

Abstract
Network biology is useful for modeling complex biological phenomena; it has attracted attention with the advent of novel graph-based machine learning methods. However, biological applications of network methods often suffer from inadequate follow-up. In this perspective, we discuss obstacles for contemporary network approaches—particularly focusing on challenges representing biological concepts, applying machine learning methods, and interpreting and validating computational findings about biology—in an effort to catalyze actionable biological discovery.
Keywords: networks, knowledge graphs, embeddings, interpretability, biological validation
Networks: A Useful But Limited Abstraction
With over 700 publicly available pathway and molecular interaction databases [4, 5], it is difficult to choose the right network. Networks can model biological systems at levels ranging from molecular to population-scale [6–13], where edges typically represent interactions between nodes corresponding to biological entities (drugs, genes, proteins, diseases, etc.; see [14, 15] for comprehensive reviews of graph theory applied to biological applications).
Biological networks are often incomplete [16, 17]. The missingness of protein–protein interaction (PPI) data is as much as 80% [18]. Even with high-throughput datasets [19–21], building accurate and comprehensive network models is a behemoth task.
The first step to ensure network quality is proper documentation of process and metadata annotation. The second step is to evaluate the network’s ability to recapitulate known interactions; manual curation is the typical gold standard [22]. A silver standard is the corroboration of interactions derived from orthogonally curated experimental sources, as done with PCNet [23]. Finally, another means of ensuring network specificity is removing potential false positive interactions due to experimental artifact. CRAPome is a contaminant repository for mass spectrometry (AP–MS) experiments used to build PPI networks that provides putative negative interaction data. Each of these approaches can increase confidence in the accuracy of new networks.
To address the issue of sparsity, networks are often aggregated from independent data sources to form a more comprehensive ‘interactome’ [24]. However, integrating heterogeneous information into a homogeneous network abstracts away biological nuance, such as cell-type specificity [8], spatial [25] and temporal [26] resolution or environmental factors [27], and so precision suffers. In addition, PPI networks are inherently biased [28, 29] by the characteristics of experimental methods as well as external factors such as funding biases—these may make heavily-studied proteins appear to have artificially high degree in networks.
One potential solution to the problem of heterogeneous data is to use attributed knowledge graphs [30]—edges are qualified by specific semantic relations between nodes and can record relevant attributes such as the ‘confidence’ in a relationship. These graphs are able to capture nuance that qualifies ‘known’ knowledge in the network [31, 32]. These techniques have not been broadly applied to molecular biology, and machine learning methods for these heterogeneous models are needed.
A host of network-based biological models can capture dynamic relationships, particularly the relationship between genes, proteins and other cellular entities in gene regulatory networks (GRNs; [33]). GRNs are flexible and enable temporal representation of node states that incorporate uncertainty in stochastic (as opposed to deterministic) models, thus making them amenable to Boolean [34, 35] and Bayesian network approaches [36]. These networks have been used to model dynamic cellular behavior [37–41]. Other architectures for dynamic models include differential equations [42], neural nets [43] and information theory-based approaches [44], all of which use gene expression data under differing experimental conditions to capture a system’s behavior in response to perturbations. The number of perturbational datasets and parameters required to accurately recapitulate a system is a combinatorial optimization problem [37], making it computationally difficult to kinetically model full-scale networks. Increasing computing power and the proliferation of large-scale sequencing datasets may enable more tractable modeling of the dynamics of biological systems at scale.
Furthering Biologically Principled Inference Over Networks
A major force driving the explosion of network biology is the availability of network-based machine learning methods to biological problems [1, 2, 45–47]. These have often been framed as the tasks of link prediction, community detection and network alignment [48]; comprehensive reviews [49, 50] survey applications of these network inference methods. Without diligence, however, the mapping from biological questions to neat network methods may be unprincipled and suffer from inadequate biological follow-up [3].
