

Abstract
High-throughput next-generation sequencing now makes it possible to generate a vast amount of multi-omics data for various applications. These data have revolutionized biomedical research by providing a more comprehensive understanding of the biological systems and molecular mechanisms of disease development. Recently, deep learning (DL) algorithms have become one of the most promising methods in multi-omics data analysis, due to their predictive performance and capability of capturing nonlinear and hierarchical features. While integrating and translating multi-omics data into useful functional insights remain the biggest bottleneck, there is a clear trend towards incorporating multi-omics analysis in biomedical research to help explain the complex relationships between molecular layers. Multi-omics data have a role to improve prevention, early detection and prediction; monitor progression; interpret patterns and endotyping; and design personalized treatments. In this review, we outline a roadmap of multi-omics integration using DL and offer a practical perspective into the advantages, challenges and barriers to the implementation of DL in multi-omics data.
Keywords: deep learning, multi-omics, data integration, imputation, missing value, harmonization, risk prediction, precision medicine
Graphical Abstract
Graphical Abstract.
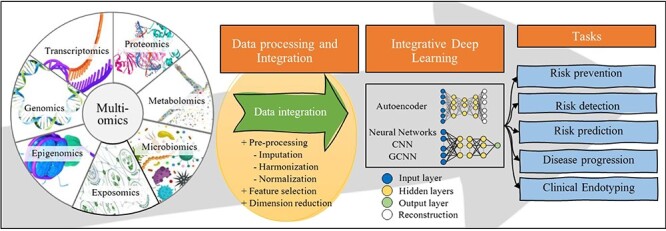
Introduction
Technological advances of high-throughput assays allow for hundreds of thousands of experimental samples to be processed simultaneously generating millions and billions of data points in various fields, including biology. The pathogenesis of complex diseases involves several cascades of events at various levels of omics, including the transcriptomics of gene expression and epigenomics of gene regulation, as well as proteomics and metabolomics, which may have direct effects on disease endotypes. To date, however, these different types of data have been analyzed independently and account for only a fraction of the estimated disease heritability. In addition, despite considerable effort towards the statistical integration of these data types [1, 2], most multi-omics analyses are based on conventional statistical approaches, such as logistic regression and support vector machine methods [1–5], and few studies have addressed the role of multi-omics resources to investigate complex diseases (Figure 1).
Figure 1.
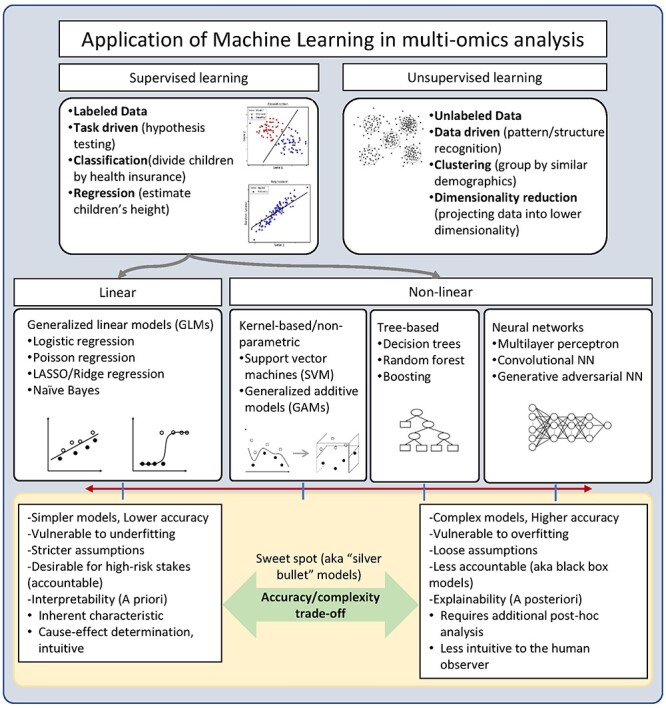
Application of machine learning in multi-omics analysis. Machine learning algorithms mainly consist of supervised learning and unsupervised learning, based on the availability of labeling on data. Linear and nonlinear patterns of multi-omics data can be captured in various machine learning algorithms.
The amount of available biological data has increased exponentially since the emergence of high-throughput technologies, such as microarrays and next-generation sequencing [6]. The generation of such large amounts of data in biomedicine requires the application of advanced informatics techniques in order to extract new insights and expand current knowledge about diseases, as well as to improve diagnosis and design personalized treatments. In this context, DL algorithms have become one of the most promising methods in the area [7].
Most statistical integrative analyses of large-scale biological data are either meta-analyses of the same type of data from different sources or analyses of different types of data from the same source, with consecutive pipelines that analyze each type of omics data independently (or in a cascading manner), to identify significant factors and then combine them for the final analysis [8]. These often fail to capture any nonlinearly associated multi-omics factors or interaction effects among multi-omics data. Advanced machine learning-based multi-omics approaches, on the other hand, exploit the synergism in multi-omics data by (1) identifying the complex interaction of multi-omics data using network analysis; (2) predicting clinical outcomes with high accuracy; (3) inferring the high-level biological representations of canonical variables of multi-omics using matrix factorization, partial least squares and canonical correlation analysis and (4) discovering disease subtypes using clustering and classification methods.
Recently, advances in machine learning algorithms have led deep learning (DL) [also called deep neural network, or artificial neural network (ANN)]. DL is a subset of machine learning, in which multi-layered neural networks learn from vast amount of data. DL algorithms not only analyze each omics type separately but also have the opportunity to integrate different omics layers, including data from clinical or health records, with great sensitivity, specificity and efficiency [9]. DL is a self-teaching artificial intelligence method, which does not rely on fixed mathematical formulas or programming IF statements to predict. DL uses larger numbers of hidden layers, whereas traditional ANNs can normally only afford one or two hidden layers. The deeper the layer, the more it can learn complex patterns and be accurate in making predictions. DL can encode and learn from heterogeneous and complex data, in both supervised and unsupervised settings. In recent years, DL has been the method of choice in various machine learning communities, such as image analysis, speech recognition and natural language processing.
In the context of biomedical research, there has been increasing interest in DL applications in omics data analysis. Omics data analysis is frequently impeded by low signal to noise ratios, as well as datasets with large number of variables and relatively small number of samples or large analytical variance. In this context, DL techniques have already outperformed previous statistical and non-DL methods in terms of sensitivity, specificity and efficiency [9]. In addition, DL algorithms not only have the capacity to analyze each data type separately but also to integrate different omics types, or even other sources of information, such as medical images and clinical health records. DL algorithms implement various integration strategies by allowing one to design network architectures in a flexible and explicit manner. This big data analysis and integration is fueling the implementation of personalized medicine approaches, allowing for the early detection and classification of diseases, or personalized therapies for each patient depending on their biochemical backgrounds.
There are several features that make the DL method a potential approach for multi-omics data analysis, and DL has already been shown to improve predictive performance in several supervised and unsupervised learning problems, including feature selection/reduction, clinical outcome prediction, survival analysis and disease subtyping. The capability of DL to capture nonlinear features without kernel tricks, along with interaction effects and hierarchical representations through multi-layered neural network architectures, are some of DL’s main benefits prevalently observed in biological systems [10–12]. In this review, we introduce multi-omics data types, as well as integration analyses with in-depth biological understanding, and recent advances in multi-omics integration, using DL models on multi-omics data, as well as discuss future research directions. The remainder of this review is structured as follows: (1) a brief overview of multi-omics data is introduced in Multi-omics Data; (2) several integrative DL models are explored in Multi-omics Data Integrative Analysis Using Deep Learning; (3) the challenges and opportunities of DL in a multi-omics data framework are addressed in Challenges and Opportunities and (4) finally, we outline DL methods to omics data analysis, with a focus on the types of analyses, as well as challenges and opportunities in precision medicine.
Multi-omics data
Multi-omics data include genome, transcriptome, epigenome, proteome, exposome and microbiome (Box 1 and Figure 2). Genomics is the study of the complete set of genes of an organism. The focus of genomics is to identify the genetic variants associated with a disease at the genome scale. Transcriptomics involves the study of RNA expression from specific tissues, developmental stages or diseases. This offers insight into cell- and tissue-specific gene expression. Epigenomes focus on the genome-wide characterization of DNA methylation (DNAm) or post-translational modifications of histones, along with chromatin conformation, and the non-coding RNAs, capable of imposing stable and heritable changes in a gene without a change in the DNA sequence. Metabolomics provides a snapshot of the metabolic state of an organism or tissue, which together with gene and protein abundance constitutes its molecular phenome. Proteomics is the qualitative and quantitative study of the proteome, and it connects the genes with their functionally diverse protein products. The exposomes are the non-genetic drivers of health and disease and represent the totality of environmental exposure over the life course, with exposure timings ranging from prenatal to postnatal periods. Microbiomes are the microorganisms, including bacteria, viruses and fungi, that colonize the human skin, mucosal surfaces and gut. Small microbial molecules and metabolites affect the physiology of an individual.
Figure 2.
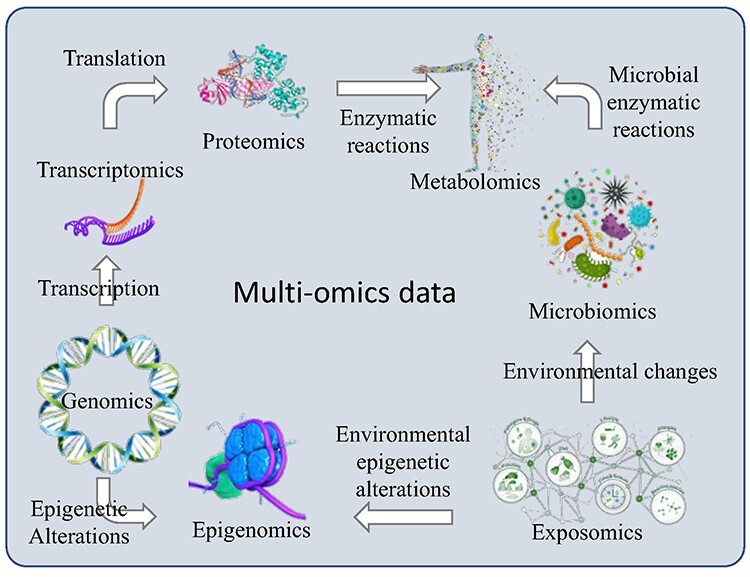
Overview of multi-omics data. Multi-omics data include genome, transcriptome, epigenome, proteome, exposome and microbiome.
