

Abstract
RNA molecules can fold into complex two- and three-dimensional shapes that are critical for their function. Chemical probes have long been utilized to interrogate RNA structure and are now considered invaluable resources in the goal of relating structure to function. Recently, the power of deep sequencing and careful chemical probe design have merged, permitting researchers to obtain a holistic understanding of how RNA structure can be utilized to control RNA biology transcriptome-wide. Within this review, we outline the recent advancements in chemical probe design for interrogating RNA structures inside cells and discuss the recent advances in our understanding of RNA biology through the lens of chemical probing.
Keywords: LASER, probing, RNA, SHAPE, structure
Introduction
RNA molecules perform a diverse array of functions in cells. Functional RNAs have been linked to controlling chromatin state and gene regulation,[1,2] the localization of diverse biomolecules,[3] and the regulation of cellular state and structure.[4] Further, many of the most pressing diseases, including neurodegenerative disorders[5] and many cancers,[6–8] have been intimately linked to misregulation of normal RNA functions. As the diverse array of critical functions performed by RNAs continues to expand, so does the need to understand the mechanistic basis of their function at the molecular level.
The functional role of RNA molecules is tightly linked to their structure.[9] Functional RNA structure elements serve as landing pads for proteins, trans-acting RNAs, and even small molecules.[10] Characterizing these structures and understanding how changes to the cellular environment manipulate them has been a long-standing goal in RNA molecular biology. Such challenges have been met through the development of highly specific chemical reagents to measure aspects of RNA structure through their chemical reactivity with functional groups in folded RNA molecules.[8]
A current challenge in the field is to extend the principles learned from studying individual RNAs in isolation to the transcriptome-wide level.[11] These endeavors are critically important as they potentially offer understanding of how different large groups of RNA populations take advantage of their structure to regulate their biology in concert, resulting in a specific cellular phenotype through large-scale gene regulation. These efforts have made significant headway in two ways. First, they have resulted in new insights into chemical design through sophisticated development of novel reagents to measure RNA structure and make such measurements amenable to large-scale analysis. Second, the intersection of structure measurements with other transcriptome-wide measurements allows investigation into global regulation of RNA structure state and gene regulation. The parallel expansion of these efforts has together given the field novel insight into how chemistry and transcriptomics can be merged to understand RNA-based regulation in the cellular environment.
Herein, we review the progress of the field in expanding the chemical scope of RNA structure probing reagents. We also detail how these efforts have been merged with RNA sequencing. These efforts have ushered in a new era in our understanding of RNA structure and its role in regulating many pathways of cellular function.
Chemical Reagents To Measure RNA Structure
Several types of RNA structure reagents have been developed, with the goal of controlled and selective chemical reactions with specific functional groups. Chemical reactivity is related to the conformational structure of a specific functional group of RNA in solution. That is, the local environment of the reactive group in an RNA controls its reactivity with structure probes, as discussed below. We first highlight the types of chemical probes with focus on the RNA region where reactivity occurs.
Probably the most widely used structure probes are those that react with the Watson-Crick (W-C) faces of different nucleobases (Figure 1). Dimethylsulfate (DMS; Figure 1a), first used in the 1980s, is ubiquitously used to identify sites that are not in base pairs.[12–14] These alkylation reactions are used to identify single-stranded sites of an RNA structure. Kethoxal (and other 1,2-dicarbonyl compounds; Figure 1b) is also used to mark single-stranded guanosine residues. Notable differences have been observed with glyoxal derivatives in cells, where methyl and phenylglyoxal derivatives work robustly in cells, while the parent molecule, kethoxal, is much less reactive in cells.[15] The capability of probes designed to mark unpaired residues has been extended through the use of carbodiimide reagents, such as CMC and EDC (Figure 1c). Reactivity with carbodiimides is notable, because the reagent itself can actas a base to deprotonate the N—H group on uridine and guanosine, activating the nucleobase as a nucleophile to react with a protonated carbodiimide. Such reactions have selectivity for single-stranded uridine nucleobases (and EDC has been observed to react with guanosine, although at a much slower rate[16]).[16,17] These reagents enable researchers to understand and identify specific structure states of nucleobases themselves, as they are highly biased in their reactivity for single-stranded positions.
Figure 1.
