

Abstract
Combination therapy has shown an obvious efficacy on complex diseases and can greatly reduce the development of drug resistance. However, even with high-throughput screens, experimental methods are insufficient to explore novel drug combinations. In order to reduce the search space of drug combinations, there is an urgent need to develop more efficient computational methods to predict novel drug combinations. In recent decades, more and more machine learning (ML) algorithms have been applied to improve the predictive performance. The object of this study is to introduce and discuss the recent applications of ML methods and the widely used databases in drug combination prediction. In this study, we first describe the concept and controversy of synergism between drug combinations. Then, we investigate various publicly available data resources and tools for prediction tasks. Next, ML methods including classic ML and deep learning methods applied in drug combination prediction are introduced. Finally, we summarize the challenges to ML methods in prediction tasks and provide a discussion on future work.
Keywords: drug combination prediction, machine learning, drug combination database, deep learning, synergy
Introduction
In recent decades, with the discovery and application of new therapeutic targets, many advances have been made in drug development [1–4]. However, due to the biological complexity of cancer, cardiovascular diseases and other diseases, multiple target genes are involved and their protein products play key roles in controlling abnormal pathways and networks [5]. The monotherapy usually cannot break down the entire disease pathways and networks [6]. The redundancy and complexity of pathways often lead to compensation and resistance to monotherapy [7]. In many cases, drug resistance is a major obstacle for the effective treatment, which is often caused by the heterogeneity of complex diseases [8–11]. The specific mechanisms of drug resistance include increased efflux of drugs [12], mutations of target proteins [13] and activation of disease alternative pathways [14]. In order to overcome the limitations of monotherapy, combination therapy is presented as a promising therapy to achieve more disease control [15]. The theoretical basis of combination therapy is that multiple drugs can be used to target multiple targets, pathways or cellular processes involved in the pathogenesis of a specific disease type [15, 16]. Compared with monotherapy, combination therapy can greatly increase the therapeutic effect, decrease the dosage to avoid toxicity and slow down the development of drug resistance [17]. Thus, combination therapy has become a standard clinical treatment strategy for many complex diseases such as cancer, acquired immune deficiency syndrome, asthma, diabetes and bacterial infection [18, 19].
Clinical experience is obviously not an efficient method to identify a large number of effective drug combinations [20]. Subsequently, systematic large-scale screening techniques are conducted, including high-throughput screening (HTS) method [21] and multiplex screening for interacting compounds method [22]. Relying on these large-scale screening methods, a large number of available drug combination databases have been accumulated. These databases greatly accelerate the discovery of drug combinations. However, as the number of drugs increases, the search space exponentially explodes, thus it is impractical to screen all possible drug combinations for all possible indications [23–25]. Therefore, computational methods are urgently needed to reduce the search space of drug combinations.
In the past decades, computational methods have been widely used in the prediction of drug combination, including systems biology methods, kinetic models, mathematical methods, stochastic search algorithms and machine learning (ML) methods. Systems biology methods focus on the control and analysis of biological networks, in which biological knowledge is needed [26]. The limitations in biological knowledge make it difficult to large-scale drug combination discovery. Kinetic modeling methods simulate the dynamic changes of nodes in realistic biological networks using kinetic equations [27]. However, the kinetics of most biological networks cannot be well-defined. In the mathematical methods, one or more direct mathematical models [28] and statistical tests [29] are applied. The success rates of them mainly depend on the quality of the assumptions behind the models. In stochastic search algorithms, drugs are iteratively combined and measured, in effect searching through the vast space of possibilities [30]. However, due to time and space costs of the calculation, they are only computational accessible to small datasets. Some studies of these methods and their brief descriptions are listed in Table 1. Different from the above hypothesis-driven methods, which are limited by prior knowledge and difficult to processing larger dataset, ML methods are more data-driven prediction. Given a certain amount of training data, they can learn the complex nonlinear relationships between input attributes data (such as chemical structure) and the associated output (such as synergy score) [31, 32]. ML methods have been increasingly applied to drug combination prediction due to their high predictive performance and large prediction range [16, 33].
Table 1.
A list and brief description of some computational methods reviewed in this manuscript
| Category of methods | Abbreviation | Algorithms | Description |
|---|---|---|---|
| Systems biology methods | MTOI | Multiple target optimal intervention | MTOI identifies potential drug targets and suggests optimal combinations of the target intervention that best restore the network to a normal state [128]. |
| Network proximity calculation | The topology relationships between drug modules (subnetworks of drug target proteins) and disease modules (subnetworks of disease proteins) were calculated according to the distance between protein nodes [129]. | ||
| TIMMA | Target inhibition interaction using maximization and minimization averaging |
TIMMA searches for optimal set of cancer-specific targets of each drug based on drug-target network [130]. | |
| Kinetic models | Kinetic model | A kinetic model for several small networks with typical crosstalk modules can investigate crosstalk in inducing drug resistance [27]. | |
| Mathematical methods | CDA | Combinatorial drug assembler | CDA makes hyper-geometric tests for signaling pathway gene set enrichment analysis to perform signaling pathway expression pattern analysis and drug set pattern analysis [29]. |
| Stochastic search algorithms | MACS | Medicinal algorithmic combinatorial screen | A novel fitness function based on the level of inhibition and the number of drugs is applied with search algorithms [30]. |
| Classic machine learning methods | SVM | Support vector machine | SVM constructs a hyperplane or a set of hyperplanes to classify data points [82, 131]. |
| NB | Naïve Bayes | Naïve Bayes is a statistical classification method based on the Bayes rule of conditional probability [82, 90]. | |
| LR | Logistic regression | LR assumes that the data obey the Bernoulli distribution (binomial distribution). Use the maximum likelihood function to solve the parameters, so as to achieve the purpose of data classification [132, 133]. | |
| RF | Random forest | An integrated classifier consisting of multiple decision trees [36, 81, 97–103] . | |
| ANN | Artificial neural network | An ANN is based on a collection of connected units or nodes called artificial neurons, which are aggregated into layers [93]. | |
| SGB | Stochastic gradient boosting | SGB [87] uses an ensemble of weak classifiers to construct a prediction model, typically decision trees that incorporate randomization into the procedure [82]. | |
| XGBoost | Extreme gradient boosting | XGBoost is a tree learning algorithm for sparse data processing in the framework of gradient boosting and provides parallel tree promotion [36, 80, 135]. | |
| GTB (GBM/GBRT) | Gradient tree boosting (gradient boosting machine or gradient boosted regression tree) | GTB algorithm is based on regression tree. Each tree is learned from the residual of all previous trees using the negative gradient value of loss function in the current model [136]. | |
| FM | Factorization machine | FM models pairwise interactions via inner products of respective feature latent vectors using the matrix factorization method [94, 137]. | |
| Deep learning methods | FNN | Feedforward neural network | A FNN is an ANN with multiple fully connected layers between the input and output layers. |
| DBN | Deep belief network | DBN is composed of stacked RBM. RBM is a generative stochastic ANN with a bipartite structure. | |
| AE | Autoencoder | AE is a neural network consisted of an encoder and a decoder, which has an internal (hidden) layer that describes a code used to represent the input. | |
| GCN | Graph convolutional network | The GCN used a convolutional neural network to do graph embedding, and thus solved a link prediction task. |
Moreover, Dialogue for Reverse Engineering Assessments and Methods consortium (DREAM) (www.dreamchallenges.org) has launched two community challenges to promote the development of innovative drug combination prediction methods. The first challenge, NCI-DREAM [28], is to predict 91 drug combinations in a single cell line (OCI-LY3) launched in 2012. ML methods are not applicable due to less training data. The best performing method, DIGRE, uses a mathematical model. The other challenge is AstraZeneca-Sanger Drug Combination prediction (AZ-DREAM) [34], which is sponsored by AstraZeneca and the Sanger Institute in 2015. This challenge provides 11 576 combinations in 85 cancer cell lines. There are 160 teams participating and MLs are more competitive to this dataset. The winning method is based on Random Forest (RF) [35] method, an ensemble learning-based ML method.
The rest of the review is divided into six sections, starting with section Machine learning workflow for drug combination prediction, which summarizes the ML workflow for drug combination prediction. Section Definition of synergistic effect in prediction task introduces the concept and controversy of synergism and antagonism between drug combinations. Section Databases, web servers and software tools provides the information about related databases, including drug combination databases and other related databases. Section Machine learning methods used in drug combination prediction summarizes the ML methods for drug combination prediction, including classic ML methods and deep learning (DL) methods. Section Challenges and future work concludes the challenges and future work.
Machine learning workflow for drug combination prediction
In ML methods, the problem of drug combination prediction is usually formulated as a multi-class classification or a regression task. Combination effects could be classified as synergistic, additive and antagonistic effect according to the definition. While most classification studies classify drug combinations into two categories, synergy and non-synergy. Non-synergy involves slight synergy, additive effect and antagonism. The regression task is to predict the quantitative synergistic score of drug combination. In the input data for training, the synergy/non-synergy categories or synergy scores are considered as label data, while the various properties about drugs, drugs’ target proteins, or cancer cell lines are regarded as feature data. There are three main ML approaches applied in the task of drug combination prediction, i.e. supervised learning [36], unsupervised learning [37] and semi-supervised learning [38, 39]. In supervised learning, the training data are composed of input features and output labels of samples. Different models are used to learn a function that maps an input to an output. An optimal model can determine the labels for unseen instances correctly. The purpose of unsupervised learning is to learn the hidden patterns from unlabeled input data. For example, clustering is the most popular technique in unsupervised learning [40]. In semi-supervised learning, the training data consist of a small number of labeled samples and a large number of unlabeled samples. The labeled samples are used to predict the unlabeled samples through the applied algorithms in semi-supervised learning, such as manifold ranking algorithms [38]. So far, most of the methods for predicting drug combinations are supervised learning. Therefore, this review mainly focuses on the application of supervised ML.
The workflow for predicting drug combinations using ML methods is shown in Figure 1. Firstly, the samples, labels and features are retrieved from drug combination databases and other related databases. In a sample, each instance (i.e. drug and/or cancer cell line) is represented by a feature vector, which reflects structural and/or other properties of the corresponding instances. Then the feature vectors of each instance are concatenated or fused to obtain the feature vector of each sample. Next, these feature vectors and corresponding labels can be fed into classic ML or DL models for training. The parameters of the ML models can be optimized during the training stage. Then various metrices are used to evaluate performance of these models and to select the optimal one. After training stage, these models can be applied to an external test set to evaluate the generalizability or to predict novel drug combinations. Biological experiments or literatures searching are conducted to verify the prediction results. Finally, some interpretable analysis would be performed to explore the related biological mechanism.
Figure 1.
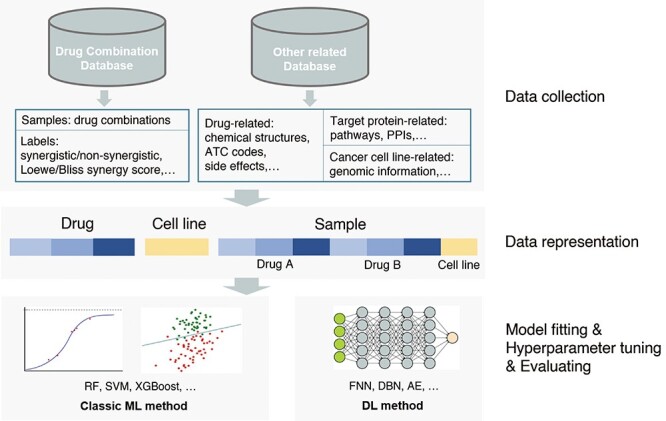
Workflow of ML methods used in drug combination prediction.
