

SUMMARY
Metastatic tumors remain lethal due to primary/acquired resistance to therapy or cancer stem cell (CSC)-mediated repopulation. We show that a fasting-mimicking diet (FMD) activates starvation escape pathways in triple-negative breast cancer (TNBC) cells, which can be identified and targeted by drugs. In CSCs, FMD lowers glucose-dependent protein kinase A signaling and stemness markers to reduce cell number and increase mouse survival. Accordingly, metastatic TNBC patients with lower glycemia survive longer than those with higher baseline glycemia. By contrast, in differentiated cancer cells, FMD activates PI3K-AKT, mTOR, and CDK4/6 as survival/growth pathways, which can be targeted by drugs to promote tumor regression. FMD cycles also prevent hyperglycemia and other toxicities caused by these drugs. These data indicate that FMD has wide and differential effects on normal, cancer, and CSCs, allowing the rapid identification and targeting of starvation escape pathways and providing a method potentially applicable to many malignancies.
In brief
Salvadori et al. report that a fasting-mimicking diet (FMD) can reduce TNBC stem cells by lowering their glucose-dependent PKA signaling. In contrast, in differentiated cancer cells, FMD induces the activation of starvation escape pathways, including PI3K/AKT, mTOR, and CDK4/6, which can be blocked by drugs, leading to tumor regression with low toxicity.
Graphical Abstract
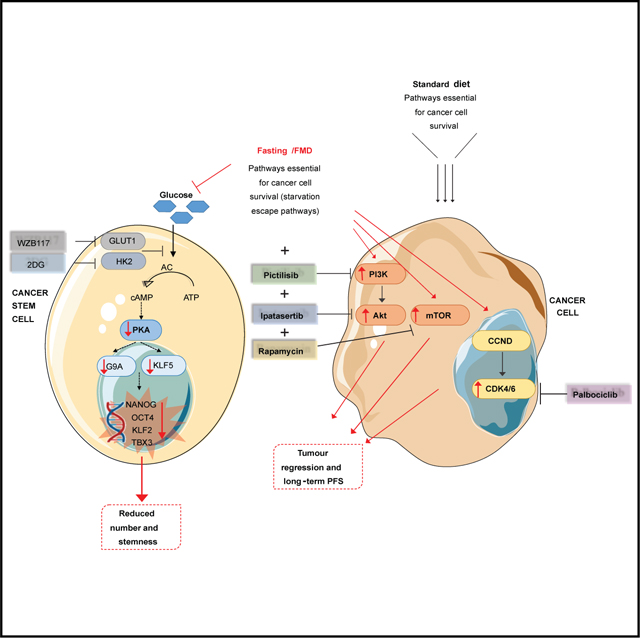
INTRODUCTION
Triple-negative breast cancer (TNBC), the most aggressive BC subtype, is characterized by high clinical aggressiveness and high recurrence rates when compared to other BC subtypes (Bianchini et al., 2016). Recently approved chemotherapy-immunotherapy combinations for the treatment of both limited-stage (in the neoadjuvant setting) and metastatic TNBC have improved patient prognosis in all disease settings (Cortes et al., 2020; Schmid et al., 2018, 2020). However, the prognosis of the vast majority of advanced TNBC patients remains poor because of primary/acquired tumor resistance to treatments and of the presence of cancer stem cells (CSCs), which are responsible for tumor re-population after initial regression (Charafe-Jauffret et al., 2009). CSCs are a subset of slow cycling cancer cells characterized by low levels of intracellular reactive oxygen species (ROS), which contribute to their self-renewal potential and resistance to chemotherapy- or radiotherapy-induced damage to DNA and other macromolecules. CSCs can arise and survive under hypoxic conditions as a result of hypoxia inducible factor (HIF-1)-mediated upregulation of the expression of glucose transporters (GLUT) and glycolysis enzymes, such as hexokinase (HK) (Das et al., 2008; Li et al., 2009). This metabolic plasticity allows CSCs to survive in an unfavorable tumor microenvironment, thus making them the most resistant cancer cell subset.
In recent years, our laboratories have shown that cycles of fasting/fasting-mimicking diet (FMD), based on severe calorie restriction of 50% or more, low levels of proteins and sugars, and relatively high fat content, enhance the efficacy of standard therapies in different types of cancer, including BC, while inducing the protection of normal cells from treatment-induced toxicities (Brandhorst et al., 2015; Caffa et al., 2020; Di Biase et al., 2016; Di Tano et al., 2020; Lee et al., 2010, 2012; Longo and Mattson, 2014; Raffaghello et al., 2008). These effects are partly mediated by fasting/FMD-induced reduction of circulating insulin growth factor 1 (IGF-1), glucose, leptin, and insulin levels, which differentially modulate genes involved in cellular protection, including antioxidant activity and DNA repair, in normal and cancer cells. Previous fasting/FMD-based interventions against TNBC, which involved chemotherapy-FMD combinations, slowed tumor progression but did not achieve long-term, progression-free survival and reduced, but did not completely prevent, the damage to normal cells and systems (Di Biase et al., 2016; Lee et al., 2012). Because CSCs play a crucial role in promoting tumor initiation, progression, and resistance to treatments, we investigated the effect of FMD on CSC survival and how this may affect TNBC growth and response to targeted therapies.
RESULTS
Fasting/FMD reduces human TNBC stem cells by lowering glucose levels
In the in vitro human TNBC SUM159 model, fasting/FMD-mimicking conditions (i.e., low-serum, low-glucose conditions, referred to as short-term starvation, STS) decreased mammosphere generation and volume, thus indicating an impact on TNBC self-renewal capacity (Figure S1A). When compared to CTR conditions, STS also reduced the proportion of CD44+ CD24− cells, which are enriched for TNBC CSCs (Figure S1B). Consistent with our in vitro results, cycles of FMD significantly slowed down tumor growth, reduced tumor size, and caused an increased expression of intratumor Caspase3 (Figures S1C and S1D), thus suggesting the activation of apoptosis. When compared to ad libitum (AL) diet, FMD cycles decreased the number of ex vivo primary mammospheres, reducing their serial propagation, as well as the percentage of cells expressing aldehyde dehydrogenase 1 (ALDH1), an enzyme highly expressed in TNBC and associated with CSC properties. (Figures 1A and 1B). To further assess the in vivo impact of the FMD on CSCs, we performed limiting dilution assays by injecting tumor cells derived from in vivo xenografts (from donor mice fed AL diet or FMD) in recipient mice, at different cell dilutions. All recipient mice were fed an AL diet, regardless of the diet to which the donor mice had been subjected. Notably, FMD decreased the frequency of cells able to promote TNBC, increasing cancer progression-free survival, when compared to AL (Figure S1E).
Figure 1. FMD reduces mammosphere growth, the expression of CSC markers, and stem cell frequency in SUM159 human triple-negative breast cancer (TNBC), and its effect is potentiated by 2-deoxy-D-glucose.

(A) SUM159 tumor masses were processed for ex vivo primary mammosphere (n = 7–9) and ex vivo serial sphere-forming assay (n = 4).
(B) Aldefluor analyses were performed by flow cytometry using the ALDEFLUOR kit to measure ALDH1 expression in SUM159 tumor masses (n = 5).
(C) Growth of SUM159 xenografts in 8-week-old female NOD scid (NSG) mice treated with AL diet or 5 cycles of FMD alone or plus 3% glucose supplementation in drinking water (n = 9).
(D) Tumor masses were excised and processed for ex vivo primary mammosphere assay (n = 9).
(E) Growth of SUM159 xenografts in 8-week-old female NOD scid (NSG) mice treated with AL diet or 5 cycles of FMD alone or combined with WZB117 (10 mg/kg) once a day intraperitoneally (i.p.) (n = 6).
(F) Tumor masses were excised and processed for ex vivo primary mammosphere assay (n = 6).
(G) Growth of SUM159 xenografts in 8-week-old female NOD scid (NSG) mice treated with AL diet or 5 cycles of FMD alone or combined with 2DG (200 mg/kg) once a day i.p. (n = 15).
(H) Tumor masses were excised and processed for ex vivo primary mammosphere assay (n = 10–15) and serial mammosphere assay (n = 5).
(I) SUM159 tumor cells derived from in vivo xenografts were injected in recipient mice at different dilutions to perform the limiting dilution assay (n = 6–14). p values were determined by log-rank (Mantel-Cox)test (100,000 cells: ad libitum versus FMD, p =0.0024; ad libitum versus 2DG, p = 0.0660; ad libitum versus FMD + 2DG, p = 0.0008; FMD versus 2DG, p = 0.1007; FMD versus FMD + 2DG, p = 0.1657; 2DG versus FMD + 2DG, p = 0.0177; 10,000 cells: ad libitum versus FMD, p = 0.0011; ad libitum versus 2DG, p = 0.0003; ad libitum versus FMD + 2DG, p < 0.0001; FMD versus 2DG, p = 0.0428; FMD versus FMD + 2DG, p = 0.3120; 2DG versus FMD + 2DG, p = 0.0192; 1,000 cells: ad libitum versus FMD, p = 0.0891; ad libitum versus 2DG, p = 0.3981; ad libitum versus FMD + 2DG, p = 0.0001; FMD versus 2DG, p = 0.4714; FMD versus FMD + 2DG, p = 0.0123; 2DG versus FMD + 2DG, p = 0.0067). The stem cell frequency was calculated using ELDA software. Data are represented as mean ± SEM. p values were determined by two-tailed unpaired t test (A and B) and one-way ANOVA (C–H). See also Figures S1 and S2.
