in VAF per year and change in blood parameters as a result of change in VAF, eg, in patient 5, with increasing PPM1D clone (in purple), there was a notable change in HgB, WBC, and platelet count (all decreased; the y-axis in (A) depicts changes in blood parameters over time). Different colors indicate different gene mutations. (B) Different colors indicate different patients and depict change in VAFs over time. Seen in example of patient 5 (purple), dramatic changes in VAF were noted over time. In the Data Supplement, additional associated changes in blood parameters are depicted, which highlight the considerable drop in all three blood parameters, particularly patient 5. HgB, hemoglobin; PRRT, peptide receptor radionuclide therapy; VAF, variant allele frequency. PRRT exposure leads to mutant TP53, CHEK2, and PPM1D expansion in blood, contributing to prolonged cytopenias.
Case 1: Patient 2
A 59-year-old individual with metastatic intermediate-grade small bowel NET with liver and bony metastases presented initially with carcinoid syndrome 4 years ago. Before initiating treatment with a somatostatin analog, they had initial blood sampling. Subsequent therapies included SIR-spheres to the right and left hepatic arteries, palliative radiation to the thoracic spine at T4-T7, treatment on a clinical trial with nintedanib, everolimus, and temozolomide, and subsequently four cycles of PRRT in 2018. The most recent treatment was hepatic arterial embolization. This patient did not develop cytopenias during or after PRRT treatment.
Clonal trajectory.
In 2015, at the time of initial sample collection, this patient had mutational events involving the TET2 and DDX41 genes with a VAF of about 2.5%. In 2018, after exposure to PRRT, both clones diminished in size to undetectable or very low levels. Subsequent sampling from 2020 demonstrated re-emergence of the TET2-mutant clone.
Case 2: Patient 4
A 77-year-old individual with low-grade NET of the small bowel presented with liver, lymph nodes, and bony metastasis 2 years ago. A baseline peripheral blood sample for the biorepository was collected before treatment with a somatostatin analog. The patient was subsequently treated for progressive disease with four cycles of PRRT. There were no cytopenias before PRRT initiation; however, during and after PRRT, the patient developed mild thrombocytopenia (range, 100-130 × 109/L), which persists to date.
Clonal trajectory.
At the time of initial sample collection, this patient had mutations in TP53 (two variants, VAFs 1.78% and 1.67%) and ASXL1 at a VAF of 1% (Table 3). After exposure to PRRT, the two TP53 variants expanded to VAFs of 6.33% and 6.12%, respectively, whereas the mutant ASXL1 clone diminished in size to a level undetectable by our assay. A mutation in CHEK2 was detected after therapy that has persisted. During and after exposure to PRRT, this patient developed new thrombocytopenia (118 × 109/L) that has persisted long term. These findings suggest that the mutant TP53 and CHEK2 clones (and not ASXL1) were selectively expanded after exposure to PRRT, with ensuing cytopenia related to expansion of primarily TP53-mutant population.22,23
Case 3: Patient 5
A 74-year-old individual with stage IV low-grade pancreatic-NET with widespread metastases diagnosed initially 15 years ago presented with a pancreatic mass and synchronous liver metastasis. Treatment was initiated with a somatostatin analog and interferon before initial peripheral blood sampling for our biorepository. This patient had a long disease course marked by treatments with temozolomide and capecitabine followed by surgical resection with stable disease for a few years, after which everolimus, an mTOR inhibitor, was used for 6 months. At further disease progression, a blood sample for the biorepository was obtained before four cycles of PRRT in 2018. Before PRRT, the patient had normal blood cell counts and an unremarkable differential. At the end of treatment with PRRT, the hemoglobin and platelet counts were decreased to 10.9 g/dL and 94 × 109/L, respectively. Another sample was obtained in follow-up at which point the patient demonstrated evidence of progressive cytopenias.
Clonal trajectory.