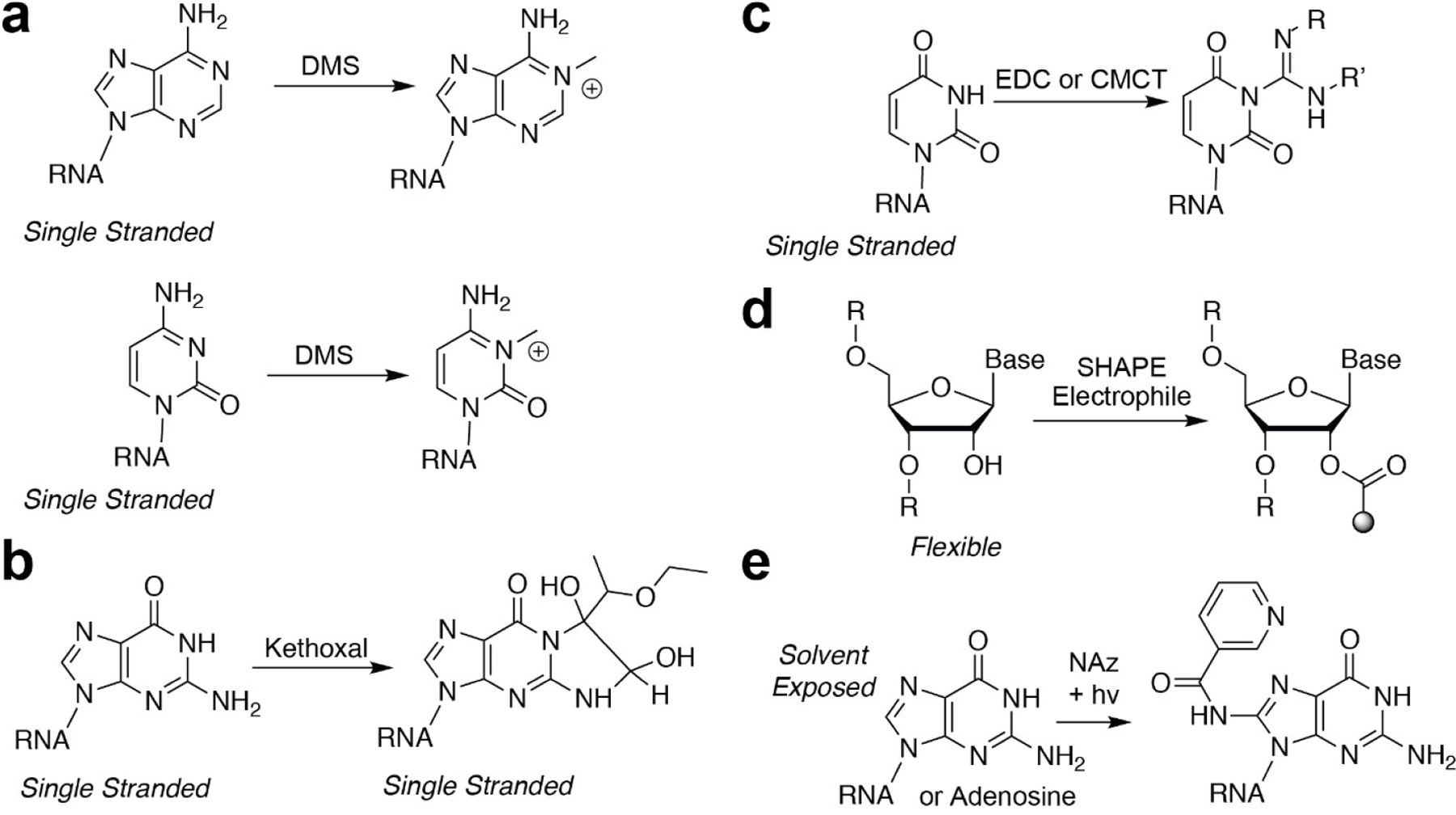
Chemical structures and reactions of structure-specific probes used to interrogate RNA structure. a) DMS reactions that measure adenosine and cytosine single-stranded positions. b) Kethoxal reaction that measures single-stranded guanosine positions. c) Carbodiimide chemical reactions used to identify single-stranded uridine positions in RNA. d) SHAPE carbonyl electrophiles for interrogating flexible positions. e) NAz aroyl azide, when exposed to light, reacts with solvent exposed C-8 guanosine and adenosine residues in RNA.
Chemical methods that extend beyond the nucleobases have also been recently expanded. The chemicals mentioned above have biases for their reactivity, depending on the nucleobase they react with. More generalized methods would be ideal, as they could be used in less selective experiments to understand the structured state of each position in a RNA in fewer experiments. Selective hydroxyl acylation analyzed by primer extension (SHAPE; Figure 1d) is now widely used to probe RNA structure.[18–20] SHAPE electrophiles work by selectively reacting with 2’-hydroxyl residues that reside in flexible regions on an RNA sequence. Internucleotide flexibility is viewed as a proxy for single-strandedness, and as such SHAPE is incredibly powerful for analyzing and identifying RNA-RNA interactions, RNA–protein interactions, and RNA-ligand interactions that can alter the flexibility.
Another generalizable aspect of RNA structure is solvent accessibility. Reagents designed to measure solvent accessibility give insight into important RNA structures that extend beyond W-C pairing or internucleotide flexibility. Traditionally, solvent accessibility has been measured through the use of Fenton reagent-generated hydroxyl radicals.[21] OH radicals are high-energy intermediates that can perform hydrogen abstraction from the C3’- or C4’-position on the ribose ring, resulting in strand cleavage. The fast rate of OH radical hydrogen abstraction enables this technology to be employed to interrogate RNA structure folding and more dynamic aspects of RNA-protein interactions.
Another more recent development has been the utility of light-activated aroyl azide reagents to interrogate nucleobase solvent accessibility. In contrast to the electrophilic reagents discussed above, which interrogate W-C base pairing, light-activated aroyl azides, such as nicotinoyl azide (NAz; Figure 1e) react with the C-8 position of electron-rich purines.[22] Once activated by long-wavelength UV light, NAz transitions to a highly reactive and hard electrophilic nitrenium ion. Nitrenium ions can react with heteroaromatic arenes through electrophilic aromatic substitution to form amidated products, in this case, C-8 amidation of adenosine and guanosine. This newer methodology, termed light activated structural examination of RNA, or LASER, can be robustly employed in living cells to examine solvent accessible regions of RNA and investigate RNA-protein interactions in their native cellular environment. These reagents and their utility to measure RNA structure, as mentioned above, have been extremely valuable to the RNA community. Combining different methods and reagents can enable a holistic understanding of a given RNA and its structured interactions through intramolecular folding or binding events with trans RNAs or proteins.
Biochemical Methods To Identify Chemical Reactivity Positions within an RNA Sequence
In parallel with the development of these novel chemical methods has been the expansion of biochemical methods to identify the sites of RNA-chemical reactions. All chemical methods mentioned above work through forming covalent adducts with RNA, which mark the site of chemical reactivity. The next key step is to identify the sites of adduct formation.
The traditional method for identifying sites of RNA-adducts is to utilize reverse transcription (RT). When reverse transcriptase enzymes encounter a site of adduct formation (Figure 2a), they stall and dissociate from the RNA-cDNA hybrid. The length and sequence of the truncated and full-length cDNA can be analyzed by denaturing gel electrophoresis, whereby a radio- (or fluorescent-) labeled cDNA primer is resolved on the gel and mapped back to the primary sequence.[23,24]
Figure 2.