We then investigated the role of STS/FMD-induced reduction of extracellular glucose or growth factors on CSC sensitization. As shown in Figure S1F, STS-mediated sensitization of CSCs was almost entirely rescued by the addition of physiological concentration (1g/L) of extracellular glucose, while the supplementation of fetal bovine serum (FBS) or its main components (IGF1, EGF, and insulin) did not significantly affect mammosphere growth (Figure S1F). We also evaluated the effect of in vivo glucose supplementation by adding 3% of glucose to the drinking water of mice fed the FMD cycles, based on the normal concentration of sugar assumed daily by mice through the standard diet. Consistent with results of in vitro experiments, glucose supplementation partially reversed FMD-induced slowing down of tumor progression and completely rescued FMD-dependent reduction of mammospheres (Figures 1C, 1D, and S1G).
To confirm the impact of glucose metabolism on CSCs, we evaluated the effect of WZB117, a specific inhibitor of the glucose transporter GLUT1 (frequently overexpressed in TNBC). In mice fed AL diet, WZB117 reduced tumor progression, even though the effect of cyclic FMD was more pronounced (Figure 1E). In addition, WZB117 did not potentiate the anti-tumor activity of FMD, in agreement with the effect of FMD alone in decreasing the expression of GLUT1, suggesting that the effects of WZB117 and FMD are overlapping (Figures 1E, S2A, and S2B). Interestingly, WZB117 mimicked the effect of FMD on reducing the number of sphere formation, confirming our hypothesis that lowering intracellular glucose levels (through reduced extracellular levels or reduced uptake) reduces CSC survival (Figure 1F). Then, we investigated the effect of compounds affecting glucose metabolism downstream of glucose uptake, namely 2-deoxy-D-glucose (2DG), an inhibitor of the glycolysis enzyme hexokinase, or the antidiabetic compound metformin, which reduces blood glucose levels and results in the inhibition of mitochondrial oxidative phosphorylation (OXPHOS). Interestingly, 2DG potentiated the effect of FMD both in terms of delaying tumor progression and in decreasing the number of mammospheres derived by tumor masses, even after a multiple serial propagation, while it did not have any effect under AL conditions (Figures 1G, 1H, S2C, and S2D). Furthermore, in limiting dilution in vivo assays, 2DG potentiated the effect of FMD in increasing mice survival compared to 2DG and FMD alone, and the 2DG-FMD combination prevented tumor formation for more than 150 days at the 1,000-cell dilution level (Figure 1I). On the other hand, metformin, which reduced tumor growth and sphere formation when compared with the control (AL diet), likely by reducing blood glucose concentration, did not show any additive or synergistic antitumor effect when combined with the FMD, thus suggesting that FMD and metformin have redundant effects on blood glucose levels, and that the effect of metformin in modulating OXPHOS does not play a relevant role in TNBC (Figures S2E–S2G). Collectively, these data indicate that CSCs are sensitive to glucose deprivation mediated by FMD and that pharmacological inhibition of glycolysis downstream of glucose uptake potentiates FMD toxicity against TNBC CSCs.
FMD reduces TNBC CSCs and delays tumor progression in syngeneic TNBC models
To confirm our data in a syngeneic mouse TNBC model, we tested the effect of fasting/FMD in 4T1 mouse transplants in immuno-competent BALB/c female mice. When compared to AL conditions, four FMD cycles delayed tumor progression and reduced mammosphere number as well as the percentage of CD44+CD24− and ALDH1+ cells, thus confirming the anti-CSC effect previously reported in the human SUM159 model (Figures 2A and S3A–S3D). Moreover, consistent with data in human TNBC, supplementing 1 g/L glucose to STS cell growth medium reversed the STS effect on 4T1 sphere formation (Figure 2B). Finally, 2DG potentiated the effect of FMD both in terms of delayed tumor progression and CSC reduction, thus confirming the results obtained with the SUM159 model (Figures 2C–2E and S3E).
Figure 2. FMD acts independently of the immune system and reduces metastasis in syngeneic TNBC models.

(A) Growth of 4T1-luc cells in the mammary fat pad of 6-week-old female Balb-c mice treated with AL diet or 4 cycles of FMD. Tumor masses were excised and processed for ex vivo primary mammosphere-forming assay (n = 7).
(B) 4T1 cells were grown under CTR (1 g/L glucose, 10% FBS), STS (0.5 g/L, 1% FBS), and STS + 1 g/L glucose conditions for a total of 48 h. Cells were then plated to perform the in vitro sphere-forming assay (n = 4 biological replicates).
(C) Growth of 4T1-luc cells in the mammary fat pad of 6-week-old female Balb-c mice treated with AL diet or 4 cycles of FMD alone or combined with 2DG (200 mg/kg) once a day i.p. (n = 15 per group).
(D) Fluorescence-activated cell sorting (FACS) analysis was performed to measure CD44 and CD24 expression in 4T1 TNBC in vivo (n = 8).
(E) Aldefluor analysis was performed by flow cytometry using the ALDEFLUOR kit to measure ALDH1 expression in 4T1 tumor masses (n = 4–5).
(F) Growth of 4T1-luc cells intravenously in 6-week-old female Balb-c mice treated with AL diet or 4 cycles of FMD alone or combined with 2DG (200 mg/kg) oncea day i.p. Tumor progression was monitored with bioluminescent imaging 17 days post-cell injection (n = 5 per group).
Data are represented as mean ± SEM. p values were determined by two-tailed unpaired t test (A) and one-way ANOVA (B–E). See also Figure S3.
Since metastases are the major cause of death in TNBC patients, and CSCs play a crucial role in sustaining metastatic spread, we evaluated the effect of cyclic FMD, alone or combined with 2DG, on metastasis formation in mice injected intravenously with 4T1-luc cells (a model of metastatic TNBC). We found that FMD cycles delay metastasis formation and prevent them from being established in a number of organs. In particular, the majority of mice fed AL plus/minus 2DG showed metastasis formation at 10 days post-cell injection, while metastases were only evident in a minority of mice undergoing the FMD (Figure S3F). At later time points (17 days after cell injection), 80% of FMD-treated mice only displayed metastasis in the lungs, while in mice fed with the normal diet metastases were much more common in the liver, spleen, ovaries, and lymph nodes (Figure 2F). Furthermore, at day 17 we observed an even more effective prevention of metastasis formation in animals treated with a combination of cyclic FMD and 2DG (Figures 2F and S3G).
Low baseline glycemia is associated with increased survival of patients with metastatic TNBC
To determine whether our findings in mice may be relevant to TNBC progression in humans, we investigated the relevance of baseline blood glucose concentration on overall survival (OS) in patients with advanced TNBC. For this purpose, we evaluated a retrospective series of advanced BC patients treated at Fondazione IRCCS Istituto Nazionale dei Tumori between October 2006 and January 2020 with platinum-based chemotherapy (Figure S4A). Out of 418 patients with different BC subtypes, we selected a homogeneous cohort of 81 consecutive patients with advanced TNBC treated with first-line carboplatin-paclitaxel or carboplatin-gemcitabine (Table S1). In these patients, we assessed the impact of baseline glucose concentration on patient OS at both univariate and multivariable analysis. Patients were considered as normoglycemic if they had baseline blood glucose levels below 100 mg/dL. Notably, hyperglycemic patients had significantly lower OS when compared with normoglycemic patients (Figure 3A). Univariate analysis aimed at identifying other prognostic factors revealed the following covariates as associated with an increased risk of patient death: lower ECOG PS (1 versus 0), higher number of metastatic sites (>3 versus 1–3), and presence of lung, liver, or bone metastasis (Table S2). Multivariable model adjusting the potential impact of hyperglycemia for those covariates confirmed an independent negative prognostic role of high blood glucose concentration on patient OS (HR in hyperglycemic versus normoglycemic patients, 1.95; 95% CI, 1.05–3.64; p = 0.035) (Figure 3B). Notably, worse ECOG PS (1 versus 0) and presence of liver and/or lung metastasis, which are well-acknowledged negative prognostic factors in advanced TNBC patients, were the only other variables significantly associated with worse OS in the multivariable model.
Figure 3. Hyperglycemia is associated with worse overall survival in advanced TNBC patients.

(A) Kaplan-Meier curves for overall survival (OS) according to baseline blood glucose levels in advanced TNBC patients treated with first-line platinum-based doublet chemotherapy. Normoglycemia and hyperglycemia were defined according to 100 mg/dL threshold. The + symbol in Kaplan Meier curves indicates patients who were censored at the time of data cut-off analysis.
(B) Multivariable analysis adjusting the impact of hyperglycemia on OS according to clinical characteristics previously selected on the basis of univariate analysis. For each covariate the hazard ratio (HR), 95% confidence interval (CI), and p value are indicated. CIs not crossing the value of 1 indicate a statistically significant impact of that variable on patient OS. The p value is indicated in bold when statistically significant.
See also Figure S4.