In 2006, at the time of initial sample collection, the patient had no detectable clonal events. In 2018, after exposure to multiple other therapies, new clonal events were detected with the appearance of a PPM1D mutation with a VAF of about 7.5%. During and after exposure to PRRT therapy, the patient was noted to develop cytopenias. As seen in Figure 2A, cytopenias were associated with a steep rise in the PPM1D-mutant VAF to 20%. These findings suggest that the worsening cytopenias after PRRT exposure arose because of repopulation of the hematopoietic compartment with less functional mutant PPM1D stem cells.
Case 4: Patient 10
An 82-year-old individual with low-grade metastatic NET with synchronous primaries in the midgut and pancreas presented with metastases to liver and lymph nodes 6 years ago. After initial surgery for a small bowel obstruction, she was treated with a somatostatin analog, chemoembolization of multiple right liver lobe lesions, and subsequently PRRT. This patient had baseline anemia (hemoglobin 10 g/dL), with no other abnormalities of the hemogram. During and after PRRT treatment, she had an ongoing anemia with the development of mild leukopenia (3-4 × 109/L) and thrombocytopenia (80-120 × 109/L range).
Clonal trajectory.
Samples drawn shortly after completion of PRRT demonstrated distinct mutational events in PPM1D gene at low initial VAFs. Six and 12 months after exposure to PRRT, initially identified PPM1D clones had expanded to higher VAFs and in addition, few additional PPM1D variants were also identified (Fig 2A). Mutation in CHEK2 showed clonal expansion as well. These findings reinforce the suggestions that PPM1D and CHEK2 clones (both DDR pathway genes) expand with PRRT exposure and can populate the hematopoietic compartment contributing to the development of cytopenias.
In summary, we identified recurrent mutations in TP53, PPM1D, and CHEK2 in three of the above four cases with NETs irrespective of prior exposure to genotoxic stressors, ie, chemotherapy and radiation. These mutations appear to opportunistically populate the hematopoietic compartment in response to the stress induced by PRRT, particularly in older individuals.