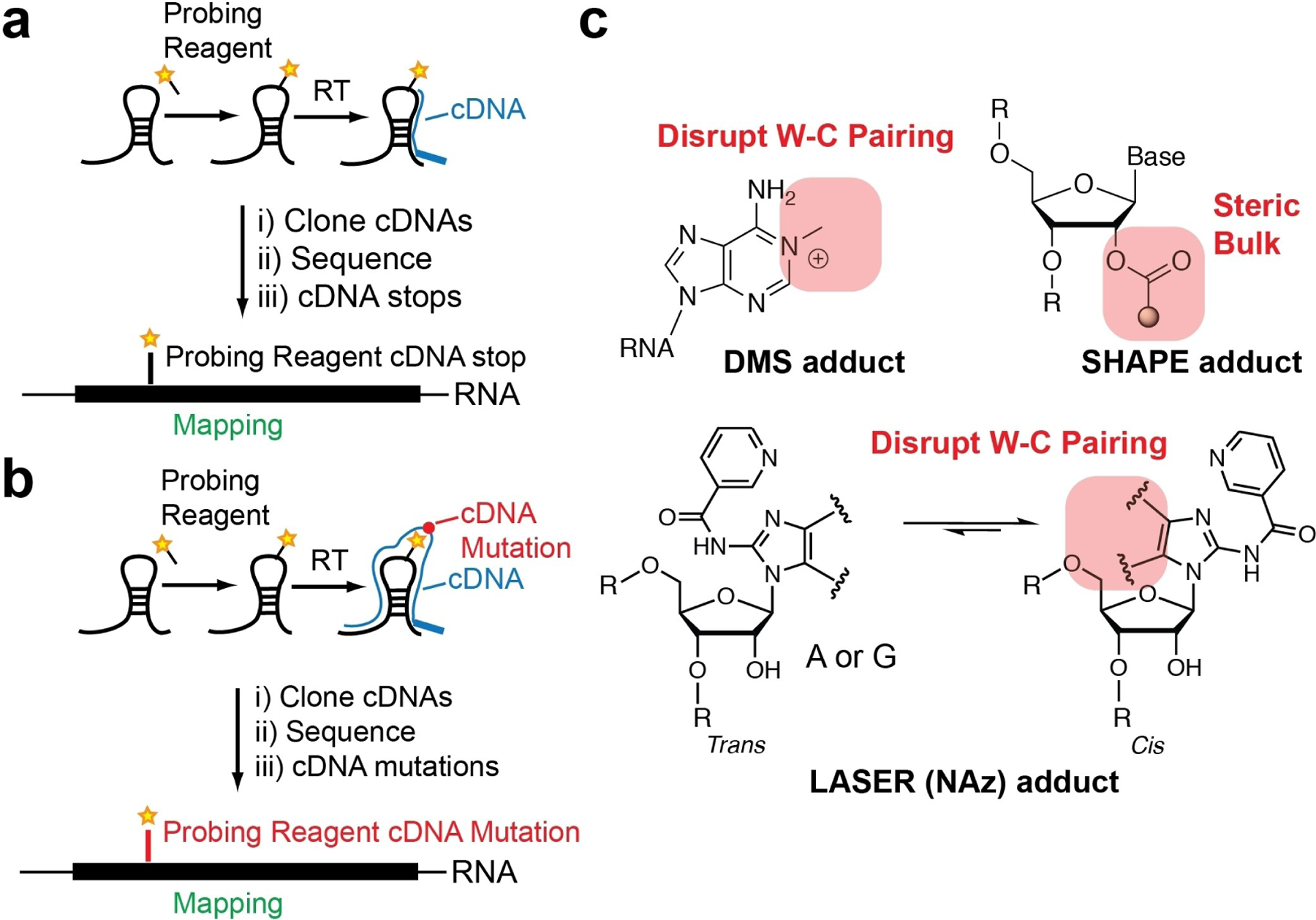
Strategies to identify sites of probing reagent adduct formation. Schematics of identifying probe adducts through reverse transcription and a) cDNA truncations or b) cDNA mutations. c) Chemical structures of adducts and how they are hypothesized to disrupt reverse transcription to result in cDNA mutations
The concept of cDNA mapping to RNA sequence has been extended to a full-transcriptome analysis. In this case, the extended cDNAs can be mapped to a reference transcriptome to obtain the reactivity profiles at each position. Full transcriptome-wide analysis has been performed in this way with DMS and SHAPE probing (discussed more below).[25–29]
In addition to RT-stop mapping, mutational profiling (MaP; Figure 2b and c) is an emergent methodology that takes advantage of mutations introduced during RT and is beginning to be employed to identify sites of chemical reactivity.[30–32] RT enzymes can have very high processivity, and under certain reaction conditions can process over a chemical adduct. When this takes place, the chemical modification can induce a mismatch between the modified RNA site and the nascent cDNA. Mutation rates are correlated with the number of adducts at a given position in the RNA. The key advantage of this approach is that on the same read, the mutation frequency can be normalized to the surrounding residues that are not mutated; as such, there is a built-in normalization to these experiments.
Data analysis of RT-stops can be more challenging and requires control RNA sequencing libraries (no reagent controls) to be used as input for normalization.[33] RT-stop frequency is compared against the reference input RNA sequencing libraries to calculate the rate of RT-stop and thus adduct formation. Lastly, the ability to identify multiple cDNA mutations in a single read (multiple reagent adduct positions) can be employed for correlative structure probing between two sites and the identification of reagent adduct sites on a single RNA molecule.[34,35] As such, MaP methods have dramatically increased the flexibility and data richness of structure probing methods, and their interpretation helps better understand RNA structure.