To assess the consistency of our results, we repeated the same analyses by using the 110 mg/dL threshold to discriminate hyperglycemic versus normoglycemic patients. With this threshold, hyperglycemia was still associated with worse patient OS (Figure S4B), thus recapitulating results observed with the 100 mg/dL threshold. Although in multivariable analysis statistical significance was not reached (likely due to the smaller number of hyperglycemic patients according to the new threshold), we still observed a trend toward an independent association between hyperglycemia and worse patient OS (HR 1.83; 95% CI, 0.91–3.68; p = 0.092) (Figure S4C).
Overall, clinical data are in line with results of preclinical experiments, i.e., they show that blood glucose concentration might affect TNBC sensitivity to standard anticancer therapies and long-term patient survival.
PKA activation reverses STS-dependent mammosphere reduction
Next, we investigated the mechanism through which glucose depletion sensitizes CSCs. We have previously shown that prolonged fasting reduces the activity of protein kinase A (PKA) in different types of normal cells (Brandhorst et al., 2015; Cheng et al., 2014), an effect mediated in part by reduced glucose availability (Di Biase et al., 2017). Interestingly, PKA inhibition resulted in the downregulation of KLF5, a potential therapeutic target for TNBC (Shi et al., 2017), through glycogen synthase kinase-3β (GSK3β) phosphorylation. Consistent with the relevance of PKA activity for CSC survival, PKA is reported to be more highly phosphorylated in BCSCs than in non-BSCSs (Chan et al., 2020). Thus, we investigated the involvement of PKA axis in FMD-dependent CSC reduction. We measured PKA activity by examining the phosphorylation of CREB, a classical downstream substrate of PKA, and KLF5 expression in SUM159 and 4T1 tumor masses. Notably, both pCREB and KLF5 were downregulated in tumor masses in both mouse models undergoing the FMD, thus indicating PKA pathway inhibition (Figure 4A). Moreover, RNA sequencing (RNA-seq) analysis performed in SUM159 tumor masses, previously sorted for CD44CD24 human antibodies, revealed that FMD-induced inhibition of the PKA pathway selectively occurs in the CD44+CD24− staminal population, but not in the CD44+CD24+ differentiated cells (Figure 4B).
Figure 4. PKA is downregulated by FMD selectively in CSCs.

(A) Detection of phosphorylated CREB levels and total CREB, KLF5, and VINCULIN, as loading control, in SUM159 and 4T1 tumor masses (n = 5).
(B) Volcano plot showing the significance versus the log2 fold change between CD44+CD24– and CD44+CD24+. Up- and downregulated genes (|log2FC| > 0.58 and adj. p < 0.05) are displayed in red and green, respectively. Deregulated genes involved in the PKA pathway are highlighted.
(C) Detection of phosphorylated CREB levels and total CREB, KLF5, and VINCULIN, as loading control, in SUM159 CD44+CD24+ and CD44+CD24– subpopulations.
(D) Detection of phosphorylated CREB levels and total CREB, KLF5, and VINCULIN, as loading control, in 4T1 CD44+CD24+ and CD44+CD24– subpopulations. Data are represented as mean ± SEM. p values were determined by two-tailed unpaired t test (A) and multiple unpaired t test (C and D). See also Figure S5.
To further evaluate FMD-induced regulation of PKA in different tumor cell subsets, we assessed the expression of proteins involved in PKA signaling, including pCREB and KLF5, both in CD44+ CD24− and CD44+CD24+ cells. In either SUM159 or 4T1 TNBC cells, the expression of pCREB and KLF5 did not significantly change between CSCs and differentiated cancer cells under CTR conditions, while both proteins were selectively downregulated in CSCs under STS conditions (Figures 4C and 4D). These results suggest the selective involvement of PKA axis in affecting CSCs. In fact, the PKA activator 8-bromoadenosine 3′,5′-cyclic mono-phosphate (8-Br-cAMP) reversed fasting/STS-dependent sphere reduction and fasting/STS-dependent lowering of CD44+CD24− cells both in the SUM159 and 4T1 models, confirming the role of PKA inhibition in stemness regulation (Figures 5A, 5B, and S5A).
Figure 5. PKA activation through 8Br-cAMP reverses STS-dependent sphere reduction and CD44+CD24– cell lowering.

(A and B) SUM159 and 4T1 cells were grown under CTR (1 g/L glucose, 10% FBS) and STS (0.5 g/L, 1% FBS) conditions for a total of 48 h. At 24 h cells were treated with 8-Br-cAMP. Cells (A) were then plated to perform the in vitro sphere-forming assay (n = 4–6 biological replicates) or (B) were used to perform flow cytometry analysis to measure the percentage of CD44+CD24+ and CD44+CD24– cells in SUM159 or 4T1 TNBC models (n = 6–9 biological replicates).
(C) Detection of G9A, H3meK9 levels, and VINCULIN, as loading control, in SUM159 and 4T1 tumors (n = 7–8).
Data are represented as mean ± SEM. p values were determined by two-tailed unpaired t test (C) and one-way ANOVA (A and B).
PKA also regulates the H3K9 methyltranferase G9A, which is implicated in the pathogenesis of several cancer types, including ovarian, lung, liver, breast, and bladder cancers (Bai et al., 2016; Hua et al., 2014; Kondo et al., 2008; Li et al., 2013). In particular, G9A-induced demethylation of H3K9 represses the expression of several tumor suppressor genes and adhesion molecules, promoting invasion and metastasis (Chen et al., 2010; Kondo et al., 2007; Wozniak et al., 2007). Both G9A and H3K9me2 levels were downregulated by FMD in SUM159 and 4T1 tumor masses (Figure 5C). Notably, exogenous reactivation of PKA pathway through 8Br-cAMP reversed STS/FMD-dependent downregulation of both KLF5 and H3K9me2 proteins, increasing their expression when compared to STS conditions alone (Figure S5B). In SUM159 tumors, cyclic FMD also reduced the mRNA of KLF5 (Parisi et al., 2008) and G9A (Luo et al., 2017) stemness-associated genes NANOG and OCT4, and KLF2 and TBX3, which are regulated by the transcriptional activity of OCT4 (Figure S5C). Together, these data indicate that FMD-induced depletion of TNBC CSCs is at least mediated by glucose reduction and the inhibition of the PKA pathway.
Combining FMD cycles with PI3K/AKT/mTOR inhibitors results in long-term animal survival and reduces treatment-induced side effects
Even though CSCs play a crucial role in tumor formation and progression, differentiated cells, which contribute to the volume bulk of tumor masses, are fundamental as well. Indeed, cyclic FMD alone, which results in a significant depletion of CSCs, only delays tumor growth, but it does not result in cancer-free survival or long-term progression-free survival.
To identify potential druggable targets that may allow differentiated cancer cells to survive under fasting/FMD conditions, and which contribute to tumor resistance to treatments, we performed RNA-seq analysis in SUM159 tumor masses from animals fed with AL diet or FMD. Notably, the FMD resulted in an increased expression of pro-apoptotic molecules, such as BIM, and ASK1, a critical cellular stress sensor frequently activated by ROS, whose production was previously shown to be increased by the FMD (Lee et al., 2012). RNA-seq analysis also indicated that FMD cycles upregulate PI3K-AKT and mTOR pathways and downregulate CCNB-CDK1 while upregulating CCND-CDK4/6 signaling axes (Figures 6A and S6A). Interestingly, FMD-induced activation of survival factors only occurred in differentiated cells, but not in CSCs (Figure S6B).
Figure 6. FMD activates starvation escape mechanisms that can be targeted by drugs.

(A) Volcano plot showing the significance versus the log2 fold change in SUM159 tumor masses, by comparing FMD versus AL. Up- and downregulated genes (|log2FC| > 0.58 and adj. p < 0.05) are displayed in red and green, respectively. Deregulated genes involved in PI3K-AKT-mTOR and CycD-CDK4/6 pathways are highlighted. Significantly deregulated genes are reported in bold.
(B) Growth of SUM159 xenografts in 8-week-old female NOD scid (NSG) mice treated with AL diet or FMD, alone or combined with pictilisib (100 mg/kg, 5 consecutive days a week by oral gavage), ipatasertib (75 mg/kg, 5 consecutive days a week by oral gavage), or rapamycin (2 mg/kg, every other day i.p.), as single or double treatments (n = 8–12). Survival curves are reported.
p values were determined by log-rank (Mantel-Cox) test (B). See also Figure S6.
Then, we investigated whether pharmacological inhibition of the PI3K/AKT and mTOR pathways can potentiate the effect of the FMD by blocking “starvation escape pathways” (SEPs), which could promote the survival of differentiated TNBC cells during FMD. In in vitro experiments, combining STS with pictilisib, ipatasertib, and rapamycin, selective inhibitors for PI3K, AKT, and mTOR, respectively, resulted in enhanced cancer cell death and reduction of mammosphere numbers in SUM159 cells, partly due to a synergistic effect on the PKA signaling pathway (Figures S6C and S6D). Indeed, the triple combination of drugs combined to STS further reduces the expression of both KLF5 and G9A proteins, when compared to STS alone (Figure S6E). In mouse xenografts of SUM159, weekly cycles of FMD significantly improved the anti-tumor activity of each of these pharmacological inhibitors, leading to a significative increase in mice survival (Figures 6B and S7A). However, the most striking effect was observed when combining all three drugs with FMD cycles. In particular, treatment with the 3 drugs targeting PI3K/AKT and mTOR showed a strong effect on progression-free survival in mice fed with standard diet, but the addition of FMD cycles caused a major enhancement of the anti-tumor activity of the triple combination of PI3K/AKT and mTOR pathway inhibitors, preventing tumor growth for more than 150 days in 85% of mice (Figure 7A). Also in this case, the observed antitumor effect could be partly mediated by a strong inhibition of the PKA signaling pathway. In fact, we found that triple pictilisib + ipatasertib + rapamycin treatment combined with STS, but not single (pictilisib) or double (pictilisib + ipatasertib) treatments, strongly downregulated the expression of KLF5 and H3K9me2 proteins, which, in addition to directly regulating the expression of stemness-associated genes, are reported to promote TNBC cell growth (Chen et al., 2017; Curry et al., 2015; Li et al., 2017, Liu et al., 2018a, 2018b, 2020; Tang et al., 2018) (Figure S7B).