DISCUSSION
The goal of cancer therapies is to create an environment conducive to favor cancer cell death. The extrinsic perturbations by cancer therapies affect HSCs (mutated and wildtype) and create a bone marrow microenvironment suited to select for mutant clones that are able to survive the stress event. Cell-intrinsic factors, perhaps acquisition of fitter somatic mutations, increase resistance to cell death from external stressors (in this case, PRRT) and allow for the emergence of clonally dominant mutant HSCs, thereby contributing to the fate of the individual HSCs. This clonal dominance and expansion in aging individuals underscore the findings that in addition to cell-intrinsic properties of mutant clones, cell-extrinsic factors such as an aging marrow environment may lead to alterations in hematopoiesis.
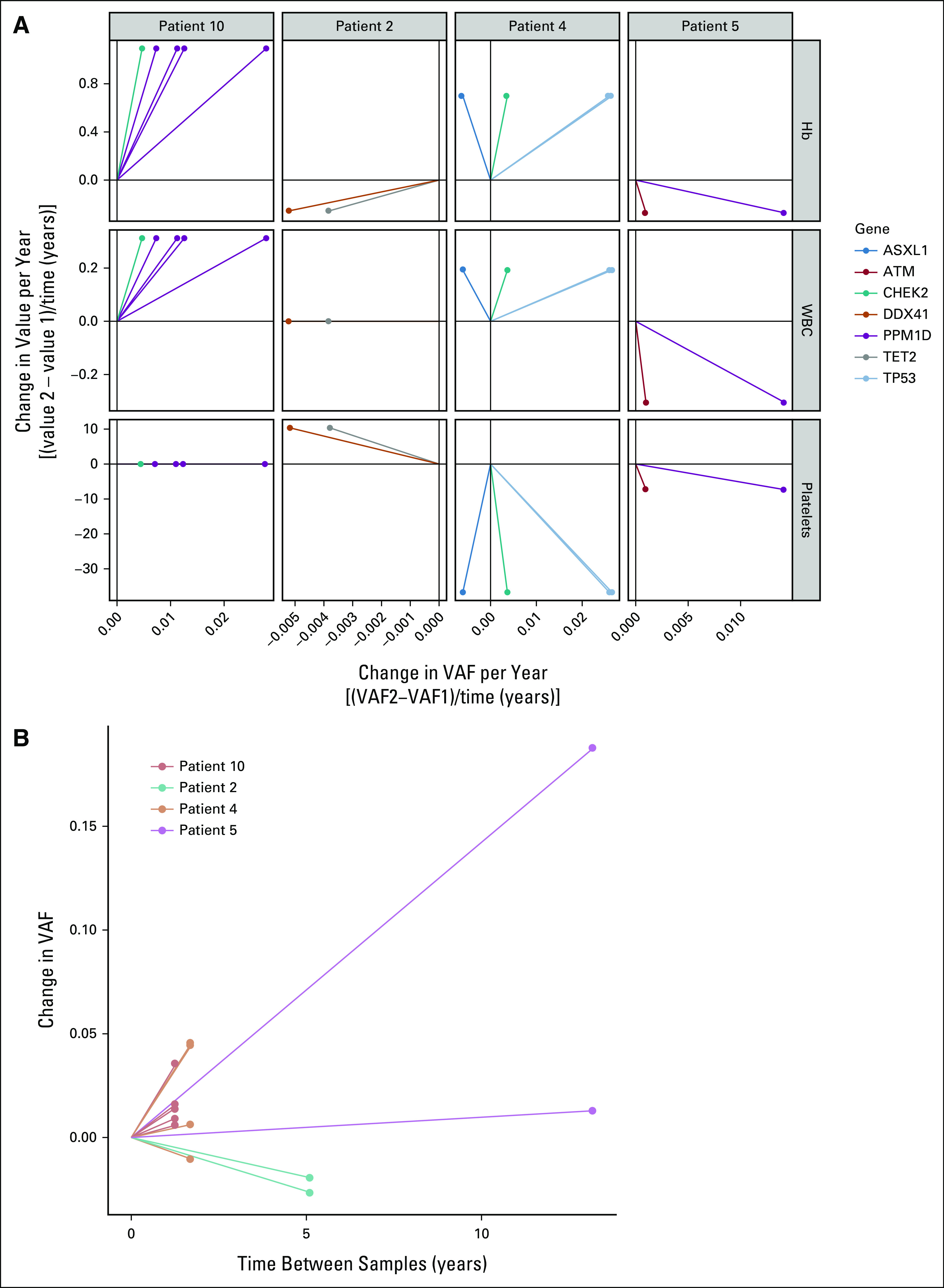
Here, to our knowledge, for the first time, we report a high baseline prevalence of CH in patients with NETs, in the setting of minimal prior chemotherapy and radiotherapy exposure. Furthermore, we demonstrate the clonal expansion of hematopoietic cells with PPM1D and TP53 mutations in response to radionuclide therapy, thereby corroborating on the growing evidence that pretreatment somatic genetic alterations may contribute to malignant hematopoiesis in a more dynamic way (Fig 3) rather than the previously thought linear phenomenon (Data Supplement). After this cytotoxic therapy, we observed expansion of clones carrying mutations in genes involved in the DDR. Many healthy individuals have low-VAF mutant DDR clones that can remain stable for many years without clinical consequences. Additional selective pressures or external perturbations are therefore likely essential for progression to t-MN as a majority of individuals with CH do not develop an overt hematologic malignancy.24
FIG 3.
