

Abstract
Circular RNAs (circRNAs) are a large class of covalently closed RNA molecules originating by a process called back-splicing. CircRNAs are emerging as functional RNAs involved in the regulation of biological processes as well as in disease and cancer mechanisms. Current computational methods for circRNA identification from RNA-seq experiments are characterized by low discovery rates and performance dependent on the analysed data set. We developed CirComPara2 (https://github.com/egaffo/CirComPara2), a new automated computational pipeline for circRNA discovery and quantification, which consistently achieves high recall rates without losing precision by combining multiple circRNA detection methods. In our benchmark analysis, CirComPara2 outperformed state-of-the-art circRNA discovery tools and proved to be a reliable and robust method for comprehensive transcriptome characterization.
Keywords: circRNAs, bioinformatics, computational pipeline, RNA-seq
Introduction
Recent research uncovered that eukaryotic transcriptomes comprise thousands of stable circular RNAs (circRNAs) originating by a process called back-splicing, where the transcript 3′ and 5′ ends are covalently joined [1]. Rather than being transcriptional by-products, circRNA molecules exert critical functions in cell biology through different mechanisms [2]. By interacting with microRNAs, circRNAs can regulate gene expression and govern important oncogenic axes [3]; moreover, similar to long non-coding RNAs, they can control diverse cellular processes by decoying RNA-binding proteins and scaffolding molecular complexes [4]. CircRNAs can also function as templates for translation to encode functional peptides [1, 5] and regulate the transcription of their parental gene [6]. Nowadays, circRNAs are considered key players that can impact all cancer hallmarks [7, 8]. The discovery of circRNA regulatory roles and their potential as biomarkers given by higher stability compared to linear RNAs [9] has actuated the integration of circRNA investigation in conventional transcriptomics, especially in cancer research and studies of pathological conditions [10, 11], including viral infections [12].
Studies of circRNAs rapidly increased in pace thanks to the development of bioinformatics tools that identify the sequences spanning circRNA back-splice junctions from total RNA-seq data. To date, several methods for circRNA identification have been developed [13]; most of them select the back-splice junction reads (BJR) by screening the output of read aligner tools that allow chimeric spliced read mappings to the reference genome, such as TopHat-Fusion [14] (which is embedded in the TopHat2 package), STAR [15], BWA-MEM [16], Segemehl [17] and MapSplice [18]. Other strategies, including machine learning and sequence feature-based ones, have been implemented through the years and were recently reviewed by Jakobi and Dieterich [19], Chen et al. [13] and Jiao et al. [20]. Still, none of the methods outperforms the others since they all provide either highly sensitive or highly precise predictions and highly variable performance across benchmark data sets [13, 21–23]. Interestingly, Hansen [24] observed that circRNA detection methods largely agreed on true predictions. In contrast, circRNAs identified by single methods were enriched in false-positive guesses (FPs) and suggested selecting circRNAs commonly predicted by two or more methods to obtain dependable results.
We formerly implemented CirComPara [25], an automated computational pipeline combining four circRNA detection methods, including CIRCexplorer [26], CIRI2 [27], Findcirc [28] and Segemehl [17]. CirComPara controlled the FP number by considering only the circRNAs commonly detected by two or more methods.
In CirComPara2, we have considerably improved our tool by: (i) including five additional circRNA detection methods, (ii) updating the software of the already integrated tools, (iii) implementing a more accurate counting of the back-spliced reads, (iv) increasing the analysis pipeline flexibility and (v) including the procedure to calculate the linear expression related to the circRNAs.
In this work, we first show that nine widely used circRNA detection tools could miss circRNAs of interest. Then, we confirm that CirComPara2 correctly reports circRNAs overlooked by other methods and achieves significantly higher sensitivity with no loss of precision. Moreover, assessment on simulated data and 142 public RNA-seq samples demonstrated the consistent higher performance of CirComPara2 compared with state-of-the-art methods. Further, we show that the CirComPara2 expression estimates highly correlate with the true circRNA abundance of simulated data. Finally, we discuss the computational cost of the CirComPara2 approach.
