

Abstract
Host cells initiate cell death programs to limit pathogen infection. Inhibition of transforming growth factor-β-activated kinase 1 (TAK1) by pathogenic Yersinia in macrophages triggers receptor-interacting serine/threonine-protein kinase 1 (RIPK1)-dependent caspase-8 cleavage of gasdermin D (GSDMD) and inflammatory cell death (pyroptosis). A genome-wide clustered regularly interspaced short palindromic repeats (CRISPR) screen to uncover mediators of caspase-8-dependent pyroptosis identified an unexpected role of the lysosomal FLCN-FNIP2-Rag-Ragulator supercomplex, which regulates metabolic signalling and the mechanistic target of rapamycin complex 1 (mTORC1). In response to Yersinia infection, FADD, RIPK1 and caspase-8 were recruited to Rag-Ragulator, causing RIPK1 phosphorylation and caspase-8 activation. Pyroptosis activation depended on Rag GTPase activity and lysosomal tethering of Rag-Ragulator, but not mTORC1. Thus, the lysosomal metabolic regulator Rag-Ragulator instructs the inflammatory response to Yersinia.
When mucosal and immune sentinel cells sense invasive infection or other danger signals, they activate the assembly of large multiprotein complexes, called inflammasomes, that recruit inflammatory caspases (caspase-1/-4/-5/-11), which become auto-activated by proximity (1–3). The activated pro-inflammatory caspases then cleave gasdermin D (GSDMD) in the cytosol to liberate an N-terminal fragment that assembles into cell membrane pores to cause an inflammatory cell death, termed pyroptosis, in which the cell membrane is rapidly permeabilized to release inflammatory mediators (4–9). Caspase-1 also processes the proinflammatory IL-1 family cytokines, which lack a conventional secretion signal, and the processed inflammatory cytokines are then released through GSDMD pores (10, 11). Pyroptosis and IL-1 family cytokines recruit and activate immune cells to the site of infection to orchestrate host defense against invading pathogens. During pathogenic Yersinia infection, the Yersinia effector protein YopJ inhibits transforming growth factor-β-activated kinase 1 (TAK1) or IκB kinase (IKK) to trigger an alternate pyroptotic pathway mediated by Toll-like receptors (TLRs) or death receptors that instigate the formation of a complex involving the adaptor Fas-associated death domain (FADD) and receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and recruitment and activation of caspase-8, which cleaves GSDMD (12, 13). Blockade of TAK1 kinase activity with the specific inhibitor 5z-7-oxozeaneol (5z7) mimics the effect of YopJ and pathogenic Yersinia infection to cause RIPK1-caspase-8-dependent GSDMD-mediated pyroptosis in the presence of TLR ligands or TNFα. However, not much is known about how this alternative caspase-8-dependent pyroptotic pathway is activated or regulated.
To dissect the molecular mechanisms underlying activation of RIPK1-caspase-8-dependent pyroptosis during Yersinia infection, we performed an unbiased genome-wide clustered regularly interspaced short palindromic repeats (CRISPR) screen. We infected Cas9-expressing immortalized mouse bone marrow-derived macrophages (iBMDMs) with lentiviruses encoding a library of sgRNAs and treated them with extracellular LPS/5z7 to trigger caspase-8-dependent pyroptosis or electroporated them with LPS to trigger caspase-11-mediated GSDMD cleavage for comparison (Fig. 1A). Surviving cells were isolated and retreated twice more to enrich for pyroptosis-resistant cells. The genomes of resistant cells were sequenced to identify enriched sgRNAs that knocked out genes required for pyroptosis. The screen identified multiple genes in the lysosomal membrane-anchored Folliculin (Flcn)-Folliculin-interacting protein 2 (Fnip2)-Rag-Ragulator complex as necessary for caspase-8-mediated pyroptosis, but not caspase-11-mediated pyroptosis.
Fig. 1. A genome wide CRISPR screen identifies multiple components of the FLCN-FNIP2-Rag-Ragulator as needed for caspase-8-mediated pyroptosis.
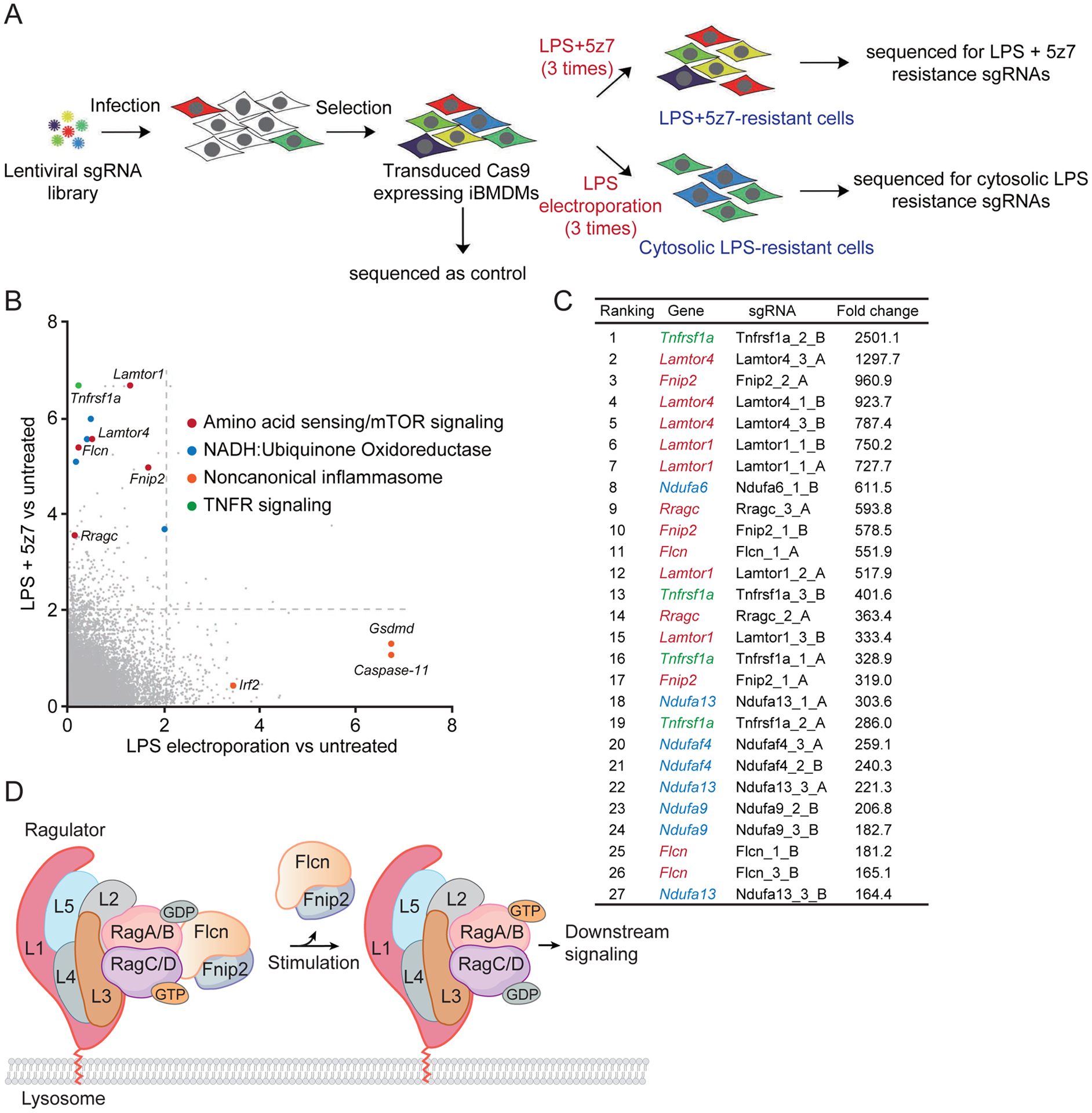
(A) Schematic workflow of the genome-wide CRISPR screening procedure in iBMDMs. (B) -lgRRA values calculated by the MAGeCK algorithm indicate the relative enrichment of genes after treatment with LPS electroporation (x axis) or LPS/5z7 (y axis) relative to controls in positive selection. The dotted lines represent p-value of 0.01. (C) List of sgRNA hits from the screening in iBMDMs treated with LPS/5z7. Corresponding genes targeted with multiple sgRNAs and fold enrichments are shown. Color code: genes encoding for TNF receptor, green; FLCN-FNIP2-Rag-Ragulator complex, red; mitochondrial electron transport complex I, blue. (D) Diagram of FLCN-FNIP2-Rag-Ragulator complex.