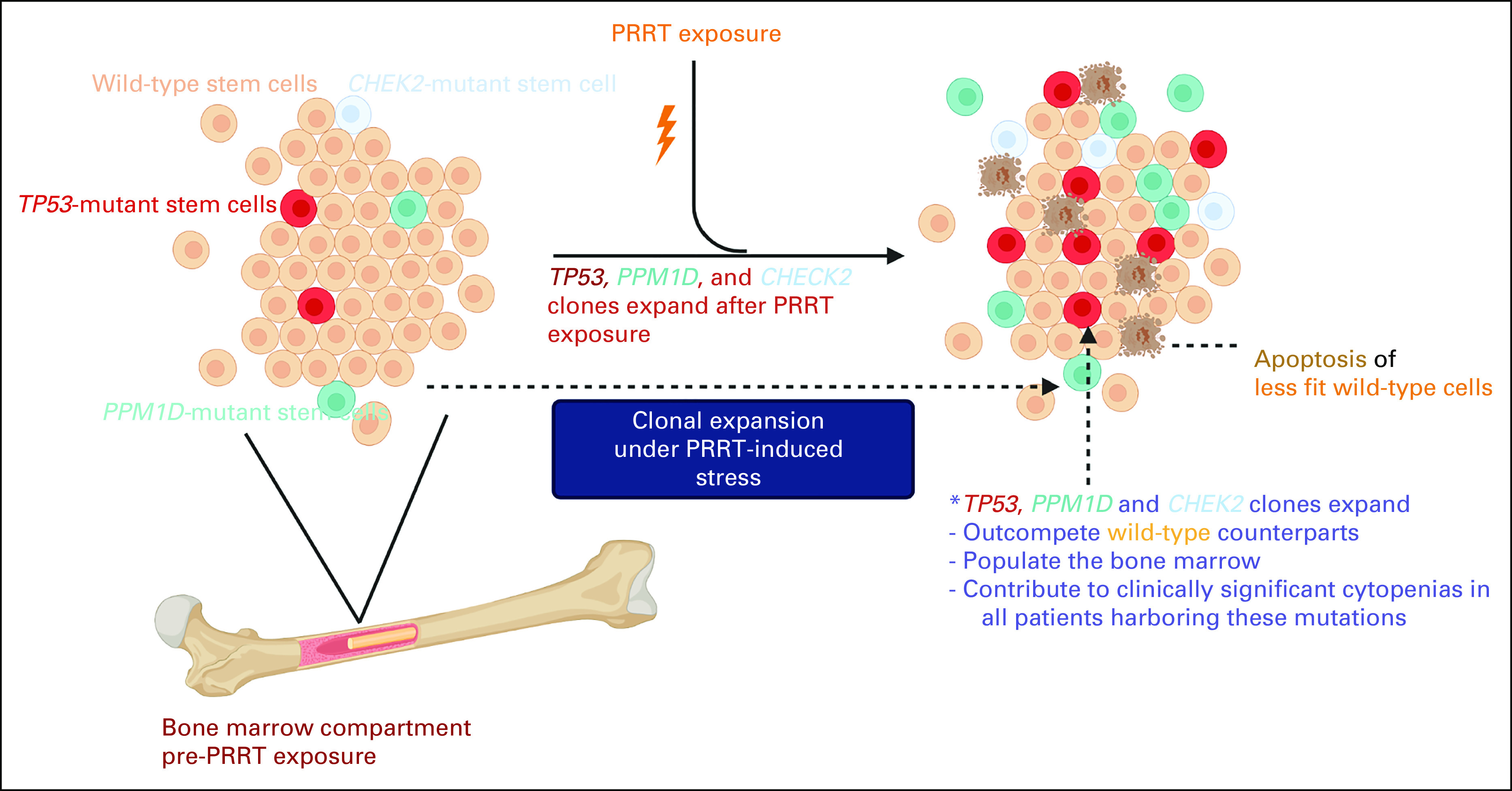
Dynamic model of clonal evolution and PRRT-induced carcinogenesis. The image was generated using BioRender. PRRT, peptide receptor radionuclide therapy.