MaP approaches have found utility when used with many types of chemical reagents. A recent survey performed by us and others suggests that chemical modifications to the nucleobases themselves are more prone to induce mutations versus SHAPE probing, which reacts with the 2’OH position.[32] DMS probing can result in ~ 1.5% mutational frequency, SHAPE adducts are ~ 1%, and LASER adducts result in ~ 2% mutational frequency.[32,36,37] It is worth mentioning that different SHAPE electrophiles have similar mutational patterns, but smaller electrophiles, such as NAI and FAI, have higher mutational frequencies in comparison to 1 M7, which may be due to 1 M7’s larger RNA adduct.[38] The increased mutational frequency with nucleobase-modifying probes is likely due to the fact that chemical modifications on the nucleobases force the RT enzyme to form an incorrect pair when the W-C pairing is imperfect due to bulky adducts. In contrast, the 2’-OH modifications are farther away from the base pairing sites and will not result in a mutation (Figure 2b and c). Overall, these data suggest that MaP approaches are extremely powerful for identifying sites of probing adduct formation and data analysis. Using such approaches may be more straightforward for analyses of RNA structure.
Over time, more traditional cornerstone methods for chemical probing of RNA have been extended to transcriptome-wide analyses. Below, we outline some of the more exciting aspects of RNA structure probing that have been employed to gain insight into RNA regulation.
Chemical Approaches for Enriching Sites of Adduct Formation with Probing Reagents
Extending RNA structure probing methodologies from single RNA measurements to a full transcriptome introduces practical challenges that chemistry can help address. One major obstacle is the lack of chemical modification along transcripts. Single-hit modification rates of reagents onto probed RNAs are proposed to result in one modification per 300 nucleotides. Approximately 10–15% of all RNAs in a complex pool are actually modified (Figure 3). As most protocols rely on RT and identification of modification sites either by RT-stop or mutational analysis, such low hit rate would result in many of the RT-stop sites as being diluted from spurious RT-stops due to RT fall off. One way this has been overcome is through gel selection of truncated RT-stops in comparison to full-length RT extensions in negative control samples.[29] However, this approach still can suffer from signal to noise issues. As a result, recent efforts have focused on developing bifunctional reagents to probe RNA structure and enrich their sites of adduct formation.
Figure 3.
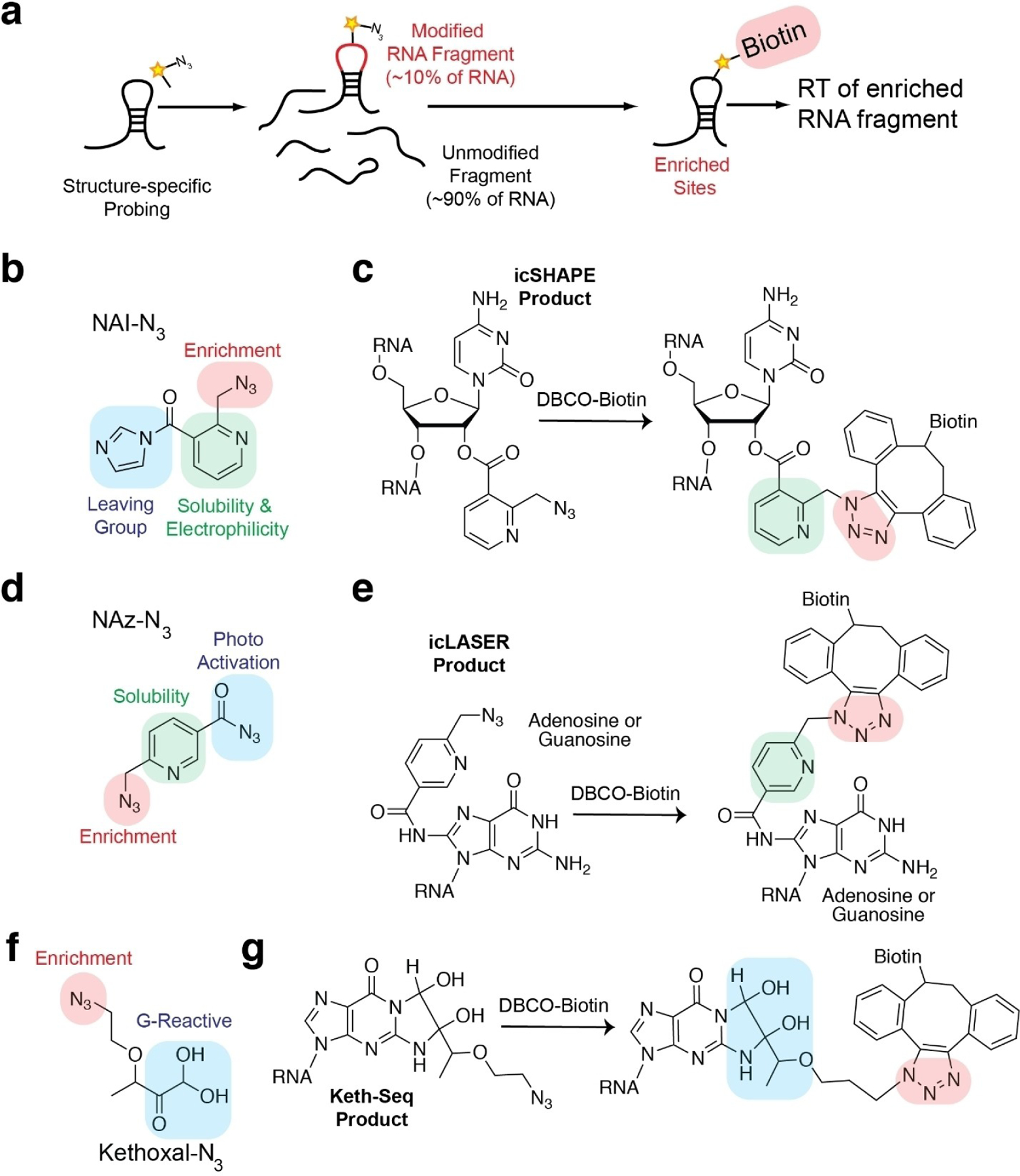
Chemical approaches to enrich modified sites in a complex RNA pool. a) Schematic of chemical probing of RNA structure and enrichment through the use of bi-functional probing reagents. b) Chemical structure of NAI-N3, a bi-functional reagent used for icSHAPE probing. c) Chemical structure of biotinylation reaction with NAI-N3 and DBCO-biotin for biotinylation of icSHAPE adduct sites. d) Chemical structure of NAz-N3, a bi-functional reagent used for icLASER probing. e) Chemical structure of biotinylation reaction with NAz-N3 and with DBCO-biotin for biotinylation of icLASER adduct sites. f) Chemical structure of kethoxal-N3, a bi-functional reagent used for icLASER probing. g) Chemical structure of biotinylation reaction with kethoxal-N3 and DBCO-biotin for biotinylation of keth-seq adduct sites.