Definition of synergistic effect in prediction task
As mentioned in above section, the prediction task of drug combination is usually divided into classification and regression task (Figure 2). In classification task, the combination effect of drugs is usually classified as synergistic, additive and antagonistic effect. Generally speaking, the synergistic, additive or antagonistic effect means that the effect of two drugs is greater than, equal to or less than the sum of the effects of individual drugs. The additive effect is mainly used as a boundary to distinguish synergistic and antagonistic effect [17]. The effect or response of a drug is usually measured by the inhibition or viability of cell proliferation [41, 42].
Figure 2.
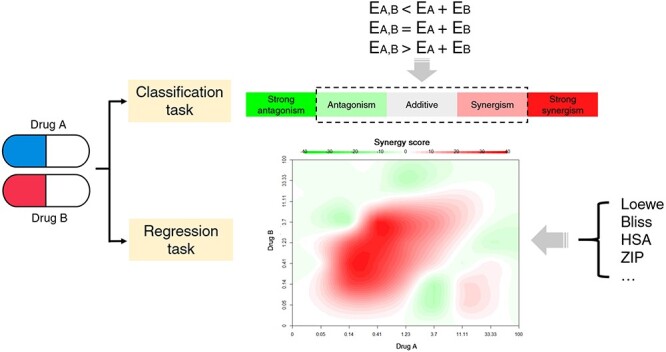
Classification and regression task in drug combination prediction. The color figure is made using R SynergyFinder package [70]. Note: EA,B, EA, EB refer to the effect of treatment with drug combination (A + B), drug A and drug B respectively. Loewe: Loewe additivity model. Bliss: Bliss independence model. HAS: Highest Single Agent model. ZIP: Zero Interaction Potency model.
In addition to the above classification methods for drug combination effects, Chou and Talalay et al. [17] subdivided the degree of synergy into additive, slight synergism, moderate synergism, synergism, strong synergism and very strong synergism. The degree of antagonism was also divided in the same way. Tang et al. [43] divided the drug combination into five categories: strong antagonism, weak antagonism, non-interaction, weak synergy and strong synergy.
In regression task, the synergism or antagonism of drug combination is quantified based on different models. The basic assumptions of these models are different. In previous reviews [17, 43–45], some definitions of synergy quantification were described in detail. In the past 20 years, researchers have not reached a consensus on the quantification and accurate definition of synergism and antagonism, which remains one of the biggest challenges in this field [16]. In these quantification models, the two most widely used reference models for calculating the synergistic and antagonistic effects of drug combinations are the Loewe additivity model (Loewe) [46] and the Bliss independence model (Bliss) [47].
Loewe additivity model defines the effect of the combination of a compound with itself as additive effect. For the combination of drug 1 and drug 2, let d1 and d2 be the dosage of drug 1 and drug 2 in combination, Di represents the dosage of single drug required when the drug is used alone to achieve the combination effect ELoewe, and E1 and E2 refer to the effects obtained by using drug 1 and drug 2 alone, respectively. According to Loewe, when there is no interaction between drug A and drug B, the Loewe additivity model of this combination is defined as:
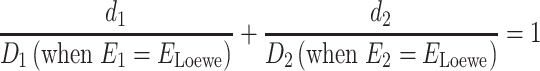 |
(1) |
Then Chow-Talalay combination index (CI) [17] for Loewe model assigns a quantitative measure to any given drug combination:
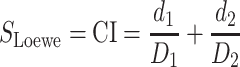 |
(2) |
If CI is less than (or greater than) 1, the combination is considered to be synergistic (or antagonistic).
The Bliss independence model takes probabilistic view and considers that the two drugs of a combination have probability independence. E1 and E2 are considered as the effects obtained by using drug 1 and drug 2 alone, and EBLISS is considered as the combination effect. When drug 1 and drug 2 act independently, this combination can achieve the following equation:
When EBLISS is greater than or less than the right-hand side of the equation, there is a synergistic or antagonistic effect.
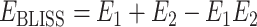 |
(3) |
There are many studies comparing Bliss with Loewe models [43, 45, 48–51]. In these comparisons, Loewe model is more consistent with the expected combination effect of two drugs acting on the same target or pathway, while Bliss model aims to non-interactive drug combinations, which means the two drugs independently act on different targets or pathways. Loewe model requires a dose–response curve for a single drug, but Bliss model does not. Moreover, Bliss model does not have a definition of ‘additive,’ because when a drug is tested in combination with itself, it will not seem to be ‘independent.’
In recent years, a variety of models have been developed to evaluate the effect of drug combination. Most of the models are derived from the variants of these two models [52, 53]. In addition, median-effect [54], highest single agent (HSA) model [55] and zero interaction potency (ZIP) model [56], etc. are also mentioned in our review. It should be noted that the synergy of drug combination depends on the specific dosage of drugs. Therefore, after determining the synergistic drug combinations, further experiments are needed to determine the synergistic dose range.
Databases, web servers and software tools
The databases, web servers and software tools available in drug combination prediction including drug combination related- and other related-databases, which provide label (sample) and feature data, respectively. The statistics and links of the databases are listed in Table 2.
Table 2.
Statistics of drug combination databases, software tools and other related databases reviewed in this paper
| Database | Statistics | Website | Latest update | ||||
|---|---|---|---|---|---|---|---|
| Drugs/ compounds |
Combination experiments | Target proteins | Interactions | ||||
| Drug combination database | DrugComb_v1.5 [57] | 8397 | 739 964 | https://drugcomb.fimm.fi/ | March 2021 | ||
| DrugCombDB [59] | 5350 | 498 865 | http://drugcombdb.denglab.org/ | May 2019 | |||
| SYNERGxDB [61] | 1977 | 536 596 | https://www.synergxdb.ca/ | 2019 | |||
| NCI-ALMANAC [42] | 104 | 304 549 | https://dtp.cancer.gov/ncialmanac | December 2017 | |||
| O’Neil et al. study [62] | 38 | 22 737 | March 2016 | ||||
| ASDCD [63] | 135 | 548 | 1225 (DTI) | http://asdcd.amss.ac.cn/ | May 2018 | ||
| AZ-DREAM [34] | 118 | 11 576 | https://www.synapse.org/#!Synapse:syn4231880/wiki/235649 | June 2016 | |||
| DCDB_v2.0 [65] | 904 | 1813 |
http://www.cls.zju.edu.cn/dcdb/
(dose not respond now) |
2014 | |||
| TTD [66] | 37 316 | 119 | 3419 | 6975 (PPI) | http://db.idrblab.net/ttd/ | June 2020 | |
| DrugR+ [67] | http://www.drugr.ir (dose not respond now) | ||||||
| Software tools | SynergyFinder [68] | https://synergyfinder.fimm.fi | 2020 | ||||
| Synergy [71] | https://pypi.org/project/synergy | June 2021 | |||||
| CompuSyn [72] | https://www.combosyn.com/ | 2005 | |||||
| Combenefit [73] | https://sourceforge.net/projects/combenefit/ | 2016 | |||||
| Other related databases | DrugBank_v5.1.6 [138] | 13 570 | 5251 | 365 984 (DDI) | http://www.drugbank.ca/ | January 2021 | |
| Chemical checker [139] | 778 531 | https://chemicalchecker.org | May 2020 | ||||
| STITCH_v5.0 [140] | >500 000 | >9.6 × 106 | >1.6 × 109 (DTI) | http://stitch.embl.de/ | 2016 | ||
| KEGG [141–143] | 30 507 | https://www.kegg.jp/ | August 2021 | ||||
| SIDER_v4.1 [144] | 1430 | http://sideeffects.embl.de/ | October 2015 | ||||
| Offsides and Twosides_v0.1 [145] | 3394 | 63 000 | 5.7 × 106 (DDI: SE) | http://tatonettilab.org/offsides/ | November 2019 | ||
| PubChem [146] | 96 157 016 | 79 622 | https://pubchem.ncbi.nlm.nih.gov/ | 2019 | |||
| Uniprot [147] | >1.2 × 108 | https://www.uniprot.org/ | February 2020 | ||||
| SuperTarget [148] | 195 770 | 6219 | 332 828 (DTI) | http://insilico.charite.de/supertarget/index.php | 2011 | ||
| BioGRID_v3.5 [149] | 1 814 182 (PPI) | https://thebiogrid.org/ | May 2020 | ||||
| HPRD [150] | 30 047 | 41,327 (PPI) | http://www.hprd.org/ | 2010 | |||
| LINCS_v2.0 [151] | 41 847 | 1469 | http://www.lincsproject.org/LINCS/ | June 2020 | |||
| ChEMBL [152] | 1 961 462 | 13 382 | https://www.ebi.ac.uk/chembl/ | May 2020 | |||
| DTC [153] | 4276 | 1007 | 204 901 (DTI) | http://drugtargetcommons.fimm.fi | 2018 | ||
| TDR targets [154] | ~2 000 000 | ~5300 | http://tdrtargets.org/ | March 2021 | |||
| The IUPHAR/BPS Guide to PHARMACOLOGY [155] | 10 905 | 2989 | http://www.guidetopharmacology.org/ | ||||
| CancerDR [156] | 148 | 116 | https://webs.iiitd.edu.in/raghava/cancerdr/ | 2012 | |||
| BindingDB [157] | 995 797 | 8561 | 2 303 972 (DTI) | http://www.bindingdb.org/bind | July 2021 | ||
Note: DTI: drug/compound–target interaction. DDI: drug–drug interaction.
ASDCD: Antifungal synergistic drug combination database. DCDB: Drug combination database. TTD: Therapeutic target database. STITCH: Search Tool for Interacting Chemicals. KEGG: Kyoto Encyclopedia of Genes and Genomes. UniProt: Universal Protein. BioGRID: Biological General Repository for Interaction Datasets. HPRD: Human Protein Reference Database. LINCS: Library of Integrated Network-Based Cellular Signatures. DTC: Drug Target Commons.
Drug combination databases are mainly established to collect drug combination information. In this review, we list nine databases, six web servers and four software tools. Different criteria are used to classify and quantify drug combinations in these databases, which can be used as the benchmarks for advanced ML methods to implement the classification or regression tasks. For classification task, the drug combinations are often classified as ‘synergistic/additive/antagonistic’ or ‘efficacious/non-efficacious’ in these databases. For regression tasks, these databases provide various synergy scores to quantify the interaction between the drugs in combinations.
Among these databases, DrugComb, DrugCombDB, SYNERGxDB, NCI-ALMANAC, O’Neil et al. study and AZ-DREAM mainly focus on anti-cancer drug combinations. Figure 3 shows the number of overlapping drugs, combination experiments between three databases, DrugComb, DrugCombDB and SYNERGxDB. In terms of the statistics, there is some overlap between the three databases since part of the data in these databases were collected from NCI-ALMANAC and O’Neil et al. study. In addition to a variety of cancers, DrugComb also provided data for other diseases such as malaria and COVID-19. In addition, ASDCD was constructed for antifungal drug combinations. DCDB and TTD databases collect many kinds of drug combinations for a variety of human diseases.
Figure 3.
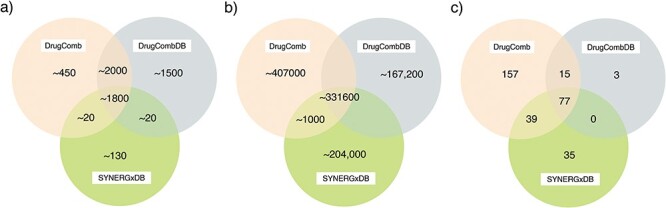
The number of overlapping (A) drugs, (B) combination experiments and (C) cell lines between three anti-cancer drug combination databases, i.e. DrugComb, DrugCombDB and SynergxDB. The number of drugs and cell lines only includes those involved in drug combination data. The statistical results of drugs and combination experiments are the approximate number of estimates since there are different identifies of drugs provided by the three databases, and the situation of synonyms is taken into account.