Figure 7. FMD reverts late-stage TNBC progression by potentiating the effect of kinase inhibitors and reversing the lethality caused by hyperglycemia.

(A) Growth of SUM 159 xenografts in 8-week-old female NOD scid (NSG) mice treated with AL dietor FMD, alone or combined with pictilisib (100 mg/kg, 5 consecutive days a week by oral gavage), ipatasertib (75 mg/kg, 5 consecutive days a week by oral gavage), and rapamycin (2 mg/kg, every other day i.p.) (n = 10–13). Progression-free survival was monitored over time and causes of death are reported.
(B) Blood glucose level was determined through Accu chek guide instrument.
(C) Growth of SUM159 xenografts in 8-week-old female NOD scid (NSG) mice treated with AL diet or FMD, alone or combined with palbociclib (62.5 mg/kg, byoral gavage, every other day) and pictilisib (100 mg/kg, by oral gavage, 5 consecutive days a week) (n = 10). Tumor masses were used to perform the ex vivo sphere-forming assay (n = 5).
(D) Growth of SUM159 xenografts in 8-week-old female NOD scid (NSG) mice treated with AL diet or FMD. Thirty-five days post-cell injection, mice started to be treated with pictilisib (100 mg/kg, 5 consecutive days a week by oral gavage), ipatasertib (75 mg/kg, 5 consecutive days a week by oral gavage), and rapamycin (2 mg/kg, every other day i.p.) plus FMD (n = 15).
(E) Putative model for the effect of FMD on SUM159 TNBC cells and CSCs.
Data are represented as mean ± SEM. p values were determined by two-tailed unpaired t test (C). See also Figure S7.
We previously showed that the hyperglycemia caused by the mTOR inhibitor rapamycin sensitizes normal cells and healthy mice to the toxic effects of chemotherapy (Di Biase et al., 2017). Hyperglycemia caused by PI3K inhibitors is also a major cause of adverse events in patients with advanced BC (Baselga et al., 2017; Bendell et al., 2012; Juric et al., 2017; Mayer et al., 2017; Patnaik et al., 2016). Thus, we investigated whether fasting/FMD-induced prevention of hyperglycemia might contribute to the observed prolongation of animal survival. Interestingly, FMD cycles protected mice from PI3K/AKT and mTOR inhibitor-induced hyperglycemia (Figure 7B), which in some animals was directly responsible for mouse death independently of tumor growth (Figure 7A). FMD cycles restored glycemia to normal levels shortly after the drug administration, thus allowing glucose levels to be normal during the great majority of the drug treatment (Figure 7B). Based on RNA-seq results, and on previous data showing that CDK4/6 inhibitors sensitize PI3KCA-mutated TNBCs to PI3K inhibitors (Vora et al., 2014), we evaluated the therapeutic effect of combining the CDK4/6 inhibitor palbociclib with pictilisib. While palbociclib + pictilisib significantly delayed tumor progression, the addition of cyclic FMD to this pharmacological combination further increased their anti-tumor activity and delayed the acquisition of drug resistance (Figures 7C and S7C). In addition, the FMD-palbociclib-pictilisib triple treatment also reduced the formation of spheres compared to pharmacologic treatment alone after 8 weeks of treatment (Figure 7C). We observed similar results in 4T1 mouse transplants, where the palbociclib + pictilisib combination delayed tumor progression, while adding the FMD to palbociclib + pictilisib further delayed acquired tumor resistance (Figure S7D).
Finally, we investigated whether FMD cycles combined with targeted drugs can induce the reversal of late-stage tumor progression in human TNBC xenografts. For this experiment, we hypothesized that FMD-induced depletion of TNBC CSCs before the administration of the quadruple FMD-pictilisib-ipatasertib-rapamycin treatment might result in long-term tumor control and prevent acquired resistance by cancer cells. To test this hypothesis, SUM159-xenograft-bearing mice were injected with TNBC cells and either subjected to 4 cycles of FMD or fed with standard diet. Thirty-five days after tumor cell injection, when tumors were in an advanced growth stage, we treated mice from the 2 groups with the 3 inhibitors plus FMD cycles.
The FMD-pictilisib-ipatasertib-rapamycin caused the regression of advanced tumors within the first week of treatment regardless of prior diet (AL or FMD). Notably, pre-treatment of mice with FMD cycles, prior to the start of the FMD + 3 drugs treatment, prevented acquisition of drug resistance at a later stage, leading to complete tumor shrinkage and long-term progression-free survival (Figures 7D and S7E). By contrast, tumor masses from mice fed with AL diet prior to FMD-pictilisib-ipatasertib-rapamycin treatment showed acquired resistance to the combination therapy (Figure 7D). Together with the results described earlier in this study, this experiment suggests that the decrease of CSCs caused by FMD cycles prior to the addition of the 3 drugs is essential in preventing long-term acquisition of drug resistance and tumor growth.
DISCUSSION
The failure of most anti-TNBC treatments in the advanced disease setting could be caused by the accumulation of a high number of treatment-resistant CSCs, which can repopulate tumor masses after initial treatment-induced tumor debulking. Therefore, our results indicate that FMD-induced depletion of TNBC CSCs when tumors are in a less advanced stage could strongly enhance the efficacy of subsequent treatments targeting both CSCs (such as the FMD) and more differentiated cancer cells (such as PI3K/AKT/mTORC1 inhibitors) in late-stage cancers.
In this study, RNA-seq analysis in tumors from animals undergoing FMD was crucial to identify druggable SEPs in differentiated cancer cells, thus suggesting new therapeutic combinations of the FMD and pharmacological inhibitors of SEPs. However, these results also underline the “wild card” property of FMD cycles, which, in addition to causing the activation of the druggable PI3K-AKT, mTOR, and CCND-CDK4/6 targets, also reduced long-term hyperglycemia and other side effects and deaths caused by inhibitors of these pathways (Figures 7A and 7B). This ability of FMD to protect from hyperglycemia, a side effect that is limiting the use of PI3K- and mTOR-inhibiting drugs in the clinic, could provide benefits in the clinical setting in combination with a wide range of drugs well established to raise glucose levels, including mTOR inhibitors rapamycin and everolimus, as well as PI3K inhibitors (Baselga et al., 2017; Bendell et al., 2012; Juric et al., 2017; Mayer et al., 2017; Patnaik et al., 2016). Notably, high glucose levels could also promote high insulin levels, well known to promote cancer growth (Hopkins et al., 2018). Although in our in vitro experiments insulin does not appear to be driving CSC growth, this does not rule out an effect of insulin on differentiated cancer cells, since their growth is also affected by high glucose (Figure 1C), in agreement with Yun et al. (2015).
Notably, recent studies have shown that cyclic FMD is safe and feasible in healthy humans, but also when combined with standard anti-tumor therapies in cancer patients (Caffa et al., 2020; de Groot et al., 2020; Wei et al., 2017). Results from these clinical studies showed that FMD reduces risk factors and metabolic markers including blood glucose levels, and one study showed that these effects on glucose reduction persist for at least 3 months after the last FMD cycle, suggesting that FMD effects on glycemia are long-lasting (Caffa et al., 2020; Wei et al., 2017). Moreover, one clinical trial reported increased clinical and pathological efficacy of chemotherapy when administered to a group that included TNBC patients (de Groot et al., 2020). Results of our study pave the way for using FMD cycles to reveal druggable SEP in different tumor types and also for testing cyclic FMD in combination with inhibitors of the PI3K/AKT/mTORC1 and CDK4/6 axes in TNBC patients.
Limitations of study
Our experiments were performed on two TNBC cell lines. Our data indicate that the effects and mechanisms shown for these two TNBC cell lines are similar. However, additional studies are needed to confirm this hypothesis and determine whether these methods could be potentially applicable to other cancer types, in addition to TNBC. Moreover, the study is focused on the importance of blood glucose level reduction mediated by fasting/FMD to reduce CSCs in mice. In different clinical trials, we and others have shown that in humans 5 days of FMD reduced blood glucose levels by ∼15%, different from studies in rodents in which cyclic FMD led to an ∼40% decrease in blood glucose levels. Therefore, a reduction in blood glucose levels in humans equivalent to that caused by FMD in mice may require a longer fasting period. Notably, despite this limitation, we observed that low baseline glycemia is associated with increased survival of patients with metastatic TNBC.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Valter D. Longo, vlongo@usc.edu.