The first approach in this area was through the development of a bi-functional SHAPE reagent, NAI-N3 (Figure 3b and c).[28] This approach was dubbed in vivo click SHAPE (icSHAPE), as NAI-N3 was designed with an acyl imidazole reactive site, a pyridine ring to increase electrophilicity and preserve the electrophilic center at the carbonyl, and an alkyl azide for subsequent enrichment. For enrichment, strain-promoted azide-alkyne cycloaddition (SPAAC) reactions were employed. SPAAC is preferred over more commonly used copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC), as CuAAC is known to produce radicals, which can result in RNA degradation. Following SPAAC, NAI-N3 enriched sites were also demonstrated to correspond to un-”clicked” NAI-N3 sites, further suggesting bi-functional modified RNA can be enriched and reverse transcribed for cDNA molecules, which can be sequenced for hydroxyl acylation analysis. Following the successful implementation of icSHAPE, two additional probes have been developed that work through similar post-probing protocols. These two probes measure purine nucleobase solvent accessibility and guanosine W-C base pairing.
Extension of LASER probing (C-8 purine solvent accessibility) has been accomplished using a new reagent, NAz-N3 (https://www.biorxiv.org/content/10.1101/2020.03.24.006866v2.full). In similar fashion to the NAI-N3 parent molecule, despite having two azido groups, NAz-N3 can be selectively activated by long wavelength UV light, while preserving the function of the alkyl azide for SPAAC biotinylation and subsequent enrichment (Figure 3d and e). This is due to the known differences in stability of azides. Aroyl azides can be activated by long wavelength UV light to reach an excited state. In contrast, alkyl azides are much more stable and require short wavelength UV light for activation. Tuning the light wavelength enables specific azide activation. Application of NAz-N3 through in vivo click LASER (icLASER) was demonstrated to probe RNA-protein interactions and polyadenylation.
Lastly, kethoxal probing has been employed using bi-functional reagents. Azido-kethoxal (N3-kethoxal) was observed to react specifically with the N1 and N2 positions at the W–C interface of guanines in single-stranded RNA.[39] This approach is now known as keth-seq (Figure 3f and g). The key chemical advancement to enable keth-seq was the synthesis of azido-kethoxal. Kethoxal and their derivatives are well-known to be incredibly challenging and proceed through oxidation reactions to obtain intermediates. The development of azido-kethoxal proceeded through mild oxidation of diazoketone and removal of chromatography steps that can be extremely limiting, as glyoxal and its analogues are sensitive to air. The development of keth-seq demonstrates the power of challenging chemical syntheses to arrive at a highly useful probe for RNA structure. Keth-seq has been employed to measure single-stranded RNA and also profile G-quadruplex RNA structures transcriptome-wide.
Biological Insight Garnered from Transcriptome-Wide Measurements of RNA Structure
Transcriptome-wide measurements of RNA structure have given unique biological insights into the transcriptome and how RNA structure contributes to its regulation. Several manuscripts have been published that take advantage of chemical probing to provide new or additional insight into the role of RNA structure in important biological problems. For example, there is growing interest in RNA molecules’ role in cellular complex phase transition.[40–42] RNA structure and changes in RNA structure due to such transitions have recently been explored with SHAPE, in which it was observed that structure-based RNA-RNA interactions promote assembly of distinct droplets and protein-driven, conformational dynamics of the RNA maintain this identity.[43] In-cell RNA structure probing of HIV-1 RNA genome with DMS-MaP-Seq probing has also shed light on the complexity of viral RNA processing and the ability of RNA structures to adopt alternative confirmations.[34] Through the development of novel computational methods to analyze RNA structure data and model alternative conformations, it was observed that heterogeneity in RNA conformation regulates splice-site use and viral gene expression. Finally, extending SHAPE-MaP analysis to the E. coli transcriptome also revealed how complex comparatively simple transcriptomes can be in terms of biological regulation.[44] SHAPE data revealed that mRNA structure remains similar between in-cell and cell-free environments and that RNA structure elements are highly conserved, indicating that RNA structure is responsible for regulating every gene in a complex transcript pool. These results establish the exciting utility of highly focused RNA structure probing and the incredibly important biological value such explorations add. In the following parts of this section, we will highlight additional new insights gathered from analyses of complex whole transcriptomes, and how unique aspects of RNA regulation have come to light due to structure methods.