Multi-omics data integrative analysis using DL
Multi-omics integration is the process of combining the information of multiple omics layers to get more insight into the disease process. Each omics data type typically provides a list of differential factors potentially associated with the disease. These data can be useful as disease markers, while providing insight as to which biological pathways or processes are different between the disease and control groups. However, analysis of only one omics data type is limited to correlations and provides only a partial view of the biological system. Integrating different omics data types could help to elucidate the potential causative changes that lead to disease or can be used to identify potential therapeutic targets for further molecular studies. A large number of publicly available tools have been developed for omics data integration [13].
The outstanding predictive performance of DL is mainly achieved by its capability to automatically capture nonlinear and hierarchical representative features through multi-layered neural network architectures. DL consists of multiple layers that involve nonlinear activation functions (e.g. sigmoid, tanh and rectifier linear units) of a linear regression form:
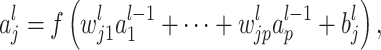 |
where 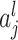 and
and 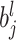 are neuron value and a bias in the
are neuron value and a bias in the 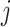 th neuron of the
th neuron of the 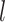 th layer, respectively;
th layer, respectively; 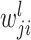 is a weight from the
is a weight from the 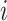 th neuron in the (
th neuron in the (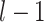 )th layer to the
)th layer to the 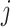 th neuron in the
th neuron in the 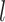 th layer and
th layer and 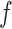 is an activation function. An activation,
is an activation function. An activation, 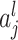 , produces a new variable that represents a nonlinear association, which is optimized to reproduce output values. The process can be considered as feature extraction without an explicitly predefined nonlinear function, such as a kernel [12].
, produces a new variable that represents a nonlinear association, which is optimized to reproduce output values. The process can be considered as feature extraction without an explicitly predefined nonlinear function, such as a kernel [12].
It is worthy to note that most conventional machine learning methods use a kernel trick for nonlinear patterns. However, the kernel trick needs to specify a kernel function that may represent the nonlinearity, which is heuristically chosen. In DL, the optimization processes of feature extraction, with multiple activations, are hierarchically performed:
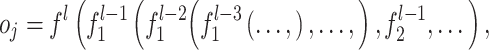 |
where  is an output value in the
is an output value in the  th neuron of the output layer.
th neuron of the output layer.
Such a hierarchical feature extraction process can capture complex nonlinear associations in a multi-layered manner. For instance, in face recognition from facial images, the input layer contains pixel values of facial images; the following layer describes primitive patterns, such as circles and lines; the deeper layer may capture facial components, e.g. nose and eyes, which are combinations of the primitive patterns; and the next layer may recognize larger facial components. In this manner, DL with biological data can potentially represent the hierarchical processes of multilayered biological systems, where a strand of DNA, for instance, is copied into messenger RNA (mRNA), and mRNA is translated into protein.
Taking advantage of outperformance in prediction, most DL-based approaches in biomedical research have handled classification and association problems, such as protein structure prediction, gene expression regulations, protein classification and genome-wide association studies. Most breakthrough DL models are with large-scale data [7, 14, 15]. For instance, SpliceAI predicts RNA alternative splicing by computing predicted scores for acceptor or donor, neither with pre-mRNA nucleotide sequences [16]. SpliceAI trains the DL model using large genomic databases in Genotype-Tissue Expression (GTEx) and GENCODE. DeepEC classifies enzyme commission numbers from protein sequence data [17]. DeepEC trains the model using the supervised data from Swiss-Prot and TrEMBL databases, which includes millions of samples.
On the other hand, multi-omics data analyses involve multiple types of extremely high-dimensional biological data but relatively smaller data sizes, which cause a severe overfitting issue when training models. Moreover, other challenges are developing effective analytic data integration approach for high-dimensional multi-omics data that can capture interaction effects, as well as biological interpretation.
This research introduces recent DL-based data integration studies using multi-omics data in the following categories, where most related research belongs: (1) feature selection/reduction, (2) clinical outcome prediction, (3) survival analysis and (4) clustering for subtype discovery. To summarize, most DL models for multi-omics data analyses follow a common pipeline (Figure 3): (1) complete or incomplete multi-omics data are cleaned by preprocessing; (2) feature selection or dimensionality reduction is applied to reduce the number of multi-omics variables using conventional feature selection techniques or feature reduction methods [e.g. principal component analysis (PCA), autoencoder); (3) multi-omics variables are concatenated into a large dataset for data integration; (4) further feature selection or reduction techniques are applied and (5) finally, the integrated data are analyzed for desired tasks, such as classification, regression and clustering. Current state-of-the art DL-based methods are introduced in the following subsections.
Figure 3.
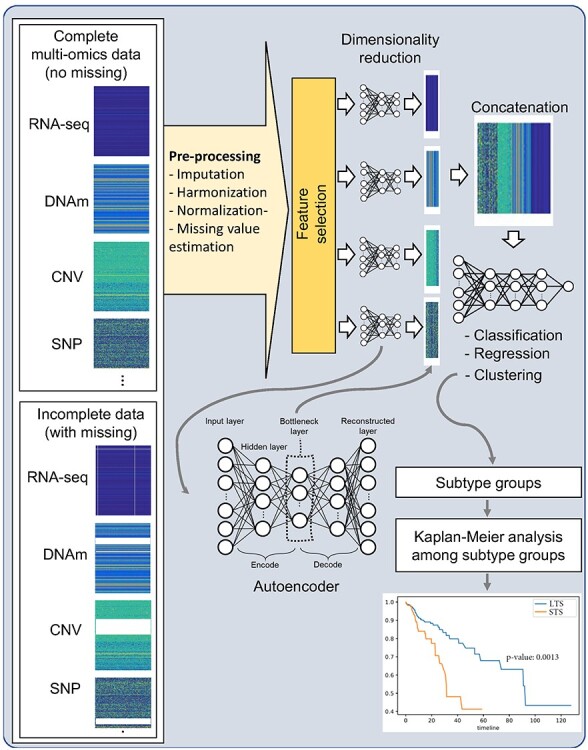
Pipeline of multi-omics data integration analyses: (1) complete or incomplete multi-omics data are cleaned by preprocessing; feature selection or dimensionality reduction is applied to reduce numbers of multi-omics variables; (3) autoencoder is a DL model that extracts low-ranked latent variables of the input data in a bottleneck layer; (4) multi-omics variables are concatenated into a large dataset for data integration; (5) further feature selection or reduction techniques are applied; (6) the integrated data are analyzed for desired tasks, such as classification, regression and clustering and (7) finally, subtypes discovered by clustering analysis re evaluated with the Kaplan-Meier analysis.
Feature selection and dimensionality reduction
One of the challenges in multi-omics data analyses is high-dimensional, low sample size (HDLSS) data. Multi-omics data are composed of several types of high-dimensional omics data, each of which is challenging to analyze, due to large feature size. Data concatenation of multi-omics data into a large input matrix is one of the conventional approaches to integrate multi-omics data, and it makes the data dimensionality much larger. DL models with higher-dimensional data involve an exponentially increasing number of model parameters, and training the nonlinear models with a relatively smaller number of samples than the parameters often causes severe overfitting problems. Therefore, reducing data dimensionality by using feature selection, or dimensionality reduction methods, helps to train robust DL models with HDLSS data and, consequently, improves DL models with high predictive performance for bioinformatics problems [18].
The preprocessing of feature selection and dimensionality reduction are conventional approaches to reduce the dimensionality of input data, as well as identify a set of meaningful features. Feature selection is used to identify a subset of relevant features for use in model construction or improving a task’s performance, whereas dimensionality reduction is used to transform features into a lower dimension. Although many traditional feature selection (e.g. univariate/multivariate) and dimensionality reduction (e.g. PCA, subsampled randomized Hadamard transform (SRHT) [19], count-min sketch [20], canonical correlation analysis [21–23]) techniques can be used in DL, such techniques are linear-based and mainly consider the main effects of variables, which may not fully take advantage of DL models. Although there are nonlinear-based feature selection methods [24], nonlinearity has various forms and patterns, which are not easy to define, unlike linearity. Thus, the identification of nonlinear features by different types of nonlinear-based feature selection may not help to improve performance in DL.
DL-based feature selection approaches have a strong capability to identify sets of features for nonlinear and interactive relationships. DL-based feature selection methods can be categorized into supervised/unsupervised approaches, depending on the availability of labels on samples, as in conventional feature selection. Deep feature selection (DFS) selects a discriminative feature subset in a DL model [25]. Although DFS is not the optimal solution with low-sample size data, DFS has shown that DL can detect a subset of informative and discriminative features of nonlinearity effects through multiple layers with high-dimensional data. Then, Deep Neural Pursuit (DNP) improves the solution of feature selection in DL, taking the HDLSS data problem into account [26]. DNP iteratively augments features in the input layer by performing multiple training with dropouts. The multiple dropouts grant the ability to train one small-sized subnetwork at a time and to compute gradients with low variance to alleviate the overfitting problem. A CancelOut layer has been suggested, in which each neuron in the input layer is solely connected to a neuron in the CancelOut layer to select important features with the corresponding weights in neural networks [27].