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
The RNA-seq data generated during the current study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO: GSE184452.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture conditions
The human triple negative breast cancer (TNBC) cell line SUM159 was purchased from Asterand; the murine TNBC cell line 4T1 was purchased from ATCC. SUM159 cells were cultured in Ham’s F-12 medium (GIBCO, Cat. #: Cat. #: 11–765-054) supplemented with 5% FBS NA, 5 μg/mL insulin (Sigma-Aldrich, Cat. #: 11376497001), 1 μg/mL hydrocortisone (Sigma-Aldrich, Cat. #: H4001) and 1% penicillin/streptomycin (Biowest, Cat. # L0022). 4T1 cells were cultured in RPMI 1640 medium (Biowest, Cat #: L0500) supplemented with 10% FBS (Biowest, Cat. #: S1810) and 1%penicillin/streptomycin. All cells were tested for mycoplasma contamination routinely. Cells were maintained in a humidified, 5% CO2 atmosphere at 37°C.
In vitro, Short-Term starvation medium (STS) consists in a DMEM medium without glucose (DMEM no glucose, Life Technologies, Cat. #: 11966025) supplemented with 0.5 g/l glucose (Sigma-Aldrich, Cat. #: G8769) and 1% FBS. Control medium (CTR) consists in a DMEM no glucose medium supplemented with 1 g/l glucose and 10% FBS. For rescuing experiments with glucose/FBS, cells were cultured under CTR, STS, STS + 1g/l glucose-1%FBS, STS + 10%FBS-0.5g/l glucose and STS + 0.5g/l glucose-1%FBS and single FBS components at CTR concentration level (IGF1: 250ng/mL (Peprotech, Cat. #: 100–11), EGF: 200ng/mL (Biomol, Cat # BPS-90201–3), Insulin: 200ng/mL (Sigma-Aldrich, Cat. #: 11376497001)) for a total of 48 h.
Mouse models
The animals were housed under specific pathogen-free conditions at 22 ± 2°C with 55 ± 10% relative humidity and with 12 h day/light cycles. All mice used in experiments throughout the study exhibited normal health. All experiments were performed in accordance with the guidelines established in the Principles of Laboratory Animal Care (directive 86/609/EEC) and were approved by the Italian Ministry of Health. For xenograft experiments, 8-weeks old female NOD scid gamma (NSG, Charles River) were subcutaneously injected with 1.5×106 SUM159 cells resuspended in 100 μL of PBS. For syngeneic model, 6-weeks old female Balbc/Ola Hsd mice (Envigo) were injected in the mammary fat pad with 2×104 4T1-luc cells resuspended in 20 μL of PBS. When tumors were palpable (approximately 7 days for SUM159 and 3 days for 4T1 after inoculation), mice were randomly divided in the different experimental groups. Body weights were recorded daily, and tumor volumes were measured twice a week by a digital caliper according to the following equation: tumor volume (mm3) = length x width x thickness x 0.5. For metastasis evaluation experiment, 6-weeks old female Balbc/Ola Hsd mice (Envigo) were injected intravenously with 2×104 4T1-luc cells resuspended in 100 μL of PBS. Body weights were recorded daily and tumor progression was monitored with bioluminescent imaging. At the end of the experiments, mice were euthanized by using CO2.
METHOD DETAILS
Reagent’s preparation
WZB117
Glucose transporter inhibitor IV, WZB117, was purchased from MERK (Cat. #: 400036) and was dissolved in DMSO. Stock solutions of 70mg/mL were prepared for in vivo experiment and were stored at −80°C.
Metformin
Metformin was purchased from Sigma-Aldrich (Cat. #: D150959) and was dissolved in sterile water to a final concentration of 1M (stock solution). Stock solutions were store at +4°C.
2-Deoxy-D-Glucose
2-Deoxy-D-Glucose was purchased from Sigma-Aldrich (Cat. #: D-6134) and was dissolved in sterile water to a final concentration of 2M (stock solution). Stock solutions were stored at +4°C
8-Bromoadenosine 3′,5′-cyclic mono-phosphates (8-Br-cAMP)
8-Br-cAMP was purchased from Cayman (Cat. #: 14431) and was dissolved in sterile water. Stock solutions of 25mg/mL were prepared and stored at −20°C.
Alpelisib
Alpelisib was purchased from MedchemTronica (Cat. #: HY-15244) and was dissolved in DMSO. Stock solutions of 10mM were prepared for in vitro experiments and stored at −80°C.
Rapamycin
Rapamycin was purchased from MedchemTronica (Cat. #: HY-10219) and was dissolved in DMSO. Stock solutions of 20mM and 20mg/mL were prepared for in vitro and in vivo experiments, respectively, and stored at −80°C.
Pictilisib
Pictilisb was purchased from MedchemTronica (Cat. #: HY-50094) and was dissolved in DMSO. Stock solutions of 10mM and 200mg/mL were prepared for in vitro and in vivo experiments, respectively, and stored at −80°C.
Ipatasertib
Ipatasertib was purchased from MedchemTronica (Cat. #: HY-15186) and was dissolved in DMSO. Stock solutions of 10mM and 200mg/mL were prepared for in vitro and in vivo experiments, respectively, and stored at −80°C.
Erythrosin B exclusion assay
Cells were seeded in 12-well plates in their maintenance media for 24 hours. The next days, cells were washed with PBS and grown in CTR/STS media for a total of 48 h. After 24 hours under CTR/STS conditions cells were treated with drugs or vehicle for the next 24 h. In particular, for the experiment with PI3K-AKT-mTOR inhibitors, cells were treated with 10 μM rapamycin, 10 μM pictilisib, 20 μM alpelisib and 20 μM ipatasertib for a total of 24 hours. To perform the erythrosin B exclusion assay, cells were suspended 1:1 with erythrosin B 0.1% in PBS (Sigma-Aldrich, Cat. #: 200964) and counted in a Burker chamber. The percentage of cell death was calculated as the ratio of erythrosine B-positive cells with the total number of cells.
Mammosphere forming assay
For mammosphere formation assay in vitro, SUM159 and 4T1 cells were seeded in 12-well plates and grown in CTR/STS media for a total of 48 h. At 24 h, cells were treated with 5mM metformin or 4mM 2-Deoxy-D-Glucose. For rescuing experiments with PKA activator, cells were treated with 0.5mM 8-Br-cAMP, for a total of 24 hours. To perform the mammosphere forming assay cells were digested into single cells using a 21G needle and then were plated in ultra-low attachment plates at a density of 500 or 1500 cells per well. Cells were cultured in a mammary epithelial basal medium (MEMB, Lonza Cat. #: CC-3151) and methylcellulose (Sigma-Aldrich Cat. #: M0512) for 8/10 days. MEMB was previously supplemented with heparin (1U/mL), hydrocortisone (0.5 μg/mL), insulin (5 μg/mL), 1% L-glutamine (Biowest, Cat. #: X0550), 1% penicillin/streptomycin, B-27 (40 μl/mL, GIBCO Cat. #: 17504044), epidermal growth factor (EGF, 40ng/mL ) and fibroblast growth factor (FGF, 40ng/mL, Peprotech Cat. #: 100–18B). For ex vivo mammosphere formation assay, tumor masses were collected the last day of FMD cycle, before refeeding period. Tumors were excised from the flank/mammary fat pad of the mice and chopped in small parts with a scalpel and then digested enzymatically in DMEM medium supplemented with hyaluronidase (10mg/mL, Sigma-Aldrich Cat. #: H4272) and collagenase (2000U/mL, Sigma-Aldrich Cat. #: C2674) for 3 h, 5%CO2 at 37°C. Cells obtained were filtered on cell strainer (100–70-40 μM) in order to achieve single cells and re-suspended in red blood cell lysing buffer hybrid-max (Sigma-Aldrich Cat. #: R7757) for 30 s/1min. Finally, cells were plated in ultra-low attachment plates at a density of 1500 cells per well. To perform the serial sphere forming assay, mammospheres obtained were mechanically dissociated in single cells and re-plated to form secondary and tertiary spheres for 3 passages. 8/10 days after being plated, mammospheres with a diameter > 60 μm were counted.
CD44CD24 flow cytometer
CD44CD24 staining was performed both on SUM159 cells and on 4T1 cells and tumor masses. 4T1 tumor tissues were collected the last day of FMD cycle, before refeeding period, and were enzymatically digested to obtain a single-cell suspension as previously described. Cells were harvested and washed in PBS 1% BSA and pellets were resuspended in blocking buffer (PBS 10% BSA) and incubated for 30 min at 4°C light protected. After a wash in PBS 1% BSA, cells were incubated with antibodies solution (200 μl/1×106 cells) for 45min at 4°C. In particular, 4T1 cells were stained with FITC-conjugated anti-murine CD24 antibody (Miltenyi Cat. #: 130–102-731, RRID: AB 2656573) and PE-conjugated anti-murine CD44 antibody (Miltenyi Cat. #: 130–102-606, RRID: AB 2658181), while SUM159 cells were stained with FITC-conjugated anti-human CD24 antibody (Miltenyi Cat. #: 130–112-844, RRID: AB2656554) and vioblue-conjugated anti-human CD44 antibody (Miltenyi Cat. #: 130–113-899, RRID: AB 2726390). For rescuing experiments with PKA activator, cells were previously treated with 0.5mM 8-Br-cAMP, for a total of 24 hours. SUM159 samples were analyzed by flow cytometry with Attune NxT flow cytometer and data were processed by Kaluza analysis software (Beckman coulter, version 2.0) or FlowJo software (version 10.7.1). 4T1 samples were analyzed by flow cytometry with FACSCalibur (BD) and data were processed by FlowJo software.