Cleavage and polyadenylation of Pol II transcripts are critical for transcription termination, cytoplasmic localization, RNA stability, and translation. Selection of 3’-ends depends on the position of the polyadenylation sequence (PAS), which is typically AAUAAA or AUUAAA, and a downstream U/G-rich or U-rich motif. It is often thought that efficient processing requires a narrow range of 10–30 nt between the PAS and the cleavage-and-polyadenylation site (poly(A) site), often CA. However, analysis of mRNA sequences has demonstrated that some human mRNAs appear to use a PAS that falls > 30 nt upstream of the poly(A) site. A prevailing hypothesis from these analyses was that folded RNA structures brought the longer primary sequences of RNA into close proximity by folding into intricate three-dimensional RNA structures. To test this, Wu et al. utilized DMS structure probing followed by mutational analysis, or DMS-MaP (Figure 4a).[45] By priming the cDNA from the poly(A) tail, the authors were able to obtain high-quality analysis of RNA structures near the very 3’-end of the RNAs. DMS-MaP probing demonstrated that extended 3’-end lengths within the PAS region resulted in more-folded structure elements that condense the distance between the PAS site and poly(A) tail priming position (Figure 4b). This key insight, brought about by RNA structure probing, provided excellent evidence that mRNAs contain complex structure elements which contribute to RNA processing and the selection of poly(A) sites and cleavage.
Figure 4.
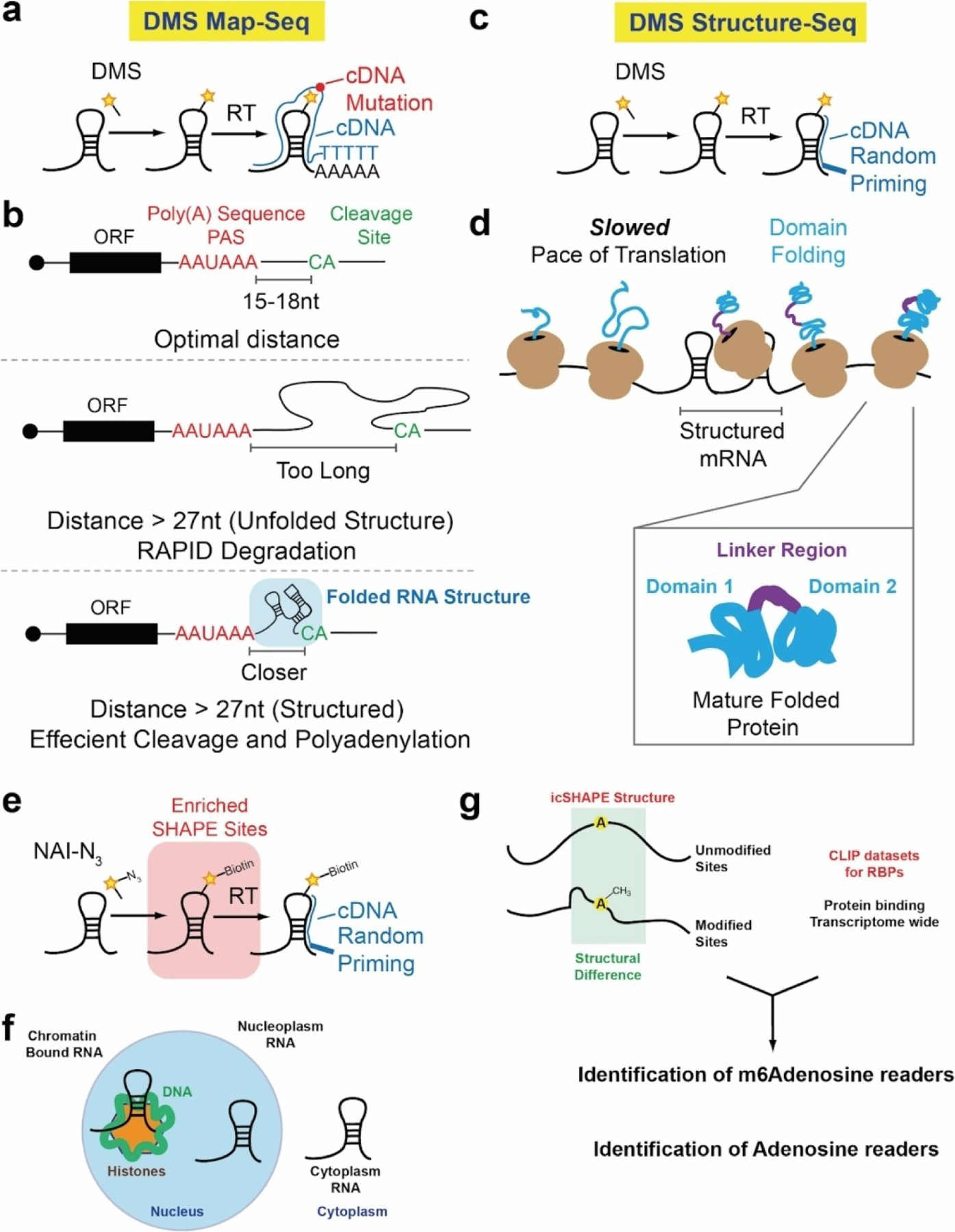
Biological insights gathered from transcriptome-wide analysis of RNA structure. a) DMS MaP-seq schematic. b) DMS MaP-seq reveals that folded RNA structures are responsible for bringing cleavage site (CA) and poly(A) sequences (PAS) into close proximity for efficient cleavage and polyadenylation. c) Schematic of DMS structure-seq. d) DMS structure-seq reveals that mRNA sequences associated with inter-domain regions in proteins have high structural complexity and integrity, which is hypothesized to slow down the rate of translation. This slow down enables the domains to fold correctly. e) Schematic of icSHAPE probing. f) icSHAPE was used to probe RNA structure in different parts of the cell. g) icSHAPE was used in combination with iCLIP datasets. iCLIP datasets profile transcriptome-wide protein occupancy. When used in combination at m6 A sites, icSHAPE was able to identify m6 A readers and adenosine readers specifically, based solely on the structure profile in combination with the CLIP data.