A key issue when using network inference methods is the quantity and quality of data used for training; however, systematic evaluations of the sensitivity of results to these parameters are rare. Huang et al. [23] studied the ability of different network topologies to recapitulate known disease gene sets using a network propagation approach [51]. They concluded that larger networks, such as STRINGdb [32], yield the best performance but observe diminishing returns in the size of the network. In addition, Menche et al. [18] used percolation theory (which describes the behavior of clustered components in networks as one randomly adds or removes edges) to draw connections between network sparsity and utility for biological tasks; they proposed heuristics about which disease gene sets might form identifiable modules in the network and their potential utility for applications.
Machine learning methodologies that use vectorized representations of graphs present opportunities and challenges when ported to biology. Recently, network embedding methods, whereby low-dimensional representations of network structures are learned, have become popular in network biology due to their power and flexibility [52]. In addition, graph-based representation learning has become popular in deep learning-based frameworks for inference over networks [53]. These methods, however, have limitations. First, the network embedding strategy must be relevant in the context of a biological question. For instance, if nodes are embedded based on local network topology, then the biological problem should depend strongly on topology alone, since other features are not captured in this embedding. Second, embedding methods usually include simplifying assumptions, for example regarding transitive and semantic matching [54], which may limit their ability to capture symmetric, inversion and compositional properties, all of which may be biologically relevant. Finally, many embedding methods offer no biological interpretation to explain predictions, which limits their broader utility to biologists, although work in this space is emerging [55, 56].
Closing the Loop with Biological Validation
In the machine learning community, validation typically entails data partitioning followed by testing on a held-out dataset containing gold standard interactions. Although this can lead to reproducible results, it has drawbacks. First, in network theory, the idea of a truly isolated, held-out partition of data is difficult to implement. Cross-validation via edge removal across the network removes key network structural features, thus biasing algorithmic evaluation [57]. Second, biological gold standard data are incomplete, and ‘truly negative’ relationships are difficult to define [58]. Therefore, it is critical to validate on a variety of sources and use metrics that are robust to the level of missing data. Cross-validation across multiple networks may reduce specific network bias. However, given that networks often share a common underlying structure and content, purely computational validation may not distinguish true biological discovery from sensitive informational retrieval. In biology, independent and prospective experimental validation remains the only generally agreed-upon gold standard.
Indeed, the strongest form of validation comes from experimental and/or clinical evidence that support network-generated hypotheses. Drug repurposing studies propose drugs that can be examined by subject matter experts and validated by in vitro drug screens or even clinical trials [22]. However, these efforts are rare due to cost (time and money). Case studies can demonstrate biological applicability [59], but these studies can only provide incremental evidence of biological validity.
Biologists routinely expect that computational models produce inference that are mechanistically grounded and experimentally confirmable. ‘Interpretable machine learning’ seems desirable but is ill-defined [60]. For network biology, interpretability has two facets. ‘Representational interpretability’ is the ease of mapping biological abstractions to computational abstractions. It defines the scope of information represented by the network; capturing nuance such as cell-type, dynamics and directionality yields representations that are more faithful to underlying biology [61–64]. ‘Algorithmic interpretability’ is the ability to generate traceable features sets that support a biological hypothesis. For instance, in link prediction tasks over knowledge graphs, the capacity to find paths of known biological relations might serve as a form of deductive reasoning to support generated hypotheses [46, 65].
The pipeline from computational exploration to biological validation is not a linear path but rather an iterative process, wherein each step must be closely aligned with fundamental biological principles (Figure 1). We are optimistic that by first ensuring robust and relevant mappings to biological concepts, network methods will generate impactful insights that will accelerate progress in biological discovery.
Figure 1 .
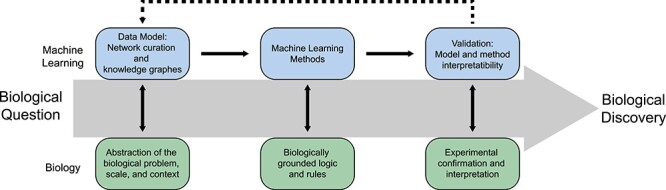
A harmonious research pipeline for network methods in machine learning applied to biology.