Our analysis suggests that cell-extrinsic perturbation conferred by PRRT selects for HSCs harboring DDR mutations such as PPM1D and TP53, because of their ability to preferentially withstand the stress induced by PRRT. It is known that detection of CH after chemotherapy or radiotherapy for a nonmyeloid neoplasm can increase the risk of tMNs, especially if the emergent clones bear a TP53 or PPM1D mutation.5,25,26 It is, therefore, conceivable that these surviving HSCs preferentially repopulate the hematopoietic compartment, leading to dysfunctional hematopoiesis and ultimately an increased risk for transformation to t-MN (although we did not report the latter in our cohort because of smaller sample size and shorter follow-up). Previous genomic analyses of samples from patients with t-MNs and analysis of samples years before the overt development of t-MN have identified mutations in TP53.23,27 In addition, studies have reported an increased incidence of CH in cancer cohorts by virtue of previous exposure to genotoxic stressors.5,26,28,29 The presence of therapy-related CH has been implicated as a mean of predicting an increased risk of hematologic malignances and inferior survival.28,29 Growing knowledge suggests that t-MNs are often driven by expansion of pre-existing mutant clones in response to chemotherapy.5,26,30 These studies in addition to our findings here provide robust evidence that specific mutant clones can be selected for various cancer therapies.
PRRT remains an excellent option for treatment of patients with progressive NETs, not only prolonging survival but also improving quality of life. These patients unfortunately do seem to be at particular risk for the development of t-MN.31 As patients treated with PRRT have extended survival, the risk of clonal expansion from PRRT-induced selective pressures will become evident as overt development of t-MNs. It is therefore imperative to develop screening or surveillance strategies for such individuals to evaluate and mitigate risks of harmful clonal expansion and subsequent t-MN development. Devising chemoprevention research strategies to mitigate the lethal adverse consequences of promising life-prolonging therapies is the next key step in therapeutic development.
Limitations to our study include the small sample size and lack of serial samples on all 13 patients. Although a smaller sample, acquisition of baseline and serial samples with adequate follow-up was feasible because of our center's expertise in treating NETs and early adoption of PRRT since its approval. In addition, certain mutations seen in our analysis, such as those in the genes DDX41 or CHEK2, could be germline mutational events or related to the primary NET. Unfortunately, their origin cannot be clearly delineated in our patients because of a lack of paired tumor and germline sample sequencing. However, the presence of these mutations at lower VAFs and appearance of CHEK2 mutation only after exposure to PRRT would argue against that.
In conclusion, we have identified baseline DDR pathway gene defects in patients with NETs that are known to increase the risk for development of future hematologic malignancy. TP53-, CHEK2-, and PPM1D-mutant clonal expansion was temporally associated with exposure to PRRT, suggestive of positive selection of these clones in response to PRRT. These mutations likely provide a survival advantage to HSCs, which harbor them and preferentially repopulate the hematopoietic compartment. An improved understanding of the impact of baseline CH and expansion in response to genotoxic stressors will inform future studies to evaluate importance of screening, surveillance, and early detection to decrease the risk of t-MN in patients undergoing cancer treatments. There is a pressing need for recommendations to guide our management of these patients undergoing PRRT, who harbor mutations in DDR genes.