Results
CircRNA detection methods could miss abundant circRNAs
We simulated RNA-seq expression data of 5680 circRNAs from the whole human genome (‘simulated data set’; see Methods) to evaluate the characteristics of circRNA detection method false-negative predictions (FNs), i.e. true circRNAs not identified as such. We applied nine widely used computational pipelines for circRNA discovery, including circRNA_finder (CF) [29], CIRI2 [27], DCC [30], Findcirc (FC) [28], Segemehl (SE) and CIRCexplorer2 [31]. CIRCexplorer2 was coupled to each of BWA (C2BW), Segemehl (C2SE), STAR (C2ST) and TopHat-Fusion (C2TH) aligners, thus composing four different pipelines.
On average, 49% of FNs detected by each method showed higher expression than the overall circRNA median expression (Figure 1A), suggesting that nearly half of the missed circRNAs had a considerable expression level no matter which method was applied. Besides, the expression distribution of the FNs was similar to the correctly identified circRNAs, i.e. the true-positive findings (TPs), whereas the false-positive (FP) expression was generally low.
Figure 1 .
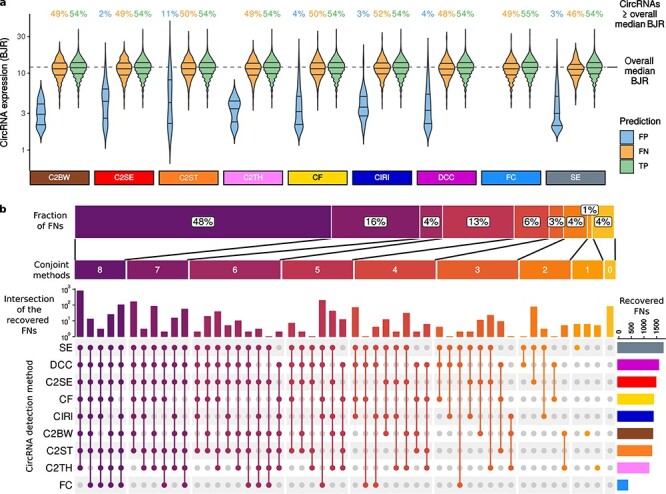
Commonly used circRNA detection methods may overlook some highly expressed circRNAs. A, The expression level of predicted circRNAs. BJR: back-splice junction read counts; C2BW: CIRCexplorer2 on BWA; C2SE: CIRCexplorer2 on Segemehl; C2ST: CIRCexplorer2 on STAR; C2TH: CIRCexplorer2 on TopHat-Fusion; CF: circRNA_finder; CIRI: CIRI2; FC: Findcirc; SE: Segemehl; FP: false-positive; FN: false-negative; TP: true-positive. B, Number of methods detecting circRNAs not detected by other methods and the number of the FNs detected. Colour refers to the number of methods conjointly detecting circRNAs missed by other tools. The vertical bars show the number of FN circRNAs detected by the methods indicated in the coloured dots below the bars. The bars denote disjoint circRNA sets. Grey dots indicate the methods failing to detect the circRNAs considered in the bar chart on the top. The horizontal bars on the right represent the overall number of FN circRNAs detected by the methods. The horizontal bar shows the percentage of detected FNs by grouping method combinations according to the number of combined methods.
Multiple method combinations increase the detection sensitivity
We further examined the 1945 circRNAs undetected by one or more tools, referred to as the ‘FN set’ from now on, by counting how many circRNAs in the FN set each method could detect. Interestingly, only 4% circRNAs of the FN set were undetected by all methods (Figure 1B), whereas 96% could be identified at least by one among the nine tools. Specifically, 1% FNs were detected individually by Segemehl, C2BW and C2TH, and 95% were commonly identified by various combinations of two or more tools. Almost half the FNs (48%) were conjointly detected by eight out of nine methods, with Segemehl, DCC and C2SE providing the most inclusive predictions. Instead, Findcirc showed the least number of recovered FNs. However, no method entirely covered the predictions of Findcirc, indicating some specificity of its algorithm.
Overall, this analysis suggested that algorithms with possibly different and complementary features can compensate for each other’s weak points and improve the detection rate if applied together.
Workflow and features of CirComPara2
Following the observation reported in the previous paragraph, we enhanced CirComPara by including more circRNA detection methods to improve its sensitivity. Moreover, we introduced new features that made CirComPara2 more flexible, computationally efficient and resilient.