On the other hand, few unsupervised feature selections have been studied for DL models. Deep-FS proposed a feature selection strategy to remove irrelevant features for deep Boltzmann machines in the unsupervised setting [28]. However, most unsupervised DL studies (e.g. clustering and data representation) have applied conventional unsupervised feature selection approaches (e.g. correlation or variance-based), which are linear- and univariate-based feature selection. Thus, novel DL-based unsupervised feature selections are desirable to investigate further, as future studies.
Several DL-based analyses with multi-omics data have transformed the high-dimensionality of multi-omics data into low-ranked latent variables using autoencoder to tackle the HDLSS problem. Low-ranked latent variables are extracted from a bottleneck layer of autoencoder as new features. Autoencoder is a nonlinear factorization technique with multi-layer neural network structures to learn data representation by reducing dimensionality in an unsupervised manner. Autoencoder consists of neural network layers for the encoder and decoder, where the encoder learns the latent variables of the input data, whereas the decoder reconstructs the input data from the latent variables. The bottleneck is the layer in the middle, between encoder and decoder in autoencoder, and contains the latent variables of the input data. The latent variables from autoencoder can take advantage of neural networks that can capture nonlinear features, as well as reduce dimensionality.
Following feature selection and dimensionality reduction, clustering, clinical outcome predictions and functional analyses can be conducted with the low-ranked latent variables from autoencoder. In survival analysis studies [29, 30], low-ranked latent variables were constructed by autoencoder from a large single matrix of concatenated multi-omics data. The latent variables were introduced to a univariate Cox-PH model, as a conventional survival analysis had done. Then, survival-associated features were identified by performing univariate cox-PH models [31, 32]. The feature set was clustered to identify cancer subtypes by K-means clustering. Functional analyses were performed with well-known biomarkers (e.g. TP53 mutation in hepatocellular carcinoma) to assess the discovered subtypes. Differential expression analysis and enriched pathway analysis were also performed to identify differentially expressed genes between the subtype groups [32]. However, these post-analyses, after feature selection and dimensionality reduction, were performed directly with the nonlinearly associated multi-omics variables of the subtype groups.
Incorporation of prior biological knowledge can improve feature selection performance by imposing well-known biological components. Prior knowledge of biological interaction networks was incorporated as interaction network regularization on low-rank nonlinear features learned by autoencoder [33]. In the study, autoencoder tackled the limitation of lower representation power that simple matrix factorization models of shallow linear structure produce. Furthermore, latent variables of time-series types of multi-omics data were proposed. Typically, time-series data are useful to identify causality between variables. Latent variables of temporal multi-omics data were extracted from long short-term memory-based Variational AutoEncoder [34], which showed the potential of feature extraction for temporal multi-omics data.
Multi-omics datasets possess high dimensionality, which is difficult to handle. High predictive powers have often been reported with high-level representations in reducing dimensionality by autoencoder with multi-layered neural network architectures. However, there are pros and cons to this effort. The advantages of dimension reduction include the ability to estimate probabilities in high-dimensional data without losing its inference; a dramatic reduction in data size, which allows faster processing and computational time; smaller storage; and removal or redundant features. The disadvantages of dimensional reduction are that it may lead to some amount of data loss or lack of biological interpretation. The low-ranked representations, obtained from the bottleneck layer of autoencoder, are transformed from the input layer of multi-omics data, so the low-ranked representations are combinations of all multi-omics features through multiple fully connected encoding layers. The low-ranked representations from autoencoder do not explicitly reflect biological components or processes. After feature section and dimensionality reduction, one can perform a multi-omics significance test and conduct functional analyses. However, these analyses may fail to find statistically significant differences for nonlinear multi-omics associations. This is because these analyses are based on linear models or pairwise analyses.
Prediction of clinical outcomes
DL models have shown outperformance mainly in supervised learning problems, including classification and regression tasks. In supervised learning, DL learns a hierarchically multilayered neural network based on the labeled data of input–output pairs. DL fully enjoys the flexibility of designing a neural network architecture, which can be applied for both classification and regression problems by modifying the output layer. For classification problems, the output layer of a neural network includes as many neurons as the number of classes (or labels) to classify, where, typically, each neuron shows a posterior probability that the given data belong to the corresponding class. For binary classification problems, the output layer can have only one neuron, which computes a posterior probability of a positive class. The loss function of cross entropy sums the cross-entropy between the ground truth and model prediction of each class. For multi-class classification, a softmax function, which normalizes the posterior probabilities of all classes, is typically be considered as the last activation function, whereas a logistic function is used to compute posterior probability independently for each class in multi-label classification. For regression problems, the output layer consists of one neuron, which directly produces continuous values to predict, with linear activation in the last layer and loss functions, such as mean square error and mean absolute error, for regressions.
Several supervised learning studies with multi-omics data have been conducted with DL models for cancer-type classification, drug response prediction and gene expression prediction. A DL model with multi-omics data of gene expression, micro RNA (miRNA) expression and DNAm classified cancer types among breast cancer, glioblastoma multiforme (GBM) and ovarian cancer [35] and showed better predictive performance than using single omics data. Low-ranked representation was first constructed on each single omics data by Stacked Autoencoder, and the three representations were combined; then further representations were constructed by autoencoder again. The final representations were taken into a deep flexible neural forest (DFNForest), which can automatically select the model structure of higher depth for multi-omics data analysis. A superlayered neural network (SNN) was proposed to extract any cross-correlations present in the multi-omics data [36], where the separate neural networks of each omics data shared cross-connections between multi-omics data. SNN provided biological insights to identify the most relevant genes and the interaction between multi-omics.
Drug response predictive models have been developed based on DL with multi-omics data. A multi-omics late integration (MOLI) DL model takes somatic mutation, copy number variation (CNV) and gene expression data as input and integrates them for drug response prediction [37]. Each omics involves separate subnetwork layers, and the low-ranked representation features are combined to introduce final classification layers. MOLI optimizes the model with the triplet loss function to impose similarity among responders, as well as classification loss. The Deep Neural Network Synergy model with autoencoders (AuDNNsynergy) predicted the synergy of pairwise drug combinations by integrating multi-omics data of gene expression, copy number and genetic mutation data [38]; autoencoders were trained with each single omics data separately, and the physicochemical features (e.g. solubility and passive permeability) of individual drugs, as well as the encoded omics data of individual cancer cell lines, were combined as input features of a deep neural network to predict the synergy score of given pairwise drug combinations against the specific cancer cell line.
There have been attempts to integrate multi-omics data by transforming them into image formatted data for use in convolutional neural networks (CNNs). Gene-based multi-omics data are combined as multi-channel vector images [39]. For instance, gene expression and gene-based CNV are combined into a matrix of 2 × the number of genes for each sample. Then, the multi-channel vector images are introduced to a CNN to classify cancer subtypes. However, since CNNs capture spatial patterns of images by a kernel, the order of genes in the input data is critical when applying a CNN. To tackle the issue, a gene similarity network is considered to transform multi-omics data into images. A DL-based model transforms multi-omics data into a gene similarity network, via self-organizing maps (iSOM-GSN) [40]; each type of omics data is transformed into a two-dimensional image, which is a self-organized map, where genes are organized based on a gene similarity network. Multi-omics data produce multi-channel images, which are introduced to a CNN to predict tumor stages. Multi-Omics gRaph cOnvolutional NETworks (MORONET) constructed a weighted patient similarity network for each type of omics data and took each similarity network into graph convolutional networks (GCNs) [41]. The GCNs of each type of omics data generate prediction scores of class labels, and then the prediction scores of multi-omics data are combined as cross-omics discovery tensor data, which reflect the cross-omics label correlation. The cross-omics discovery tensor data are trained by View Correlation Discovery Network for the final label prediction.
For regression problems, deep denoising auto-encoder (DDAE) has been proposed to estimate gene expression (i.e. RNA-seq) from DNAm and CNVs [42]. The concatenated data of DNAm and CNV are introduced to DDAE to generate low-rank representation features. Then, a multi-layered perceptron model predicts RNA-seq from the features.
Survival analysis using multi-omics data
Survival analysis estimates the survival distribution of a particular population, as well as investigating the associations between covariates and survival time. Survival analysis is basically for supervised regression problems, but it focuses on time-to-event outcomes, handling censored observations, in which an event was not observed during follow-up. Thus, survival analysis includes two clinical outcomes of survival time and survival status [43, 44]. One of the most conventional methods in survival analysis is the Cox proportional hazards (Cox-PH) model. Cox-PH is a semi-parametric model that consists of a baseline hazard function (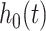 ) and an exponential function of covariate effects (
) and an exponential function of covariate effects (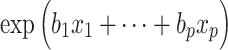 :
:
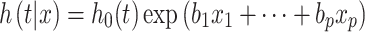 |
where 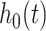 describes the risk of event over time at baseline levels of covariates, and
describes the risk of event over time at baseline levels of covariates, and 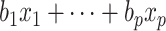 describes the effect of the predictors on the overall hazard. Conventional statistical survival analysis assumes the independence of variables, as well as linearity with the log of the hazard ratio. For high-dimensional data, feature selection techniques or Cox-PH with LASSO/elastic-net regularization [45] have often been considered to reduce covariate size.
describes the effect of the predictors on the overall hazard. Conventional statistical survival analysis assumes the independence of variables, as well as linearity with the log of the hazard ratio. For high-dimensional data, feature selection techniques or Cox-PH with LASSO/elastic-net regularization [45] have often been considered to reduce covariate size.