Aldefluor assay
ALDEFLUOR kit (STEMCELL technologies, Cat. #: 01700) is used for the identification, evaluation and isolation of stem and progenitor cells expressing high levels of ALDH. SUM159 and 4T1 tumor tissues were collected the last day of FMD cycle, before refeeding period, and were dissociated enzymatically to obtain a single-cell suspension as previously described. Mouse stromal cells were removed with EasySep Mouse Epithelial Cell Enrichment Kit (STEMCELL technologies, Cat. #19868). A specific inhibitor of ALDH, diethylaminobenzaldehyde (DEAB), was used to control for background fluorescence for each sample. Cells expressing high levels of ALDH become brightly fluorescent (ALDHbr) and were identified by flow cytometry with FACSCalibur (BD). Data were processed by FlowJo software.
Immunohistological staining
Immunohistological analysis was performed to determine the expression of Caspase-3 protein in sample tissues of SUM159 tumor masses. Paraffin sections of 3 μm thickness were baked and prepared according to the procedure. Tumor masses slides were incubated overnight (4°C) with cleaved caspase-3 antibody (Asp175) (Euroclone Cat. #: BK9661S). Images of sections were taken by microscope (Upright BX 51 Full Manual) and the number of cells positive to Caspase-3 was calculated with ImageJ plugin (version 1.50i).
Protein extraction and western blot analysis
Cells were washed in ice-cold PBS and lysates were prepared in RIPA lysis buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% deoxycholic acid, 1 mM EDTA) supplemented with protease and phosphatase inhibitors (protease inhibitor cocktail set III EDTA-free, Calbiochem, Cat. #: S39134; PhosStop, Roche Cat. #: 04906845001).
Tumor tissues were collected, as previously described, and snap frozen in liquid nitrogen immediately after mice were sacrificed, and stored in −80°C until use. For protein extraction, tumors were homogenized with Tissue lyser II (QIAGEN) in RIPA buffer supplemented with protease and phosphatase inhibitors and then ultra-centrifuged (45000 rpm using MLA-130 Beckman rotor) for 1 h. Protein concentrations were determined by BCA assay (Thermo Fisher Scientific, Cat. #: 23225). Proteins (30μg) were separated using pre-casted or home-made Acrylamid gels and transferred to 0.45μM nitrocellulose membranes or 0.2μM nitrocellulose membranes (depending on protein molecular weight). Proteins were then detected with the following antibodies: Vinculin (1:10000, Sigma-Aldrich, Cat. #: V9131, RRID: AB 477629), GLUT1 (1:10000, Cell Signaling, Cat. #: 12939, RRID: AB 2687899), CREB (1:1000, Cell Signaling, Cat. #: 4820S, RRID: AB 1903940), Phospho-CREB (1:1000, Cell Signaling, Cat. #: 9198S, RRID: AB 2561044), KLF5 (1:1000, Abcam, Cat. #: AB137676, RRID: AB 2744553), G9A/EHMT2 (1:1000, Euroclone, Cat. #: BK68851T) and histone H3dimethylK9 (1:1000, Abcam, Cat. #: AB1220, RRID: AB 449854). Next, membranes were washed with TBST (3 × 10 min) and incubated for 1 h RT with anti-Mouse (1:3000, Biorad, Cat. #: 170–6516) or anti-Rabbit (1:3000, Biorad, Cat. #: 170–6515) secondary antibodies. Specific bindings were detected by a chemiluminescence system (Thermo Scientific). Bands’ intensity was quantified with Image Lab software (version 5.2.1).
RNA extraction, RT-PCR and qRT-PCR
Total RNA was isolated using the miRNeasy Mini Kit (QIAGEN, Cat. #:217004) according to the manufacturer’s instructions. Next, 1 μg of purified RNA was retrotranscribed by using SuperScript Vilo cDNA synthesis kit (Invitrogen, Cat #:11754050). Resulting cDNA was analyzed by real-time polymerase reaction (qRT-PCR) using Quant-Studio 12K flex Real Time PCR system (Thermo Fisher). Human target gene primers for NANOG (hs02387400_g1), OCT4 (hs00742896_s1), TBX3 (hs00195612_m1) and KLF2 (Hs00360439_g1) (ThermoFisher, Cat. #: 4331182) were used.
CD44CD24 Cell sorting
SUM159 tumor masses were collected and digested as previously described. Cells were washed in PBS 1% BSA and pellets were resuspended in blocking buffer (PBS 10% BSA) and incubated for 30 min at 4°C light protected. After a wash in PBS 1% BSA, cells were incubated with antibodies solution (200 μl/1×106 cells) for 45min at 4°C. In particular, cells were stained with FITC-conjugated anti-human CD24 antibody (Miltenyi Cat. #: 130–112-844, RRID: AB 2656554) and vioblue-conjugated anti-human CD44 antibody (Miltenyi Cat. #: 130–113-899, RRID: AB 2726390). Finally, cells were washed with PBS 1% BSA and sorted with MoFlo Astrios Cell Sorter (Beckman Coulter). To select a population highly enriched in CSCs, after sorting, CD44+CD24+ cells, CD44+CD24– cells and CD44+CD24− cells were plated to perform mammosphere forming assay. CD44+CD24– population was selected to study CSCs.
RNA sequencing
Libraries for RNA sequencing were prepared following the manufacturer protocols for transcriptome sequencing with the Illumina NextSeq 550DX sequencer (ILLUMINA).
Total RNA was isolated from cells, previously sorted, using the miRNeasy Mini Kit (QIAGEN, Cat. #:217004), according to the manufacturer’s instructions. RNA abundance was measured using Nanodrop and its integrity was assessed using Agilent Bioanalyzer 2100 with Nano Rna kit (RIN > 8). mRNA-seq indexed library preparation was performed starting from 500 ng of total RNA with the TruSeq stranded mRNA (Illumina). Indexed libraries were quality controlled on Agilent Bioanalyzer 2100 with High Sensitivity DNA kit, quantified with Qubit HS DNA, normalized and pooled to perform a multiplexed sequencing run. 1% PhiX control was added to the sequencing pool, as a positive run control. Sequencing was performed in PE mode (2×75nt) on an Illumina NextSeq550Dx platform, generating on average 45 million PE reads per sample.
RNA-seq Bioinformatics Analysis
Reads were aligned to the hg38 reference genome with STAR (https://doi.org/10.1093/bioinformatics/bts635) with default settings and using the parameter quantMode GeneCounts in order to count the number reads per gene while mapping. Differential gene expression analysis between groups was performed with DESeq2 (https://doi.org/10.1186/s13059–014-0550–8). Genes with |log2FC| > 2 and adjusted p value < 0.05 were considered as significantly deregulated.
Gene Set Enrichment Analysis (GSEA) for pathways of interest was performed on the fold change rank ordered gene list with the fgsea (https://doi.org/10.1101/060012) package of Bioconductor. Volcano plots were generated with the R package ggplot2. Rendering of the fold changes on pathway graphs was achieved with the Pathview (https://doi.org/10.1093/bioinformatics/btt285) Bioconductor package, which allows to download KEGG pathway graph data and render them with the mapped data.
Animal diets and treatments
Mice were fed ad libitum with irradiated standard diet VRFI (P) diet (Special Diets Services (SDS) Cat #: 801900) containing 3.89 kcal/g of gross energy. Our FMD is based on a nutritional screen that identified ingredients that allow nourishment during periods of low-calorie consumption. The FMD diet consists of two different components designated as day 1 diet and days 2–4 diet. Day 1 diet consists of a mix of low-calorie broth powders, a vegetable powder, extra virgin olive oil and essential fatty acids; it contains 7.67 kJ/g (provided 50% of normal daily intake; 0.46 kJ/g protein, 2.2 kJ/g carbohydrate, 5.00 kJ/g fat). The day 2–3 diet is identical on all feeding days, consists of low-calorie broth powders and glycerol and contains 1.48 kJ/g (provided at 10% of normal daily intake; 0.01 kJ/g protein/fat, 1.47 kJ/g carbohydrates). Mouse weight was monitored daily and during FMD cycle weight loss did not exceed 20%.
For experiments on tumor growth, mice were fed with standard diet or underwent FMD cycles (4 consecutive days per week). Before FMD cycle was repeated, mice completely recovered their original bodyweight and also blood glucose levels return to the normal ones.
For experiments performed to mimic the effect of FMD on CSCs or further reducing blood glucose levels, mice were daily treated with WZB117 (10mg/kg in PBS) via intraperitoneal injection, with metformin (150 mg/kg in PBS) via intraperitoneal injection, with 2-Deoxy-D-Glucose (500mg/kg in PBS) via intraperitoneal injection. For the experiment with PI3K-AKT-mTOR and CDK4/6 inhibitors, mice were treated with pictilisib for 5 consecutive days per week (100mg/kg in 10%DMSO, 40% PEG300 and 50% saline) by oral gavage, palbociclib every other day (62.5mg/kg in 5%DMSO, 40%PEG300 and 55% saline) by oral gavage, ipatasertib for 5 consecutive days per week (75mg/kg in 5%DMSO, 40%PEG300 and 55% saline) by oral gavage and rapamycin every other day (2mg/kg in 2%DMSO, 40%PEG300 and 58% saline) via intraperitoneal injection.