In addition, we list six web servers containing DrugComb, DrugCombDB, SYNERGxDB, NCI-ALMANAC, DrugR+, ASDCD and SynergyFinder. The introduction of web servers is along with that of databases and soft tools. In these websites, users can search and visualize the synergy effects of drug combinations on specific tissues or cell lines. Besides, four software tools, i.e. SynergyFinder, Synergy, Combenefit and CompuSyn, are also used to calculate the synergy scores of drug combination based on some synergy models. The software tools can also provide the visualization, analysis and quantification of synergistic or antagonistic drug combination effects.
Other related databases introduced in this review can be used as the feature data of samples to be fed into ML models. When applying ML methods to make predictions, choosing appropriate feature data as input can effectively improve model prediction performance, and reduce training time and the risk of overfitting. The data stored in these databases including (1) chemical feature of the drugs, including the chemical structures, the anatomical therapeutic chemical (ATC) codes, chemical–chemical interactions (CCI); (2) feature data of drugs’ target proteins, including gene expression data, protein sequences, gene ontology (GO) terms, pathways, etc.; (3) pharmacological information, including cell line response to drugs, drug side effects (SE) and other information. Statistics and some information of these databases are shown in Table 2. The brief introduction of each other related database is shown in Supplementary file.
DrugComb
DrugComb is a web-based database published in 2019 [41] and updated in March 2021 [57]. In addition to data of drug combination synergy, the monotherapy sensitivity screening data are also provided, involving diseases such as cancers, malaria and COVID-19. The data stored in the DrugComb database are collected from 37 studies. Both combination and single drug screening data provided in this database involve 8397 drugs. Combination data stored in DrugComb (v1.5) contain 739 964 combination experiments for 4268 drugs tested in 288 cell lines. DrugComb also provides five types of synergy score for these combinations. The synergy scores involve four commonly used reference models, Bliss, HSA, Loewe, and ZIP and a novel measurement named S score. Additionally, the targets of drugs are provided in the database.
DrugComb provides a web server to analyze and visualize the synergy of drug combinations. In this website, it can provide the predicted synergy scores of a given drug combination for a cell line at the single dose level. The prediction is based on a ML model, CatBoost [58], which is a gradient boosting framework based on decision-trees. Furthermore, DrugComb also uses a drug-target network-based model to visualize the mechanisms of action of drug combinations.
DrugCombDB
DrugCombDB [59] is also a web-based database of drug combinations released in 2019. The data in DrugCombDB are from four sources, i.e. HTS assays of drug combinations, the U.S. Food and Drug Administration (FDA) Orange Book [60], manual curations from the literatures, and external databases. In total, DrugCombDB includes 498 865 combination experiments, covering 5350 drugs and 104 cancer cell lines. It also provides quantitative synergy scores based on four models, i.e. HSA, Loewe, Bliss and ZIP.
In the DrugCombDB website, the properties of the combination or component drugs are also concluded, such as the common target proteins of the combination, molecular weight and chemical structure.
SYNERGxDB
SYNERGxDB [61] is a pharmacogenomic database released in 2020. It integrates nine drug combination datasets from academic groups and pharmaceutical companies, resulting in 536 596 combination experiments for 1977 compounds tested in 151 cell lines. In addition to drug combinations, SYNERGxDB also includes metabolomics, gene expression, copy number and mutation profiles of the cancer cell lines. All the drugs and cell lines are annotated to unique identifiers. The same four synergy scores are provided, Loewe, Bliss, HSA and ZIP scores.
In the SYNERGxDB website, it provides further analysis of biomarker discovery, cell-line sensitivity analysis, tissue-specific enrichment analysis and consistency in synergy scores.
NCI-ALMANAC
The NCI-ALMANAC (a large matrix of anti neoplastic agent combinations) database [42] was published by the US National Cancer Institute (NCI) in 2017. These data come from an HTS that tested 304 549 combination experiments of 104 investigational and approved drugs across 60 cancer cell lines forming the NCI-60 panel. The synergy level is quantified by the ComboScore, a modified version of the Bliss model.
NCI-ALMANAC website allows searching the ComboScores of drug combination. In addition, the data can be visualized as a heatmap summarizing the entire dataset, as a bar plot of the ComboScores for a particular drug pair in all cell lines, or as dose response curves for one drug combination in a given cell line.
O’Neil et al. study
O’Neil et al. [62] from the Merck research laboratories conducted a large-scale oncology screening, and published the data in 2016. The database consists of 22 737 combination experiments covering 38 experimental and approved drugs in 39 cancer cell lines. Two models, i.e. HSA and Bliss are used to quantify the synergy in drug combinations.
ASDCD
Antifungal synergistic drug combination database (ASDCD) [63] focuses on synergistic drug combinations for the therapy of fungal infection. The first version of ASDCD database was released in 2011. In the latest version released in 2013, ASDCD recorded previously published synergistic antifungal drug combinations, chemical structures, target proteins, target-related signaling pathways, indications and other pertinent data. The latest version includes 548 combination experiments and 1225 drug–target interactions of 135 individual drugs from literatures and external databases.
For each entered drug, the ASDCD website provides researchers with three related links: drug target, target-related signaling pathway, and its synergistic interactions with other drugs.
AZ-DREAM
In order to accelerate the understanding and prediction of drug combination synergy, DREAM Challenges cooperated with AstraZeneca and the Sanger Institute to launch the AZ-DREAM Challenge in 2016 [34]. The AZ-DREAM Challenge database includes 11 576 combination experiments in 85 cancer cell lines. The challenge also provides additional information on drugs and molecular characterization of these cell lines.
DCDB
The first release of drug combination database (DCDB) [64] collected drug combinations from PubMed, FDA Orange Book [60]. Since the release of 2.0 version [65] in 2014, DCDB contains 1813 combination experiments, which are classified as ‘efficacious,’ ‘need further study’ or ‘non-efficacious,’ consisting of 904 distinctive drugs. 1445 combinations are annotated as ‘efficacious’ and reported in ClinicalTrials website (https://clinicaltrials.gov/) to meet their study criteria in clinical trials.
TTD
Therapeutic target database (TTD) [66] is not a drug combination database. It mainly provides the information about therapeutic protein and nucleic acid targets, and corresponding drugs against these targets. TTD contains 119 combination experiments, which are classified as synergistic, additive, antagonistic, potentiative and reductive. In addition, it also includes 1008 target combinations of drug combinations.
DrugR+
DrugR+ [67] is a computational database based on DrugBank and KEGG data published in 2019, which is organized for drug repurposing. It also provides drug combination-related information because combination therapy is considered as an alternative strategy to enhance the success rate of drug repurposing. This alternative strategy is called ‘combined drug replacement (CDR)’ in this database, which means a drug can be replaced by drugs with no/trivial adverse reactions. This database also supports drug combinations to enhance the effect of drugs. By searching existing data and mining latent information based IF-THEN rules, this database provides the drugs with the same biological targets and same/similar mechanism of action for CDR or combination therapy.
SynergyFinder
The R package SynergyFinder [68] implements four synergy scoring models, HSA, Loewe, Bliss and ZIP. The synergy scores are calculated across the tested concentration combinations, which can be visualized as either a two-dimensional or a three-dimensional landscape on the dose matrix. This landscape of drug interaction scoring is informative for specifying specific dose regions where synergistic or antagonistic drug interactions occur.
There is also a SynergyFinder web application for the preprocessing and visualization of the drug combination dose–response data released in 2017 [69] and updated in 2021 [70] (release 2.0). The tool calculates synergy scoring using four reference models, i.e. HSA, Loewe, Bliss and ZIP models. The degree of a drug combination effect can be visualized as a synergy landscape. In addition to two-drug combination, SynergyFinder 2.0 supports the similar synergy analysis of higher order drug combination data (a combination involved three or more drugs), along with automated outlier detection procedure, extended curve-fitting functionality and statistical analysis of replicate measurements.
Synergy
Synergy [71] is a python library for calculating, analyzing and visualizing drug combination synergy released in 2021. For the calculation of synergy, it provides nine synergy models, including Loewe, CI, Bliss, HSA, ZIP and other models. Moreover, it provides tools for evaluating confidence intervals and conducting power analysis. The synergy also could be visualized through heatmaps, surfaces and isosurfaces.
CompuSyn
CompuSyn [72] is a software based on the theory of the median-effect equation of mass-action law and the CI theorem for automated quantitative simulation of synergism or antagonism in drug combination studies. It was set up by Dr Dorothy Chou in 2005. It can generate graphics including dose-effect curves, median-effect plot, CI plot and isobologram for synergism or antagonism.
Combenefit
Combenefit [73] is a free software tool that enables the quantification, analysis and visualization of synergistic or antagonistic drug combinations. Combenefit is provided as a Matlab package and a standalone software for Windows OS. In this software, three synergy models (Loewe, Bliss, HSA) are used to quantify synergy effect. In Combenefit, the graphical outputs consist of the single agent or combination dose–response data and the resulting synergy distribution for a particular combination.
Machine learning methods used in drug combination prediction
The widely used ML methods can be divided into classic ML method and DL method. Classic ML methods include support vector machine (SVM) [74], decision tree (DT) [75], RF and extreme gradient boosting (XGBoost) [76]. While DL models contain feedforward neural network (FNN) [77], deep belief network (DBN) [78], autoencoder (AE) [79], etc. Due to its characteristic of multiple processing layers, DL methods require more training data, more hyperparameters, more computational resource and memory [33]. The performance of DL models would greatly improve with the increasing input data, especially large-scale dataset (more than hundreds of thousands of samples). While in the conditions of small- (hundreds of samples) and medium-scale (thousands to tens of thousands of samples) datasets, classic ML methods may perform better than DL methods with less hyperparameter tuning [80].
In the studies introduced in this manuscript, most studies of classic ML methods apply various feature types, while most DL methods only use structural and physiochemical information to represent drugs. The input feature type and other information of each method reviewed in this manuscript are listed in Table 3. Furthermore, some classic ML methods, such as tree-based algorithms (DT, RF, XGBoost, etc.), can extract important features [81, 82], and explain the individual prediction by decomposing the decision path into one component per feature. While DL model is lack of interpretability.
Table 3.