Elizabeth A. Griffiths
Honoraria: Novartis
Consulting or Advisory Role: Alexion Pharmaceuticals, Takeda, Taiho Oncology, Novartis, Genentech, Celgene/Bristol Myers Squibb, AbbVie, Otsuka US, CTI BioPharma Corp, PicnicHealth
Research Funding: Genentech (Inst), Celgene (Inst), Apellis Pharmaceuticals (Inst), Astex Pharmaceuticals (Inst), Celldex (Inst), Bristol Myers Squibb/Celgene
Open Payments Link: https://openpaymentsdata.cms.gov/physician/134906
Mark G. Faber
Stock and Other Ownership Interests: AstraZeneca/MedImmune/Spirogen
Eti Sinha
Employment: Foundation Medicine
Stock and Other Ownership Interests: Roche/Genentech
Duane C. Hassane
Employment: Tempus
Stock and Other Ownership Interests: Tempus
Research Funding: Daiichi Sankyo
Patents, Royalties, Other Intellectual Property: ddPCR assessment of minimal residual disease via NPM1 mutations, risk prediction for acute myeloid leukemia
David Neil Hayes
Leadership: GeneCentric
Stock and Other Ownership Interests: GeneCentric
Consulting or Advisory Role: GeneCentric, Merck, Turnstone Bio
Patents, Royalties, Other Intellectual Property: I hold several diagnostic patents or pending patents in the area of solid tumor diagnostics
Monica L. Guzman
Consulting or Advisory Role: SeqRx, Bridge Medicines
Research Funding: Cellectis (Inst), Bridge Medicines (Inst), Daiichi Sankyo/UCB Japan (Inst)
Renuka Iyer
Consulting or Advisory Role: Lexicon, Novartis, Eisai, Merck, Bayer, Advanced Accelerator Applications, Exelixis, Sun pharma, QED therapeutics, Ipsen, Sandoz, TerSera, AstraZeneca
Research Funding: Genentech/Roche (Inst), Ipsen (Inst), Lilly (Inst), Merck (Inst), Taiho Pharmaceutical (Inst), Taiho Pharmaceutical (Inst), Cleveland BioLabs (Inst), Novartis (Inst)
Eunice S. Wang
Consulting or Advisory Role: AbbVie, Pfizer, Jazz Pharmaceuticals, Astellas Pharma, Stemline Therapeutics, Kite/Gilead, MacroGenics, PTC Therapeutics, Celgene/Bristol Myers Squibb, GlaxoSmithKline, Novartis, Genentech, Takeda
Speakers' Bureau: Stemline Therapeutics, Pfizer, Dava Oncology
Swapna Thota
Honoraria: Blueprint Medicines, Incyte, Adelson Medical Research Foundation
Speakers' Bureau: Incyte
No other potential conflicts of interest were reported.
SUPPORT
Supported by KL2 BTC award from the University of Buffalo's CTSI (S.T.). Data Bank and BioRepository (DBBR) provided data and samples for this study. DBBR is funded by the National Cancer Institute (P30 CA016056) and is a Roswell Park Cancer Institute Cancer Center Support Grant shared resource.
AUTHOR CONTRIBUTIONS
Conception and design: Abhay Singh, Duane C. Hassane, Swapna Thota
Administrative support: David Neil Hayes, Eunice S. Wang, Swapna Thota
Provision of study materials or patients: Renuka Iyer
Collection and assembly of data: Abhay Singh, Mahesh Swaminathan, Matthew Gravina, Rutaba Tajammal, LunBiao Yan, Eti Sinha, Duane C. Hassane, Renuka Iyer, Eunice S. Wang
Data analysis and interpretation: Abhay Singh, Nuria Mencia-Trinchant, Elizabeth A. Griffiths, Alaa Altahan, Medhavi Gupta, Rutaba Tajammal, Mark G. Faber, Duane C. Hassane, David Neil Hayes, Monica L. Guzman, Renuka Iyer, Eunice S. Wang, Swapna Thota
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Elizabeth A. Griffiths
Honoraria: Novartis
Consulting or Advisory Role: Alexion Pharmaceuticals, Takeda, Taiho Oncology, Novartis, Genentech, Celgene/Bristol Myers Squibb, AbbVie, Otsuka US, CTI BioPharma Corp, PicnicHealth
Research Funding: Genentech (Inst), Celgene (Inst), Apellis Pharmaceuticals (Inst), Astex Pharmaceuticals (Inst), Celldex (Inst), Bristol Myers Squibb/Celgene
Open Payments Link: https://openpaymentsdata.cms.gov/physician/134906
Mark G. Faber
Stock and Other Ownership Interests: AstraZeneca/MedImmune/Spirogen
Eti Sinha
Employment: Foundation Medicine
Stock and Other Ownership Interests: Roche/Genentech
Duane C. Hassane
Employment: Tempus
Stock and Other Ownership Interests: Tempus