Limiting dilution assay
For the limiting dilution assay, 8-weeks old female NOD scid gamma mice (NSG, Charles River) were subcutaneously injected with 1.5×106 SUM159 cells resuspended in 100 μL of PBS. Mice were fed with standard diet or underwent FMD cycles (4 consecutive days per week) and were daily treated with 2DG (500mg/kg, via intraperitoneal injection) or vehicle. Body weights were recorded daily, and tumor volumes were measured twice a week by a digital caliper. After 5 weeks, donor mice were sacrificed and tumor masses were excised and digested, as previously described. Tumor cells derived from donor mice were re-injected at different dilution (100.000, 10.000, 1000 cells) in recipient 8-weeks old female NOD scid gamma mice. Recipient mice were always fed with standard diet and any drug wasn’t administered; curves were performed considering the percentage of tumor palpability. Tumor initiating cell frequency was calculated with ELDA software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Survival analyses of TNBC patients
To evaluate the impact of blood glucose levels on overall survival (OS), advanced TNBC patients’ data treated at “Fondazione IRCCS Istituto Nazionale dei Tumori” were retrospectively collected. Out of 418 patients with different BC subtypes, 81 patients with advanced TNBC were retrospectively collected. Eligibility criteria were: 1) pathologically or cytologically confirmed diagnosis of clinical stage IV TNBC; 2) patients treated with at least one cycle of first-line, platinum-based doublet chemotherapy between October 2006 and January 2020; 3) available data regarding baseline (i.e., within 30 days from treatment initiation) plasma glucose concentration; 4) available information about patients’ outcome. All patients were female, median age was 56 years (range 34–80). The detailed characteristics are reported in Table S1. Platinum-based chemotherapy consisted of carboplatin-paclitaxel or carboplatin-gemcitabine combination. Carboplatin-paclitaxel doublet consisted of carboplatin at an Area Under the Curve (AUC) of 2 combined with paclitaxel 80 mg/mq, both administered intravenously on days 1 and 8 of every-three-week cycles. Carboplatin-gemcitabine consisted of carboplatin at an AUC of 2 combined with gemcitabine 800 mg/mq, both administered intravenously on days 1 and 8 of every-three-week cycles. OS was defined as the time between chemotherapy start and death from any cause. Patient data were collected according to the ethical principles for medical research involving human subjects adopted in the Declaration of Helsinki. Patients alive at the time of data collection and/or analyses signed an informed consent for the use of their personal data for research purposes.
Statistical analysis
GraphPad Prism V9 was used for the analysis of the data and graphic representations. Comparisons between two groups were performed with two-tailed unpaired Student’s t test. Comparison among more than two groups were performed with ANOVA analysis followed by Tukey’s test. Comparison of survival curves were performed with Log-rank (Mantel-Cox) test. P values ≤ 0.05 were considered significant.
Survival curves of patients with TNBC were estimated by Kaplan Meier method and compared with the log-rank (Mantel-Cox) test. Patients who had not undergone death at the time of data cut-off and analysis were censored at their last disease evaluation. The impact of clinical variables (e.g., baseline glycemia, age, ECOG PS, body mass index, de novo metastatic disease, previous taxane and anthracycline, disease free interval, number of metastatic sites, sites of metastasis) on patient OS was assessed by univariate analysis. Factors significantly associated with the risk of death, with a threshold of significance of 0.1, were included in the Cox proportional hazard model in order to assess their independent association with survival. A threshold of significance of 0.05 was set for others statistical evaluations. Survival analyses of patient data were performed using the software R (version 4.0.2 (2020–06-22)).
All the statistical parameters can be found in the figures and figures legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| FITC-conjugated anti-murine CD24 antibody | Miltenyi | Cat. #: 130–102-731; RRID: AB 2656573 |
| PE-conjugated anti-murine CD44 antibody | Miltenyi | Cat. #: 130–102-606; RRID: AB 2658181 |
| FITC-conjugated anti-human CD24 antibody | Miltenyi | Cat. #: 130–112-844; RRID: AB2656554 |
| Vioblue-conjugated anti-human CD44 antibody | Miltenyi | Cat. #: 130–113-899; RRID: AB 2726390 |
| Cleaved caspase-3 antibody (Asp175) | Euroclone | Cat. #: BK9661S |
| Mouse Anti-Vinculin Monoclonal Antibody 1:10000 |
Sigma-Aldrich | Cat. #: V9131; RRID: AB 477629 |
| Rabbit anti-GLUT1 monoclonal antibody 1:10000 |
Cell Signaling | Cat. #: 12939; RRID: AB 2687899 |
| Rabbit anti-CREB monoclonal antibody 1:1000 | Cell Signaling | Cat. #: 4820S; RRID: AB 1903940 |
| Rabbit anti-phosphoCREB monoclonal antibody 1:1000 | Cell Signaling | Cat. #: 9198S; RRID: AB 2561044 |
| Rabbit anti-KLF5 polyclonal antibody 1:1000 | Abcam | Cat. #: AB137676; RRID: AB 2744553 |
| Rabbit anti-G9A/EHMT2 monoclonal antibody 1:1000 | Euroclone | Cat. #: BK68851T |
| Mouse anti-histone H3dimethylK9 monoclonal antibody 1:1000 | Abcam | Cat. #: AB1220; RRID: AB 449854 |
| Goat Anti-Mouse IgG 1:3000 | Biorad | Cat. #: 170–6516 |
| Goat Anti-Rabbit IgG 1:3000 | Biorad | Cat. #: 170–6515 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Glucose transporter inhibitor IV, WZB117 | MERK | Cat. #: 400036 |
| Metformin | Sigma-Aldrich | Cat. #: D150959 |
| 2-Deoxy-D-Glucose | Sigma-Aldrich | Cat. #: D-6134 |
| 8-Bromoadenosine 3′,5′-cyclic monophosphates (8-Br-cAMP) | Cayman | Cat. #: 14431 |
| Alpelisib | MedchemTronica | Cat. #: HY-15244 |
| Rapamycin | MedchemTronica | Cat. #: HY-10219 |
| Pictilisb | MedchemTronica | Cat. #: HY-50094 |
| Ipatasertib | MedchemTronica | Cat. #: HY-15186 |
| Erythrosin B | Sigma-Aldrich | Cat. #: 200964 |
| Mammary epithelial basal medium (MEMB) | Lonza | Cat. #: CC-3151 |
| Methylcellulose | Sigma-Aldrich | Cat. #: M0512 |
| B-27 | GIBCO | Cat. #: 17504044 |
| Epidermal growth factor (EGF) | Biomol | Cat # BPS-90201–3 |
| Fibroblast growth factor (FGF) | Peprotech | Cat. #: 100–18B |
| Insulin growth factor 1 (IGF-1) | Peprotech | Cat. #: 100–11 |
| Hyaluronidase | Sigma-Aldrich | Cat. #: H4272 |
| Collagenase | Sigma-Aldrich | Cat. #: C2674 |
| Red blood cell lysing buffer hybrid-max | Sigma-Aldrich | Cat. #: R7757 |
| Protease inhibitor cocktail set III EDTA-free | Calbiochem | Cat. #: S39134 |
| PhosStop | Roche | Cat. #: 04906845001 |
| 1%penicillin/streptomycin | Biowest | Cat. #: L0022 |
| RPMI 1640 medium | Biowest | Cat #: L0500 |
| Ham’s F-12 medium | GIBCO | Cat. #: 11–765-054 |
| FBS | Biowest | Cat. #: S1810 |
| DMEM no glucose | Life Technologies | Cat. #: 11966025 |
| Glucose | Sigma-Aldrich | Cat. #: G8769 |
| Hydrocortisone | Sigma-Aldrich | Cat. #: H4001 |
| Insulin | Sigma-Aldrich | Cat. #: 11376497001 |
| L-glutamine | Biowest | Cat. #: X0550 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ALDEFLUOR kit | STEMCELL technologies | Cat. #: 01700 |
| EasySep Mouse Epithelial Cell Enrichment Kit |
STEMCELL technologies | Cat. #:19868 |
| BCA assay | Thermo Fisher Scientific | Cat. #: 23225 |
| miRNeasy Mini Kit | QIAGEN | Cat. #:217004 |
| SuperScript Vilo cDNA synthesis kit | Invitrogen | Cat. #:11754050 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq data | Gene Expression Omnibus (GEO) | GEO: GSE184452 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| SUM159 PT | Asterand | N/A |
| 4T1 | ATCC | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NOD scid gamma mice | Charles River | N/A |
| Balbc/Ola Hsd mice | Envigo | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer for NANOG (hs02387400_g1) | ThermoFisher | Cat. #: 4331182 |
| Primer for OCT4 (hs00742896_s1 | ThermoFisher | Cat. #: 4331182 |
| Primer for TBX3 (hs00195612_m1) | ThermoFisher | Cat. #: 4331182 |
| Primer for KLF2 (Hs00360439_g1) | ThermoFisher | Cat. #: 4331182 |
|
| ||
| Software and algorithms | ||
|
| ||
| Kaluza analysis software (Beckman coulter, version 2.0) | Kaluza Software | N/A |
| FlowJo (version 10.7.1) | FlowJo software | N/A |
| ImageJ (version 1.50i). | ImageJ software | N/A |
| Image Lab (version 5.2.1) | Image Lab software | N/A |
| ELDA | ELDA software | N/A |
| GraphPad Prism V9 | GraphPad software | N/A |
| R (version 4.0.2) | R software | N/A |
|
| ||
| Other | ||
|
| ||
| Standard diet (VRFI (P)) | Special Diets Services (SDS) | Cat #: 801900 |
Highlights.