The control over gene expression, from RNA to protein, and the relationship between RNA structure and protein folding and/or translation rates have been topics of research for decades.[46,47] Transcriptome-wide measurements of RNA structure probing are starting to provide more insight into this relationship and have offered a prevailing model for how RNA structure controls protein synthesis: RNA structure is inversely related to protein structure. For example, the full transcriptome structure of HIV RNA viral genome revealed high levels of RNA structure and sequences that encode inter-domain loops in HIV proteins. This correlation suggests that RNA structure modulates ribosome elongation to promote native protein folding.[48] Extending these findings to a full transcriptome resulted in a very similar observation. Transcriptome-wide DMS structure probing (Figure 4c) was used to derive a relationship between mRNA structure and protein structure.[21] Regions of individual mRNAs that code for protein domains generally have higher reactivity to DMS (single-stranded) than regions that encode protein domain junctions. This relationship is prominent for proteins annotated for catalytic activity and reversed in proteins annotated for binding and transcription regulatory activity. From these results, it was postulated that decreased DMS reactivity of RNA regions that encode protein domain junctions may reflect increased RNA structure that may slow translation, allowing time for the nascent protein domain or ordered region of the protein to fold, thereby reducing protein misfolding (Figure 4d). This example highlights the use of structure probing to derive relationships between mRNAs and their encoded proteins may have evolved to allow efficient and accurate protein folding.
mRNA molecules traverse through many parts of the cell, from their birth in the nucleus to their processing and translation in the cytoplasm. During these processes they can interact with different proteins and fold into different RNA structure states. Conventional methods to probe RNA structure are done on a whole-cell level and cannot distinguish between different RNA structures in unique cellular compartments. Using icSHAPE, Sun et al.[49] (Figure 4e) probed the RNA structures in three cellular compartments: chromatin, nucleoplasm, and cytoplasm. The cytotropic structures substantially expand RNA structural information and enable detailed investigation of the central role of RNA structure in linking transcription, translation, and RNA decay – aspects of an RNA lifetime that are unique to an RNA’s position in a cell (Figure 4f). In addition, this approach identified unique RNA binding proteins that are responsible for regulating m6 A RNA modification depending on an RNA’s position in the cell. This example highlights the use of transcriptome-wide SHAPE probing to understand dynamic RNA structures and its functional importance in gene regulation (Figure 4g).
Conclusion
Chemical methods to measure RNA structure have been maturing since the mid-1980s. Recent efforts have been focused on extending the chemical reactions which can be utilized to probe RNA structure, with the goal of ultimately transitioning those approaches to the natural environment of the cell. In parallel, extending one-RNA-at-a-time approaches to whole-transcriptome analyses is being pursued, with changing methods to identify RNA-chemical adducts and ways to process and analyze adducts in the context of RNA folding. Exciting insights into RNA structure and its role in controlling RNA biology have been made, but there is still much work to be done. For example, intersecting RNA structure measurements with whole-transcriptome CLIP (protein-RNA crosslinking)[50] data would be invaluable for predicting the RNA-protein interface for many RNAs in parallel. In addition, integrating RNA structure probing experiments of many types could complement experiments investigating aspects of RNA processing, such as transcription, polyadenylation, and translation initiation, providing structural context for these measurements. The continuing advancement of chemical methods merged with more accurate and comprehensive transcriptome-wide RNA structure probing measurements is sure to extend our understanding of RNA structure and function in normal cells and disease.
Biography

Whitney E. EnglandDr. England received her Ph.D in Microbiology from the University of Illinois at Urbana-Champaign in 2016, where she was an NIH T32 trainee studying infection biology in microbial populations with Dr. Rachel Whitaker. She became a research specialist at UC Irvine and joined the labs of Dr. Katrine Whiteson, where she studied microbial populations in cystic fibrosis patients, and Dr. Robert Spitale, where she studies RNA biology. Her research currently involves applying bioinformatics to investigate diverse aspects of RNA biology, including novel chemical techniques for RNA structure determination and RNA in transplanted human microglia.

Chely M. Garfio completed her bachelor’s degree in biochemistry with a minor in philosophy at the University of California, Los Angeles. Her undergraduate research focused on microRNA biogenesis. Soon after, she obtained her master’s degree in Biochemistry at California State University, Los Angeles. During the program, she studied the molecular mechanism of protein enhancers. Currently, she is a graduate student in the Spitale lab at the University of California, Irvine. Her current work focuses on using chemical probes as tools for RNA structure probing methods.