Genome-wide CRISPR screen identifies the Rag-Ragulator complex as critical for pyroptosis induced by LPS/5z7 treatment
We identified several sgRNAs enriched in cells that survived three sequential exposures to stimuli that triggered caspase-8 or caspase-11 dependent pyroptosis, compared to medium-treated control cells (Fig. 1, A–C, and fig. S1). Only a handful of sgRNA target genes were enriched in cells that survived both caspase-8 and caspase-11 mediated pyroptosis. Cells that survived LPS transfection were significantly enriched for sgRNAs designed to knockout Gsdmd, Casp-11 and Irf2, all known genes implicated in the noncanonical inflammasome pathway, including the gene encoding Irf2, which transcriptionally activates Gsdmd expression (14). Caspase-8-induced pyroptosis-resistant cells were highly enriched for sgRNAs targeting genes involved in tumor necrosis factor receptor (TNFR) signalling, mitochondrial electron transport chain (ETC) complex I and the lysosome-associated Rag-Ragulator complex. The 27 most enriched sgRNAs, which were increased 164–2500-fold compared to mock-treated cells, included 4 sgRNAs targeting the gene encoding TNFR1; 8 sgRNAs targeting 4 genes encoding subunits of ETC complex I (Ndufa6, Ndufa13, Ndufaf4, Ndufa9), and 15 sgRNAs targeting genes encoding 5 components of the Rag-Ragulator supercomplex (Lamtor1, Lamtor4, RagC, Flcn and Fnip2), which serves as a platform for recruiting Raptor-the mechanistic target of rapamycin (mTOR) to the lysosomal surface and regulating its activation (15). The recovery of TNFR1 sgRNAs in LPS/5z7-treated, but not LPS-transfected, surviving cells confirmed the selectivity of the screen. The identification of ETC complex I gene sgRNAs was not surprising because mitochondrial ROS, for which ETC complex I is a major source, have been implicated in augmenting GSDM pore formation (16, 17). In comparison, neither Casp-8- nor Gsdmd-targeting guides were enriched, likely because deficiency of caspase-8 or Gsdmd could lead to Ripk3-dependent or Gsdme-directed cell death (18–20). The Rag-Ragulator supercomplex is composed of 9 subunits – Ragulator, a pentameric scaffold (Lamtor1–5) that tethers the complex to the lysosomal membrane via lipidation of Lamtor1 (21, 22); 2 Rag GTPases (GTP-loaded RagA or RagB and GDP-loaded RagC or RagD); and FLCN complexed with its binding partners, Fnip1 or Fnip2 (FLCN-interacting protein 1/2), which facilitates the activation of the Rag heterodimer by catalyzing GTP hydrolysis of RagC or RagD (Fig. 1D). Because the identification of the lysosomal Rag-Ragulator complex was unexpected and dominated the list of most enriched sgRNAs, subsequent experiments focused on examining the putative role of Rag-Ragulator in caspase-8-mediated pyroptosis.
Ragulator complex is required for caspase-8-mediated, but not inflammasome-activated, pyroptosis
To verify the contribution of the Rag-Ragulator top gene hits to LPS/5z7-induced pyroptosis, we used CRISPR/Cas9-mediated gene editing to knockout Lamtor1, Lamtor4, Rragc, Flcn and Fnip2 in iBMDMs. For each gene, two clones transduced with distinct gRNAs that target different regions of each gene were obtained. Gene editing was verified by DNA sequencing and immunoblot (Fig. 2A, and fig. S2). Concurrent treatment of wild-type (WT) iBMDMs with LPS and 5z7 resulted in rapid pyroptotic cell death, as assessed by lactate dehydrogenase (LDH) release, but iBMDMs deficient in each of the Rag-Ragulator genes tested were highly resistant to LPS/5z7-induced pyroptosis (Fig. 2A, and fig. S3A). Consistent with the dependency of caspase-8-mediated pyroptosis on these genes, caspase-8 maturation and GSDMD cleavage to p30 and p20 fragments, which were detected by immunoblot in LPS and 5z7-treated WT iBMDMs, were not detected in the knockout cells (Fig. 2A, and fig. S3B). iBMDMs knocked out for the same set of Rag-Ragulator genes also survived treatment with an alternate stimulus of caspase-8-dependent pyroptosis (TNFα plus 5z7) (12) and showed no sign of GSDMD cleavage (Fig. 2, B–C). Caspase-3/7 cleavage, occurring downstream of caspase-8 activation (13, 23), was blunted by Rag-Ragulator deficiency upon LPS/5z7 treatment (fig. S3B). By contrast, Rag-Ragulator deficiency had no effect on caspase-3/7 cleavage and activation in etoposide-induced apoptosis (fig. S3C), indicating the specific role of Rag-Ragulator in LPS/5z7-induced pyroptosis. To determine whether the Rag-Ragulator complex was also involved in inflammasome and inflammatory caspase-mediated pyroptosis, WT and knockout clones of iBMDMs were treated with two stimuli of canonical inflammasomes and caspase-1-mediated pyroptosis (LPS plus nigericin to activate the NLRP3 inflammasome or anthrax protective antigen plus lethal factor-flagellin fusion protein (PA + LFN-Fla) to activate the NLRC4 inflammasome) or were electroporated with LPS to induce the noncanonical caspase-11 inflammasome and pyroptosis (Fig. 2, D–G, and fig. S4). WT iBMDMs and iBMDMs knocked out for the 5 Rag-Ragulator genes were all similarly killed by activating the canonical or noncanonical inflammasomes. As positive controls and as expected, iBMDMs genetically deficient in Gsdmd were resistant to electroporated LPS, iBMDMs deficient in Nlrp3 were resistant to LPS plus nigericin and iBMDMs deficient in NIrc4 were resistant to PA plus LFN-Fla. In these experiments cell death was assessed either by an ATP assay measuring surviving cells or an LDH assay that measures pyroptosis. GSDMD cleavage, assessed by immunoblot, correlated with cell death, as expected. Thus, Lamtor1, Lamtor4, Rragc, Flcn, and Fnip2 are selectively required for RIPK1-caspase-8-dependent pyroptosis, but not for inflammasome-mediated pyroptosis.
Fig. 2. FLCN-FNIP2-Rag-Ragulator gene deficient cells are resistant to LPS/5z7 treatment, but sensitive to inflammasome-induced pyroptosis.
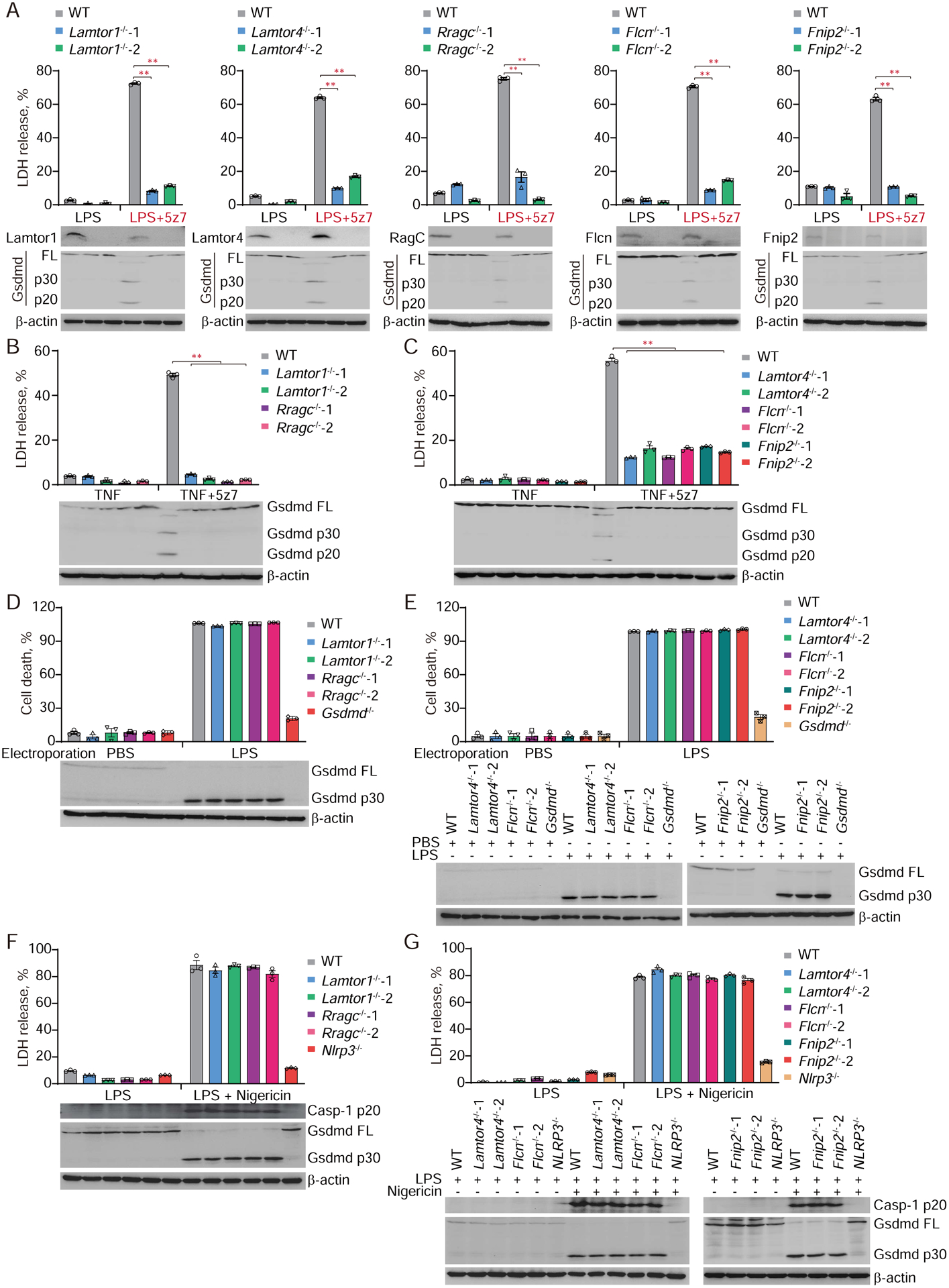
(A) The indicated iBMDMs were challenged with LPS or LPS/5z7. Cell death was measured by LDH release after 2.5 hr. Specific gene ablation was confirmed and Gsdmd cleavage was examined by immunoblot, shown in the bottom panel. (B-C, F-G) The indicated iBMDMs were treated with TNF, TNF/5z7 (B-C) or LPS/Nigericin (F-G). Cell death was measured by LDH release after 2.5 hr (B-C) or 1.5 hr (F-G). (D-E) The indicated iBMDMs were electroporated with LPS. Cell death was assessed and calculated by measuring ATP level after 2.5 hr. Gsdmd cleavage and caspase-1 processing (during canonical inflammasome activation) were examined by immunoblot. Graphs show mean ± SEM of triplicate wells. Data are representative of at least three independent experiments. Data were analyzed using a two-tailed Student’s t test. **P < 0.01.