CirComPara2 implements a fully automated computational pipeline for circRNA detection, quantification, annotation and integration with linear gene expression data (Figure 2A). Several parameters are available to customize the analysis workflow and the integrated methods. The minimal input consists of the RNA-seq reads in FASTQ format and a reference genome in FASTA format. Optionally, the user can also provide the gene annotation in GTF format. The software will then build the genome indexes for each read aligner and perform the necessary file format conversions. Previously computed indexes can be reused as input to save computing time.
Figure 2 .
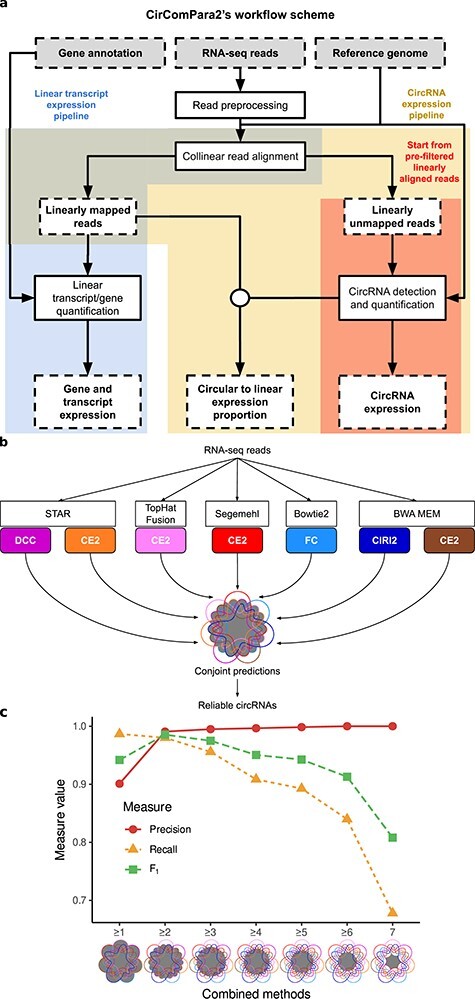
The CirComPara2 workflow and approach. A, Boxes with dashed contour indicate input and output data; boxes with solid-line contour indicate computing tasks; the circular connector indicates merging data. Background colours highlight the different pipeline branches (linear transcript analysis in blue, full circRNA analysis in yellow and strict circRNA analysis in red). B, A detail of the CirComPara2 strategy (‘CircRNA detection and quantification’ box in a) showing the embedded circRNA detection methods (coloured rounded corner boxes) with the respective chimeric read aligners (white boxes). The central Venn diagram represents the optimized combination of method prediction intersections implemented by CirComPara2 with circRNAs conjointly detected by two or more methods (grey filled intersections) retained as default. Method abbreviations as in Figure 1. C, Precision (red), recall (yellow) and F1-score (green) for different numbers of methods that conjointly identified the circRNAs: ≥1 indicates that at least one method identified the circRNAs, or, in other words, it represents the union of all single methods predictions; ≥2 indicates the circRNAs conjointly predicted by two or more methods; ≥3 indicates the circRNAs commonly identified by any combination set of at least three methods; similar is for the larger number of methods sharing the predictions. The ‘7’ indicates circRNAs commonly predicted by all the methods. The grey-filled parts in Venn diagrams show the intersections considered by the conjoint method combinations.
The CirComPara2 workflow comprises an optional pre-processing of the input raw reads by Trimmomatic [32] to trim or discard low-quality reads. Statistics of the pre-processing steps are produced with the FastQC tool [33]. Next, the reads are aligned collinearly to the reference genome using HISAT2 [34] to (i) identify the reads that are later used for linear transcript analysis (Figure 2A) and (ii) extract the reads not collinearly aligned, which are used to detect back-splices. The linear gene and transcript expression analysis is performed with StringTie [35] and produces files that can be easily imported into packages for downstream expression analysis, such as tximport [36] and tximeta [37]. The circRNA analysis aligns the collinearly unmapped reads independently with five methods allowing chimeric alignments, namely Bowtie2 [38], BWA-MEM, Segemehl, STAR and TopHat-Fusion (Figure 2B). The chimeric aligner outputs are subsequently parsed by six circRNA detection tools, which compose the nine different circRNA detection sub-pipelines presented in the previous paragraph. Of note, the computationally expensive chimeric alignment step is performed only once per aligner and reused by multiple circRNA detection tools, boosting CirComPara2 efficiency. For instance, the same alignments from STAR are passed to CIRCexplorer2, circRNA_finder and DCC (Figure 2B). The outputs of the various tools are automatically handled, converting them into a standard format to compare the predicted back-splices. Moreover, the identifiers of the back-spliced reads are collected while keeping track of the predicting method to obtain non-redundant read counts for each circRNA. Finally, the linear expression of circRNA host genes is evaluated by counting the reads collinearly mapped at each back-splice junction using bedtools [39], GNU parallel [40] and custom scripts.