Key Points
The promise of network tools for biological discovery is great, albeit the field is filled with addressable computational and validation challenges.
Heterogenous network models, such as knowledge graphs, are needed to capture the growing number of literature-based and structured biological datasets and can provide context and metadata for properly qualifying our biological models.
The availability of more computationally powerful hardware allows cross-validating and testing on multiple networks and thus reduces specific network bias while enabling better empirical ‘null’ models used to assess significance within methods.
Machine learning methods for more complex, heterogenous network models are still needed.
Acknowledgements
We would like to thank members of the Altman lab for their helpful discussion.
Margaret Guo is an MD–PhD Candidate in Biomedical Informatics.
Daniel Sosa is a PhD Candidate in Biomedical Informatics at Stanford University.
Russ Altman is the Kenneth Fong Professor of Bioengineering, Genetics, Medicine, Biomedical Data Science and (by courtesy) Computer Science at Stanford University.
Contributor Information
Margaret G Guo, Stanford Program in Biomedical Informatics, Stanford University, Stanford, CA, USA; Program in Epithelial Biology, Stanford University, Stanford, CA, USA.
Daniel N Sosa, Stanford Program in Biomedical Informatics, Stanford University, Stanford, CA, USA.
Russ B Altman, Department of Bioengineering, Stanford University, Stanford, CA, USA; Department of Genetics, Stanford University, Stanford, CA, USA.
Conflict of Interest
Authors declare that they have no competing interests.
Funding
This work was supported by the National Institutes of Health (grant numbers GM102365, LM007033 and GM007365).
References
- 1. Niepert M, Ahmad M, Kutzkov K. Learning convolutional neural networks for graphs. 33rd Int Conf Mach Learn ICML 2016;2016:4. [Google Scholar]
- 2. Grover A, Leskovec J. node2vec: Scalable Feature Learning for Networks, 2016. [DOI] [PMC free article] [PubMed]
- 3. Nelder JA. Statistics, science and technology. J R Stat Soc Ser A 1986;149:109–21. [Google Scholar]
- 4. Bader GD, Cary MP, Sander C. Pathguide: a pathway resource list. Nucleic Acids Res 2006;34:D504–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bader G, Donaldson S. Pathguide: the pathway resource list, 2013.
- 6. Aghamirzaie D, Collakova E, Li S, et al. CoSpliceNet: a framework for co-splicing network inference from transcriptomics data. BMC Genomics 2016;17:845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beltrao P, Cagney G, Krogan NJ. Quantitative genetic interactions reveal biological modularity. Cell 2010;141:739–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greene CS, Krishnan A, Wong AK, et al. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet 2015;47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guven-Maiorov E, Tsai CJ, Nussinov R. Structural host-microbiota interaction networks. PLoS Comput Biol 2017;13:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lei X, Wang F, Wu FX, et al. Protein complex identification through Markov clustering with firefly algorithm on dynamic protein-protein interaction networks. Inf Sci (Ny) 2016;329:303–16. [Google Scholar]
- 11. Maulik U, Basu S, Ray S. Identifying protein complexes in PPI network using non-cooperative sequential game. Sci Rep 2017;7:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mehla J, Caufield JH, Uetz P. The yeast two-hybrid system: a tool for mapping protein-protein interactions. Cold Spring Harb Protoc 2015;2015:425–30. [DOI] [PubMed] [Google Scholar]
- 13. Teichmann SA, Babu MM. Gene regulatory network growth by duplication. Nat Genet 2004;36:492–6. [DOI] [PubMed] [Google Scholar]
- 14. Pavlopoulos GA, Secrier M, Moschopoulos CN, et al. Using graph theory to analyze biological networks. BioData Min 2011;4:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koutrouli M, Karatzas E, Paez-Espino D, et al. A guide to conquer the biological network era using graph theory. Front Bioeng Biotechnol 2020;0:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee TI, Rinaldi NJ, Robert F, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 2002;298:799–804. [DOI] [PubMed] [Google Scholar]
- 17. Schuster S, Fell DA, Dandekar T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat Biotechnol 2000;18:326–32. [DOI] [PubMed] [Google Scholar]