The Rag-Ragulator complex recruits RIPK1 and caspase-8 to the lysosomal membrane
Because the Rag-Ragulator complex is localized to the lysosomal membrane and required for caspase-8 mediated pyroptosis, we postulated that RIPK1 and caspase-8 might be recruited to and activated on the lysosomal membrane after LPS/5z7 treatment. In the absence of cFLIPL, TLR4 signalling typically causes the assembly of a complex containing a death domain-containing protein FADD and RIPK1 that recruit and activate caspase-8 (24). The complex also assembles in response to LPS/5z7 (24). Consistently, within 100 minutes of adding LPS/5z7, but not LPS on its own, FADD immunoprecipitated with RIPK1, procaspase-8 and the p43 subunit of activated caspase-8, indicating the formation of a FADD-RIPK1-caspase-8-containing complex that activated caspase-8 (Fig. 3A). Although cleaved caspase-8 levels increased by 150 min, the RIPK1 band was strongly diminished and the interaction of FADD with both RIPK1 and caspase-8 became undetectable, suggesting that the complex was transient and disintegrated, potentially because of RIPK1 degradation. To determine if the complex depended on Rag-Ragulator, the experiment was repeated in iBMDMs knocked out for Rragc. In RagC-deficient cells treated with LPS/5z7, FADD did not pulldown RIPK1 or caspase-8, RIPK1 was not degraded, and the p43 activated caspase-8 band was barely detected (Fig. 3A). These data suggest that assembly of the FADD-RIPK1-caspase-8 complex depends on Rag-Ragulator and might be recruited to lysosomes for subsequent function. Indeed, treatment with LPS/5z7 significantly and specifically induced co-localization of caspase-8 and RIPK1 with lysosomes, but not endoplasmic reticulum (ER), mitochondria or early endosomes, which was dependent on Lamtor1 (Fig. 3B–D, and fig. S5). To confirm that RIPK1 and caspase-8 colocalize with Rag-Ragulator after LPS/5z7 stimulation, HA-tagged RIPK1 (Fig. 3E) and caspase-8 (Fig. 3F) were co-expressed with Flag-tagged Lamtor1, RagC, Flcn or Fnip2 in HEK293T cells and co-immunoprecipitation was assessed by anti-HA immunoprecipitation followed by anti-Flag immunoblot. Co-immunoprecipitation of both RIPK1 and caspase-8 with RagC and Flcn was detected but not with Lamtor1 or Fnip2 (Fig. 3, E and F). Moreover, interaction of endogenous RIPK1 and caspase-8 with RagC could both be detected in iBMDMs treated with LPS/5z7 (Fig. 3G). Compared to GTP-loaded inactive RagC (Q119L), GDP-loaded active RagC (S74N) showed stronger binding to RIPK1/caspase-8 (Fig. 3, H and I). Mapping of interactive domains via truncation revealed that the kinase domain (KD) of RIPK1 and the caspase domain (CD) of caspase-8 were required and sufficient for RagC binding (fig. S6). These domains are distinct from the domains implicated in RIPK1 binding to caspase-8 and FADD via a death domain (DD)-death effector domain (DED) interaction and DD-DD interaction, respectively (25–27) and in caspase-8 binding to FADD through a DED-DED interaction (28, 29). Because caspase-8-mediated pyroptosis depended on Lamtor1 and Fnip2, we suspect that RIPK1 and caspase-8 associate with the large supercomplex, but their interaction with these other components could be indirect. This result suggested that RIPK1 and caspase-8 directly interacted with RagC and Flcn, which are in contact with each other in the cryo-EM structure of the human Rag-Ragulator supercomplex (30). Thus, Rag-Ragulator acts as a lysosomal platform for recruiting RIPK1 and procaspase-8 and activating caspase-8.
Fig. 3. Recruitment and activation of an LPS/5z7-induced FADD-RIPK1-caspase-8 complex to lysosomes depends of binding to RagC and Flcn in the Rag-Ragulator complex.
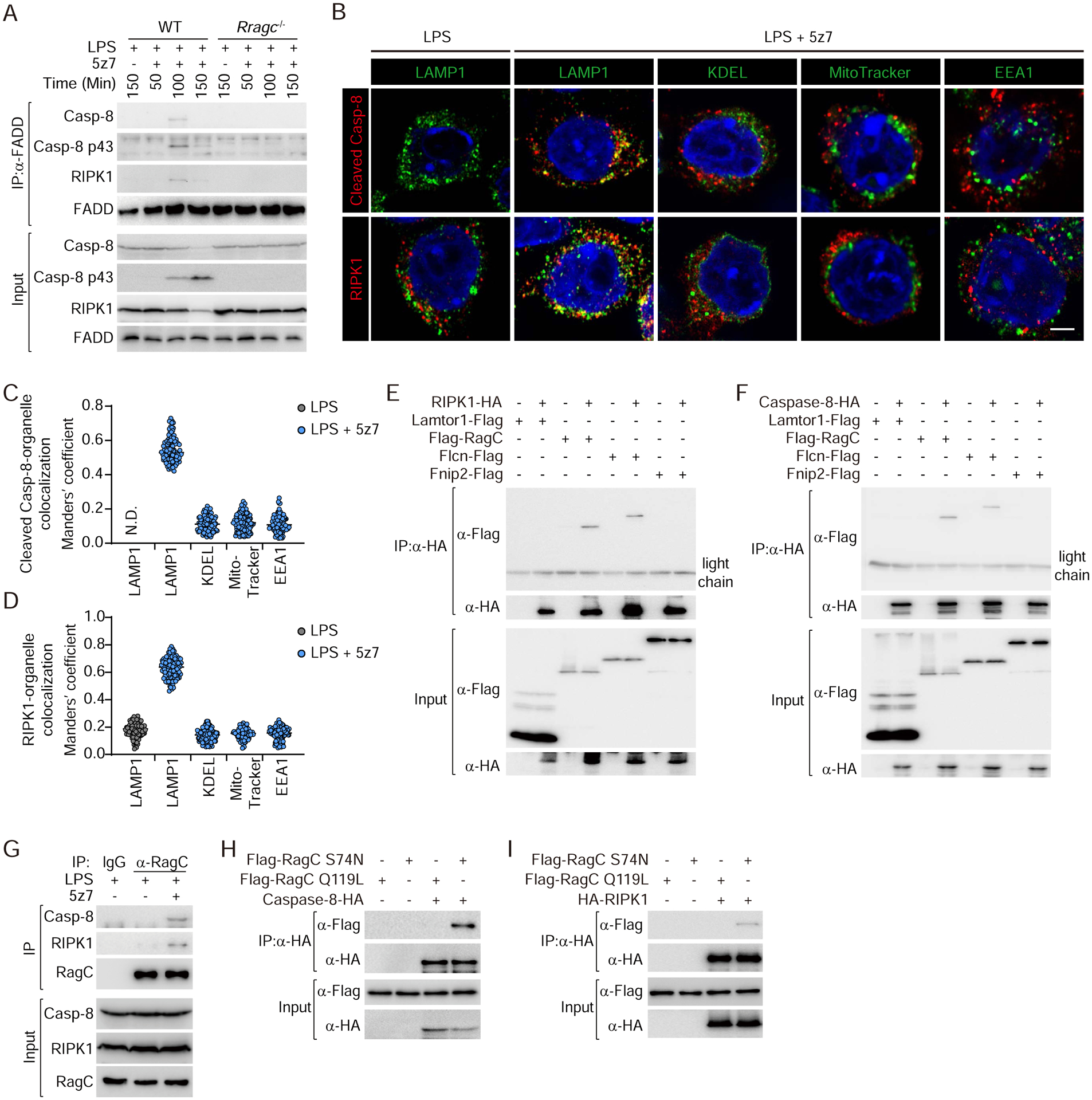
(A) WT and Rragc−/− iBMDMs were treated with LPS/5z7 for the indicated times. Endogenous FADD complex was immunoprecipitated with anti-FADD antibody and analyzed by immunoblot with the indicated antibodies. (B) Representative confocal fluorescence images of LPS or LPS plus 5z7 treated iBMDMs co-stained for cleaved caspase-8 or RIPK1 with the indicated organelle markers (LAMP1, lysosomes; KDEL, ER; MitoTracker, mitochondria; EEA1, early endosomes) and DAPI. Scale bars: 2 μm. (C and D) Quantification of colocalization of cleaved caspase-8 (C) and RIPK1 (D) with organelles in multiple confocal images (as in (B)) by calculating Manders’ overlap coefficient (each point represents a single cell). N.D., not detected. (E-F, H-I) Lysates of HEK293T cells transfected with the indicated plasmids were assayed for immunoprecipitation with anti-HA antibody and analyzed by immunoblot probed with the indicated antibodies. (G) Lysates of iBMDMs treated with LPS or LPS/5z7 were assayed for immunoprecipitation with anti-RagC antibody and analyzed by immunoblot probed with the indicated antibodies. Data are representative of at least three independent experiments.