CirComPara2 has a modular and highly parallelized implementation that makes it computationally efficient and resilient. By using custom parameters, CirComPara2 allows skipping computation tasks that are not of interest to the user. For instance, the user can select to run only the pipeline branch computing the linear or the circular transcript analysis, the collinear alignments (for instance, if they were previously computed), or both the collinear alignment and the linear transcript pipeline branch, therefore performing only the circRNA detection from pre-filtered reads. Plus, the Scons (www.scons.org) engine is leveraged to run independent tasks in parallel, resume an interrupted analysis by performing only uncompleted tasks and compute only the tasks dependent on the modified parameters if the user changed some parameters from a previous run.
CirComPara2 is available as stand-alone software (https://github.com/egaffo/circompara2) and Docker image (https://hub.docker.com/r/egaffo/circompara2), which facilitates installation, portability and reproducibility of the analysis.
Optimal method combination for circRNA detection
Concurrently to the improvement of CirComPara2 sensitivity, we wanted to control the number of the introduced FPs to preserve a high precision. As observed in a previous study [24], the combination of specific methods does not ameliorate the discovery of true circRNAs. Accordingly, our method combination assessment on the simulated data showed that combining circRNA_finder or Segemehl with other methods increased the FP number (Supplementary Figure S1 available online at http://bib.oxfordjournals.org/). For this reason, circRNA_finder and Segemehl were excluded in the CirComPara2 default method combination (Figure 2B); nevertheless, these two methods can be included if enabled by the user.
Further, Hansen [24] observed that circRNAs predicted by multiple methods were enriched in reliable findings and suggested using the shared output from two (or more) algorithms. To determine an optimal default setting to use in CirComPara2, we evaluated the amount of the recovered FNs against the introduced FPs in relation to the number of methods sharing the predictions. As expected, considering the predictions from all methods resulted in the highest recall (0.99) and the lowest precision (0.90) among the combination strategies (Figure 2C). Further, excluding predictions from single methods, i.e. selecting only the circRNAs commonly detected by two or more tools, showed a slightly reduced recall (0.98) with a substantially increased precision (0.99). This large precision gain indicated that most of the FPs were predicted by single methods. Further increasing the number of conjoint methods (i.e. from three-or-more to all-seven conjoint methods) led to a considerable decrease of recall (0.96–0.68) with only a modest gain in precision (0.99–1.00). In summary, we confirmed that the larger the number of methods sharing the output circRNAs, the more reliable the findings but also observed that the amount of circRNAs discarded increased accordingly.
To evenly weigh recall and precision in ranking the method combination strategies, we calculated the F1-score for each combination. The best trade-off between recall and precision, indicated by the highest F1-score (0.99), was obtained with the two-or-more method strategy (Figure 2C). Therefore, the default CirComPara2 parameters have been set to simultaneously use seven circRNA detection methods (C2BW, C2SE, C2ST, C2TH, CIRI2, DCC and FC) and later discard the circRNAs not shared between at least two of these methods.
CirComPara2 outperforms other methods in simulated data
We next set CirComPara2 with the two-or-more method strategy and compared it with the single methods on the simulated data plus CIRI-full [41] (CIRFU) and the former implementation of CirComPara (CCP1) (Figure 3A). CirComPara2 obtained the highest F1-score (0.99) by achieving the highest recall (0.98) while holding a precision comparable to or higher than the other algorithms (0.99 versus 0.92–1.00), confirming that CirComPara2 rectified the true circRNAs missed by the other methods. Notably, CirComPara2 identified 7% additional true circRNAs compared to its former implementation (Supplementary Figure S2 available online at http://bib.oxfordjournals.org/).
Figure 3 .