Dr. Spitale received his Ph.D. degree in Chemistry at the University of Rochester in 2009 as an Elon Huntington Hooker Fellow. He then transitioned to postdoctoral studies at Stanford University and was awarded the A.P. Giannini Fellowship to support his research. Dr. Spitale joined UCI Pharmaceutical Sciences department in 2014, being promoted to Professor this year. His research focuses on developing novel chemical and bioinformatic approaches toward understanding the role of RNA structure and function in normal biology as well as disease. He has received numerous awards for his research contributions, including: the NIH Director’s New Innovator Award, Pew Biomedical Research Fellowship, an American Cancer Society IRG Award, a W.M. Keck Medical Research Grant Award (w/Professor John Chaput), the Ono Pharmaceuticals Breakthrough Science Award, and was recently named the Rising Star by the International Chemical Biology Society.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Yin Y, Lu JY, Zhang X, Shao W, Xu Y, Li P, Hong Y, Cui L, Shan G, Tian B, Zhang QC, Shen X, Nature 2020, 580, 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sun X, Wang Z, Hall JM, Perez-Cervantes C, Ruthenburg AJ, Moskowitz IP, Gribskov M, Yang XH, PLoS Comput. Biol 2020, 16, e1007119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Adereth Y, Dammai V, Kose N, Li R, Hsu T, Nat. Cell Biol 2005, 7, 1240–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sun Q, Hao Q, Prasanth KV, Trends Genet 2018, 34, 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Renoux AJ, Todd PK, Prog. Neurobiol 2012, 97, 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhou W, Pan B, Liu L, Oncol. Lett 2019, 18, 3705–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Do H, Kim W, Genomics Inform 2018, 16, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Png KJ, Halberg N, Yoshida M, Tavazoie SF, Nature 2011, 481, 190–194. [DOI] [PubMed] [Google Scholar]
- [9].Miao Z, Westhof E, Annu. Rev. Biophys 2017, 46, 483–503. [DOI] [PubMed] [Google Scholar]
- [10].Bevilacqua PC, Ritchey LE, Su Z, Assmann SM, Annu. Rev. Genet 2016, 50, 235–266. [DOI] [PubMed] [Google Scholar]
- [11].Strobel EJ, Yu AM, Lucks JB, Nat. Rev. Genet 2018, 19, 615–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tijerina P, Mohr S, Russell R, Nat. Protoc 2007, 2, 2608–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Moazed D, Stern S, Noller HF, J. Mol. Biol 1986, 187, 399–416. [DOI] [PubMed] [Google Scholar]
- [14].Maxam AM, Gilbert W, Proc. Natl. Acad. Sci. U. S. A 1977, 74, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mitchell D 3rd, Ritchey LE, Park H, Babitzke P, Assmann SM, Bevilacqua PC, RNA 2018, 24, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mitchell D 3rd, Renda AJ, Douds CA, Babitzke P, Assmann SM, Bevilacqua PC, RNA 2019, 25, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang PY, Sexton AN, Culligan WJ, Simon MD, RNA 2019, 25, 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wilkinson KA, Merino EJ, Weeks KM, Nat. Protoc 2006, 1, 1610–1616. [DOI] [PubMed] [Google Scholar]
- [19].Chan D, Feng C, Zhen Y, Flynn RA, Spitale RC, Biochemistry 2017, 56, 1811–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY, Nat. Chem. Biol 2013, 9, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ingle S, Azad RN, Jain SS, Tullius TD, Nucleic Acids Res 2014, 42, 12758–12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Feng C, Chan D, Joseph J, Muuronen M, Coldren WH, Dai N, Corrêa IR Jr., Furche F, Hadad CM, Spitale RC, Nat. Chem. Biol 2018, 14, 325. [DOI] [PubMed] [Google Scholar]
- [23].Ziehler WA, Engelke DR, Curr. Protoc. Nucleic Acid Chem 2001, 0, 6.1.1–6.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Woodson SA, Koculi E, Methods Enzymol 2009, 469, 189–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Incarnato D, Neri F, Anselmi F, Oliviero S, Genome Biol 2014, 15, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ritchey LE, Su Z, Assmann SM, Bevilacqua PC, Methods Mol. Biol 2019, 1933, 305–341. [DOI] [PubMed] [Google Scholar]
- [27].Ding Y, Kwok CK, Tang Y, Bevilacqua PC, Assmann SM, Nat. Protoc 2015, 10, 1050–1066. [DOI] [PubMed] [Google Scholar]
- [28].Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, Chang HY, Nature 2015, 519, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS, Nature 2014, 505, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Smola MJ, Weeks KM, Nat. Protoc 2018, 13, 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Smola MJ, Rice GM, Busan S, Siegfried NA, Weeks KM, Nat. Protoc 2015, 10, 1643–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zinshteyn B, Chan D, England W, Feng C, Green R, Spitale RC, Nucleic Acids Res 2019, 47, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ritchey LE, Su Z, Tang Y, Tack DC, Assmann SM, Bevilacqua PC, Nucleic Acids Res 2017, 45, e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tomezsko PJ, Corbin VDA, Gupta P, Swaminathan H, Glasgow M, Persad S, Edwards MD, Mcintosh L, Papenfuss AT, Emery A, Swanstrom R, Zang T, Lan TCT, Bieniasz P, Kuritzkes DR, Tsibris A, Rouskin S, Nature 2020, 582, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Krokhotin A, Mustoe AM, Weeks KM, Dokholyan NV, RNA 2017, 23, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Siegfried NA, Busan S, Rice GM, Nelson JAE, Weeks KM, Nat. Methods 2014, 11, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, Rouskin S, Nat. Methods 2017, 14, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Guo LT, Adams RL, Wan H, Huston NC, Potapova O, Olson S, Gallardo CM, Graveley BR, Torbett BE, Pyle AM, J. Mol. Biol 2020, 432, 3338–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Weng X, Gong J, Chen Y, Wu T, Wang F, Yang S, Yuan Y, Luo G, Chen K, Hu L, Ma H, Wang P, Zhang QC, Zhou X, He C, Nat. Chem. Biol 2020, 16, 489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jacobs WM, Frenkel D, Biophys. J 2017, 112, 683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shin Y, Brangwynne CP, Science 2017, 357, eaaf4382. [DOI] [PubMed] [Google Scholar]
- [42].Choi JM, Holehouse AS, Pappu RV, Annu. Rev. Biophys 2020, 49, 107–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Langdon EM, Qiu Y, Niaki AG, McLaughlin GA, Weidmann CA, Gerbich TM, Smith JA, Crutchley JM, Termini CM, Weeks KM, Myong S, Gladfelter AS, Science 2018, 360, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mustoe AM, Busan S, Rice GM, Hajdin CE, Peterson BK, Ruda VM, Kubica N, Baryza JL, Weeks KM, Cell 2018, 173, 181–195.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wu X, Bartel DP, Cell 2017, 169, 905–917.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kozak M, J. Biol. Chem 1991, 266, 19867–19870. [PubMed] [Google Scholar]
- [47].Gebauer F, Hentze MW, Nat. Rev. Mol. Cell Biol 2004, 5, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr., Swanstorm R, Burch CL, Weeks KM, Nature 2009, 460, 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sun L, Fazal FM, Li P, Broughton JP, Lee B, Tang L, Huang W, Kool ET, Chang HY, Zhang QC, Nat. Struct. Mol. Biol 2019, 26, 322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, Stanton R, Rigo F, Guttman M, Yeo GW, Nat. Methods 2016, 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]