Neither mTOR activity nor Tfeb/Tfe3 are involved in caspase-8 or inflammasome-induced pyroptosis
Because the Rag-Ragulator complex regulates mTORC1 activation, we next examined whether caspase-8-mediated pyroptosis is regulated by mTORC1. To inhibit mTORC1, iBMDMs were treated with rapamycin, a potent allosteric mTORC1 inhibitor or with Torin 1, a selective ATP-competitive mTORC1/2 inhibitor (31). Both inhibitors inhibited mTORC1 kinase activity in iBMDMs at the doses used and caused dephosphorylation of the mTORC1 target p70 S6 kinase (fig. S7A). However, incubation of iBMDMs with rapamycin or Torin 1 before treatment with LPS/5z7 had no effect on GSDMD cleavage or on the extent or kinetics of pyroptotic cell death, assessed by propidium iodide (PI) uptake (Fig. 4, A–B). Furthermore, treatment with 5-aminoimidizole-4-carboxamide riboside (AICAR), which blocks mTOR signalling by activating AMP-activated protein kinase (AMPK), did not affect LPS/5z7-triggered GSDMD cleavage and pyroptosis (Fig. 4C, and fig. S7B). Thus, mTOR kinase activity does not affect caspase-8-induced pyroptosis although the Rag-Ragulator complex is required. These mTORC1 inhibitors also did not affect electroporated LPS-induced noncanonical inflammasome or LPS plus nigericin induced canonical inflammasome activation of GSDMD cleavage and pyroptosis (Fig. 4, D and E). Thus, mTOR activity is not involved in pyroptosis triggered by TAK1 inhibition or inflammasome signalling. In addition, simultaneous knockdown of the MiT/TFE family transcription factors, Tfeb and Tfe3, master regulators of lysosomal biogenesis and autophagy in response to various stresses, showed no effect on LPS/5z7-triggered pyroptosis of Ragulator/Rag/Flcn-deficient cells (fig. S8, A–D). Forced nuclear localization and activation of Tfeb (S141, 210A) also did not affect LPS/5z7-induced pyroptosis (fig. S8, E and F), ruling out the possibility that the defects of Flcn-Rag-Ragulator-knockout cells in LPS/5z7-triggered pyroptosis resulted from constitutive nuclear localization of Tfeb/Tfe3 and subsequent induction of downstream autophagy-lysosomal gene expression (32, 33).
Fig. 4. mTORC1 activity is not required for LPS/5z7 or inflammasome-triggered pyroptosis.
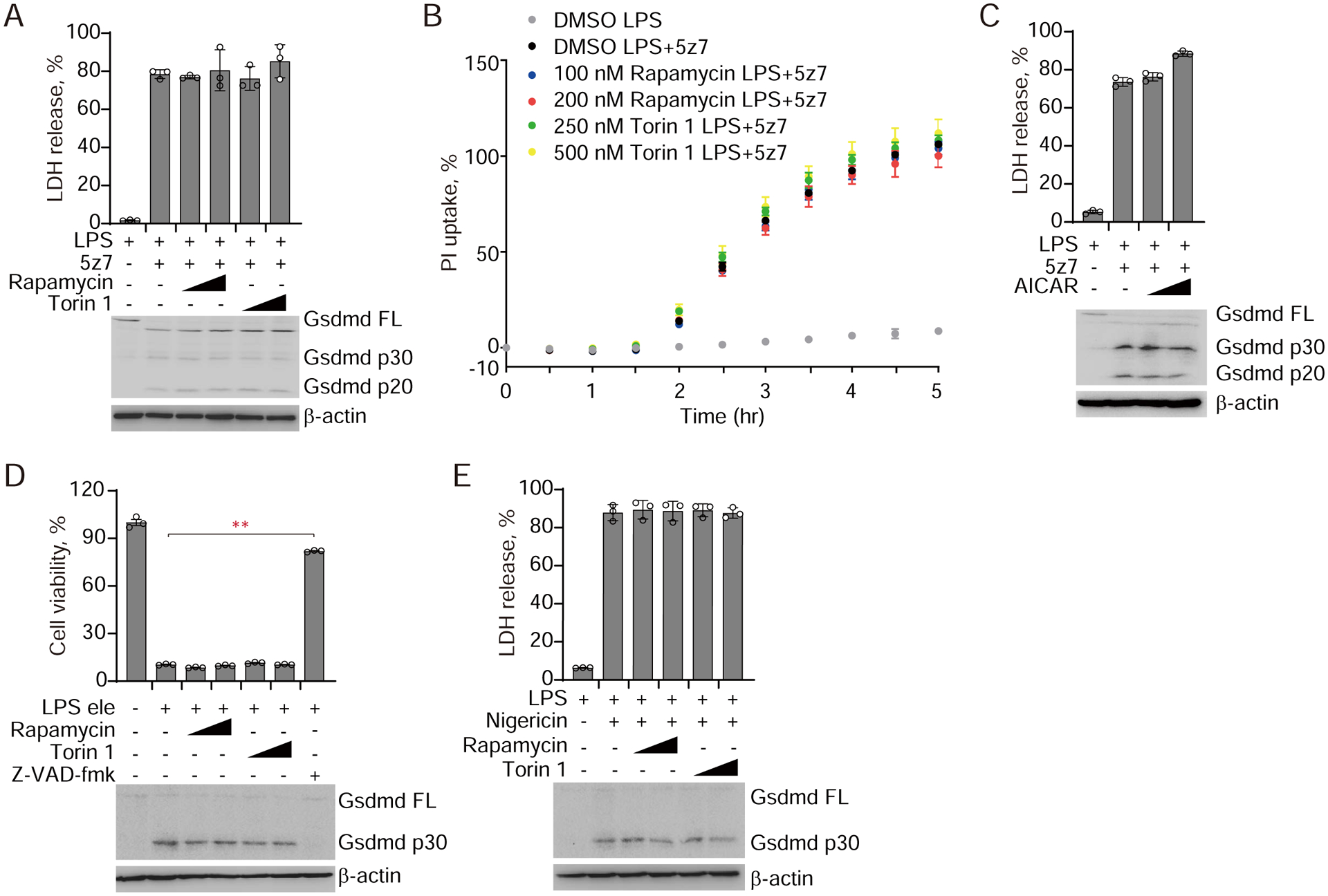
(A-B) iBMDMs pre-incubated with Rapamycin or Torin 1 for 2 hr were treated with LPS or LPS/5z7. Cell death was measured by LDH release after 2.5 hr (A), or by entry of PI into cells (B). (C) iBMDMs pre-incubated with AICAR for 2 hr were treated with LPS or LPS/5z7. Cell death was measured by LDH release 2.5 hr later. (D) iBMDMs pre-incubated with Rapamycin or Torin 1 for 2 hr were electroporated with LPS. Cell viability was assessed by measuring ATP level after 2.5 hr. (E) iBMDMs pre-incubated with Rapamycin or Torin 1 for 2 hr were treated with LPS or LPS/Nigericin and cell death was measured by LDH release 1.5 hr later. Gsdmd cleavage was examined by immunoblot. Graphs show mean ± SEM of triplicate wells. Data are representative of at least three independent experiments. Data were analyzed using a two-tailed Student’s t test. **P < 0.01.