- 18. Menche J, Sharma A, Kitsak M, et al. Uncovering disease-disease relationships through the incomplete interactome. Science (80-) 2015;347:1257601–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis CA, Hitz BC, Sloan CA, et al. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 2018;46:D794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, et al. . Genetic effects on gene expression across human tissues. Nature 2017;550:204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Wspolczesna Onkol 2015;1A:A68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thorn CF, Klein TE, Altman RB. PharmGKB: the pharmacogenomics Knowledge Base. Methods Mol Biol 2013;1015:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. JK H, DE C, MK Y, et al. Systematic evaluation of molecular networks for discovery of disease genes. Cell Syst 2018;6:484–495.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vidal M, Cusick ME, Barabási AL. Interactome networks and human disease. Cell 2011;144:986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Secrier M, Schneider R. Visualizing time-related data in biology, a review. Brief Bioinform 2013;15:771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Decalf J, Albert ML, Ziai J. New tools for pathology: a user’s review of a highly multiplexed method for in situ analysis of protein and RNA expression in tissue. J Pathol 2019;247:650–61. [DOI] [PubMed] [Google Scholar]
- 27. Tagkopoulos I, Liu Y-C, Tavazoie S. Predictive behavior within microbial genetic networks. Science (80-) 2008;320:1313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gillis J, Ballouz S, Pavlidis P. Bias tradeoffs in the creation and analysis of protein-protein interaction networks. J Proteomics 2014;100:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Skinnider MA, Stacey RG, Foster LJ. Genomic data integration systematically biases interactome mapping. PLoS Comput Biol 2018;14:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan J, Wang C, Cheng W, et al. A retrospective of knowledge graphs. Front Comput Sci 2018;12:1–20. [Google Scholar]
- 31. Percha B, Altman RB. A global network of biomedical relationships derived from text. Bioinformatics 2018;34:2614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen L, Kulasiri D, Samarasinghe S. A novel data-driven Boolean model for genetic regulatory networks. Front Physiol 2018;0:1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schwab JD, Kühlwein SD, Ikonomi N, et al. Concepts in Boolean network modeling: what do they all mean? Comput Struct Biotechnol J 2020;18:571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kauffman SA. Metabolic stability and epigenesis in randomly constructed genetic nets. J Theor Biol 1969;22:437–67. [DOI] [PubMed] [Google Scholar]
- 36. N F, M L, I N, et al. Using Bayesian networks to analyze expression data. J Comput Biol 2000;7:601–20. [DOI] [PubMed] [Google Scholar]
- 37. Hecker M, Lambeck S, Toepfer S, et al. Gene regulatory network inference: data integration in dynamic models—a review. Biosystems 2009;96:86–103. [DOI] [PubMed] [Google Scholar]
- 38. Albert R. Network inference, analysis, and modeling in systems biology. Plant Cell 2007;19:3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klein C, Marino A, Sagot M-F, et al. Structural and dynamical analysis of biological networks. Brief Funct Genomics 2012;11:420–33. [DOI] [PubMed] [Google Scholar]
- 40. Chai LE, Loh SK, Low ST, et al. A review on the computational approaches for gene regulatory network construction. Comput Biol Med 2014;48:55–65. [DOI] [PubMed] [Google Scholar]
- 41. Zhao M, He W, Tang J, et al. A comprehensive overview and critical evaluation of gene regulatory network inference technologies. Brief Bioinform 2021;2021:1–15. [DOI] [PubMed] [Google Scholar]
- 42. Gebert J, Radde N, Weber GW. Modeling gene regulatory networks with piecewise linear differential equations. Eur J Oper Res 2007;181:1148–65. [Google Scholar]
- 43. Vohradsky J. Neural network model of gene expression. FASEB J 2001;15:846–54. [DOI] [PubMed] [Google Scholar]
- 44. Song L, Langfelder P, Horvath S. Comparison of co-expression measures: mutual information, correlation, and model based indices. BMC Bioinforma 2012;13:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Das R, Dhuliawala S, Zaheer M, et al. Go for a walk and arrive at the answer: Reasoning over paths in knowledge bases using reinforcement learning. In: 6th Int. Conf. Learn. Represent. ICLR 2018- Conf. Track Proc, 2018.