In DL-based survival analyses, the linear combination of covariates in the exponential function has often been replaced with neural networks to take the interaction effects, as well as the nonlinearity of covariates, into account. DL-based survival models, such as Cox-nnet [46], SurvivalNet [47] and Cox-PASNet [48, 49], take covariates into the input layer of a neural network and produce a prognostic index in the output layer, with the cost function of partial log-likelihood for survival analysis with censored data. The prognostic index is computed by a linear combination of low-ranked high-level representations, which are nodes in the last hidden layers, without activation functions. Thus, the nodes of the last hidden layers in survival neural networks can be considered as new representations of the input covariates. For model interpretation and prognostic factor identification, the importance of each node in survival neural networks is computed by partial derivatives of risk, with respect to each input variable:
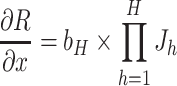 |
where 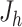 is the Jacobian matrix of the hth hidden layer with respect to its input variable,
is the Jacobian matrix of the hth hidden layer with respect to its input variable, 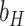 is the weights between the final hidden layer and the output layer and
is the weights between the final hidden layer and the output layer and 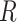 is the risk cost function.
is the risk cost function.
As the most typical DL approach for survival analysis with multi-omics data, Survival Analysis Learning with Multi-Omics Neural Networks (SALMON) reduce the dimensionality of multi-omics data using an eigengene matrix through gene co-expression analysis on each type of omics data separately, and the combined eigengene matrix, as well as copy number burden, tumor mutation burden and demographical/clinical variables (e.g. age, ER status, and PR status), is introduced to a neural network that integrates Cox-PH [50]. A DL-based survival analysis has proposed a cross-modality autoencoder (CrossAE) to integrate multi-omics by performing similar cross-modality reconstruction [51]. The element-wise average low-rank representations in the bottleneck layer of CrossAE are input into a neural network with Cox-PH. Similarly, the low-rank representations of each type of omics data from autoencoder are combined with consensus constrains (using cosine similarity) to generate modality-invariant representations, followed by the output layer for survival analysis [52]. To improve the predictive performance with robust models, an ensemble-based DL model, DeepProg, has been proposed [53]. DeepProg iteratively builds DL models with a subset of features, followed by feature transformation, and a univariate Cox-PH model for further feature selection. The results of multiple DL models are combined for the final prediction.
Multi-omics data have been integrated based on a gene in DL models, by incorporating biological knowledge. Multi-omics features of the same gene were grouped by gene annotation, so that either interaction or group effects were mainly considered. Multi-omics features of RNA-seq, CNV, RPPA and somatic mutation were grouped by a gene, and a DL model, Group lasso regularized DL for cancer Prognosis (GDP), was trained with group lasso regularization in the objective function of the partial log-likelihood of a CPH model [54]. The group regularization for the gene-based multi-modal features, by the incorporation of the biological knowledge, reduced the number of parameters to train, which tackled overfitting issues. A gene- and pathway-based deep neural network (MiNet) grouped multi-omics features by genes, taking their interaction effects, and produced representations of canonical gene expression [55]. The canonical gene expression were grouped by biological pathways and introduced to a sparse neural network to predict a cancer patient’s survival. The neural network architecture of MiNet reflects the biological processes of the interaction effects of multi-omics data and pathways, as well as the hierarchical interactions of pathways, which enables biological model interpretation.
Clustering for subtype discovery
Identification of subtypes plays a critical role in improving treatment modalities and clinical outcomes in several diseases [56]. The cellular origin of cancer subtypes can be comprehensively characterized by the pathological morphologies and molecular profiles of multi-omics data [57]. For instance, the four molecular subtypes of (1) luminal A, (2) luminal B, (3) human epidermal growth factor receptor 2 (HER2)-enriched (HER2-positive) and (4) basal-like breast cancer are well-known subtypes in breast cancer, showing significant differences in responses to treatment, as well as disease progression and survival [58]. The discovery of unknown cancer subtypes has been done computationally by grouping or stratifying patient populations based on multilayered biological profiles, including genome, proteome, transcriptome, metabolome and epigenome [59–61].
Most DL-based clustering methods have focused on capturing nonlinearly associated multi-omics features by autoencoder and pairwise feature selection coupled with clustering algorithms. Subtypes have been evaluated with the Kaplan–Meier analysis. Each omics data of miRNA, mRNA, DNAm, somatic mutation, CNV and RPPA were compressed by autoencoder, and statistically significant features among the transformed features were further identified by a univariate Cox-PH [62]. The selected features were grouped by K-means clustering to identify subtypes. Similarly, all multi-omics data were concatenated into a large input matrix and transformed into low-ranked representations by autoencoder [63]. A significance test was performed on the transformed features by univariate Cox-PH and LASSO regression, followed by K-means clustering.
Prior knowledge of well-known biological pathway databases has been incorporated in cancer subtyping. PathME proposed a multi-modal sparse denoising autoencoder framework with sparse non-negative matrix factorization (sNMF) for robust integrative clustering [64]. PathME computed a score per pathway from multi-omics data by mapping each type of omics data into a pathway via a sparse autoencoder with group lasso regularization, in which multi-omics data grouped by their gene annotations were transformed to a pathway score. Finally, the patient data of pathway scores were clustered by consensus sNMF. The study interpreted the model based on pathways.
Challenges and opportunities
While DL methods highlight the potential of omics-based data integration to drive innovative in biomedical research, DL-based approaches have the following challenges to take into consideration: (1) training with high-dimensional, low sample size data; (2) missing value imputation; (3) model interpretation and (4) integrating clinical and environmental exposure data.
High-dimensionality and small sample size
DL’s capability to automatically identify nonlinear and hierarchical features requires a large amount of training data, as well as validation data, to find a generalized model. Nonlinear patterns are typically infeasible to formulate in high-dimensional data, in contrast with linear patterns, which often cause overfitting issues. Multi-omics data intrinsically involve the large features, small samples' problem, and multi-omics HDLSS data make it even more challenging to training DL models that consist of a huge number of parameters, avoiding overfitting and bias. For instance, a simple fully connected neural network, which is composed of an input layer of 1000 nodes, a hidden layer of 100 nodes and an output layer of 2 nodes, requires training 100,302 parameters. However, there are only a few biological databases that include more than 100,000 samples, and multi-omics databases have many fewer samples available. Particularly, backpropagation gradients in neural networks are of high variance on HDLSS data, which, consequently, cause model overfit [26].
In order to tackle the HDLSS problem in training DL models, a leave-one-out approach can be used to avoid the overfitting problem in the backpropagation phase [65]. The risk of overfitting can be examined with validation data by the leave-one-out approach, and training can be terminated early when overfitting occurs. While tracing the overfitting risk with validation, a small learning rate, as well as high dropout regularization with higher epoch size, is often encouraged in HDLSS, which reduces the backpropagation gradients of high variance on HDLSS data. As an alternative solution, an attempt to reduce the dimensionality of the input space to a feasible size has been made, by adding a random project layer in front of the network [66]. Dimension reduction techniques, such as SRHT and count sketch-base construction, were utilized to reduce the dimensional size of the input data. Then, the data projected into the lower space were introduced to a neural network for training.
However, the optimization of generalized DL models with HDLSS data is still extremely challenging. Potential solutions may include incorporating prior biomedical knowledge and sparse learning. Prior biomedical knowledge can reduce model complexity with constraints to a model’s architecture although such intervention to a model is against the fundamental concept of DL, which automatically identifies useful patterns without predefinition. Constraints obtained from prior domain knowledge can prevent the model from exploring unnecessary hypothesis spaces, which may not be matched to the domain’s knowledge. For instance, candidate genes of objective traits are prioritized by a CNN by integrating multiple biological databases, such as genomic data, transcriptome data and quantitative trait-associated gene/nucleotide data [67]. The gene prioritization can reduce false positives by imposing well-known biological knowledge into the model. As another example, PASNet incorporates biological pathway databases to design a neural network architecture [68], where the network between the gene layer and the pathway layer explicitly models the relationships of genes and pathways with sparse connections. The sparse network dramatically reduces the number of parameters; PASNet contains only 0.01% connections of the fully connected layers. Sparse coding finds a parsimonious neural network architecture with sparse connections but may preserve or maximize its predictive performance. Sparse coding can reduce the number of parameters to train, while increasing model interpretability.
The limited sample sizes of most multi-omics datasets are still one of the biggest obstacles to consider DL-based integration, compared to conventional integration methods (e.g. sparsity-regularized integration). However, the advantages of DL’s capabilities, which identify the nonlinear/interactive patterns of multiple features, have attracted DL-based integration. Advanced optimization strategies (e.g. dropout, regularization and bootstrapping) with HDLSS make applying DL for multi-omics data analysis promising. Furthermore, the rapid advances of sequencing technologies and their dramatic price decrease will provide more samples to train robust DL in the near future.
Missing data and data heterogeneity
In multi-omics data, it is common for some individuals to be represented by some omics data only, but not all, which is problematic since most statistical and machine learning analyses require complete datasets. There are two types of missing values in multi-omics data: omics-wise and sample-wise missing values. Omics-wise missing values refer different sample sizes of omics data. Some samples include more available omics data, but others have less. For instance, there are 434 available samples of DNAm, whereas 578 samples of CNV are available in the GBM of The Cancer Genome Atlas (TCGA) project. This indicates that at least 141 samples do not have features of DNAm. Sample-wise missing values refer to conventional missing values occasionally observed in a sample, due to technical limitations and various experimental constraints.
Data cleaning, missing value imputation and harmonization across data sets are conventional preprocessing techniques to handle sample-wise missing values. In data cleaning, omics features (or samples) with missing values (features) of more than a certain percentage have been excluded in many multi-omics data analyses (Figure 4; Supplementary Table 1 available online at http://bib.oxfordjournals.org/). Outliers with the lowest variance and means can be further removed. Conventional missing value imputation techniques, such as K-nearest neighbors and mean value imputation, have been widely used to handle missing values.
Figure 4.