Lysosomal tethering of Ragulator is required for TAK1 inhibition-induced pyroptosis
We next asked whether lysosomal localization of Rag-Ragulator is required for TAK1 inhibition-induced pyroptosis. Rag-Ragulator is tethered to the cytosolic side of the lysosomal membrane by lipidation of Lamtor1 at its N-terminal G2 and C3C4 residues (21, 22). To address this question, we ectopically expressed WT or a Lamtor1 mutant of its three N-terminal lipidation sites to alanine (Lamtor1 3A) in Lamtor1−/− iBMDMs. Lamtor1 3A was no longer tethered to the lysosome and redistributed to the cytosol (Fig. 5A). Rescue of Lamtor1−/− by ectopic expression of WT Lamtor1 in iBMDMs restored LPS/5z7-induced pyroptosis, assessed by measuring LDH release, PI uptake, and pyroptotic morphological changes (ballooning cell membrane) (Fig. 5, B–D). By contrast, Lamtor1 3A did not rescue LPS/5z7-induced pyroptosis, indicating that Ragulator needs to be tethered to the lysosome to promote LPS/5z7-induced pyroptosis. Overexpression of Lamtor1 3A disrupted the cleavage and lysosomal localization of caspase-8 after LPS/5z7 treatment (fig. S9). Next, we used immunoblotting to examine which step of RIPK1-caspase-8-GSDM-mediated pyroptosis was regulated by Ragulator (Fig. 5E). In WT iBMDMs, phosphorylation of RIPK1 at Ser166 was detected within 100 min of adding LPS/5z7, which was followed by detection of RIPK1 cleavage and cleavage of caspase-8 and GSDMD to their active fragments at 150 min. GSDME was also cleaved concurrently to produce a p30 active N-terminal fragment. The detection of activated caspase-8, GSDMD and GSDME fragments coincided with the kinetics of membrane disruption recorded by PI uptake in WT iBMDMs (Fig. 5C). By contrast, Lamtor1 deficient iBMDMs showed severely blunted RIPK1 phosphorylation and cleavage and caspase-8, GSDMD and GSDME activation (Fig. 5E). Re-expression of Lamtor1 restored the defects in RIPK1 and caspase-8 activation as well as GSDM cleavage in Lamtor1-deficient cells. Thus, the activation of RIPK1 and the downstream caspase-8-GSDM pyroptotic pathway only occurs when the RIPK1-caspase-8-containing complex is anchored to the lysosomal membrane by Ragulator.
Fig. 5. Pyroptosis activated by LPS/5z7 or pathogenic Yersinia depends on lipidated Lamtor1.
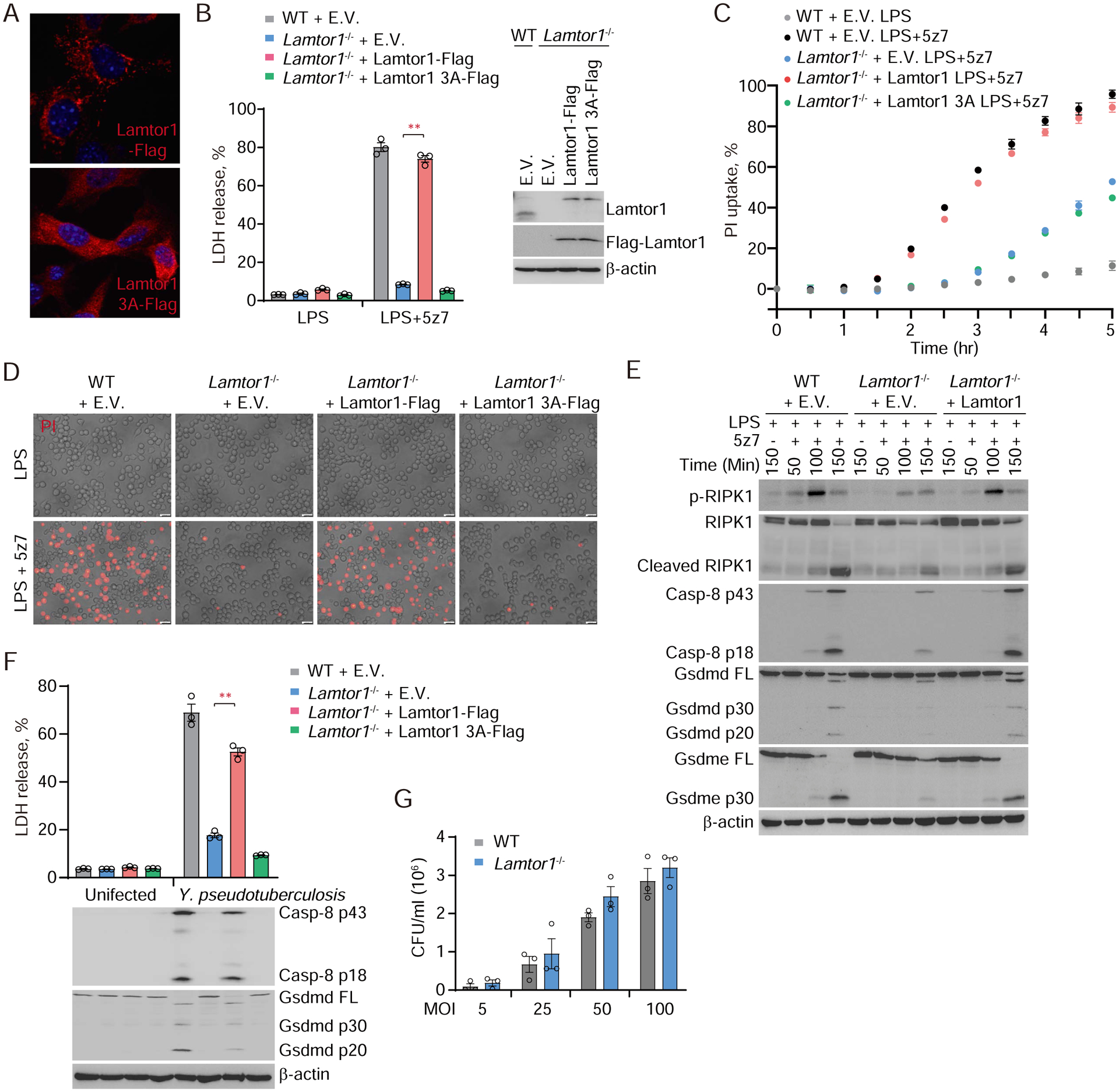
(A) The subcellular localization of Lamtor1 and Lamtor1 3A mutant was imaged by confocal microscopy. (B, C, D and F) The indicated iBMDMs were treated with LPS, LPS/5z7 (B to D) or Yersinia (F). Cell death was measured by LDH release after 2.5 hr (B) or 5 hr (F), or measured by entry of PI into cells in real-time (C), or observed by phase-contrast fluorescence microscopy (D). (E) Full-length and cleaved products of caspase-8, RIPK1, Gsdmd and Gsdme from whole-cell lysates of the indicated iBMDMs treated with LPS or LPS/5z7 for the indicated times. (G) The number of Yersinia that have been taken up by WT or Lamtor1−/− iBMDMs was quantified by counting colony forming units (CFU). Graphs in B, C, F and G show mean ± SEM of triplicate wells. Data are representative of at least three independent experiments. Data were analyzed using a two-tailed Student’s t test. **P < 0.01. E.V., empty vector.
Next we examined the role of lysosomal Ragulator in pyroptosis induced by Y. pseudotuberculosis. Like LPS/5z7 treatment, Yersinia infection in WT iBMDMs triggered massive pyroptosis, as assessed by LDH release, which, as expected, was associated with caspase-8 and GSDMD cleavage to their active forms (Fig. 5F). For the first 60 min after infection, before pyroptosis occurred, bacterial replication as measured by colony counts was comparable in WT and Lamtor1−/− iBMDMs (Fig. 5G). Lamtor1 deficiency also did not affect the secretion and activity of YopJ (fig. S10). However, cell death and caspase-8 and GSDMD processing were strongly reduced in Lamtor1−/− iBMDMs (Fig. 5F). Pyroptosis was rescued by ectopic expression of Flag-tagged WT Lamtor1, but not Lamtor1 3A. Thus, lysosomal targeting of Lamtor1 is also prerequisite for Yersinia-triggered pyroptosis.