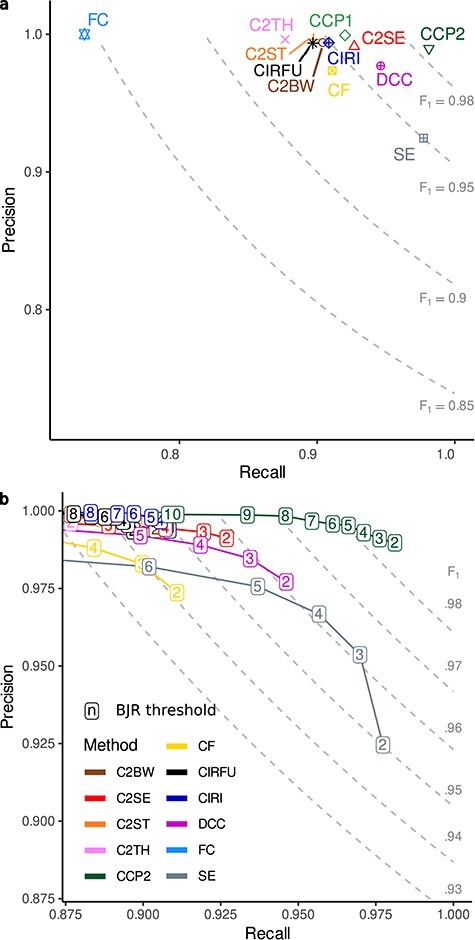
Performance of circRNA detection methods on the simulated data set. A, Precision and recall of the circRNA detection methods (labels as in Figure 1), including CirComPara2 (CCP2; green dot), CirComPara (CCP1; light green), and CIRI-full (CIRFU; black); the dashed-line curves display plot areas for 0.85, 0.90, 0.95 and 0.98 F1-scores. B, CircRNA detection methods’ performance upon application of filters from 2 to 10 minimum circRNA raw expression estimates (BJR: back-splice junction read counts); dashed-line curves delimit F1-scores plot areas from 0.92 to 0.98 with 0.01 increase steps; method colours as in (A). CCP1 was not included because it gives normalized expression estimates that are not compatible with the filtering on BJR counts.
To assess the extent of the annotation-guided method contribution to CirComPara2 predictions, we performed the analysis also with a pruned gene annotation input to the algorithms (see Methods). As expected, the CIRCexplorer2 pipelines showed a dramatic reduction (up to −0.14) of the recall and F1-scores (Supplementary Figure S3 available online at http://bib.oxfordjournals.org/). Instead, CirComPara2 maintained the highest F1-score (0.98), suggesting that it can be efficient when applied to RNA-seq data of organisms with incomplete or poor genome annotation by leveraging the embedded annotation-independent tools.
A typical circRNA expression analysis usually involves post-detection data cleaning to remove background noise signals or mapping errors and poorly expressed circRNAs of little interest [42]. Consequently, the circRNAs with small back-splice junction read counts (BJRs) are routinely filtered out. As shown in Figure 1A, FPs generally have small BJRs, suggesting that more reliable circRNAs can be retained by simply filtering according to expression abundance. We applied this procedure to our data, progressively filtering out circRNAs with less than 2 up to 10 BJRs. We stress that removing circRNAs with ≤10 BJRs was an extreme filter since it was close to the overall median BJR (Figure 1A). As expected, by increasing the minimum-BJR filter threshold, all methods scored higher precision but with a corresponding reduced recall, indicating that true circRNAs were discarded as well (Figure 3B; Supplementary Figure S4 available online at http://bib.oxfordjournals.org/). Nevertheless, CirComPara2 maintained the highest recall compared to other methods at equal precision (Figure 3B; Supplementary Tables S1 available online at http://bib.oxfordjournals.org/) and obtained the highest area under the precision–recall curve (Supplementary Table S2 available online at http://bib.oxfordjournals.org/), suggesting that the circRNAs recovered by CirComPara2 were valid findings with a considerable abundance that may represent relevant circRNAs in actual experiments.