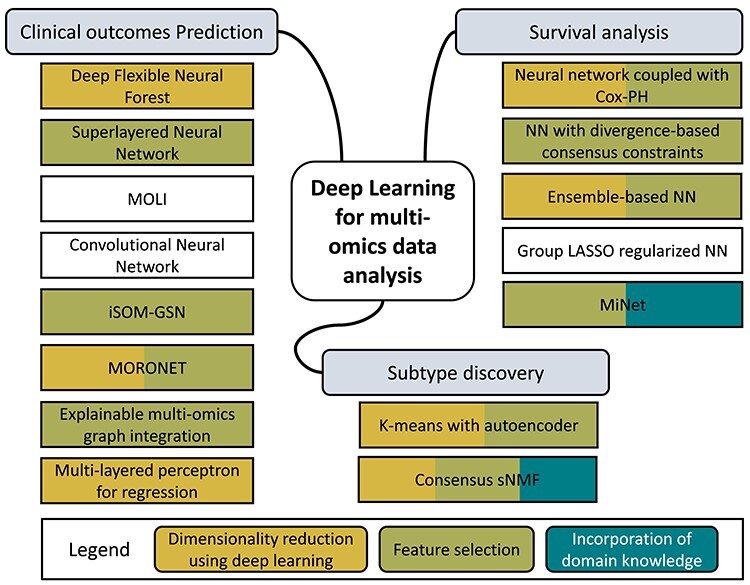
Overview of DL methods for multi-omics data analysis. The methods are groups based on their applications and colored as their pipeline components of dimensionality reduction, using DL (e.g. autoencoder), feature selection and domain knowledge incorporation (e.g. pathway databases). MOLI, multi-omics late integration; iSOM-GSN, self-organizing maps with gene similarity network; MORONET, multi-omics graph convolutional networks; MINet, a gene- and pathway-based deep neural network.
Although most imputation techniques have been optimally developed for each type of single omics data separately, there are several integrative imputation methods for multi-omics data, which are mainly categorized 3-fold: (1) machine learning-based regression strategy, (2) transfer learning-based and (3) multi-view matrix factorization-based imputation [69]. Expressional missing values are imputed by their relationship with other omics data. Regression models have often been used to find correlations between multi-omics datasets to impute missing values [70–72]; for instance, gene expression data are inferred from SNPs or epigenomics data. Transfer learning leverages information from reference databases to the query dataset to impute data [73, 74]. The reference annotations and databases include GTEx, the Roadmap Epigenomics Project, the CommonMind Consortium and TCGA. The transfer learning-based approach provides a global inference for missing values without bias to given local datasets. Additionally, multi-view matrix factorization-based imputation is a robust, unsupervised approach that reconstructs incomplete datasets. As an extended version of matrix factorization techniques, with single view data for missing value imputation [75], the multi-view approach handles at least two modality datasets, where it finds low-dimensional common factors for all datasets [76, 77].
In other ways, NEighborhood-based Multi-Omics clustering (NEMO) integrates multi-omics data based on inter-patient similarity, where it completes similarity for incomplete data without missing imputation by using an average of observed values [60]. A study considered common and uncommon data sets, where the common dataset referred to samples including all multi-omics data types, and the uncommon dataset lacked a certain type of multi-omics data [62]; the common data were used to optimize the model, with autoencoder and feature selection with a univariate Cox-PH, while the uncommon dataset was used as validation for the final task.
Although most multi-omics integrative methods assume that multi-omics data are structured and obtained from the same sources (e.g. mRNA and CNV from a same individual), there are often unstructured multi-omics data types (e.g. DNA sequence, protein sequence) and samples obtained from multiple sources (e.g. mRNA from one group, but CNV from another group). Feature extraction and data encoding can convert unstructured data into structured data. For instance, structured radiomics features were extracted from unstructured medical imaging data [78, 79]. Amino acid sequence data were analyzed by recurrent neural networks [80] and converted into image looking data by one-hot encoding followed by CNN [17]. For the multi-omics data that do not share data sources, obtaining high-level representation (e.g. by using autoencoder) of single omics independently, as well as their integration, may be a potential solution. However, this approach may not characterize the interactive effects among multi-omics data, which may not fully take advantage of multi-omics integration.
Model interpretability
Biomedical research often stresses model interpretation to understand biological mechanisms (e.g. by performing biomarker identification and correlation/causality/regulatory relationship inferences between biological components) rather than simply predicting biological or clinical outcomes (e.g. survival rate, disease status and risk score). Although DL has the strength of outstanding predictive performance by capturing nonlinear and hierarchical representations through multi-layered neural network architectures, the most common structure of fully connected neural networks lacks in interpreting what the hierarchical representation features describe biologically, since the high-level representations are products involving all features.
There are two strategies for model interpretation in DL: (1) intrinsic model interpretation and (2) post hoc interpretation [81, 82]. Intrinsic model interpretation is interpreting directly from the model’s construction, as well as the optimal parameters of DL models, to understand relationships between the biological components from the data. Neural networks, whose hierarchical multi-layered architectures are intrinsically inspired from biological neural systems, may be able to explicitly model biological systems of interest, and a model component of the neural network (e.g. a neuron in neural networks) may be associated to a biological component or a biological process. For instance, sparsely connected neural networks have shown the potential capability of intrinsic model interpretation, identifying hierarchical relationships among a subset of biological components in multi-layered biological processes by incorporating biological pathway databases into their models [55, 68]. Biological pathways are explicitly embedded into the pathway layer, with given sparse connections between the gene layer and the pathway layer, where the sparse connections refer to the membership of genes in a pathway. Important sets of biological pathways and genes could be identified directly from the models by ranking nodes in the pathway layer and the gene layer by partial derivatives. Data representation of pathway-associated gene sets has produced a latent value of pathway expression [64]. A sparse autoencoder transformed pathway-associated genes for each pathway, and the latent values of the pathways were analyzed to rank pathways for cancer subtyping.
Post hoc interpretation is extracting various forms of information after training a model. Post hoc interpretation often falls into two categories: (1) global and (2) local interpretation [82]. Global interpretation demonstrates the averaged behaviors of a model, which are often shown as expected values based on the distribution of the entire samples. Global interpretation is useful for learning general relationships associated to target outcomes. One of the traditional global interpretation methods is feature importance [83], which is useful to find potential biomarkers. On the other hand, local interpretation focuses on how a model predicts individual outcomes. Local interpretation provides insights into predictive mechanisms shown in subgroups of interest, instead of the overall mechanisms of the entire population. The most popular local interpretation approaches include Local Interpretable Model-agnostic Explanations (LIME) [84] and SHapley Additive exPlanations (SHAP) [85]. LIME and SHAP are model-agnostic methods, which generate a unified interpretation model that can be mapped from any machine learning models for interpretation. LIME reconstructs input data and trains an interpretation model by capturing the patterns of neighbors [84]. SHAP transfers any machine learning models to a linear-based interpretation model for characterizing the contribution of each variable to an outcome [85]. A variable’s contribution to the prediction is called its SHAP values, and the sum of SHAP values corresponds to the predication score.
Integrating multi-omics and non-omics data to improve predictive performance
Although multi-omics data have proved to be a powerful predictor of various traits and diseases, there are other factors, such as clinical and environmental factors, that can play an important part [86, 87]. In addition, with the growing digital healthcare industry, clinical and electronic health record (EHR) data are expected to become more widely available. In particular, the integration between multi-omics data and pathological images is promising. Pathological images are considered as clinical gold standards for diagnosis. Association studies between multi-omics data and pathological image data can help researchers to understand how multilayered biological processes (represented by multi-omics data) cause morphological differences in pathological images. Pathological images have been used to predict the RNA-seq expression of tumors [88], as well as DNAm [89]. The performance of survival analysis and risk prediction has been improved by integrating pathological images on genomic data or multi-omics data [90–94]. Challenges in integration with pathological images includes: (1) the large sizes of pathological images and (2) small sample sizes. Pathological images are gigapixel images, which makes it difficult to introduce a whole-slide image into a model. Patch-wise analyses are conventional for pathological images. Thus, a whole slide image consists of a number of patch images, whereas multi-omics data are well-structured data, which create a challenge for integration. Pathological images of gigapixel size increase the dimensionality of the data to analyze. Besides, various types of imaging data have been integrated with multi-omics data. Structured radiomics features, obtained from magnetic resonance imaging data, have been integrated with multi-omics data for phenotype prediction [78, 79]. Similarly, radiomics features have been extracted from positron emission tomography, by the Image Biomarker Standardization Initiative’s protocol, and analyzed with multi-omics data for actuarial outcome prediction [95].
However, available sample numbers are still limited. In addition, integration of EHR data on multi-omics data is also promising for clinical diagnosis and decision support systems in precision medicine. Although EHR data have, to date, been solely analyzed to make clinical decisions, its integration with multi-omics data could provide thoroughly personalized solutions to predict the most appropriate action for patients with complex diseases like cancer [96, 97]. However, most EHR data are time-series data (but unevenly distributed over time), and the data source is very heterogenous, which is extremely challenging to integrate with multi-omics data. Nevertheless, the importance of integration with non-omics data, such as clinical and environmental exposure data, is understood, and an increasing number of multi-source databases and projects, e.g. All of Us (https://allofus.nih.gov), are shedding light on more active research on integration.