Research Funding: Daiichi Sankyo
Patents, Royalties, Other Intellectual Property: ddPCR assessment of minimal residual disease via NPM1 mutations, risk prediction for acute myeloid leukemia
David Neil Hayes
Leadership: GeneCentric
Stock and Other Ownership Interests: GeneCentric
Consulting or Advisory Role: GeneCentric, Merck, Turnstone Bio
Patents, Royalties, Other Intellectual Property: I hold several diagnostic patents or pending patents in the area of solid tumor diagnostics
Monica L. Guzman
Consulting or Advisory Role: SeqRx, Bridge Medicines
Research Funding: Cellectis (Inst), Bridge Medicines (Inst), Daiichi Sankyo/UCB Japan (Inst)
Renuka Iyer
Consulting or Advisory Role: Lexicon, Novartis, Eisai, Merck, Bayer, Advanced Accelerator Applications, Exelixis, Sun pharma, QED therapeutics, Ipsen, Sandoz, TerSera, AstraZeneca
Research Funding: Genentech/Roche (Inst), Ipsen (Inst), Lilly (Inst), Merck (Inst), Taiho Pharmaceutical (Inst), Taiho Pharmaceutical (Inst), Cleveland BioLabs (Inst), Novartis (Inst)
Eunice S. Wang
Consulting or Advisory Role: AbbVie, Pfizer, Jazz Pharmaceuticals, Astellas Pharma, Stemline Therapeutics, Kite/Gilead, MacroGenics, PTC Therapeutics, Celgene/Bristol Myers Squibb, GlaxoSmithKline, Novartis, Genentech, Takeda
Speakers' Bureau: Stemline Therapeutics, Pfizer, Dava Oncology
Swapna Thota
Honoraria: Blueprint Medicines, Incyte, Adelson Medical Research Foundation
Speakers' Bureau: Incyte
No other potential conflicts of interest were reported.
REFERENCES
- 1.American Cancer Society : Facts & figures 2020 reports largest one-year drop in cancer mortality, 2020. https://www.cancer.org/latest-news/facts-and-figures-2020.html [Google Scholar]
- 2.McNerney ME, Godley LA, Le Beau MM: Therapy-related myeloid neoplasms: When genetics and environment collide. Nat Rev Cancer 17:513-527, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Granfeldt Ostgard LS, Medeiros BC, Sengelov H, et al. : Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: A national population-based cohort study. J Clin Oncol 33:3641-3649, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Chung J, Sallman DA, Padron E: TP53 and therapy-related myeloid neoplasms. Best Pract Res Clin Haematol 32:98-103, 2019 [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, Wang F, Kantarjian H, et al. : Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: A case-control study. Lancet Oncol 18:100-111, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Shea T, Druce M: When should genetic testing be performed in patients with neuroendocrine tumours? Rev Endocr Metab Disord 18:499-515, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guidetti A, Carlo-Stella C, Ruella M, et al. : Myeloablative doses of yttrium-90-ibritumomab tiuxetan and the risk of secondary myelodysplasia/acute myelogenous leukemia. Cancer 117:5074-5084, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Baum RP, Kulkarni HR, Singh A, et al. : Results and adverse events of personalized peptide receptor radionuclide therapy with (90)Yttrium and (177)Lutetium in 1048 patients with neuroendocrine neoplasms. Oncotarget 9:16932-16950, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonbol MB, Halfdanarson TR, Hilal T: Assessment of therapy-related myeloid neoplasms in patients with neuroendocrine tumors after peptide receptor radionuclide therapy: A systematic review. JAMA Oncol 6:1086-1092, 2020 [DOI] [PubMed] [Google Scholar]
- 10.Molenaar RJ, Sidana S, Radivoyevitch T, et al. : Risk of hematologic malignancies after radioiodine treatment of well-differentiated thyroid cancer. J Clin Oncol 36:1831-1839, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergsma H, van Lom K, Raaijmakers MH, et al. : Persistent hematologic dysfunction after peptide receptor radionuclide therapy with 177Lu-DOTATATE: Incidence, course, and predicting factors in patients with gastroenteropancreatic neuroendocrine tumors. J Nucl Med 59:452-458, 2018 [DOI] [PubMed] [Google Scholar]