Summary of some studies involved in this review
| Study | Published year | Algorithms | Category of methods | Drug combination data set | Input data types | Program code |
|---|---|---|---|---|---|---|
| Tang et al. [130] | 2013 | TIMMA | Systems biology methods | Experiments | Target profiles | https://cran.r-project.org/web/packages/timma/index.html [158] |
| Pivetta et al. [93] | 2013 | ANN | Classic ML methods | Experiments | Concentrations of drugs | |
| Chen et al. [98] | 2013 | RF | Classic ML methods | Zhao et al. [23] | DDIs, PPIs, targets-enriched pathways | |
| Sun et al. [90] | 2014 | One-class SVM | Classic ML methods | DCDB | Gene expression profiles | |
| Huang et al. [132] | 2014 | LR | Classic ML methods | DCDB | Side effects | |
| Li et al. [159] | 2015 | PEA | Systems biology methods | DCDB, TTD, Literatures | Fingerprints, ATC codes, side effects, targets’ sequences, PPIs, targets’ GO terms | |
| Sun et al. [38] | 2015 | RACS | Systems biology methods | DCDB, Literatures, NCI-DREAM, TTD | GO-based mutual information entropy, topological features in drug/target networks | https://github.com/DrugCombination/RACS |
| Wildenhain et al. [97] | 2015 | SONAR | Classic ML methods | Experiments. | Chemical-genetic interactions, fingerprints | |
| Chen et al. [39] | 2016 | NLLSS | Systems biology methods | Literatures | Structural information, DTIs | |
| Gayvert et al. [81] | 2017 | RF | Classic ML methods | Held et al. [160] study. | Single drug dose response | |
| Li et al. [100] | 2017 | RF | Classic ML methods | AZ-DREAM | Structural information, target networks, drug induced gene expression data | |
| Xu et al. [82] | 2017 | SGB | Classic ML methods | DCDB | Fingerprints, ATC codes, PPIs, CCIs, and disease pathways. | |
| Shi et al. [108] | 2017 | TLMCS | Classic ML methods | DCDB | ATC codes, DDIs, DTIs, targets’ GO terms, SEs | |
| Shi et al. [133] | 2017 | LR, ensemble learning | Classic ML methods | DCDB | DDIs, DTIs, SEs, ATC codes | https://github.com/JustinShi2016/Drug-Drug-Interactions/tree/master/ISBRA2016 |
| KalantarMotamedi et al. [99] | 2018 | RF | Classic ML methods | NCATS [161] | Targets, targets’ pathways | |
| Preuer et al. [20] | 2018 | DeepSynergy | Deep learning methods | O’neil et al. study. | Fingerprints, physicochemical, toxicophore features, cell lines’ gene expression levels | www.bioinf.jku.at/software/DeepSynergy |
| Janizek et al. [80] | 2018 | TreeCombo | Classic ML methods | O’Neil et al. study. | Fingerprints, toxicophore structures, cell lines’ gene expression levels | |
| He et al. [102] | 2018 | DCPT | Classic ML methods | Experiments | Exome-seq, RNA-seq and target profiles | |
| Chen et al. [116] | 2018 | DBN | Deep learning methods | AZ-DREAM | Ontology fingerprints, cell lines’ gene expression, targets’ pathways | |
| Cheng et al. [129] | 2019 | Proximity | Systems biology methods | DCDB, TTD | PPIs | https://github.com/emreg00/toolbox |
| Liu et al. [136] | 2019 | RWR, GTB | Classic ML methods | DCDB | DTIs, CCIs, targets’ sequences, targets’ GOs | https://github.com/hliu2016/SynerDrug |
| Sidorov et al. [36] | 2019 | RF, XGBoost | Classic ML methods | NCI-ALMANAC | structure features, physicochemical properties | http://ballester.marseille.inserm.fr/NCI-Alm-Predictors.zip |
| Andrew et al. [103] | 2019 | RF | Classic ML methods | Commercial data. | Clinical trial features | |
| Lanevski et al. [18] | 2019 | DECREASE | Classic ML methods | Experiments, O’Neil et al. study. | Dose–response matrix |
http://decrease.fimm.fi
https://github.com/IanevskiAleksandr/DECREASE/tree/master/210_Novel_Anticancer_combinations |
| Zhang et al. [137] | 2019 | FFM | Classic ML methods | DCDB, NData [2] | Targets, enzymes, ATC codes, | |
| Julkunen et al. [94] | 2020 | comboFM | Classic ML methods | NCI-ALMANAC, etc. | Fingerprints, cell lines’ gene expression, drugs’ concentrations | https://doi.org/10.5281/zenodo.4129688 |
| Jiang et al. [121] | 2020 | GCN | Deep learning methods | O’Neil et al. Study | PPIs, DTIs | |
| Kuru et al. | 2021 | MatchMaker | Deep learning methods | DrugComb. | Structural and physiochemical, cells’ gene expression | |
| Zhang et al. [117] | 2021 | AuDNNsynergy | Deep learning methods | O’neil et al. study | Fingerprints, physicochemical properties, cell lines’ gene expression, mutation, copy number variation |
Notes: TIMMA: Target inhibition interaction using maximization and minimization averaging. PEA: Probability ensemble approach. RACS: Ranking-system of anti-cancer synergy. NLLSS: Network-based Laplacian regularized least-square synergistic drug combination prediction. GTB: Gradient tree boosting. cNMF: Composite non-negative matrix factorization. FFM: Field-aware factorization machines. FM: Factorization machine.
The performance and validation scheme of each method are listed in Table 4. It should be noted that different sets of performance metrics should be applied in different conditions. In binary classification tasks, the most used metrics are receiver operating characteristic (ROC) curve and the area under the ROC curve (AUROC) [83]. However, the high AUROC may not indicate a good predictor under imbalanced dataset condition [84]. In the case of class imbalance, the precision-recall (PR) curve and the area under the PR curve (AUPR) should be applied. When we care only about positive samples in imbalanced dataset, F1-score and positive predictive value should be used. If the large number of true negatives cannot be ignored, balanced accuracy (BACC) and Matthew’s correlation coefficients (MCC) would be more suitable. In regression tasks, mean squared error (MSE) and root mean squared error (RMSE) could not be compared in different dataset since different data distribution would obtain different score range.
Table 4.
Performance scores and validation scheme of some methods involved in this review
| Study | Algorithms | Validation scheme | Classification performance | Regression performance | Remarks | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUROC | AUPR | ACC | F1 | MCC | Recall | Pre | Kappa | MSE | RMSE | SCC | PCC | R 2 | ||||
| Chen et al. [98] | RF | 0.880 | 0.915 | |||||||||||||
| Sun et al. [90] | One-class SVM | 10-fold CV | 0.684 | 0.670 | ||||||||||||
| Huang et al. [132] | LR | 10-fold CV | 0.92 | 0.86 | ||||||||||||
| Li et al. [159] | PEA | 10-fold CV | 0.90 | |||||||||||||
| Sun et al. [38] | RACS | 0.85 | ||||||||||||||
| Wildenhain et al. [97] | SONAR | LOOCV | 0.91 | 0.56 | ||||||||||||
| Chen et al. [39] | NLLSS | LOOCV | 0.905 | |||||||||||||
| Gayvert et al. [81] | RF | 10-fold CV | 0.866 | 0.821 | ||||||||||||
| Li et al. [100] | RF | 0.89 | ||||||||||||||
| Xu et al. [82] | SGB | 10-fold CV | 0.952 | 0.898 | 0.805 | 0.869 | 0.929 | |||||||||
| Shi et al. [108] | TLMCS | 10-fold CV | 0.824 | 0.372 | ||||||||||||
| Shi et al. [133] | LR, Ensemble learning | 10-fold CV | 0.954 | 0.821 | ||||||||||||
| Preuer et al. [20] | DeepSynergy | 5-fold CV | 0.90 | 0.59 | 0.92 | 0.56 | 0.51 | 255.5 | 15.91 | 0.73 | ||||||
| Janizek et al. [80] | TreeCombo | 5-fold CV | 0.519 | 0.70 | ||||||||||||
| Chen et al. [116] | DBN | LOOCV | 0.654 | 0.602 | 0.715 | |||||||||||
| Cheng et al. [129] | Proximity | 0.589 | ||||||||||||||
| Liu et al. [136] | GTB | 10-fold CV | 0.949 | 0.884 | 0.772 | 0.872 | 0.897 | |||||||||
| Sidorov et al. [36] | RF, XGBoost | Leave-one-drug-out CV | 35.6–45.0 | 0.39–0.81 | 0.43–0.86 | 0.17–0.74 | Performance in different cell line | |||||||||
| Andrew et al. [103] | RF | 5 or 10-fold CV | 0.81 | |||||||||||||
| Lanevski et al. [18] | DECREASE | 5-fold CV | 0.82–0.91 | Dose–response matrix prediction | ||||||||||||
| Zhang et al. [137] | FFM | 5-fold CV | 0.925 | 0.934 | 0.761 | |||||||||||
| Julkunen et al. [94] | comboFM | 10 × 5 nested CV |
9.86–13.04 | 0.88–0.91 | 0.95–0.97 | Dose–response matrix prediction | ||||||||||
| Jiang et al. [121] | GCN | 10-fold CV | 0.892 | 0.794 | 0.919 | 0.584 | ||||||||||
| Kuru et al. [113] | MatchMaker | Leave-drug combination-out CV | 0.97 | 0.85 | 267.9 | 0.69 | 0.69 | |||||||||
| Zhang et al. [117] | AuDNNsynergy | 5-fold CV | 0.91 | 0.63 | 0.93 | 0.72 | 0.51 | |||||||||
CV: Cross validation. LOOCV: Leave-one-out cross validation.
Classic ML methods
This section focuses on the advanced or special ML algorithms applied in drug combination prediction, which can achieve high prediction accuracy (ACC) in small- and medium-scale data sets, including artificial neural network (ANN) [85], factorization machine (FM) [86], RF, stochastic gradient boosting (SGB) [87], XGBoost and other ensemble learning methods. Due to the relatively low prediction ACC, some classical ML algorithms are mostly used as baselines, such as Naïve Bayes (NB) [88], logistic regression (LR) [89] and SVM [18, 20, 82, 90, 91]. As shown in Figure 4, some studies used only one classic ML model (classifier or regressor) for prediction (e.g., DT), while other studies integrating a series of classifiers or regressors through ensemble learning (e.g., RF). Among ensemble learning models, bagging (bootstrap aggregating) and boosting are the two main methods [92]. Table 1 lists classic ML algorithms mentioned in this section and provides brief descriptions for these algorithms.
Figure 4.
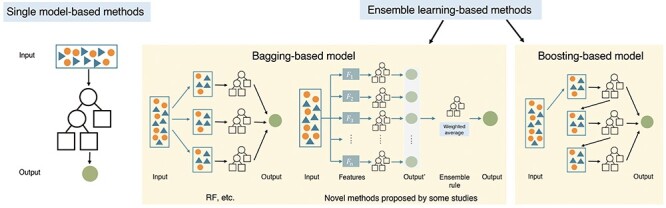
Frameworks of the classic ML methods introduced in this review.
Single model-based methods
Since most single models are used as baseline models (SVM, NB, LR, etc.) to show the performance improvement of ensemble learning-based or DL methods. We would only introduce two studies using advanced single model-based methods, ANN and FM. Different from other studies, both studies do not predict synergistic drug combinations using these models directly. The ANN-based study predicts the cytotoxicity of drug pairs firstly, while the FM-based study predicts complete dose–response matrix of drug combinations.
ANN. Pivetta et al. [93] proposed an algorithm based on ANN and experimental design. To construct training dataset, they first obtained the experiment results of cytotoxicity when the drugs were used alone or in combination within a fixed concentration range. Then an ANN was used to predict the cytotoxicity of drug pairs on the entire concentration space within the selected concentration range. According to the predicted cytotoxicity values, they further calculated the synergistic effects of drug pairs through a defined mathematical equation. The prediction results were validated in experiment. Unlike other methods, the authors found out synergistic drug combinations through the analysis of the calculated cytotoxicity surface. Drug combinations with desired cytotoxicity and the related lowest dose of the drugs can be found.
comboFM. Julkunen et al. [94] presented comboFM to obtain the synergistic drug pairs by predicting the complete dose–response matrix. They applied FM to learn the five-order tensors of drug pairs. The input features of comboFM contained five groups, two molecular fingerprints of the drugs, concentration values of both drugs, and gene expression profiles of cancer cell lines. Then an FM was applied to achieve the regression task. At last, they applied NCI ComboScore to quantify the synergism of drug pairs according to the dose–response matrix. Compared with RF, FM achieved better performance with Pearson’s correlation coefficient (PCC) of 0.95–0.97. In order to support the prediction results, experimental validation was further performed on 16 predicting drug pairs. The predicted dose–response matrix took the concentration information into account, which would be an effective strategy for drug pair synergy prediction.
Ensemble learning-based methods
In supervised learning algorithms, ensemble learning-based methods could have a better prediction performance than single model-based methods by integrating the predictions of multiple models into a single output [95]. As shown in Figure 4, two main methods of ensemble learning are bagging and boosting. Bagging [96] applies a set of ML algorithms, which are trained separately with random samples from the training set and produce the final prediction through a voting or averaging approach. The idea of boosting is to iteratively fit models so that the training of model at a given step depends on the models fitted in the previous steps. For the typical algorithms in bagging and boosting introduced in this section (RF and gradient boosting), there may be few improvements to the algorithms in these studies, but we believe the feature data and workflows they used are illuminating.