Benchmark real data sets
The gold standard for evaluating circRNA detection methods on real RNA-seq data is to compare ribosomal RNA-depleted (ribo−) with circRNA-enriched sequencing libraries. The most used technique is the additional treatment of the ribo− library with RNase R to exploit the exonuclease degradation resistance of circRNAs derived by their lack of a single-stranded 3′ end [22–24, 27, 43]. Accordingly, we collected a total of 142 public real RNA-seq data samples for which sample-matched ribo− and RNase R-treated libraries were available (Table 1; Supplementary Table S3 available online at http://bib.oxfordjournals.org/). This validation data set comprised samples of human cell lines and various human, Rhesus macaque and mice tissues from six independent studies.
Table 1.
Real data sets of rRNA- and rRNA-/RNase R-treated matched pairs of samples used to benchmark the circRNA detection methods
| SRA/GEO/BIGD ID | Tissue(s)/cell line(s) | Study reference |
|---|---|---|
| SRR3476956, SRR1636985, SRR1636986; SRR3476958, SRR1637089, SRR1637090. SRR3479244, SRR3479243 |
HeLa, HEK293 | [27] [44] |
| GSE130905 (10 samples) | HeLa | [45]* |
| CRA001838 (8 samples) | HeLa | [46] |
| SRR444655, SRR444975; SRR445016, SRR444974 | Hs68 | [47] |
| GSE113120 (24 samples) | 22Rv1, 42D, PC3, V16, LNCaP | [48] |
| PRJCA000751 (88 samples) | Homo sapiens (17 tissues) | [49] |
| Mus musculus (14 tissues) | ||
| Macaca mulatta (13 tissues) |
The matched RNase R-treated samples were used as a control to define the TP circRNAs (see Methods). Assuming that the RNase R treatment would deplete the linear more than the circular transcripts, we considered TPs the circRNAs having circular-to-linear expression proportion (CLP) higher or equal in the RNase R-treated compared to the ribo− matched samples according to at least one method. In this way, we accounted for different sequencing library depths between the matched samples. Moreover, each circRNA host–gene linear expression was estimated commonly to all the detection tools and independently of the circRNA abundance estimated by each method, allowing to remove possible advantages given by specific method quantification approaches.
CirComPara2 performs consistently better than single methods
The performance of the methods on the real RNA-seq data agreed with the analysis of simulated data and previous benchmark study results [23, 24]. We did not include CIRI-full because it showed inferior performance than CIRI2 in the simulated data. Segemehl showed high recall (median 0.75; Figure 4A) but the lowest precision (median 0.92; Figure 4B), whereas C2BW, as expected from annotation-based algorithms, showed the most reliable predictions (median precision 0.97; Figure 4B). Similar to the evaluation in the simulated data set, we computed the F1-scores in the real data set. According to F1-score medians, CirComPara2 had the highest (0.91) and significantly different value (q < 0.001; Figure 4C), and substantially outperformed the next best method, Segemehl (median F1-score 0.82), with a 0.09 F1-score difference (Supplementary Table S4 available online at http://bib.oxfordjournals.org/). The highest F1-score, achieved by CirComPara2, resulted from a significantly larger recall (median 0.86; q < 0.001; Figure 4A) and a negligible loss of precision compared to the other methods (0.01 median reduction to the most precise method; Figure 4B). Moreover, CirComPara2 had the narrowest interquartile range of F1-scores across the real data sets (0.11; Figure 4C; Supplementary Table S4 available online at http://bib.oxfordjournals.org/), proving that it is robust and almost unaffected by the experimental scenario. CirComPara2 showed considerable and significant improvements also compared to its former implementation (F1-score q < 0.001; precision q = 1.0; recall q < 0.001; Supplementary Table S4 available online at http://bib.oxfordjournals.org/), identifying in average 47% more true circRNAs than its former version. In light of the more challenging nature of real compared to simulated data [21, 43], these results further confirmed the advantage of CirComPara2 over the methods here assessed.
Figure 4 .
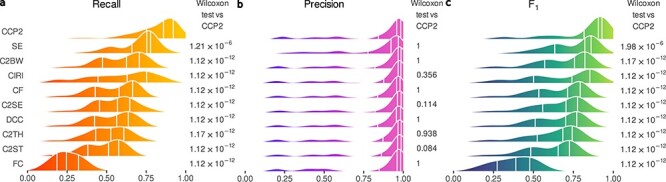
Performance of circRNA detection methods on the real RNA-seq data set. Results of the analysis on the 71 pairs of samples of the real RNA-seq data set. Density plots of (A) recall, (B) precision and (C) F1-score; distribution quartiles are indicated by white vertical lines. On the right-hand side of each plot, the Bonferroni adjusted P-values of Wilcoxon paired tests contrasting each method with CirComPara2. Methods’ abbreviations as in Figure 1; CCP2: CirComPara2.