MOVING FORWARD IN THE ERA OF PRECISION MEDICINE
With high-resolution data from next generation sequencing, differences at the genomic level, and their consequences, are becoming better understood, leading to a new medical paradigm: precision medicine. Precision medicine is a patient-specific tailored approach and goes beyond traditional medicine’s ‘one size fits all’ approach. The power of the multi-omics approaches in predictive medicine for complex diseases has recently been emphasized [98, 99]. Omics technologies in precision medicine can help identify new disease biomarkers, for diagnosis, prognosis and patient stratification, along with drug development and repurposing, including the existential threat of COVID-19. For instance, a recent study applied a DL approach to synergistically identify drug combinations for treating COVID-19. The study showed that in contrast to previous conventional data science approaches using drug–target interaction as fixed descriptors, a DL model learns to predict drug–target interaction and drug–drug combination from molecular structures and identify the right synergistic drug combinations for the rapidly spreading SARS-CoV-2 [100]. However, although omics-level studies have been very useful in understanding disease mechanisms, there have not been sufficient data nor attempts to integrate different omics data, and most omics-level data refer only to genomics and transcriptomics. Genes identified from genomic (GWAS), epigenomic and proteomic studies can be used to build disease-associated networks from each omics level, and a network-level overlap can be calculated. Integrating multi-omics data, such as microbiome, proteome, transcriptome, epigenome, exposome and genome, is largely lacking. Publicly accessible databases can serve as powerful resources of omics-level data to unravel new biological insights into the etiologies of complex diseases, as well as to confirm previously reported disease-associated genes and pathways [101].
There is a clear trend towards incorporating multi-omics analysis in biomedical research to help explain the complex relationships between the molecular layers. However, the integration of these diverse datasets remains challenging. DL approaches have become popular for disease risk stratification [102–105]. DL approaches are useful to generate hypothesis to understand patient stratification and diseases progression and for learning the biological mechanism and its contribution to disease risk in multi-omics data [106]. Advanced computational methods, such as DL, help to model risk prediction, diagnosis and therapeutic response in diverse populations. For instance, DeepProg is an example of such a program that uses DL to integrate multi-omics phenotypes, such as survival in predicting cancer prognosis [53]. Integrating multi-omics data with deep phenotyping of the cohort, using a DL approach, offers a path to deeper functional insights and precision diagnoses in complex diseases.
The availability of omics data and integrative omics approaches has created unique opportunities to unravel the molecular underpinnings of target endotypes to develop personalized risk stratifications and therapies. Additionally, statistical methods to integrate multi-omics data are emerging to provide important insights into the disease pathophysiology of allergic diseases [107]. As research moves forward, accounting for an individual’s unique race-specific lifestyle, medical imaging and environmental exposure, along with clinical and multi-omics-based data-driven DL frameworks and integration, will be crucial for discovering and designing therapeutic strategies for specific allergic endotypes. Such an approach will help develop precision treatment options tailored to distinct endotypes in allergic diseases [108, 109].
CONCLUSION
DL is an effective approach to decipher multilayered complex biological systems by capturing complex nonlinear effects and hierarchical features and their interactions in multi-omics data. The power of the multi-omics approach in personalized and predictive medicine for complex diseases has recently been emphasized by several authors [99, 110]. In particular, big data-driven and unbiased DL approaches can now gather detailed molecular information to deconvolute and identify patterns from the data and provide further insights into the biology of diseases, along with the health states of individual patients [111, 112]. However, in critically evaluating the existing literature, compared with emerging high-throughput technology, the integration of multi-omics data has been limited, and there are even fewer reports describing the multi-omics integration approach with clinical environmental data, standards or ground truth to evaluate the performance metrics of multi-omics integration methods to elucidate their role in health and disease.
However, recent progress in DL methods to integrate multi-omics data is emerging to provide important insights into the pathophysiology of complex diseases [107]. Such an increase in focus on DL method development can be expected to prompt efforts to harness the power of multi-omics data as an approach to improve patient prediction, prevention intervention, diagnosis and personalized therapy [107]. As the research moves forward, accounting for race-specific lifestyle and environmental exposure, along with a clinical and multi-omics-based data-driven DL framework with integration, will be crucial for discovering patterns, as well as risk detection and prediction strategies for specific clinical outcomes. Therefore, approaches that are fast and robust towards missing data and heterogeneity, as well as offering a balanced trade-off between performance and model interpretability (or complexity), are critical. Such approaches will help to develop precision treatment options tailored to distinct endotypes in complex diseases [108, 113].
Finally, the availability of multi-omics data and integrative DL approaches has created unique opportunities to unravel the molecular signature underpinnings of disease outcome to develop personalized risk stratification and therapies. A combined multi-omics dataset can provide synergism and molecular insight, beyond the sum of individual omics. As the research moves move forward, since interpretability is more important than accuracy in DL modeling, there are several questions to answer, including How can we select features that are interpretable? What features contribute to a certain prediction and how? and How to make biological or clinical sense of a black-box model? A comprehensive disease risk assessment and precision medicine tools using a multi-omics and non-omics data integration approach will be prevalent.
Key Points
This paper provides a comprehensive review of current cutting-edge deep learning-based multi-omics integration analysis methods.
Deep learning methods in both unsupervised and supervised learning are reviewed with several biomedical applications, including risk prevention/detection/prediction, disease progression and clinical endotyping.
We demonstrate novel viewpoints of deep learning to apply in multi-omics analysis, including harmonizing multi-omics data that offer practical perspectives into the implementation of deep learning.
We outline insights on current applications, challenges and future directions in deep learning-based analyses with multi-omics data.
Multi-omics data integration and study design should ensure a diversity of cohorts, harmonize and standardize multi-omics data and methods, and develop benchmarking in analytical tools and multi-omics integration.
Supplementary Material
Mingon Kang is an assistant professor in the Department of Computer Science at the University of Nevada, Las Vegas. His research interests include machine learning, deep learning, and bioinformatics.
Euiseong Ko is a PhD candidate in the Department of Computer Science at the University of Nevada, Las Vegas. His research interests include deep learning in bioinformatics.
Tesfaye B. Mersha is an associate professor at the Department of Pediatrics, Cincinnati Children's Hospital Medical Center and University of Cincinnati. His research combines quantitative, bioinformatics, ancestry and functional genomics to unravel genetic and non-genetic contributions to complex diseases and racial disparities in human populations, particularly asthma and asthma-related allergic disorders.
Contributor Information
Mingon Kang, Department of Computer Science at the University of Nevada, Las Vegas, NV, USA.
Euiseong Ko, Department of Computer Science at the University of Nevada, Las Vegas, NV, USA.
Tesfaye B Mersha, Department of Pediatrics, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, Cincinnati, OH, USA.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Funding
National Heart, Lung, and Blood Institute (NHLBI) (grant R01 HL132344 to T.B.M.); The Institute for Information and Communications Technology Planning and Evaluation (IITP) under Grant (2019-0-01601) at Ministry of Science and ICT (MSIT), South Korea.
References
- 1. Mirza B, Wang W, Wang J, et al. Machine learning and integrative analysis of biomedical big data. Genes (Basel) 2019;10(2):87. 10.3390/genes10020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu C, Zhou F, Ren J, et al. A selective review of multi-level omics data integration using variable selection. High Throughput 2019;8(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olivier M, Asmis R, Hawkins GA, et al. The need for multi-omics biomarker signatures in precision medicine. Int J Mol Sci 2019. 10.3390/ijms20194781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang S, Chaudhary K, Garmire LX. More is better: recent progress in multi-omics data integration methods. Front Genet 2017;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Subramanian I, Verma S, Kumar S, et al. Multi-omics data integration, interpretation, and its application. Bioinform Biol Insights 2020;14:1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rigden DJ, Fernández-Suárez XM, Galperin MY. The 2016 database issue of nucleic acids research and an updated molecular biology database collection. Nucleic Acids Res 2016;44(D1):D1–D6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grapov D, Fahrmann J, Wanichthanarak K, et al. Rise of deep learning for genomic, proteomic, and metabolomic data integration in precision medicine. Omi A J Integr Biol 2018;22(10):630–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Siva N. 1000 Genomes project. Nat Biotechnol 2008;26(3):256–7. [DOI] [PubMed] [Google Scholar]
- 9. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 2016;bbw068. 10.1093/bib/bbw068. [DOI] [PubMed] [Google Scholar]
- 10. Lecun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521(7553):436–44. [DOI] [PubMed] [Google Scholar]