Bagging-based methods
A commonly used bagging-based algorithm is RF, which is an ensemble of multiple decision trees. A certain number of RFs are used in drug combination prediction [36, 81, 97–103]. Especially in AZ-DREAM challenge, the top winning method is based on RF. One of the possible reasons is that RF is particularly suitable for processing high-dimensional data and does not require feature selection [104]. Especially in classification tasks, RF is considered to be one of the most effective ML methods [33, 105].
RF. Gayvert et al. [81] used only single drug dose response as the feature of drugs, and applied RF to predict both synergy and effectiveness of drug pairs in mutant BRAF melanomas. The single drug dose response was expressed as GI50, which was the percentage of concentration required to inhibit 50% of growth inhibition. Each sample was represented by a 54-dimensional feature vector consisting of the mean and difference between GI50 of two drugs in 27 melanoma cell lines. Synergy labels were based on CI index, and effectiveness labels were defined by setting thresholds for GI50. Then RF model was trained on this dataset and achieved good performance for predicting synergy (AUROC = 0.8663) and effectiveness (AUROC = 0.8809). They further performed experiments to validate the prediction results on novel cell lines independent of training set, and proved the generalization ability of the approach. Through one of the experiments, the authors found that synergy and effectiveness can be predicted from a relatively small subset (36 samples) based only on single drug responses. This inspires us drug responses may be effective features. However, there was no compared method, and they did not report the sizes of positive or negative samples, the metrics AUROC and ACC are not enough to evaluate the performance.
SONAR. Wildenhain et al. [97] predicted drug pairs that exhibited species-selective toxicity toward human fungal pathogens by combining RF and NB, which was called Second Order Naive Bayesian and Random Forest (SONAR). Chemical-genetic interaction matrix (CGM) data and chemical structural features are used. Firstly, a NB was built to predict genetic sensitivities using structural features and CGM data. Then the feature set contained seven parameters derived from CGM and NB-derived predicted likelihood scores, and were used as the input to RF model. This algorithm obtained an AUROC of 0.91 and a PCC of 0.56 with Bliss scores. Then the predicted results were verified by experiments. According to comparing different feature combinations, the authors found that the predicted likelihoods derived from NB slightly improved predictive power and combining the parameters from CGM greatly augmented prediction ACC. But there was no compared method.
DCPT. Non-synergistic effect in healthy controls for combination therapy should also be taken into account. He et al. [102] developed a drug combination prediction and testing (DCPT) platform to predict the patient-specific combination effects (synergy versus non-synergy) using a RF model. DCPT predict the patient-specific responses of single-compound and drug combination using exome-seq, RNA-seq and target profiles as input features, which were obtained from ex vivo testing in patient-derived samples. Then HSA model was used to calculate the synergistic effects through the response results. Additionally, the cancer-selective synergies were identified by using differential single-compound response between patient cells and healthy controls, hence reducing the likelihood of toxic combination effects. Finally, using T-cell prolymphocytic leukemia (T-PLL) as a case study, they show how the DCPT platform successfully predicted distinct synergistic combinations for each of the three T-PLL patients.
In addition to RF, some studies have designed novel bagging-based methods that combine multiple classic ML models. Two popular ensemble rules to integrate the outputs of multiple base predictors are classifier ensemble rule and weighted average rule [106, 107]. The classifier ensemble rule applies a classification function to map outputs of base predictors to a label, while the weighted average ensemble rule takes the weighted average of outputs from base predictors.
TLMCS. Shi et al. [108] combined one-class SVMs to design a two-layer multiple classifier system (TLMCS), which integrated five types of feature (Figure 5a). In the first layer of TLMCS, five SVM classifiers are regarded as the base predictors training with five feature types separately. Then the outputs of five base predictors are concatenated into a vector as the input to the SVM in the second layer. Compared with concatenated vectors of the original features and other two ML methods, TLMCS showed a better performance. This showed that TLMCS utilizes the feature vectors as fully as possible in its first layer and has no need to train the model with highly-dimensional concatenation of heterogeneous features. Additionally, the authors performed another experiment using one-class SVMs in TLMCS to discover potential drug pairs among unknown drug pairs. That is, they only used positive samples (effective drug combinations) to train TLMCS and run the trained TLMCS on the unknown drug pairs. The top predicted results were validated from literatures. The predicting power of one-class SVMs has been demonstrated from this experiment. However, there is a problem of class imbalance in this study; the number of negative samples is about 200 times than that of positive samples. The metric AUROC is not accurate to evaluate the performance of models. And the metric of AUPR is 0.372; we consider that there should be given other metrics to prove the predictive power of the models.
Figure 5.

The flowcharts of some selected typical methods reviewed in this manuscript. (A) TLMCS [108], an ensemble learning-based method. (B) DeepSynergy [20], an FNN. (C) MatchMaker [113], an FNN-based method. (D) A DBN-based method [116]. (E) AuDNNsynergy [117], an AE-based method. (F) A GCN-based method [121].
DECREASE. Ianevski et al. [18] predicted the complete dose–response matrix of drug combinations. They proposed drug combination response prediction (DECREASE), which is an ensemble of composite non-negative matrix factorization (cNMF) and XGBoost algorithms. The input of DECREASE was a single row or column or diagonal of the dose–response matrix. Then DECREASE detected outliers, and used the average of cNMF and XGBoost to predict the complete dose–response matrix. According to the predicted dose–response values, the synergism of drug pairs was computed using four synergy scores (Bliss, Loewe, HSA or ZIP). Compared with other seven classic ML algorithms, DECREASE showed better performance. The authors found that it worked the best when the diagonal of the dose–response matrix was used as input through computational experiments in various datasets. This is an efficient experimental–computational approach to identify synergistic combinations with a minimal set of measurements and could reduce the cost and time required for HTS experiments. And the only input needed is a submatrix of dose–response matrix, without other features of drugs.
Boosting-based methods
Gradient boosting method [109] is a popular method in boosting-based methods. The core idea of gradient boosting is to minimize the residuals of each model in a sequential way through the calculation of the gradient.
SGB. Xu et al. [82] proposed a computational model based on SGB. Six features were integrated, including molecular structures, structural similarities, ATC codes similarities, protein–protein interactions (PPIs), CCIs and disease pathways. To avoid overfitting, the minimum redundancy and maximum relevance method was performed to extract useful features. Three ML methods, SVM, NB and SGB, were used to predict, among which SGB-based model had the best performance. The authors also found that therapy information of drugs (ATC codes) plays an important role in prediction, and the performance of biological feature (disease pathways) was relatively lower, this may because the incompleteness of pathways or the simple features they used.
TreeCombo. Janizek et al. [80] presented an XGBoost-based approach trained on O’Neil et al. study dataset, named TreeCombo. The input features were the drug physiochemical features and cancer cell line gene expression data as used by DeepSynergy (see Feedforward neural network) [20]. According to the comparison, TreeCombo outperformed DeepSynergy, a DNN-based model. This showed the excellent performance of XGBoost was comparable to DL in the terms of medium-scale dataset (~22 000 samples). And XGBoost require less hyperparameter tuning or feature preprocessing. Moreover, TreeCombo is also interpretable. It can extract genes with well-established links to cancer as important features according to importance identified by TreeSHAP [110]. However, DNNs also can give the feature importance with the help of DeepSHAP tool [110], which should be used in future studies of DNNs.
Deep learning methods
DL is a particular type of ML with multiple data processing layers, which are based on ANNs (shallow neural network). The deep network structure makes DLs better able to capture the nonlinear and complex relationships between input and output [111, 112]. To learn the complex functions mapping the input to the output, DLs can perform automatic feature extraction from raw data and learn features at multiple levels, while classic ML models would depend more on the human-crafted features. But it is not clear that features learned directly from raw data will always be better than human-crafted features [111]. In addition to feature vectors, the input to the DL models can also be a graph constructed by integrating multiple networks, which is trained by graph neural networks.
Feedforward neural network
FNN is a deep ANN with multiple fully connected layers, and is trained with a back-propagation learning algorithm [77]. The FNN model can be regarded as the simplest DL models, which is often used as a baseline for DL methods.
DeepSynergy. Preuer et al. [20] proposed DeepSynergy, which is often used as a baseline in predicting drug combinations. It is an FNN with two hidden layers (Figure 5b). The input layer received chemical descriptors of both drugs and gene expression values of corresponding cancer cell lines. Compared with four advanced ML methods, i.e. gradient boosting machines (GBM), RF, SVM and elastic nets (EN), DeepSynergy was confirmed to have the best performance in various scoring metrics. DeepSynergy resulted in an AUROC of 0.90 in the classification task and a PCC of 0.73 in the regression task. However, it is difficult for DeepSynergy and other comparison methods to predict novel drug combinations when applying data from novel cell lines or drugs. They suggested this was due to limitations in size and diversity of the training dataset. Additionally, we noted that this classification task was class imbalanced, although they got high AUROC and ACC scores, both metrics were inappropriate to evaluate the performance of methods. The BACC score they used is more suitable in an imbalanced dataset test, while BACC of DeepSynergy (0.76) is lower than GBM (0.80).
MatchMaker. Kuru et al. [113] proposed MatchMaker in 2021 (Figure 5c). MatchMaker contains three FNNs and is trained in DrugComb database. Firstly, the chemical structure features of each drug were concatenated with gene expression features of the corresponding cell line and input to two FNNs. The representation learned by the two FNNs were then concatenated and input to a third FNN to predict the synergy scores. In both regression and classification tasks, MatchMaker showed better performance than DeepSynergy [20] and TreeCombo [80]. Especially, compared with DeepSynergy, the correlation was increased by about 20% and MSE was improved by about 40%. Matchmaker does show high prediction performance, but it still has similar problems as DespSynergy in class imbalanced classification task, which should be more explored in future studies.
Deep belief network
DBN is a less common type of DL model and composed of stacked restricted Boltzmann machine (RBM). The RBM is an undirected, generative stochastic ANN with a bipartite structure [114], which is composed of a visible input layer and a hidden layer and connections between but not within layers. This composition leads to a fast, layer-by-layer unsupervised training procedure [115].
DBN. Chen et al. [116] reported a method based on DBN to predict effective drug combination from gene expression, pathway and the ontology fingerprints of drugs (Figure 5d). They used the drug pairs from AZ-DREAM [34]. The authors claimed that according to the results published on the DREAM website, the stacked RBMs-based DBN (Figure 5c) outperformed the participating groups of the AZ-DREAM. But the feature set they used was different from the AZ-DREAM challenge, so the performance comparison results are questionable.
Autoencoder
AEs are unsupervised learning technique, which are usually used for dimensionality reduction and feature representation learning before using other ML methods for prediction [111]. An AE is a neural network consisting of an encoder and a decoder. Between encoder and decoder, there is an internal (hidden) layer that describes a code used to represent the input [79]. A good AE can accurately learn the representation of input with accurately building a reconstruction.
AuDNNsynergy. Zhang et al. [117] proposed AuDNNsynergy, in which AEs were used to extract deep representations and a FNN was used to achieve prediction task (Figure 5e). AuDNNsynergy integrated chemical structure data of drugs and multi-omics data of cell lines. First, three AEs were trained to obtain the representations of cancer cell lines from gene expression, copy number and genetic mutation data of tumor samples. The AEs consisted of six densely connected layers. Then the output of the three encoders of AEs, combined with physicochemical properties of drugs, was used as the input of FNN to predict the synergy value of given drug pairs. The comparison results showed that AuDNNsynergy outperformed four state-of-art approaches, i.e. DeepSynergy, GBM, RF and EN. The authors also found that AuDNNsynergy can be used to predict combinations in novel cell lines. This may show the generalizability of applying informative features of cell lines, which could transfer and maintain the knowledge of different cell lines’ samples to achieve robust predictions. In addition, AuDNNsynergy can be applied to conduct interpretation analysis to identify important genes as critical predictors.