Quantification of circRNA expression
Most circRNA expression quantification tools, including CIRCexplorer2, circRNA_finder, CIRI2, DCC and Findcirc, consider BJR counts to estimate the circular transcript abundance. However, different sets of BJRs may contribute to the expression estimates, especially if the tools were based on different chimeric aligners (Figure 5A). The method combination strategy, which we demonstrated to have superior detection performance, poses the question of which tool expression estimate should be considered or how to combine multiple expression estimates.
Figure 5 .
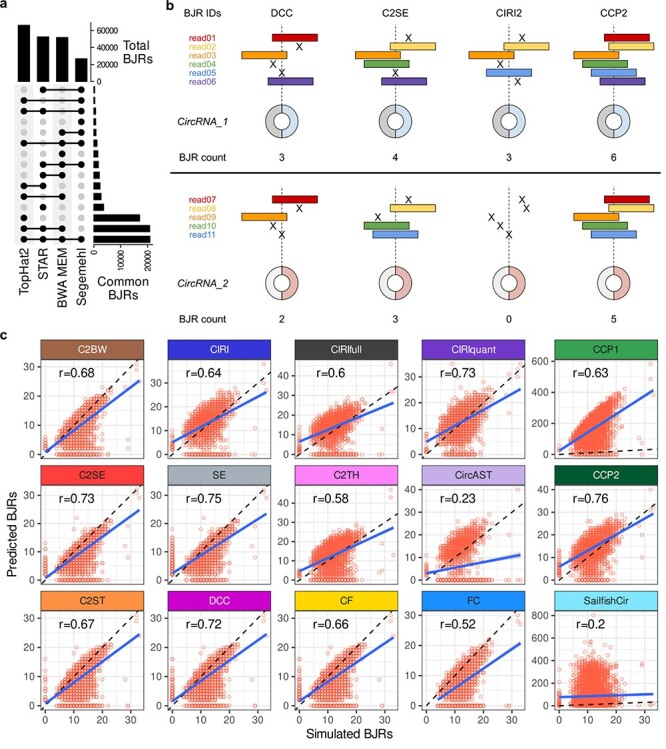
CircRNA expression estimates from CirComPara2. A, The overlap between the different sets of the back-splice junction reads (BJRs) identified by four chimeric aligners (BWA-MEM, Segemehl, STAR and TopHat-Fusion/TopHat2). B, The CirComPara2 BJR count approach: the upper panel shows the non-redundant BJR count of CirComPara2 (CCP2) compared to other methods (for simplicity, only three methods are displayed), which is based on unique BJR identifiers (BJR IDs); the lower panel highlights how CCP2 can preserve the information even when no BJRs are in common between circRNA detection methods. C, The estimated circRNA expression in BJRs compared to the true expression from the simulated data set. The Pearson correlation coefficient (r) values are reported in the boxes. CIRIquant, CircAST and Sailfish-cir were inputted with the circRNAs predicted by CirComPara2.
The former version of CirComPara estimated the circRNA abundance as the library-size normalized BJR median across the methods detecting a circRNA. Instead, CirComPara2 extracts the BJR from each method and for each circRNA to count only the non-redundant BJR identifiers (Figure 5B). By this approach, the information brought by each method is preserved.
Recent tools such as CIRIquant, Sailfish-cir and CircAST proposed a circRNA sequence reconstruction followed by read re-alignment to improve circRNA expression estimation. These methods have been devised for quantification purposes and require external tools for circRNA detection.
To evaluate the quantification accuracy of CirComPara2, we compared the true BJR counts of the simulated circRNAs with the expression estimates. We also compared the estimates from the other tools, plus CIRIquant, Sailfish-cir and CircAST. As in Zhang et al. [46], we computed the Pearson correlation coefficient between the estimated and the simulated BJR of each circRNA (Figure 5C). CirComPara2 achieved the highest correlation (r = 0.76), followed by Segemehl (r = 0.75), C2SE (r = 0.73), CIRIquant (r = 0.73) and DCC (r = 0.72). Notably, CirComPara2 largely improved over its former implementation (r = 0.63) and showed overall the most accurate expression estimates without requiring additional re-alignment steps.