- 11. Lancashire LJ, Lemetre C, Ball GR. An introduction to artificial neural networks in bioinformatics - application to complex microarray and mass spectrometry datasets in cancer studies. Brief Bioinform 2009;10(3). 10.1093/bib/bbp012. [DOI] [PubMed] [Google Scholar]
- 12. Pilario KE, Shafiee M, Cao Y, et al. A review of kernel methods for feature extraction in nonlinear process monitoring. Processes 2020;8(1):24. [Google Scholar]
- 13. Wanichthanarak K, Fahrmann JF, Grapov D. Genomic, proteomic, and metabolomic data integration strategies. Biomark Insights 2015;10(Suppl 4):1–6. 10.4137/BMI.S29511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tang B, Pan Z, Yin K, et al. Recent advances of deep learning in bioinformatics and computational biology. Front Genet 2019. 10.3389/fgene.2019.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Y, Huang C, Ding L, et al. Deep learning in bioinformatics: introduction, application, and perspective in the big data era. Methods 2019;166:4–21. [DOI] [PubMed] [Google Scholar]
- 16. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell 2019;176(3):535–48. e24. [DOI] [PubMed] [Google Scholar]
- 17. Ryu JY, Kim HU, Lee SY. Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers. Proc Natl Acad Sci U S A 2019;16(28):13996–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen Z, Pang M, Zhao Z, et al. Feature selection may improve deep neural networks for the bioinformatics problems. Bioinformatics 2020;36(5):1542–52. [DOI] [PubMed] [Google Scholar]
- 19. Tropp JA. Improved analysis of the subsampled randomized Hadamard transform. In: Advances in Adaptive Data Analysis. 2011;3(01n02):115–26. [Google Scholar]
- 20. Cormode G, Muthukrishnan S. An improved data stream summary: the count-min sketch and its applications. J Algorithms 2005. 10.1016/j.jalgor.2003.12.001. [DOI] [Google Scholar]
- 21. Kang M, Zhang B, Wu X, et al. Sparse generalized canonical correlation analysis for biological model integration: a genetic study of psychiatric disorders. 2013 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), IEEE, 2013, p. 1490–3. [DOI] [PubMed] [Google Scholar]
- 22. Kang M, Kim D-C, Liu C, et al. Multiblock discriminant analysis for integrative genomic study. Biomed Res Int 2015;2015. 10.1155/2015/783592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang M, Park J, Kim DC, et al. Multi-block bipartite graph for integrative genomic analysis. IEEE/ACM Trans Comput Biol Bioinform 2016;14(6):1350–8. [DOI] [PubMed] [Google Scholar]
- 24. Krakovska O, Christie G, Sixsmith A, et al. Performance comparison of linear and nonlinear feature selection methods for the analysis of large survey datasets. PLoS One 2019;14(3). 10.1371/journal.pone.0213584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Y, Chen C-Y, Wasserman WW. Deep feature selection: theory and application to identify enhancers and promoters. J Comput Biol 2016;23(5):322–36. [DOI] [PubMed] [Google Scholar]
- 26. Liu B, Wei Y, Zhang Y, Yang Q. Deep neural networks for high dimension, low sample size data. PIJCAI. 2017, p. 2287–93. doi: 10.24963/ijcai.2017/318. [DOI]
- 27. Borisov V, Haug J, Kasneci G. CancelOut: a layer for feature selection in deep neural networks. In: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics). 2019. doi: 10.1007/978-3-030-30484-3_6 [DOI] [Google Scholar]
- 28. Taherkhani A, Cosma G, McGinnity TM. Deep-FS: a feature selection algorithm for deep Boltzmann machines. Neurocomputing 2018;322. 10.1016/j.neucom.2018.09.040. [DOI] [Google Scholar]
- 29. Lv J, Wang J, Shang X, et al. Survival prediction in patients with colon adenocarcinoma via multiomics data integration using a deep learning algorithm. Biosci Rep 2020;40(12):BSR20201482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chai H, Zhou X, Cui Z, et al. Integrating multi-omics data with deep learning for predicting cancer prognosis. bioRxiv 2019, 807214. 10.1101/807214. [DOI] [Google Scholar]
- 31. Zhang L, Lv C, Jin Y, et al. Deep learning-based multi-omics data integration reveals two prognostic subtypes in high-risk neuroblastoma. Front Genet 2018;9:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaudhary K, Poirion OB, Lu L, et al. Deep learning-based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res 2018;24(6):1248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma T, Zhang A. Multi-view factorization autoencoder with network constraints for multi-omic integrative analysis. 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2019, p. 702–7. doi: 10.1109/BIBM.2018.8621379 [DOI]
- 34. Chung NC, Mirza B, Choi H, et al. Unsupervised classification of multi-omics data during cardiac remodeling using deep learning. Methods 2019. 10.1016/j.ymeth.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu J, Wu P, Chen Y, et al. A hierarchical integration deep flexible neural forest framework for cancer subtype classification by integrating multi-omics data. BMC Bioinformatics 201920(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bica I, Veličković P, Xiao H, Liò P. Multi-omics data integration using cross-modal neural networks. European Symposium on Artificial Neural Networks, Computational Intelligence and Machine Learning. 2018.
- 37. Sharifi-Noghabi H, Zolotareva O, Collins CC, et al. MOLI: multi-omics late integration with deep neural networks for drug response prediction. Bioinformatics 2019. 10.1093/bioinformatics/btz318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang T, Zhang L, Payne PRO, et al. Synergistic drug combination prediction by integrating multiomics data in deep learning models. Translational Bioinformatics for Therapeutic Development. New York, NY: Humana, 2021, p. 223–38. 10.1007/978-1-0716-0849-4_12. [DOI] [PubMed] [Google Scholar]
- 39. Zeng J, Cai H, Akutsu T. Breast cancer subtype by imbalanced omics data through a deep learning fusion model. Proceedings of the 2020 10th International Conference on Bioscience, Biochemistry and Bioinformatics. 2020, p. 78–83. doi: 10.1145/3386052.3386063 [DOI]
- 40. Fatima N, Rueda L. iSOM-GSN: an integrative approach for transforming multi-omic data into gene similarity networks via self-organizing maps. Bioinformatics 2020. 10.1093/bioinformatics/btaa500. [DOI] [PubMed] [Google Scholar]
- 41. Wang T, Shao W, Huang Z, et al. MORONET: multi-omics integration via graph convolutional NETworks for biomedical data classification. bioRxiv 2020. 10.1101/2020.07.02.184705. [DOI] [Google Scholar]
- 42. Seal DB, Das V, Goswami S, et al. Estimating gene expression from DNA methylation and copy number variation: a deep learning regression model for multi-omics integration. Genomics 2020. 10.1016/j.ygeno.2020.03.021. [DOI] [PubMed] [Google Scholar]
- 43. George B, Seals S, Aban I. Survival analysis and regression models. J Nucl Cardiol 2014. 10.1007/s12350-014-9908-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rao SJ. Regression modeling strategies: with applications to linear models, logistic regression, and survival analysis. J Am Stat Assoc 2003. 10.1198/jasa.2003.s263. [DOI] [Google Scholar]
- 45. Xu J. High-dimensional cox regression analysis in genetic studies with censored survival outcomes. J Probab Stat 2012;2012. 10.1155/2012/478680. [DOI] [Google Scholar]
- 46. Ching T, Zhu X, Garmire LX. Cox-nnet: an artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput Biol 2018;14(4):1–18. 10.1371/journal.pcbi.1006076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yousefi S, others. Predicting clinical outcomes from large scale cancer genomic profiles with deep survival models. Sci Rep. 2017;7. doi: 10.1038/s41598-017-11817-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hao J, Kim Y, Mallavarapu T, Oh JH, Kang M. Cox-PASNet: pathway-based sparse deep neural network for survival analysis. In: IEEE International Conference on Bioinformatics & Biomedicine (IEEE BIBM 2018). 2018.
- 49. Hao J, Kim Y, Mallavarapu T, et al. Interpretable deep neural network for cancer survival analysis by integrating genomic and clinical data. BMC Med Genet 2019. 10.1186/s12920-019-0624-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang Z, Zhan X, Xiang S, et al. Salmon: survival analysis learning with multi-omics neural networks on breast cancer. Front Genet 2019. 10.3389/fgene.2019.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tong L, Mitchel J, Chatlin K, et al. Deep learning based feature-level integration of multi-omics data for breast cancer patients survival analysis. BMC Med Inform Decis Mak 2020. 10.1186/s12911-020-01225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tong L, Wu H, Wang MD. Integrating multi-omics data by learning modality invariant representations for improved prediction of overall survival of cancer. Methods 2020. 10.1016/j.ymeth.2020.07.008. [DOI] [PubMed] [Google Scholar]
- 53. Poirion O, Chaudhary K, Huang S, et al. DeepProg: an ensemble of deep-learning and machine-learning models for prognosis prediction using multi-omics data. medRxiv 2020. 10.1101/19010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xie G, Dong C, Kong Y, et al. Group lasso regularized deep learning for cancer prognosis from multi-omics and clinical features. Genes (Basel) 2019. 10.3390/genes10030240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hao J, Masum M, Oh JH, Kang M. Gene- and pathway-based deep neural network for multi-omics data integration to predict cancer survival outcomes. In: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics). 2019. doi: 10.1007/978-3-030-20242-2_10 [DOI] [Google Scholar]
- 56. Sims AH, Howell A, Howell SJ, et al. Origins of breast cancer subtypes and therapeutic implications. Nat Clin Pract Oncol 2007. 10.1038/ncponc0908. [DOI] [PubMed] [Google Scholar]
- 57. Russnes HG, Lingjærde OC, Børresen-Dale AL, et al. Breast cancer molecular stratification: from intrinsic subtypes to integrative clusters. Am J Pathol 2017. 10.1016/j.ajpath.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 58. Johnson KS, Conant EF, Soo MS. Molecular subtypes of breast cancer: a review for breast radiologists. J Breast Imaging 2021. 10.1093/jbi/wbaa110. [DOI] [PubMed] [Google Scholar]
- 59. Vidman L, Källberg D, Rydén P. Cluster analysis on high dimensional RNA-seq data with applications to cancer research - an evaluation study. PLoS One 2019. 10.1371/journal.pone.0219102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rappoport N, Shamir R. NEMO: cancer subtyping by integration of partial multi-omic data. Bioinformatics 2019. 10.1093/bioinformatics/btz058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang D, Gu J. Integrative clustering methods of multi-omics data for molecule-based cancer classifications. Quant Biol 2016;4(1):58–67. 10.1007/s40484-016-0063-4. [Google Scholar]
- 62. Takahashi S, Asada K, Takasawa K, et al. Predicting deep learning based multi-omics parallel integration survival subtypes in lung cancer using reverse phase protein array data. Biomolecules 2020. 10.3390/biom10101460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee TY, Huang KY, Chuang CH, et al. Incorporating deep learning and multi-omics autoencoding for analysis of lung adenocarcinoma prognostication. Comput Biol Chem 2020. 10.1016/j.compbiolchem.2020.107277. [DOI] [PubMed] [Google Scholar]