Graph convolutional network
The input of graph convolutional network (GCNs) is represented as a graph, which is the network structure consisting of nodes (vertices) and edges (links). GCNs use a convolutional neural network to obtain the embeddings of nodes or the graphs [118]. Especially in biomedical studies, the graph structure data are more informative due to the complex and systematic biological interactions between different biological entities. Thus, GCNs have been popular in various drug discovery prediction tasks [119, 120].
GCN. Jiang et al. [121] proposed a cell line-specific GCN (Figure 5f) model using the database from O’Neil et al. study. In each cell line, the multimodal graph in this study was constructed by integrating the drug–drug combination, drug–protein interaction and PPI networks. The GCN model consists of an encoder and a decoder. The encoder can obtain new representations from the multimodal graph and the decoder can obtain the predicted synergy scores from representations. The performance of this method is not too much different from DeepSynergy in AUROC. There is also the similar problem of class imbalance, the number of negative samples is about 10 times than that of positive samples in this study. In addition, the authors found that the drug–protein interaction data are highly limited with the small data set. This may mask some hidden associations in cell line-specific networks.
Visible neural network
DrugCell. Kuenzi et al. [122] proposed an interpretable model, DrugCell, to predict the responses of human cancer cells to monotherapy. DrugCell uses a modular neural network design that combines visible neural network (VNN) with conventional ANN. In VNN, the hierarchy of cell subsystems is guided by GO priori knowledge to simulate human cell biology. Each subsystem contains several neurons to express multiple distinct states. Then relative local improvement in predictive power (RLIPP) scores are calculated to represent the importance of biological mechanisms simulated by subsystems in each drug response. For identifying synergistic drug combinations, the RLIPP scores of 25 drugs from DeepSynergy study are ranked. The authors pointed that drugs would be synergistic if they inhibit separate pathways of regulating common basic functions. Although this method does not perform too well in prediction performance, it provides a good inspiration in the interpretation of drug synergy mechanism.
Challenges and future work
In order to accelerate the discovery of combination therapy for complex diseases, many AI-driven biopharmaceutical companies have been committed to the development of drug combinations based on ML and network pharmacology methods. Pharnext (https://pharnext.com/), a French company, has developed disease molecular networks composed of disease targets to find low-dose synergistic drug combinations for neurodegenerative diseases. The company has developed a novel fixed-dose synergistic combination of baclofen, naltrexone and sorbitol for the treatment of Charcot–Marie–Tooth Disease Type 1A (CMT1A), and is planning to conduct an additional Phase 3 clinical trial. Lantern Pharma company (https://www.lanternpharma.com/) works on predicting drug combinations through ML methods and Genomics. The company has found that BNP7787 can substantially prevent and mitigate the severity of paclitaxel-induced neurotoxicity as well as cisplatin-induced neurotoxicity [123]. Currently, some companies, including Healx (https://healx.io/) and Innoplexus (https://www.innoplexus.com/), are committed to using their artificial intelligence platform to find effective combination therapy for COVID-19.
Although there have been many studies of academic and industry fields on the prediction of combination therapy and high prediction performance has been obtained, there are still some challenges in this field.
Limited sample data and lack of generalizability
According to the above studies, appropriate datasets seem crucial for gaining better prediction performance. Although some large publicly available databases and web servers have been developed recently, the number of cancer cell lines and drugs is limited, which may affect the model generalizability in predicting novel drug combinations. Most methods are difficult to generalize novel drugs and cell lines. Researchers should pay more attention to improving the generalizability of models in future studies, such as using external test sets and adding regularization techniques in models.
Lack of informative feature type
To represent drugs and cell lines, the state-of-the-art studies usually applied structure information, physicochemical properties of drugs and gene expression profiles of untreated cancer cell lines as feature sets to predict combinations across different cell lines. This may ignore the biological connection between drugs and cells since synergism is the response of cells to drugs. Thus, more feature types should be considered, such as multi-omics cellular response under drug perturbations. These informative feature types would also help to further explore the biological mechanism of synergistic effects.
Inconsistence of label data
Researchers have not reached a consensus on the quantification and accurate definition of synergism and antagonism yet, which is one of the most controversial concepts in this field [17, 43–45]. Many drug combination databases and software tools (DrugComb, DrugCombDB, synergy, etc.) have provided the synergy scores of a variety of quantification models. But the synergy results of multiple samples are quite inconsistent. For example, some quantification models may show a sample as strong synergy, while some would obtain an antagonistic result. In addition, there may be some experimental noises in the synergy scores [124], which makes the results inaccurate. Inaccurate label set would have a great impact on the prediction results. We hope that the future research would focus on the accurate quantification of synergism and antagonism. And researchers should screen out the accurate labels when making predictions.
The study of antagonism
Most studies report synergism but rarely report antagonism as the main topic. It should be noted that antagonism is also important in combination therapy [44]. This may be related to the study of SE, toxicity of combination therapy and can avoid lots of unnecessary clinical trials. Richards et al. [125] have revealed death kinetics as a predictive feature of antagonism since inhibitory crosstalk between cell death pathways.
Prediction of synergy on specific dose combination
In addition, the determination of synergy depends on the specific dose tested in experiments. Drug combinations are frequently found to be synergistic in one does range and antagonistic in another [51]. Rather than simply classifying whether a combination has synergistic effect, we should consider what dose range optimizes the synergy of this combination. Moreover, when using different dose combination, the response prediction of drug combination should not be ignored. It has been a major challenge to accurately predict the drug response in the era of precision medicine [111]. And there have been some studies applied in prediction of single drug response [126, 127], and drug combination [94]. These studies would help us get closer to achieving the goal of precision medicine in the clinic, which should be further explored in future research.
Class imbalanced in classification task
Drug combination prediction is more suitable for regression task since many databases have provided various synergy scores of samples. In previous studies, most carry out classification tasks, that is, drug combinations are simply classified as synergistic and non-synergistic effects. However, the training data set used in many studies has the problem of imbalanced classes. The number of synergistic (positive) samples is small (minority class), whereas the number of non-synergistic (negative) samples is large (majority class), which is generally more than 10 times than that of positive samples [121]. Thus, these methods could get high AUROC or ACC results, but AUROC metric would pay more attention to the predictive performance of majority class (negative samples) and ignore that of minority class (positive samples). While we should pay great attention to the prediction performance of minority class. The AUROC metric is not accurate to evaluate the performance of methods in imbalanced datasets. The F1-score, BACC, MCC and other more metrics would better reflect the model performance with this problem (see Section ‘5 Machine learning methods used in drug combination prediction’). Moreover, ML models should be improved to solve the problem of class imbalance and augment the prediction ACC of minority class (synergy samples).
Lack of interpretability
Furthermore, in the context of models that guide medical decision-making, interpretability is critical. Especially through DL models, although they can get relatively accurate prediction results, the potential factors affecting the prediction results cannot be understood. Some models and tools could help to extract key features from the training set, such as tree-based methods, TreeSHAP and DeepSHAP, but they still cannot explain some mechanisms behind the biological processes. Recently, Kuenzi et al. [122] attempted to develop an interpretable DL model to predict drug response of cancer cells, which can be used to suggest synergistic drug combinations. This would be a great attempt and inspire researchers to study more mechanisms of action of drug synergy through designing interpretable DL models.
Experimental validation to adjust the calculation model
Finally, after prediction by various methods, clinical trials can be carried out to further verify the selected drug combination. The experimental validation includes in vitro experiments, in vivo experiments and clinical trials. The closed loop of calculation prediction and wet test verification is conductive to drug combination prediction. The experimental results can be used to adjust the calculation model to obtain a better prediction effect.
Key Points
This manuscript reviews nine drug combination databases, six related web servers, four related software tools and other related databases, which contain input features of samples. A list of statistical information and links of all databases are provided.
ML methods including classic ML methods and DL methods are introduced, the advantages and disadvantages of these methods are discussed.
The challenges including the limited data, inconsistence of label data, class imbalanced in classification task, lack of generalizability and interpretability of models. Additionally, more informative feature type, prediction of dose combination and antagonism, and more experimental validation should be further explored in future studies.
Supplementary Material
Lianlian Wu is a Master student in Academy of Medical Engineering and Translational Medicine, Tianjin University, Tianjin, China.
Yuqi Wen is a Ph.D. candidate in Beijing Institute of Radiation Medicine, Beijing, China.
Dongjin Leng is a Ph.D. candidate in Beijing Institute of Radiation Medicine, Beijing, China.
Qinglong Zhang is a Master student in Beijing Institute of Radiation Medicine, Beijing, China.
Chong Dai is a Master student in College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, China.
Zhongming Wang is a Master student in Academy of Medical Engineering and Translational Medicine, Tianjin University, Tianjin, China.
Ziqi Liu is a Master in State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences (Beijing), Beijing Institute of Lifeomics, AMMS, Beijing, China.
Bowei Yan is a Master student in Beijing Institute of Radiation Medicine, Beijing, China.
Yixin Zhang is a postdoc in Beijing Institute of Radiation Medicine, Beijing, China.
Jing Wang is a Ph.D. candidate in School of Medicine, Tsinghua University, Beijing, China.
Song He is an associate professor in Beijing Institute of Radiation Medicine, Beijing, China.
Xiaochen Bo is a professor in Beijing Institute of Radiation Medicine, Beijing, China.
Contributor Information
Lianlian Wu, Academy of Medical Engineering and Translational Medicine, Tianjin University, Tianjin, China.
Yuqi Wen, Beijing Institute of Radiation Medicine, Beijing, China.
Dongjin Leng, Beijing Institute of Radiation Medicine, Beijing, China.
Qinglong Zhang, Beijing Institute of Radiation Medicine, Beijing, China.
Chong Dai, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, China.
Zhongming Wang, Academy of Medical Engineering and Translational Medicine, Tianjin University, Tianjin, China.
Ziqi Liu, State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences (Beijing), Beijing Institute of Lifeomics, AMMS, Beijing, China.
Bowei Yan, Beijing Institute of Radiation Medicine, Beijing, China.
Yixin Zhang, Beijing Institute of Radiation Medicine, Beijing, China.
Jing Wang, School of Medicine, Tsinghua University, Beijing, China.
Song He, Beijing Institute of Radiation Medicine, Beijing, China.
Xiaochen Bo, Beijing Institute of Radiation Medicine, Beijing, China.
Funding
This work is supported by the National Natural Science Foundation of China [http://www.nsfc.gov.cn; nos. 62103436] to Song He.