Lysosomal Rag GTPase drives RIPK1-caspase-8-induced pyroptosis
Our genetic screen and its validation (Fig. 1, 2) suggested that RagC and Flcn-Fnip, which activates RagC’s GTPase activity by converting RagC into the GDP-loaded form (22, 34, 35), were required for RIPK1-caspase-8-mediated pyroptosis. To determine which form of RagC promotes TAK1 inhibition-triggered pyroptosis, we re-introduced RagC WT, the GTP-bound form of RagC (RagC Q119L) or the GDP-bound form of RagC (RagC S74N) into RagC-deficient iBMDMs. Ectopic expression of either RagC WT or the GDP-bound form of RagC restored LPS/5z7-mediated pyroptosis to Rragc knockout iBMDMs as assessed by LDH release, PI uptake and ballooning morphology, whereas the GTP-bound form of RagC failed to do so (Fig. 6, A–C). RagC deficiency inhibited activation of RIPK1, caspase-8, GSDMD and GSDME, which could be rescued by re-expression of RagC (Fig. 6D). Similarly, ectopic expression of WT or GDP-bound RagC in RagC-deficient iBMDMs made them susceptible to pyroptosis caused by infection of pathogenic Yersinia, while the GTP-bound form of RagC did not (Fig. 6E). The presence of RagC did not affect bacterial replication at early time points or the secretion and activity of YopJ (Fig. 6F, and fig. S10). In comparison, Salmonella infection-induced caspase-1-dependent GSDMD cleavage and pyroptosis in iBMDMs were not affected by Rragc knockout or knockout of any of the other Rag-Ragulator complex genes tested (Lamtor1, Lamtor4, Flcn, Fnip2) (fig. S11). Thus, the GDP-bound form of RagC selectively mediates caspase-8-induced pyroptosis. We hypothesized that Ragulator and Flcn-Fnip function upstream of the Rag GTPase to tether it to the lysosome and maintain it in its GDP-bound state. To test this hypothesis, we performed rescue experiments in Flcn or Fnip-deficient iBMDMs by re-introducing RagC (Fig. 6, G and H). Ectopic expression of GDP-bound RagC restored susceptibility of Flcn or Fnip2 deficient iBMDMs to LPS/5z7-triggered pyroptosis, whereas the GTP-bound form of RagC did not. To determine whether Ragulator’s role was primarily to direct RagC to the lysosomal membrane, we generated a RagC construct in which the lysosomal targeting sequence of Lamtor1 was added to the N-terminus of RagC (termed Lyso-RagC). Although lysosomal localization of WT RagC was lost in Lamtor1-deficient cells, the lysosomal targeting sequence relocalized Lyso-RagC to the lysosome when Lamtor1 was lacking (Fig. 6I). Ectopic expression of Lyso-RagC made Lamtor1-deficient cells sensitive to LPS/5z7-triggered pyroptosis (Fig. 6J). In addition, LPS/5z7 treatment did not affect the subcellular localization of Flcn (fig. S12). Thus, lysosomal targeting of GDP-bound RagC is sufficient to trigger caspase-8-mediated pyroptosis.
Fig. 6. GTPase activity of lysosomal membrane-anchored RagC is required for caspase-8 activation and pyroptosis triggered by LPS/5z7 or pathogenic Yersinia.
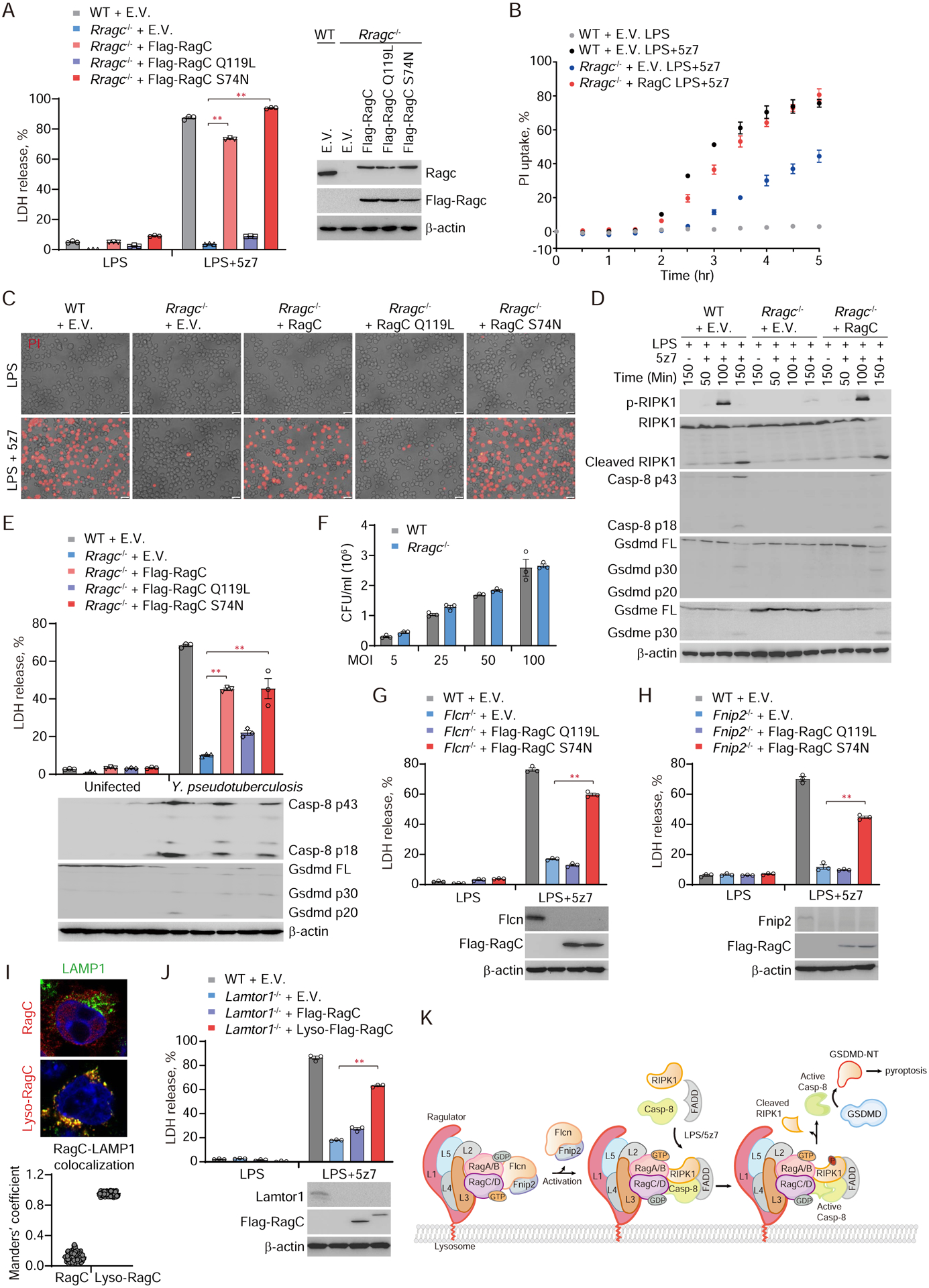
(A, B, C and E) The indicated iBMDMs were treated with LPS, LPS/5z7 or Yersinia. Cell death was measured by LDH release after 2.5 hr (A) or 5 hr (E), or by entry of PI into cells in real-time (B), or as observed by phase-contrast fluorescence microscopy (C). (D) Full-length and cleaved products of caspase-8, RIPK1, Gsdmd and Gsdme from whole-cell lysates of the indicated iBMDMs treated with LPS or LPS/5z7 for the indicated times. (F) The number of Yersinia taken up by WT or Rragc−/− iBMDMs was quantified by counting CFU. (G, H and J) WT, Flcn−/−, Fnip2−/− or Lamtor1−/− iBMDMs reintroduced with the indicated plasmids were treated with LPS/5z7 and assayed for LDH release 2.5 hr post treatment. (I) Lysosomal localization of ectopically expressed RagC and Lyso-RagC in Lamtor1−/− iBMDMs was assessed by confocal microscopy (representative images, upper panel). Colocalization of RagC with LAMP1 was analyzed by calculating Manders’ overlap coefficient (lower panel). (K) Model of RIPK1-caspase-8 activation mediated by Rag-Ragulator complex. Graphs in A, B, E-H, and J show mean ± SEM of triplicate wells. Data are representative of at least three independent experiments. Data were analyzed using a two-tailed Student’s t test. **P < 0.01. E.V., empty vector.
Discussion
Recent studies revealed an alternative inflammasome- and inflammatory caspase-independent, but caspase-8 and gasdermin-dependent, pyroptotic pathway activated by TLR/TNFα signalling during pathogenic Yersinia infection (12, 13). Here we discovered in an unbiased CRISPR screen a critical and unexpected role of the lysosomal membrane-tethered Rag-Ragulator supercomplex. Rag-Ragulator served as a platform for activating a FADD-RIPK1-caspase-8 complex formed in response to TLR/TNFR ligation and Rag GTPase activity was critical for triggering the pathway (Fig. 6K). There is still much to learn. Future kinetics, biochemistry and live-cell imaging experiments can determine whether the FADD-RIPK1-containing complex recruits caspase-8 before or after Rag-Ragulator binding and whether it contains other components or its formation is regulated by post-translational modifications of its component proteins. Our data suggest that the death-receptor-triggered complex binds to RagC and Flcn, but does the Rag-Ragulator complex remain intact after binding or do some components dissociate? Are other proteins recruited? Although we found that mTORC1 activity did not affect caspase-8 activation or pyroptosis, does the FADD-RIPK1-caspase-8 complex compete for binding to Rag-Ragulator with mTORC1? Does cellular metabolism (amino acid and ATP availability and cellular stress, which regulate mTORC1), affect the execution of this cell death pathway? One study suggests that lysosomes are disrupted during inflammasome-mediated pyroptosis (36). Do the gasdermins form pores in lysosomal membranes to disrupt lysosomes and cause their proteases to be released into the cytosol and wreak additional damage? RIPK1 phosphorylation is Rag-dependent and occurs once the complex is bound to Rag-Ragulator. Activated caspase-8 cleaved and activated both GSDMD and GSDME, as has previously been shown (12, 13), but the mechanism for GSDME cleavage is uncertain. Does caspase-8 directly cleave GSDME or is cleavage indirect via caspase-8 cleavage of caspase-3, a known activator of GSDME? The role of Rag-Ragulator in pyroptosis described here confirm the key role of Rag-Ragulator as a central hub for monitoring environmental cues. This role is now extended to include infection as well as nutrient and energy availability and helping to decide not only whether a cell proliferates, but also whether it survives. Future studies can leverage these insights to explore more mechanistic details of pyroptosis, as well as to manipulate this process for therapeutic benefits.