- 64. Lemsara A, Ouadfel S, Fröhlich H. PathME: pathway based multi-modal sparse autoencoders for clustering of patient-level multi-omics data. BMC Bioinformatics 2020. 10.1186/s12859-020-3465-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pasini A. Artificial neural networks for small dataset analysis. J Thorac Dis 2015;7(5):953–60. 10.3978/j.issn.2072-1439.2015.04.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wójcik PI, Kurdziel M. Training neural networks on high-dimensional data using random projection. Pattern Anal Applic 2018. 10.1007/s10044-018-0697-0. [DOI] [Google Scholar]
- 67. Fu Y, Xu J, Tang Z, et al. A gene prioritization method based on a swine multi-omics knowledgebase and a deep learning model. Commun Biol 2020. 10.1038/s42003-020-01233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hao J, Kim Y, Kim T, et al. PASNet: pathway-associated sparse deep neural network for prognosis prediction from high-throughput data. BMC Bioinformatics 2018. 10.1186/s12859-018-2500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Song M, Greenbaum J, Luttrell J, et al. A review of integrative imputation for multi-omics datasets. Front Genet 2020. 10.3389/fgene.2020.570255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yeung KF, Yang Y, Yang C, et al. CoMM: a collaborative mixed model that integrates GWAS and eQTL data sets to investigate the genetic architecture of complex traits. Bioinform Biol Insights 2019;13. 10.1177/1177932219881435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang W, Voloudakis G, Rajagopal VM, et al. Integrative transcriptome imputation reveals tissue-specific and shared biological mechanisms mediating susceptibility to complex traits. Nat Commun 2019;10(1). 10.1038/s41467-019-11874-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dong X, Lin L, Zhang R, et al. ToBMI: trans-omics block missing data imputation using a k-nearest neighbor weighted approach. Bioinformatics 2019;35(8). 10.1093/bioinformatics/bty796. [DOI] [PubMed] [Google Scholar]
- 73. Zhou X, Chai H, Zhao H, et al. Imputing missing RNA-sequencing data from DNA methylation by using a transfer learning-based neural network. Gigascience 2020;9(7). 10.1093/gigascience/giaa076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cheng CY, Tseng WL, Chang CF, et al. A deep learning approach for missing data imputation of rating scales assessing attention-deficit hyperactivity disorder. Front Psychiatry 2020. 10.3389/fpsyt.2020.00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lin X, Boutros PC. Optimization and expansion of non-negative matrix factorization. BMC Bioinformatics 2020;21(1). 10.1186/s12859-019-3312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu X, Zhu X, Li M, et al. Late fusion incomplete multi-view clustering. IEEE Trans Pattern Anal Mach Intell 2019;41(10). 10.1109/TPAMI.2018.2879108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Argelaguet R, Velten B, Arnol D, et al. Multi-omics factor analysis—a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol 2018;14(6). 10.15252/msb.20178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zanfardino M, Castaldo R, Pane K, et al. MuSA: a graphical user interface for multi-OMICs data integration in radiogenomic studies. Sci Rep 2021;11(1). 10.1038/s41598-021-81200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zanfardino M, Franzese M, Pane K, et al. Bringing radiomics into a multi-omics framework for a comprehensive genotype-phenotype characterization of oncological diseases. J Transl Med 2019;17(1). 10.1186/s12967-019-2073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Elabd H, Bromberg Y, Hoarfrost A, et al. Amino acid encoding for deep learning applications. BMC Bioinformatics 2020. 10.1186/s12859-020-03546-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Du M, Liu N, Hu X. Techniques for interpretable machine learning. Commun ACM 2020;63(1). 10.1145/3359786. [DOI] [Google Scholar]
- 82. Murdoch WJ, Singh C, Kumbier K, et al. Definitions, methods, and applications in interpretable machine learning. Proc Natl Acad Sci U S A 2019;116(44). 10.1073/pnas.1900654116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fisher A, Rudin C, Dominici F. All models are wrong, but many are useful: learning a variable’s importance by studying an entire class of prediction models simultaneously. J Mach Learn Res 2019;20. [PMC free article] [PubMed] [Google Scholar]
- 84. Ribeiro MT, Singh S, Guestrin C. “Why should I trust you?” Explaining the predictions of any classifier. In: Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 13–17 August 2016. 2016. doi: 10.1145/2939672.2939778 [DOI]
- 85. Lundberg SM, Lee SI. A unified approach to interpreting model predictions. In: Advances in Neural Information Processing Systems, Vol. 2017, 2017. [Google Scholar]
- 86. Ghosh D, Bernstein JA, Khurana Hershey GK, et al. Leveraging multilayered “omics” data for atopic dermatitis: a road map to precision medicine. Front Immunol 2018;9:2727. 10.3389/fimmu.2018.02727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mersha TB, Afanador Y, Johansson E, et al. Resolving clinical phenotypes into endotypes in allergy: molecular and omics approaches. Clin Rev Allergy Immunol 2021;60(2):200–19. 10.1007/s12016-020-08787-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schmauch B, Romagnoni A, Pronier E, et al. A deep learning model to predict RNA-Seq expression of tumours from whole slide images. Nat Commun 2020;11(1):3877. 10.1038/s41467-020-17678-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zheng H, Momeni A, Cedoz P-L, et al. Whole slide images reflect DNA methylation patterns of human tumors. npj Genomic Med 2020;5(1):11. 10.1038/s41525-020-0120-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen RJ, Lu MY, Wang J, et al. Pathomic fusion: an integrated framework for fusing histopathology and genomic features for cancer diagnosis and prognosis. IEEE Trans Med Imaging 2020;1. 10.1109/TMI.2020.3021387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ning Z, Pan W, Chen Y, et al. Integrative analysis of cross-modal features for the prognosis prediction of clear cell renal cell carcinoma. Bioinformatics 2020;36(9):2888–95. 10.1093/bioinformatics/btaa056. [DOI] [PubMed] [Google Scholar]
- 92. Vale Silva LA, Rohr K. Pan-cancer prognosis prediction using multimodal deep learning. In: 2020 IEEE 17th International Symposium on Biomedical Imaging (ISBI). 2020:568–71. doi: 10.1109/ISBI45749.2020.9098665 [DOI]
- 93. Wang Z, Li R, Wang M, et al. GPDBN: deep bilinear network integrating both genomic data and pathological images for breast cancer prognosis prediction. Bioinformatics 2021. 10.1093/bioinformatics/btab185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hao J, Kosaraju SC, Tsaku NZ, et al. PAGE-Net: interpretable and integrative deep learning for survival analysis using histopathological images and genomic data. Pac Symp Biocomput 2020, 2020;25. [PubMed] [Google Scholar]
- 95. Cui S, Ten Haken RK, El Naqa I. Integrating multiomics information in deep learning architectures for joint actuarial outcome prediction in non-small cell lung cancer patients after radiation therapy. Int J Radiat Oncol Biol Phys 2021;110(3). 10.1016/j.ijrobp.2021.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ahmed Z. Practicing precision medicine with intelligently integrative clinical and multi-omics data analysis. Hum Genomics 2020;14(1):35. 10.1186/s40246-020-00287-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wu P-Y, Cheng C-W, Kaddi CD, et al. Omic and electronic health record big data analytics for precision medicine. IEEE Trans Biomed Eng 2017;64(2):263–73. 10.1109/TBME.2016.2573285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bunyavanich S, Schadt EE. Systems biology of asthma and allergic diseases: a multiscale approach. J Allergy Clin Immunol 2015;135(1):31–42. 10.1016/j.jaci.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Benson M. Clinical implications of omics and systems medicine: focus on predictive and individualized treatment. J Intern Med 2015;279(3):229–40. 10.1111/joim.12412. [DOI] [PubMed] [Google Scholar]
- 100. Jin W, Stokes JM, Eastman RT, et al. Deep learning identifies synergistic drug combinations for treating COVID-19. Proc Natl Acad Sci 2021;118(39). 10.1073/pnas.2105070118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rung J, Brazma A. Reuse of public genome-wide gene expression data. Nat Rev Genet 14(2):89–99. nrg3394 [pii]10.1038/nrg3394. [DOI] [PubMed] [Google Scholar]
- 102. Aliper A, Plis S, Artemov A, et al. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharm 2016;13(7). 10.1021/acs.molpharmaceut.6b00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 2016;26(7). 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Niu X, Yang K, Zhang G, et al. A pretraining-retraining strategy of deep learning improves cell-specific enhancer predictions. Front Genet 2020;10. 10.3389/fgene.2019.01305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sigurdsson AI, Westergaard D, Winther O, et al. Deep integrative models for large-scale human genomics. bioRxiv 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhu W, Xie L, Han J, et al. The application of deep learning in cancer prognosis prediction. Cancers (Basel) 2020;12(3). 10.3390/cancers12030603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Reinke SN, Gallart-Ayala H, Gómez C, et al. Metabolomics analysis identifies different metabotypes of asthma severity. Eur Respir J 2017;49(3):1601740. 10.1183/13993003.01740-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wills-Karp M, Ewart SL. Time to draw breath: asthma-susceptibility genes are identified. Nat Rev Genet 2004;5(5):376–87. 10.1038/nrg1326. [DOI] [PubMed] [Google Scholar]
- 109. Zosky GR, Sly PD. Animal models of asthma. Clin Exp Allergy 2007;37(7):973–88. 10.1111/j.1365-2222.2007.02740.x. [DOI] [PubMed] [Google Scholar]
- 110. Crouser ED, Fingerlin TE, Yang IV, et al. Application of “Omics” and systems biology to sarcoidosis research. Ann Am Thorac Soc 2017;14. 10.1513/AnnalsATS.201707-567OT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Holzinger A, Dehmer M, Jurisica I. Knowledge discovery and interactive data mining in bioinformatics - state-of-the-art, future challenges and research directions. BMC Bioinformatics 2014. 10.1186/1471-2105-15-S6-I1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Yu KH, Snyder M. Omics profiling in precision oncology. Mol Cell Proteomics 2016. 10.1074/mcp.O116.059253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Aun MV, Bonamichi-Santos R, Arantes-Costa FM, et al. Animal models of asthma: utility and limitations. J Asthma Allergy 2018. 10.2147/JAA.S121092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.