References
- 1. Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 2019;18(6):463–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jia J, Zhu F, Ma X, et al. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov 2009;8(2):111–28. [DOI] [PubMed] [Google Scholar]
- 3. Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics 2019;20(2):273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lopez JS. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nature Nat Rev Clin Oncol 2017;14(1):57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pang K, Wan YW, Choi WT, et al. Combinatorial therapy discovery using mixed integer linear programming. Bioinformatics 2014;30(10):1456–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 2008;4(11):682–90. [DOI] [PubMed] [Google Scholar]
- 7. Ramsay RR, Popovic-Nikolic MR, Nikolic K, et al. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med 2018;7(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sheng Z, Sun Y, Yin Z, et al. Advances in computational approaches in identifying synergistic drug combinations. Brief Bioinform 2017;19(6):1172–82. [DOI] [PubMed] [Google Scholar]
- 9. Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 2017;171(7):1678–91e1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Narayan RS, Molenaar P, Teng J, et al. A cancer drug atlas enables synergistic targeting of independent drug vulnerabilities. Nat Commun 2020;11(1):2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancer 2014;6(3):1769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang G, Roth CB. Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 2001;293(5536):1793. [DOI] [PubMed] [Google Scholar]
- 13. Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45(12):1439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lovly CM, Shaw AT. Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin Cancer Res 2014;20(9):2249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem 2014;57(19):7874–87. [DOI] [PubMed] [Google Scholar]
- 16. Madani Tonekaboni SA, Soltan Ghoraie L, Manem VSK, et al. Predictive approaches for drug combination discovery in cancer. Brief Bioinform 2018;19(2):263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chou T-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006;58(3):621–81. [DOI] [PubMed] [Google Scholar]
- 18. Ianevski A, Giri AK, Gautam P, et al. Prediction of drug combination effects with a minimal set of experiments. Nat Mach Intell 2019;1(12):568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holohan C, Van Schaeybroeck S, Longley DB, et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13(10):714–26. [DOI] [PubMed] [Google Scholar]
- 20. Preuer K, Lewis RPI, Hochreiter S, et al. Deep synergy: predicting anti-cancer drug synergy with deep learning. Bioinformatics 2018;34(9):1538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leh, Aacute, R J, 2, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol 2009;27(7):659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tan X, Hu L, LuquetteLJ, 3rd, et al. Systematic identification of synergistic drug pairs targeting HIV. Nat Biotechnol 2012;30(11):1125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao X-M, Iskar M, Zeller G, et al. Prediction of drug combinations by integrating molecular and pharmacological data. PLoS Comput Biol 2011;7(12):e1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bleicher KH, Böhm H-J, Müller K, et al. Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discov 2003;2(5):369–78. [DOI] [PubMed] [Google Scholar]
- 25. Morris MK, Clarke DC, Osimiri LC, et al. Systematic analysis of quantitative logic model ensembles predicts drug combination effects on cell signaling networks. CPT Pharmacometrics Syst Pharmacol 2016;5(10):544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feala JD, Cortes J, Duxbury PM, et al. Systems approaches and algorithms for discovery of combinatorial therapies. Wiley Interdiscip Rev Syst Biol Med 2010;2(2):181–93. [DOI] [PubMed] [Google Scholar]
- 27. Sun X, Bao J, You Z. Modeling of signaling crosstalk-mediated drug resistance and its implications on drug combination. Oncotarget 2016;7(39):63995–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bansal M, Yang J, Karan C, et al. A community computational challenge to predict the activity of pairs of compounds. Nat Biotechnol 2014;32(12):1213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee J-H, Kim DG, Bae TJ, et al. CDA: combinatorial drug discovery using transcriptional response modules. Plos One 2012;7(8)::e42573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zinner RG, Barrett BL, Popova E, et al. Algorithmic guided screening of drug combinations of arbitrary size for activity against cancer cells. Mol Cancer Ther 2009;8(3):521–32. [DOI] [PubMed] [Google Scholar]
- 31. Talevi A, Morales JF, Hather G, et al. Machine learning in drug discovery and development part 1: a primer. CPT Pharmacometrics Syst Pharmacol 2020;9(3):129–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiménez-Luna J, Grisoni F, Schneider G. Drug discovery with explainable artificial intelligence. Nat Mach Intell 2020;2(10):573–84. [Google Scholar]
- 33. Tsigelny IF. Artificial intelligence in drug combination therapy. Brief Bioinform 2019;20(4):1434–48. [DOI] [PubMed] [Google Scholar]
- 34. Menden MP, Wang D, Mason MJ, et al. Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat Commun 2019;10(1):2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Breiman L. Random forests. Mach Learn 2001;45(1):5–32. [Google Scholar]
- 36. Sidorov P, Naulaerts S, Ariey-Bonnet J, et al. Predicting synergism of cancer drug combinations using NCI-ALMANAC data. Front Chem 2019;7:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang L, Li F, Sheng J, et al. Drug combo ranker: drug combination discovery based on target network analysis. Bioinformatics 2014;30(12):i228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun Y, Sheng Z, Ma C, et al. Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer. Nat Commun 2015;6:8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen X, Ren B, Chen M, et al. NLLSS: predicting synergistic drug combinations based on semi-supervised learning. PLoS Comput Biol 2016;12(7):e1004975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu D, Tian Y. A comprehensive survey of clustering algorithms. Ann Data Sci 2015;2(2):165–93. [Google Scholar]
- 41. Zagidullin B, Aldahdooh J, Zheng S, et al. Drug comb: an integrative cancer drug combination data portal. Nucleic Acids Res 2019;47(W1):W43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holbeck SL, Camalier R, Crowell JA, et al. The National Cancer Institute ALMANAC: a comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer Res 2017;77(13):3564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang J, Wennerberg K, Aittokallio T. What is synergy? The Saariselka agreement revisited. Front Pharmacol 2015;6:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chou TC. Preclinical versus clinical drug combination studies. Leuk Lymphoma 2008;49(11):2059–80. [DOI] [PubMed] [Google Scholar]
- 45. Englehardt JD, Demidenko E, Miller TW. Statistical determination of synergy based on Bliss definition of drugs independence. Plos One 2019;14(11):e0224137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 1953;3(6):285–90. [PubMed] [Google Scholar]
- 47. Bliss C. The toxicity of poisons applied jointly. Ann Appl Biol 1939;26:585–615. [Google Scholar]
- 48. Shafer SL, Hendrickx JFA, Flood P, et al. Additivity versus synergy: a theoretical analysis of implications for anesthetic mechanisms. Anesth Anal 2008;107(2):507–24. [DOI] [PubMed] [Google Scholar]
- 49. Laskey SB, Siliciano RF. A mechanistic theory to explain the efficacy of antiretroviral therapy. Nat Rev Microbiol 2014;12(11):772–80. [DOI] [PubMed] [Google Scholar]
- 50. Chevereau G, Bollenbach T. Systematic discovery of drug interaction mechanisms. Mol Syst Biol 2015;11(4):807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Keith CT, Borisy AA, Stockwell BR. Multicomponent therapeutics for networked systems. Nat Rev Drug Discov 2005;4(1):71–8. [DOI] [PubMed] [Google Scholar]
- 52. Tallarida RJ. An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther 2006;319(1):1. [DOI] [PubMed] [Google Scholar]
- 53. Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev 1995;47(2):331. [PubMed] [Google Scholar]
- 54. Chou T-C, Talalay P. Analysis of combined drug effects: a new look at a very old problem. Trends Pharmacol Sci 1983;4:450–4. [Google Scholar]
- 55. Borisy AA, Elliott PJ, Hurst NW, et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci U S A 2003;100(13):7977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yadav B, Wennerberg K, Aittokallio T, et al. Searching for drug synergy in complex dose–response landscapes using an interaction potency model. Comput Struct Biotechnol J 2015;13:504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zheng S, Aldahdooh J, Shadbahr T, et al. Drug comb update: a more comprehensive drug sensitivity data repository and analysis portal. Nucleic Acids Res 2021;1:gkab438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hancock JT, Khoshgoftaar TM. CatBoost for big data: an interdisciplinary review. J Big Data 2020;7(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu H, Zhang W, Zou B, et al. DrugCombDB: a comprehensive database of drug combinations toward the discovery of combinatorial therapy. Nucleic Acids Res 2019;48(D1):D871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Giordano S, Petrelli A. From single- to multi-target drugs in cancer therapy: when aspecificity becomes an advantage. Curr Med Chem 2008;15(5):422–32. [DOI] [PubMed] [Google Scholar]
- 61. Haibe-Kains B, Madani Tonekaboni Seyed A, Rezaie A, et al. SYNERGxDB: an integrative pharmacogenomic portal to identify synergistic drug combinations for precision oncology. Nucleic Acids Res 2020;48(W1):W494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. O'neil J, Benita Y, Feldman I, et al. An unbiased oncology compound screen to identify novel combination strategies. Mol Cancer Ther 2016;15(6):1155–62. [DOI] [PubMed] [Google Scholar]
- 63. Chen X, Ren B, et al. ASDCD: antifungal synergistic drug combination database. PLoS One 2014;9(1):e86499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu Y, Hu B, Fu C, et al. DCDB: drug combination database. Bioinformatics 2010;26(4):587–8. [DOI] [PubMed] [Google Scholar]
- 65. Liu Y, Wei Q, Yu G, et al. DCDB 2.0: a major update of the drug combination database. Database 2014;2014:bau124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhu F, Chen YZ, Chen Z, et al. Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res 2018;46(D1):D1121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Masoudi-Sobhanzadeh Y, Omidi Y, Amanlou M, et al. DrugR+: a comprehensive relational database for drug repurposing, combination therapy, and replacement therapy. Comput Biol Med 2019;109:254–62. [DOI] [PubMed] [Google Scholar]
- 68. He L, Kulesskiy E, Saarela J, et al. Methods for high-throughput drug combination screening and synergy scoring. Methods Mol Biol 1711;2018:351–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ianevski A, He L, Aittokallio T, et al. SynergyFinder: a web application for analyzing drug combination dose–response matrix data. Bioinformatics 2017;33(15):2413–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Aittokallio T, Giri AK, Ianevski A. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 2020;48(W1):W488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wooten DJ, Albert R. Synergy: a python library for calculating, analyzing and visualizing drug combination synergy. Bioinformatics 2021;37(10):1473–4. [DOI] [PubMed] [Google Scholar]
- 72. Chou T-C, Martin N. The mass-action law-based new computer software, CompuSyn, for automated simulation of synergism and antagonism in drug combination studies. Cancer Res 2007;67(9 Supplement):637. [Google Scholar]
- 73. Di Veroli GY, Fornari C, Wang D, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016;32(18):2866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Noble WS. What is a support vector machine? Nat Biotechnol 2006;24(12):1565–7. [DOI] [PubMed] [Google Scholar]
- 75. Safavian SR, Landgrebe D. A survey of decision tree classifier methodology. IEEE Trans Syst Man Cybern 1991;21(3):660–74. [Google Scholar]
- 76. Chen TQ, Guestrin C. XGBoost: a scalable tree boosting system. Kdd'16: Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining 2016;785–94.
- 77. Schmidhuber J. Deep learning in neural networks: an overview. Neural Netw 2015;61:85–117. [DOI] [PubMed] [Google Scholar]
- 78. Yuming H, Junhai G, Hua Z. Deep belief networks and deep learning: Proceedings of 2015 International Conference on Intelligent Computing and Internet of Things, F 17-18 2015.
- 79. Kramer MA. Nonlinear principal component analysis using autoassociative neural networks. AIChE J 1991;37(2):233–43. [Google Scholar]
- 80. Janizek JD, Celik S, Lee S-I. Explainable machine learning prediction of synergistic drug combinations for precision cancer medicine. bioRxiv 2018. [Google Scholar]
- 81. Gayvert KM, Aly O, Platt J, et al. A computational approach for identifying synergistic drug combinations. PLoS Comput Biol 2017;13(1):e1005308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xu Q, Xiong Y, Dai H, et al. PDC-SGB: prediction of effective drug combinations using a stochastic gradient boosting algorithm. J Theor Biol 2017;417:1–7. [DOI] [PubMed] [Google Scholar]
- 83. Rifaioglu AS, Atas H, Martin MJ, et al. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform 2019;20(5):1878–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jiao Y, Du P. Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quant Biol 2016;4(4):320–30. [Google Scholar]
- 85. Dreiseitl S, Ohno-Machado L. Logistic regression and artificial neural network classification models: a methodology review. J Biomed Inform 2002;35(5):352–9. [DOI] [PubMed] [Google Scholar]
- 86. Rendle S. Factorization machines. 2010 IEEE International Conference on Data Mining. 2010;995–1000.