Materials and Methods
Plasmids, antibodies and reagents
psPAX2 (#12260), pMD2.G (#12259), lentiCas9-Blast (#52962), lentiCRISPR v2 (#52961), pFastBacDual with LFn + Fla (#84866) and pFastBacDual with PA+His tag (#84870) constructs were obtained from Addgene. Mouse Ripk1, Casp-8, Lamtor1, Rragc, Flcn, Fnip2 and Tfeb were amplified by PCR from the mouse cDNA library and cloned into mammalian expression vectors (pHAGE-BSD-Flag, pHAGE-Ble, pcDNA3-NHA or pcDNA3-CHA). All point mutations and truncations were generated using the overlap PCR method. For shRNA cloning, annealed shRNA oligos were ligated into pLKO.1 (Addgene #8453). The oligo sequences for targeting the genes of interest are as follows: negative control (5’-CCT AAG GTT AAG TCG CCC TCG-3’), Tfeb (5’-GCA GGC TGT CAT GCA TTA TAT-3’), Tfe3 (5’-GCC TAA CAT CAA ACG CGA GAT-3’). All plasmids were verified by sequencing.
Monoclonal anti-Flag antibody (F3165) and anti-β-actin antibody (A1978) were from Sigma. Antibodies against HA (#3724, #2367), Flag (#14793), TFEB (#32361), TFE3 (#14779), EEA1 (#48453), p70 S6 Kinase (#2708), Phospho-p70 S6 Kinase (#9234), p38 (#9212), Phospho-p38 (#4511), Erk (#4695), Phospho-Erk (#4370), cleaved caspase-8 (#8592), caspase-8 (#4927), caspase-3 (#9665), caspase-7 (#12827), RIPK1 (#3493), Phospho-RIPK1 (#31122), RagC (#3360, #9480), Lamtor1 (#8975), Lamtor4 (#12284) and FLCN (#3697) were from Cell Signaling Technology. Monoclonal anti-FADD antibody (sc-166516) and monoclonal anti-KDEL antibody (sc-58774) were from Santa Cruz. Monoclonal anti-LAMP1 antibody was from Developmental Studies Hybridoma Bank (1D4B). Monoclonal anti-GSDMD antibody (ab209845), monoclonal anti-GSDME antibody (ab215191), polyclonal anti-FNIP2 antibody (ab106611), monoclonal anti-pro caspase-8 antibody (ab108333), monoclonal anti-LAMP1 antibody (ab208943) and monoclonal anti-FADD antibody (ab124812) were from Abcam. Anti-YopJ antibody was kindly shared by Professor Zongmin Du (Beijing Institute of Microbiology and Epidemiology, China).
LPS (L4524), Etoposide (E1383), Polybrene (H9268), Rapamycin (V900930), 5z7 (O9890) and 5-aminoimidizole-4-carboxamide riboside (AICAR) (A9978) were obtained from Sigma-Aldrich. Recombinant murine TNFα (315-01A) was from Peprotech. Z-VAD-fmk (550377) was from BD Pharmingen. Ultrapure LPS (tlrl-3pelps), Nigericin (tlrl-nig) and Blasticidin (anti-bl) were purchased from Invivogen. Torin 1 (MB3467) was from Meilunbio. Puromycin (A1113802), Zeocin (R25005) and MitoTracker (M7510) were from Invitrogen.
Cell culture and stimulation
HEK293T, MEFs, and C57BL/6 mouse iBMDM cells, described previously (7), were cultured in Dulbeccos modified Eagle’s medium supplemented with 10% fetal bovine serum, 1×GlutaMAX (ThermoFisher, 35050061), 1×Penicillin-Streptomycin (ThermoFisher, 15140148) and 50 μM 2-Mercaptoethanol (β-ME), and verified to be free of mycoplasma contamination. To induce pyroptosis, cells were stimulated with 10 ng/mL LPS plus 250 nM 5z7, 50 ng/mL TNFα plus 250 nM 5z7, 1 μg/mL LPS plus 20 μM Nigericin or 2 μg/mL PA plus 1 μg/mL LFn-Fla. For noncanonical inflammasome activation, 2 million iBMDMs were mixed with 2 μg ultrapure LPS in 10 μL of buffer R, and electroporated using the Neon Transfection System (ThermoFisher) with parameters (1400 V, 10 ms, and 3 pulses). To induce apoptosis, iBMDMs were treated with 50 μM Etoposide. Transient transfection of HEK293T cells was performed with Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. For stable transfection, lentiCas9-Blast, pHAGE-BSD-Flag or pHAGE-Ble containing the gene of interest together with packaging plasmids psPAX2 and pMD2.G in a ratio of 5:3:2, were transiently transfected into HEK293T cells. 72 hr post transfection, the cell culture supernatant containing lentivirus was collected and filtered through a 0.45-μm membrane (Millipore, SLHV033RB) to remove cell debris. iBMDMs were then infected with the lentivirus for 3 days before adding 10 μg/ml Blasticidin or 400 μg/ml Zeocin for stable transfection selection.
Genome-wide CRISPR/Cas9 screen
The mouse CRISPR knockout pooled library for genome-wide CRISPR/Cas9 screen was obtained from Addgene (#1000000053) and amplified according to the protocol provided by Addgene. For lentivirus production, mouse GeCKO A and B library plasmids were mixed equally and transfected into HEK293T cells together with the packaging plasmids psPAX2 and pMD2.G. 72 hr later, lentivirus was collected and the viral titer was measured by using the QuickTiter™ Lentivirus Titer kit (Cell Biolabs, VPK-107). For the genome-wide screen, Cas9 stably expressing iBMDMs were seeded in 10 cm dishes (2 × 106 cells/dish) and a total of 7 × 107 cells was infected with the lentivirus containing sgRNA library at MOI of 0.3. 60 hr later, cells were treated with puromycin to remove uninfected cells. 6 days later, the transduced cells were seeded in 40 × 10 cm dishes (8 × 106 cells/dish) and treated with 10 ng/ml LPS plus 125 nM 5z7 for 6 hr. 3 days later, the surviving cells were reseeded and treated with 10 ng/ml LPS plus 250 nM 5z7 overnight. 2 days later, surviving cells were again treated with 20 ng/ml LPS plus 250 nM 5z7 overnight before surviving cells were harvested. For the noncanonical inflammasome screen, 3 × 108 transduced cells were electroporated with ultra LPS for 3 rounds. Surviving cells and untreated transduced cells (as the control sample) were harvested and lysed in the SNET buffer (20 mM Tris-HCL pH 8.0, 5 mM EDTA, 400 mM NaCl, 1% SDS and 400 μg/ml Proteinase K (NEB, P8107S)). Genomic DNAs were prepared by using phenol-chloroform extraction and isopropanol precipitation and amplified by two-step PCR using the 2 × Hieff Canace® Gold PCR Master Mix (Yeasen, 10149ES01). The samples were quantified and sequenced on a HiSeq 2500 (Illumina) by the GENEWIZ company. Sequencing data were further processed and analyzed using the MAGeCK algorithm(37). MAGeCK built a linear model to estimate the variance of gRNA read counts, evaluated the gRNA abundance changes between control and treatment conditions, and assigned P values for positive and negative selection.
Generation of knockout iBMDMs by CRISPR/Cas9
HEK293T cells were transfected with sgRNA-expressing lentiCRISPR v2 together with psPAX2 and pMD2.G in a ratio of 5:3:2. 72 hr later, the supernatant containing lentivirus was collected and filtered through a 0.45-μm membrane (Millipore) for subsequent iBMDMs infection. 3 days post infection, cells were treated with 5 μg/ml Puromycin before diluting to single clones cultured in 96-well plates. The candidate knockout clones were verified by sequencing of genomic DNA and immunoblot. sgRNA sequences for targeting the genes of interest are as follows: Lamtor1 (5’-TGG ACC GGG CAA GGC AGT AC-3’/5’-GCT CTT CTT TCG CAT CCA CG-3’), Lamtor4 (5’-AGC CAG TGC CAT CTC GGA GT-3’/5’-TAG ACT TCC GCA CTG ACC CA-3’), Rragc (5’-TTT CTG TAC CAC CTT ACT GA-3’/5’-TCA TAA GAC TGC ATA TCC AC-3’), Flcn (5’-GGC TGC CGG TCA CTT GCC GT-3’/5’-GCC TGC TAC CGC ATG CCT TC-3’), Fnip2 (5’-ACC GTA TGT AGT GTA TCT TC-3’/5’-ACT TTA CTA ATC ATC AGT TG-3’), Gsdmd (5’-AGC ATC CTG GCA TTC CGA G-3’), Nlrp3 (5’-GAA GAT TAC CCG CCC GAG AA-3’), Nlrc4 (5’-TGT TTC GAA TAG TCC CCC CC-3’).