- 12.Strosberg J, El-Haddad G, Wolin E, et al. : Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med 376:125-135, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bodei L, Kidd M, Paganelli G, et al. : Long-term tolerability of PRRT in 807 patients with neuroendocrine tumours: The value and limitations of clinical factors. Eur J Nucl Med Mol Imaging 42:5-19, 2015 [DOI] [PubMed] [Google Scholar]
- 14.Kesavan M, Turner JH: Myelotoxicity of peptide receptor radionuclide therapy of neuroendocrine tumors: A decade of experience. Cancer Biother Radiopharm 31:189-198, 2016 [DOI] [PubMed] [Google Scholar]
- 15.Goncalves I, Burbury K, Michael M, et al. : Characteristics and outcomes of therapy-related myeloid neoplasms after peptide receptor radionuclide/chemoradionuclide therapy (PRRT/PRCRT) for metastatic neuroendocrine neoplasia: A single-institution series. Eur J Nucl Med Mol Imaging 46:1902-1910, 2019 [DOI] [PubMed] [Google Scholar]
- 16.Oronsky B, Ma PC, Morgensztern D, et al. : Nothing but NET: A review of neuroendocrine tumors and carcinomas. Neoplasia 19:991-1002, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith SM, Le Beau MM, Huo D, et al. : Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago Series. Blood 102:43-52, 2003 [DOI] [PubMed] [Google Scholar]
- 18.R: A language and environment for statistical computing. Vienna, Austria, R Foundation for Statistical Computing. https://www.R-project.org [Google Scholar]
- 19.Hadley W: Ggplot2: Elegrant Graphics for Data Analysis. New York, NY, Springer, 2016 [Google Scholar]
- 20.Gu Z, Eils R, Schlesner M: Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32:2847-2849, 2016 [DOI] [PubMed] [Google Scholar]
- 21.Busque L, Buscarlet M, Mollica L, et al. : Concise review: Age-related clonal hematopoiesis: Stem cells tempting the devil. Stem Cells 36:1287-1294, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindsley RC, Saber W, Mar BG, et al. : Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med 376:536-547, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong TN, Ramsingh G, Young AL, et al. : Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 518:552-555, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bewersdorf JP, Ardasheva A, Podoltsev NA, et al. : From clonal hematopoiesis to myeloid leukemia and what happens in between: Will improved understanding lead to new therapeutic and preventive opportunities? Blood Rev 37:100587, 2019 [DOI] [PubMed] [Google Scholar]
- 25.Steensma DP: Clinical implications of clonal hematopoiesis. Mayo Clin Proc 93:1122-1130, 2018 [DOI] [PubMed] [Google Scholar]
- 26.Gillis NK, Ball M, Zhang Q, et al. : Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: A proof-of-concept, case-control study. Lancet Oncol 18:112-121, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schulz E, Kashofer K, Heitzer E, et al. : Preexisting TP53 mutation in therapy-related acute myeloid leukemia. Ann Hematol 94:527-529, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coombs CC, Zehir A, Devlin SM, et al. : Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21:374-382.e374, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibson CJ, Lindsley RC, Tchekmedyian V, et al. : Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol 35:1598-1605, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu JI, Dayaram T, Tovy A, et al. : PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell 23:700-713.e706, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strosberg JR, Wolin EM, Chasen BA, et al. : First update on overall survival, progression-free survival, and health-related time-to-deterioration quality of life from the NETTER-1 study: 177Lu-Dotatate vs. high dose octreotide in progressive midgut neuroendocrine tumors. J Clin Oncol 36, 2018. (suppl 15; abstr 4099) [DOI] [PMC free article] [PubMed] [Google Scholar]