- 87. Friedman JH. Stochastic gradient boosting. Comput Stat Data Anal 2002;38(4):367–78. [Google Scholar]
- 88. Webb GI. Naïve Bayes. Encyclopedia of Machine Learning. Boston, MA: Springer US, 2010, 713–4. [Google Scholar]
- 89. Wright RE. Logistic Regression. Reading and Understanding Multivariate Statistics. Washington, DC, US: American Psychological Association, 1995, 217–44. [Google Scholar]
- 90. Sun Y, Xiong Y, Xu Q, et al. A hadoop-based method to predict potential effective drug combination. Biomed Res Int 2014;2014:196858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li J, Tong XY, Zhu LD, et al. A machine learning method for drug combination prediction. Front Genet 2020;11:1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhou Z-H. Ensemble Methods: Foundations and Algorithms. Hall/CRC, 2012. [Google Scholar]
- 93. Pivetta T, Isaia F, Trudu F, et al. Development and validation of a general approach to predict and quantify the synergism of anti-cancer drugs using experimental design and artificial neural networks. Talanta 2013;115:84–93. [DOI] [PubMed] [Google Scholar]
- 94. Julkunen H, Cichonska A, Gautam P, et al. Leveraging multiway interactions for systematic prediction of pre-clinical drug combination effects. Nat Commun 2020;11:6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Polikar R. Ensemble based systems in decision making. IEEE Circuits Syst Mag 2006;6(3):21–45. [Google Scholar]
- 96. Breiman L. Bagging predictors. Machine Learning 1996;24(2):123–40. [Google Scholar]
- 97. Wildenhain J, Spitzer M, Dolma S, et al. Prediction of synergism from chemical-genetic interactions by machine learning. Cell Syst 2015;1(6):383–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen L, Li B-Q, Zheng M-Y, et al. Prediction of effective drug combinations by chemical interaction, protein interaction and target enrichment of KEGG pathways. Biomed Res Int 2013;2013:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kalantarmotamedi Y, Eastman RT, Guha R, et al. A systematic and prospectively validated approach for identifying synergistic drug combinations against malaria. Malar J 2018;17(1):160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li X, Xu Y, Cui H, et al. Prediction of synergistic anti-cancer drug combinations based on drug target network and drug induced gene expression profiles. Artif Intell Med 2017;83:35–43. [DOI] [PubMed] [Google Scholar]
- 101. Ma S, Jaipalli S, Larkins-Ford J, et al. Transcriptomic signatures predict regulators of drug synergy and clinical regimen efficacy against tuberculosis. MBio 2019;10(6):e02627–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. He L, Tang J, Andersson EI, et al. Patient-customized drug combination prediction and testing for T-cell prolymphocytic leukemia patients. Cancer Res 2018;78(9):2407–18. [DOI] [PubMed] [Google Scholar]
- 103. Lo AW, Siah KW, Wong CH. Machine learning with statistical imputation for predicting drug approval. Harvard Data Sci Rev 2019;1(1). [Google Scholar]
- 104. Degenhardt F, Seifert S, Szymczak S. Evaluation of variable selection methods for random forests and omics data sets. Brief Bioinform 2019;20(2):492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Verikas A, Vaiciukynas E, Gelzinis A, et al. Electromyographic patterns during golf swing: activation sequence profiling and prediction of shot effectiveness. Sensors 2016;16(4):592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhang W, Chen Y, Liu F, et al. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinformatics 2017;18(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tang X, Cai L, Meng Y, et al. Indicator regularized non-negative matrix factorization method-based drug repurposing for COVID-19. Front Immunol 2020;11:603615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Shi JY, Li JX, Mao KT, et al. Predicting combinative drug pairs via multiple classifier system with positive samples only. Comput Methods Programs Biomed 2019;168:1–10. [DOI] [PubMed] [Google Scholar]
- 109. Natekin A, Knoll A. Gradient boosting machines, a tutorial. Front Neurorobot 2013;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lundberg SM, Lee SI. A unified approach to interpreting model predictions. Adv Neur In 2017;4765–74. [Google Scholar]
- 111. Baptista D, Ferreira PG, Rocha M. Deep learning for drug response prediction in cancer. Brief Bioinform 2021;22(1):360–79. [DOI] [PubMed] [Google Scholar]
- 112. Muzio G, O'bray L, Borgwardt K. Biological network analysis with deep learning. Brief Bioinform 2021;22(2):1515–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kuru HI, Tastan O, Cicek E. MatchMaker: a deep learning framework for drug synergy prediction. IEEE/ACM Trans Comput Biol Bioinform 2021;1–1. [DOI] [PubMed] [Google Scholar]
- 114. Larochelle H, Mandel M, Pascanu R, et al. Learning algorithms for the classification restricted Boltzmann machine. J Mach Learn Res 2012;13(1):643–69. [Google Scholar]
- 115. Hinton GE, Osindero S, Teh Y-W. A fast learning algorithm for deep belief nets. Neural Comput 2006;18(7):1527–54. [DOI] [PubMed] [Google Scholar]
- 116. Chen G, Tsoi A, Xu H, et al. Predict effective drug combination by deep belief network and ontology fingerprints. J Biomed Inform 2018;85:149–54. [DOI] [PubMed] [Google Scholar]
- 117. Zhang T, Zhang L, Payne PRO, et al. Synergistic drug combination prediction by integrating multiomics data in deep learning models. Methods Mol Biol 2021;2194:223–38. [DOI] [PubMed] [Google Scholar]
- 118. Kipf TN, Welling M. Semi-Supervised Classification with Graph Convolutional Networks, arXiv 2016.
- 119. Torng W, Altman RB. Graph convolutional neural networks for predicting drug-target interactions. J Chem Inf Model 2019;59:4131–49. [DOI] [PubMed] [Google Scholar]
- 120. Zitnik M, Agrawal M, Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018;34(13):i457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Jiang P, Huang S, Fu Z, et al. Deep graph embedding for prioritizing synergistic anticancer drug combinations. Comput Struct Biotechnol J 2020;18:427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kuenzi BM, Park J, Fong SH, et al. Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell 2020;38(5):672–84e676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Boven E, Westerman M, Van Groeningen CJ, et al. Phase I and pharmacokinetic study of the novel chemoprotector BNP7787 in combination with cisplatin and attempt to eliminate the hydration schedule. Br J Cancer 2005;92(9):1636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Malyutina A, Majumder MM, Wang W, et al. Drug combination sensitivity scoring facilitates the discovery of synergistic and efficacious drug combinations in cancer. PLoS Comput Biol 2019;15(5):e1006752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Richards R, Schwartz HR, Honeywell ME, et al. Drug antagonism and single-agent dominance result from differences in death kinetics. Nat Chem Biol 2020;16(7):791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Guan NN, Zhao Y, Wang CC, et al. Anticancer drug response prediction in cell lines using weighted graph regularized matrix factorization. Mol Ther Nucleic Acids 2019;17:164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Liu H, Zhao Y, Zhang L, et al. Anti-cancer drug response prediction using neighbor-based collaborative filtering with global effect removal. Mol Ther - Nucleic Acids 2018;13:303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Yang K, Bai H, Ouyang Q, et al. Finding multiple target optimal intervention in disease-related molecular network. Mol Syst Biol 2008;4(1):228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cheng F, Kovacs IA, Barabasi AL. Network-based prediction of drug combinations. Nat Commun 2019;10(1):1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tang J, Karhinen L, et al. Target inhibition networks: predicting selective combinations of Druggable targets to block cancer survival pathways. PLoS Comput Biol 2013;9(9):e1003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hu P, Li S, Niu Z. Prediction of synergistic drug combinations by learning from deep representations of multiple networks. Stud Health Technol Inform 2019;264:1482–3. [DOI] [PubMed] [Google Scholar]
- 132. Huang H, Zhang P, Qu XA, et al. Systematic prediction of drug combinations based on clinical side-effects. Sci Rep 2014;4:7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Shi JY, Li JX, Gao K, et al. Predicting combinative drug pairs towards realistic screening via integrating heterogeneous features. BMC Bioinformatics 2017;18(Suppl 12):409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sheridan RP, Wang WM, Liaw A, et al. Extreme gradient boosting as a method for quantitative structure–activity relationships. J Chem Inf Model 2016;56(12):2353–60. [DOI] [PubMed] [Google Scholar]
- 135. Ji X, Tong W, Liu Z, et al. Five-feature model for developing the classifier for synergistic vs. antagonistic drug combinations built by XGBoost. Front Genet 2019;10:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Liu H, Zhang W, Nie L, et al. Predicting effective drug combinations using gradient tree boosting based on features extracted from drug-protein heterogeneous network. BMC Bioinformatics 2019;20(1):645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhang C, Yan G. Synergistic drug combinations prediction by integrating pharmacological data. Synth Syst Biotechnol 2019;4(1):67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46(D1):D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Duran-Frigola M, Pauls E, Guitart-Pla O, et al. Extending the small-molecule similarity principle to all levels of biology with the chemical checker. Nat Biotechnol 2020;38(9):1087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Szklarczyk D, Santos A, Von mering C, et al. STITCH 5: augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res 2016;44(D1):D380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017;45(D1):D353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 2006;34(suppl_1):D354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kanehisa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008;36(suppl_1):D480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kuhn M, Letunic I, Jensen LJ, et al. The SIDER database of drugs and side effects. Nucleic Acids Res 2016;44(D1):D1075–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tatonetti NP, Ye PP, Daneshjou R, et al. Data-driven prediction of drug effects and interactions. Sci Transl Med 2012;4(125):125ra131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kim S, Chen J, Cheng T, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 2019;47(D1):D1102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Uniprot C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47(D1):D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Günther S, Kuhn M, Dunkel M, et al. SuperTarget and matador: resources for exploring drug-target relationships. Nucleic Acids Res 2008;36(Database issue):D919–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Oughtred R, Stark C, Breitkreutz B-J, et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res 2019;47(D1):D529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Keshava Prasad TS, Goel R, Kandasamy K, et al. Human protein reference database--2009 update. Nucleic Acids Res 2009;37(Database issue):D767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Stathias V, Turner J, Koleti A, et al. LINCS data portal 2.0: next generation access point for perturbation-response signatures. Nucleic Acids Res 2020;48(D1):D431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Gaulton A, Hersey A, Nowotka M, et al. The ChEMBL database in 2017. Nucleic Acids Res 2017;45(D1):D945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Tang J, Tanoli ZU, Ravikumar B, et al. Drug target commons: a community effort to build a consensus knowledge base for drug-target interactions. Cell Chem Biol 2018;25(2):224–9e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Urán landaburu L, Berenstein A J, Videla S, et al. TDR targets 6: driving drug discovery for human pathogens through intensive chemogenomic data integration. Nucleic Acids Res 2020;48(D1):D992–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Pawson AJ, Sharman JL, Benson HE, et al. The IUPHAR/BPS guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res 2014;42(D1):D1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kumar R, Chaudhary K, Gupta S, et al. CancerDR: cancer drug resistance database. Sci Rep 2013;3(1):1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Liu T, Lin Y, Wen X, et al. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res 2007;35(suppl_1):D198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. He L, Wennerberg K, Aittokallio T, et al. TIMMA-R: an R package for predicting synergistic multi-targeted drug combinations in cancer cell lines or patient-derived samples. Bioinformatics 2015;31(11):1866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Li P, Huang C, Fu Y. Large-scale exploration and analysis of drug combinations. Bioinformatics 2015;31(12):2007–16. [DOI] [PubMed] [Google Scholar]
- 160. Held MA, Langdon CG, Platt JT, et al. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov 2013;3(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mott BT, Eastman RT, Guha R, et al. High-throughput matrix screening identifies synergistic and antagonistic antimalarial drug combinations. Sci Rep 2015;5(1):13891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.