Recombinant protein purification
PA and LFn-flagellin recombinant proteins were purified from Sf9 cells. 72 hr post P3 baculovirus infection, cells were resuspended in lysis buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10 mM β-ME, 5 mM Imidazole, 1% Triton X-100) and then clarified by centrifugation at 42,000 rpm for 2 hr at 4°C. Proteins were purified using Ni-NTA agarose (QIAGEN), eluted with elution buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10 mM β-ME, 500 mM Imidazole) and further purified by Superdex 200 (10/300) gel-filtration chromatography and mono Q ion-exchange.
Bacterial strains and culture conditions
Y. pseudotuberculosis YPIII strain, a gift from Dr. Shiyun Chen (Wuhan Institute of Virology, Chinese Academy of Sciences), was grown overnight in 2×YT broth at 26°C. On the day of infection, bacteria were diluted 1:50 into 2 × YT plus 20 mM MgCl2 and 20 mM sodium oxalate and grown for 2 hr at 26°C followed by a shift to 37°C for 2 hr. Bacteria were then washed in PBS (Gibco) and added to cells at a multiplicity of infection (MOI) of 40. 100 μg/ml gentamicin was added to the cultures 2 hr after infection. To quantify the number of bacteria that have been taken up by cells, iBMDMs were infected with Y. pseudotuberculosis YPIII strain at indicated MOI. 30 min later, cells were washed with PBS for 3 times and gentamicin were added to kill extracellular bacteria. Then, intracellular bacteria were released by treating cells with 0.05% Triton X-100 before lysates were serially diluted and plated on 2×YT agar. Bacterial colonies were counted after 1 day of culture at 37°C. For Salmonella typhimurium infection assay, bacteria were grown overnight in Luria-Bertani (LB) broth at 37°C. On the day of infection, bacteria were diluted 1:50 into LB broth and grown for 4 hr at 37°C. Bacteria were then washed with PBS and added to cells at an MOI of 20. 100 μg/ml gentamicin was added to the cultures 0.5 hr after infection to kill extracellular bacteria.
Cell death assays
Cell death and viability of stimulated macrophages were determined by measuring lactate dehydrogenase (LDH) release using the CytoTox 96® assay kit (Promega, G1780) and ATP level using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7570), respectively. Luminescence and absorbance were measured on a BioTek Synergy H1 plate reader. For kinetic cytotoxicity assay via monitoring cell permeability, the cell culture medium was changed into buffer B (25 mM HEPES pH 7.4, 120 mM NaCl, 4 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 5mM glucose, 0.1% BSA) containing 2 μg/mL propidium iodide (Invitrogen, P3566) and the fluorescence (excitation: 535 nm, emission: 617 nm) was continuously recorded after treatment for 5 hr at 30 min intervals using a BioTek Synergy plate reader.
Immunoprecipitation (IP) and immunoblotting
For IP assay, cells were harvested and resuspended in lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100) supplemented with a complete protease inhibitor cocktail (Sigma). Cell extracts were then incubated with the indicated antibodies for 4 hr at 4°C before adding protein A-G agarose beads for 2 hr. The beads were extensively washed at least three times with wash buffer (50 mM Tris-HCl pH 7.4, 300 mM NaCl, 0.5% Triton X-100) and bound proteins were eluted by boiling with SDS-loading buffer. For immunoblot analysis, IP samples or whole cell lysate samples were subjected to electrophoresis through SDS-PAGE gels. The separated proteins were then transferred to a PVDF membrane (Millipore). Immunoblot was probed with the indicated antibodies. The protein bands were visualized using a SuperSignal West Pico chemiluminescence ECL kit (Pierce).
Immunostaining and confocal microscopy
iBMDMs grown on coverslips were stimulated with 10 ng/mL LPS plus 250 nM 5z7. Cells were fixed 100 min later, with 4% paraformaldehyde for 20 min before permeabilization using 0.1% Triton X-100 (10 min) and blocking with 5% BSA (1 hr). Then, cells were immunostained with the indicated primary antibodies followed by incubation with the corresponding fluorescent-conjugated secondary antibodies (Jackson ImmunoResearch). Nuclei were counterstained with DAPI (4,6-diamidino-2-phenylindole) (Cell Signaling Technology). Slides were mounted using Aqua-Poly/Mount (Dako). Images were captured using a Zeiss 880 laser scanning confocal microscope at 63× magnification and analyzed using Zeiss Zen software. Manders’ overlap coefficient was calculated using ImageJ (each point represents a single cell, 100 cells per sample). All images are representative of at least three independent experiments.
Statistics
Student’s t-test (two-tailed) was used for the statistical analysis of all experiments. P values < 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank Professor Shiyun Chen (Wuhan Institute of Virology, Chinese Academy of Sciences, China) for providing Y. pseudotuberculosis YPIII strain; Professor Zongmin Du (Beijing Institute of Microbiology and Epidemiology, China) for sharing anti-YopJ antibody.
Funding:
This work was supported by National Key R&D Program of China (2020YFA0509600), Key Research Program of the Chinese Academy of Sciences (ZDBS-LY-SM008), National Natural Science Foundation of China (31972897), Strategic Priority Research Program of Chinese Academy of Sciences (XDB29030300), Shanghai Municipal Science and Technology Major Project (2019SHZDZX02) to X.L, National Natural Science Foundation of China for the Youth (31900456) to Z.Z. and NIH R01 CA240955 to J.L.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability:
All data are available in the main text or the supplementary materials.
References and notes:
- 1.Broz P, Dixit VM, Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 2.de Vasconcelos NM, Lamkanfi M, Recent Insights on Inflammasomes, Gasdermin Pores, and Pyroptosis. Cold Spring Harb Perspect Biol 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green DR, The Coming Decade of Cell Death Research: Five Riddles. Cell 177, 1094–1107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kayagaki N et al. , Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Shi J et al. , Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Ding J et al. , Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Liu X et al. , Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sborgi L et al. , GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. The EMBO journal 35, 1766–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aglietti RA et al. , GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proceedings of the National Academy of Sciences of the United States of America 113, 7858–7863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heilig R et al. , The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol 48, 584–592 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Evavold CL et al. , The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orning P et al. , Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarhan J et al. , Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A 115, E10888–E10897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kayagaki N et al. , IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci Signal 12, (2019). [DOI] [PubMed] [Google Scholar]
- 15.Bar-Peled L, Sabatini DM, Regulation of mTORC1 by amino acids. Trends Cell Biol 24, 400–406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirst J, Mitochondrial complex I. Annu Rev Biochem 82, 551–575 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Rogers C et al. , Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10, 1689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaiser WJ et al. , RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oberst A et al. , Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vercammen D et al. , Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187, 1477–1485 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nada S et al. , The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J 28, 477–489 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sancak Y et al. , Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muendlein HI et al. , ZBP1 promotes LPS-induced cell death and IL-1beta release via RHIM-mediated interactions with RIPK1. Nat Commun 12, 86 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muendlein HI et al. , cFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science 367, 1379–1384 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsuda I et al. , The C-terminal domain of the long form of cellular FLICE-inhibitory protein (c-FLIPL) inhibits the interaction of the caspase 8 prodomain with the receptor-interacting protein 1 (RIP1) death domain and regulates caspase 8-dependent nuclear factor kappaB (NF-kappaB) activation. J Biol Chem 289, 3876–3887 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park YH, Jeong MS, Park HH, Jang SB, Formation of the death domain complex between FADD and RIP1 proteins in vitro. Biochim Biophys Acta 1834, 292–300 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Anderton H et al. , RIPK1 prevents TRADD-driven, but TNFR1 independent, apoptosis during development. Cell Death Differ 26, 877–889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muzio M et al. , FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell 85, 817–827 (1996). [DOI] [PubMed] [Google Scholar]
- 29.Fu TM et al. , Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol Cell 64, 236–250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen K et al. , Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell 179, 1319–1329 e1318 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thoreen CC et al. , An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284, 8023–8032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawrence RE et al. , Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 366, 971–977 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Napolitano G et al. , A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature 585, 597–602 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM, Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsun ZY et al. , The Folliculin Tumor Suppressor Is a GAP for the RagC/D GTPases That Signal Amino Acid Levels to mTORC1. Mol Cell 52, 495–505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M, Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 26, 146–161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W et al. , Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol 16, 281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.