- 46. Sang S, Yang Z, Wang L, et al. SemaTyP: a knowledge graph based literature mining method for drug discovery. BMC Bioinformatics 2018;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. West DB. Introduction to graph theory (2nd edition). Vaccine 2012;43:1–588. [Google Scholar]
- 48. Ideker T, Nussinov R. Network approaches and applications in biology. PLoS Comput Biol 2017;13:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu C, Ma Y, Zhao J, et al. Computational network biology: data, models, and applications. Phys Rep 2020;846:1–66. [Google Scholar]
- 50. Nelson W, Zitnik M, Wang B, et al. To embed or not: network embedding as a paradigm in computational biology. Front Genet 2019;0:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Köhler S, Bauer S, Horn D, et al. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet 2008;82:949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hamilton WL, Ying R, Leskovec J. Representation Learning on Graphs: Methods and Applications. IEEE Data Eng Bull 2017;40(3):52–74. [Google Scholar]
- 53. Li MM, Huang K, Zitnik M. Representation Learning for Networks in Biology and Medicine: Advancements, Challenges, and Opportunities. arXiv preprint arXiv:2104.04883, 2021.
- 54. Wang Q, Mao Z, Wang B, et al. Knowledge graph embedding: a survey of approaches and applications. IEEE Trans Knowl Data Eng 2017;29:2724–43. [Google Scholar]
- 55. Veličković P, Casanova A, Liò P, et al. Graph attention networks. In: 6th Int. Conf. Learn. Represent. ICLR 2018- Conf. Track Proc, 2018.
- 56. Brasoveanu A, Moodie M, Agrawal R. GNN explainer: a tool for post-hoc explanation of graph neural networks. CEUR Workshop Proc. 33rd Conference on Neural Information Processing Systems (NeurIPS 2019), Vancouver, Canada, 2020;2657. [Google Scholar]
- 57. Tabe-Bordbar S, Emad A, Zhao SD, et al. A closer look at cross-validation for assessing the accuracy of gene regulatory networks and models. Sci Rep 2018;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schrynemackers M, Küffner R, Geurts P. On protocols and measures for the validation of supervised methods for the inference of biological networks. Front Genet 2013;4:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sirota M, Dudley JT, Kim J, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med 2011;3:96ra77:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lipton ZC. The mythos of model interpretability. Commun ACM 2018;61:36–43. [Google Scholar]
- 61. Carrera J, Covert MW. Why build whole-cell models? Trends Cell Biol 2015;25:719–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Karr JR, Sanghvi JC, MacKlin DN, et al. A whole-cell computational model predicts phenotype from genotype. Cell 2012;150:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Terzer M, Maynard ND, Covert MW, et al. Genome-scale metabolic networks. Wiley Interdiscip Rev Syst Biol Med 2009;1:285–97. [DOI] [PubMed] [Google Scholar]
- 64. Covert MW, Knight EM, Reed JL, et al. Integrating high-throughput and computational data elucidates bacterial networks. Nat 2004;429:92–6. [DOI] [PubMed] [Google Scholar]
- 65. Sosa DN, Derry A, Guo M, et al. A literature-based knowledge graph embedding method for identifying drug repurposing opportunities in rare diseases. Pacific Symp Biocomput 2020;25:463–74. [PMC free article] [PubMed] [Google Scholar]