Fasting/FMD depletes TNBC stem cells by lowering glucose levels
PKA activation reverses the FMD-induced reduction in cancer stem cells
In cancer cells, FMD activates starvation escape mechanisms that can be targeted
FMD + kinase inhibitors cause TNBC regression without drug toxicity
ACKNOWLEDGMENTS
This work was supported in part by the Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG#17605 and IG#21820 to V.D.L. and MFAG#22977 to C.V.), the US National Institute on Aging-National Institutes of Health (NIA-NIH) grants AG034906 and AG20642 (to V.D.L.), and the Scientific Directorate of Fondazione IRCCS Istituto Nazionale dei Tumori (to C.V.). The authors would like to thank Dr. Maria Grazia Totaro of Imaging TDU (IFOM, FIRC Institute of Molecular Oncology, Milan, Italy) for flow cytometry service and analysis and Dr. Mirko Riboni of RNA-seq facility (IFOM, FIRC Institute of Molecular Oncology, Milan, Italy) for sequencing service.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2021.10.008.
DECLARATION OF INTERESTS
V.D.L. is a scientific advisor and has equity interest in L-Nutra, a company that develops medical food. V.D.L. and G.S. have submitted a patent related to this work. C.V. is an inventor of an FMD regimen (patent pending).
REFERENCES
- Bai K, Cao Y, Huang C, Chen J, Zhang X, and Jiang Y. (2016). Association of histone methyltransferase G9a and overall survival after liver resection of patients with hepatocellular carcinoma with a median observation of 40 months. Medicine (Baltimore) 95, e2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Im SA, Iwata H, Cortés J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, et al. (2017). Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 18, 904–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, et al. (2012). Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol 30, 282–290. [DOI] [PubMed] [Google Scholar]
- Bianchini G, Balko JM, Mayer IA, Sanders ME, and Gianni L. (2016). Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol 13, 674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, et al. (2015). A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 22, 86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffa I, Spagnolo V, Vernieri C, Valdemarin F, Becherini P, Wei M, Brandhorst S, Zucal C, Driehuis E, Ferrando L, et al. (2020). Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 583, 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YT, Lai AC, Lin RJ, Wang YH, Wang YT, Chang WW, Wu HY, Lin YJ, Chang WY, Wu JC, et al. (2020). GPER-induced signaling is essential for the survival of breast cancer stem cells. Int. J. Cancer 146, 1674–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, et al. (2009). Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 69, 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MW, Hua KT, Kao HJ, Chi CC, Wei LH, Johansson G, Shiah SG, Chen PS, Jeng YM, Cheng TY, et al. (2010). H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 70, 7830–7840. [DOI] [PubMed] [Google Scholar]
- Chen Z, Wu Q, Ding Y, Zhou W, Liu R, Chen H, Zhou J, Feng J, and Chen C. (2017). YD277 suppresses triple-negative breast cancer partially through activating the endoplasmic reticulum stress pathway. Theranostics 7, 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, Da Sacco S, Mirisola M, Quinn DI, Dorff TB, et al. (2014). Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 14, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Holgado E, et al. ; KEYNOTE-355 Investigators (2020). Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 396, 1817–1828. [DOI] [PubMed] [Google Scholar]
- Curry E, Green I, Chapman-Rothe N, Shamsaei E, Kandil S, Cherblanc FL, Payne L, Bell E, Ganesh T, Srimongkolpithak N, et al. (2015). Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenetics 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, and Yeger H. (2008). Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells 26, 1818–1830. [DOI] [PubMed] [Google Scholar]
- de Groot S, Lugtenberg RT, Cohen D, Welters MJP, Ehsan I, Vreeswijk MPG, Smit VTHBM, de Graaf H, Heijns JB, Portielje JEA, et al. ; Dutch Breast Cancer Research Group (BOOG) (2020). Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun 11, 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, Cacciottolo M, Martin-Montalvo A, de Cabo R, Wei M, et al. (2016). Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell 30, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Biase S, Shim HS, Kim KH, Vinciguerra M, Rappa F, Wei M, Brandhorst S, Cappello F, Mirzaei H, Lee C, and Longo VD (2017). Fasting regulates EGR1 and protects from glucose- and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol. 15, e2001951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tano M, Raucci F, Vernieri C, Caffa I, Buono R, Fanti M, Brandhorst S, Curigliano G, Nencioni A, de Braud F, and Longo VD (2020). Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun 11, 2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, et al. (2018). Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua KT, Wang MY, Chen MW, Wei LH, Chen CK, Ko CH, Jeng YM, Sung PL, Jan YH, Hsiao M, et al. (2014). The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 13, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juric D, Krop I, Ramanathan RK, Wilson TR, Ware JA, Sanabria Bohorquez SM, Savage HM, Sampath D, Salphati L, Lin RS, et al. (2017). Phase I dose-escalation study of taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov. 7, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, Kato H, Mizuno Y, Yokoe M, Sugauchi F, et al. (2007). Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol. Res 37, 974–983. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Ahmed S, Boumber Y, Sekido Y, Haddad BR, and Issa JP (2008). Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE 3, e2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Safdie FM, Raffaghello L, Wei M, Madia F, Parrella E, Hwang D, Cohen P, Bianchi G, and Longo VD (2010). Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 70, 1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, Pistoia V, Wei M, Hwang S, Merlino A, et al. (2012). Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med 4, 124ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. (2009). Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SF, Guo L, Qian SW, Liu Y, Zhang YY, Zhang ZC, Zhao Y, Shou JY, Tang QQ, and Li X. (2013). G9a is transactivated by C/EBPβ to facilitate mitotic clonal expansion during 3T3-L1 preadipocyte differentiation. Am. J. Physiol. Endocrinol. Metab 304, E990–E998. [DOI] [PubMed] [Google Scholar]
- Li Z, Dong J, Zou T, Du C, Li S, Chen C, Liu R, and Wang K. (2017). Dexamethasone induces docetaxel and cisplatin resistance partially through up-regulating Krüppel-like factor 5 in triple-negative breast cancer. Oncotarget 8, 11555–11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Zhi X, Zhou Z, Zhang H, Yang R, Zou T, and Chen C. (2018a). Mithramycin A suppresses basal triple-negative breast cancer cell survival partially via down-regulating Krüppel-like factor 5 transcription by Sp1. Sci. Rep 8, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XR, Zhou LH, Hu JX, Liu LM, Wan HP, and Zhang XQ (2018b). UNC0638, a G9a inhibitor, suppresses epithelial-mesenchymal transition-mediated cellular migration and invasion in triple negative breast cancer. Mol. Med. Rep 17, 2239–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Chen H, Zhao P, Chen CH, Liang H, Yang C, Zhou Z, Zhi X, Liu S, and Chen C. (2020). Mifepristone derivative FZU-00,003 suppresses triple-negative breast cancer cell growth partially via miR-153-KLF5 axis. Int. J. Biol. Sci 16, 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, and Mattson MP (2014). Fasting: molecular mechanisms and clinical applications. Cell Metab. 19, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo CW, Wang JY, Hung WC, Peng G, Tsai YL, Chang TM, Chai CY, Lin CH, and Pan MR (2017). G9a governs colon cancer stem cell phenotype and chemoradioresistance through PP2A-RPA axis-mediated DNA damage response. Radiother. Oncol 124, 395–402. [DOI] [PubMed] [Google Scholar]
- Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, Juric D, Solit D, Berger MF, Won HH, et al. (2017). A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2− metastatic breast cancer. Clin. Cancer Res 23, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi S, Passaro F, Aloia L, Manabe I, Nagai R, Pastore L, and Russo T. (2008). Klf5 is involved in self-renewal of mouse embryonic stem cells. J. Cell Sci 121, 2629–2634. [DOI] [PubMed] [Google Scholar]
- Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Weiss GJ, Sachdev JC, Chadha M, Fulk M, et al. (2016). First-in-human phase I study of copanlisib (BAY 80–6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol 27, 1928–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, and Longo VD (2008). Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 105, 8215–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, et al. ; IMpassion130 Trial Investigators (2018). Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med 379, 2108–2121. [DOI] [PubMed] [Google Scholar]
- Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N, et al. ; KEYNOTE-522 Investigators (2020). Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med 382, 810–821. [DOI] [PubMed] [Google Scholar]
- Shi P, Liu W, Tala, Wang H, Li F, Zhang H, Wu Y, Kong Y, Zhou Z, Wang C, et al. (2017). Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 3, 17010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Li Y, Sang Y, Yu B, Lv D, Zhang W, and Feng H. (2018). LncRNA PVT1 regulates triple-negative breast cancer through KLF5/beta-catenin signaling. Oncogene 37, 4723–4734. [DOI] [PubMed] [Google Scholar]
- Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, et al. (2014). CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26, 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, Groshen S, Mack WJ, Guen E, Di Biase S, et al. (2017). Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med 9, eaai8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak RJ, Klimecki WT, Lau SS, Feinstein Y, and Futscher BW (2007). 5-Aza-2′-deoxycytidine-mediated reductions in G9A histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene 26, 77–90. [DOI] [PubMed] [Google Scholar]
- Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio II, Giannopoulou EG, Rago C, et al. (2015). Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350, 1391–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
The RNA-seq data generated during the current study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO: GSE184452.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.