Computation time and memory requirements
When running the circRNA detection methods on the simulated data, we observed that the chimeric alignment steps were the most computational-resource demanding tasks of the pipelines. In contrast, parsing the chimeric alignment output required <1% of the resources used in the alignment step in both computation time and peak memory. Only CIRI2 showed more memory usage to process the alignments (~30 GB compared to the ~6 GB of BWA-MEM) because it ran with eight parallel processes.
CirComPara2 inherited from and extended the CirComPara1 parsimonious computational design that does not repeat tasks in common between the integrated methods. For instance, the linear read alignment is common to all the circRNA sub-pipelines and is performed only once. Further, the chimeric alignments of BWA-MEM are used for both CIRI2 and CIRCexplorer2. Likewise, the STAR alignments are shared by three parsers, i.e. DCC, CF and C2ST. CirComPara2 runtime and memory requirements are comparable to its former implementation since the most computational-resource demanding tools (STAR and Segemehl) were already included in CirComPara1.
When processing the simulated data set (~3.7 million paired-end reads, 100 bp long, from the whole human genome), CirComPara2 saved ~20% computation time compared to the sequential independent running of the seven method full pipelines (SeqMet; Figure 6). Besides, CirComPara2 provides concurrent running of independent tasks, including the circRNA detection methods, which can reduce the computation time. Setting CircComPara2 to run multiple (up to seven) tasks in parallel (mpCCP2) saved ~74% computing time compared to the SeqMet, and ~68% compared to CirComPara2 with no augmented parallelization (Figure 6). The mpCCP2 setting reduces the computation time up to the longest program execution (C2TH) but at the cost of higher peak memory and CPU cores requirements: 56 CPU cores and up to 120 GB of random access memory were used to run in parallel all the aligners on the human genome, and CIRI2 with eight parallel processes.
Figure 6 .
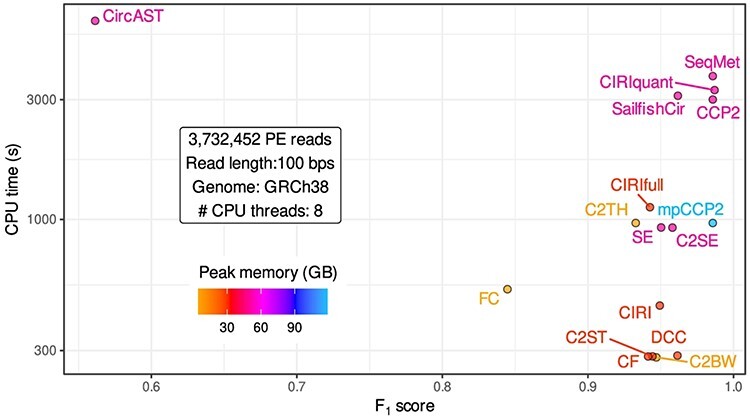
Computation time and memory requirements of circRNA detection methods. F1-scores of 14 circRNA detection pipelines compared to their computing time in seconds (s) and peak random access memory (in GigaBytes) required to process ~3.7 million paired-end (PE) reads of 100 bp length, mapped to the human genome (GRCh38) and using eight CPU threads for each tool. Plus, one hypothetical pipeline running sequentially the seven methods used in CirComPara2 (SeqMet) and CirComPara2 set with maximal parallelization settings (mpCCP2) allowing to run up to seven tasks in parallel, for peak usage of 56 CPU threads in total.
Finally, the CirComPara2 running time scaled linearly with the number of input sequencing reads (Supplementary Figure S5 available online at http://bib.oxfordjournals.org/). CirComPara2, set with the mpCCP2 parameters and applied to a multiple-sample batch, showed a runtime bounded only by the analysis of the sample having the largest input size (Supplementary Figure S5 available online at http://bib.oxfordjournals.org/). This result demonstrates that the CirComPara2 augmented parallelization harnessed the system’s computational power by consuming multiple short tasks concurrently to longer ones also from different samples, eventually saving computation time.
Discussion
In the early days after circRNA discovery, bioinformaticians put significant effort into developing circRNA detection methods with highly precise predictions. To this aim, the current circRNA detection algorithms apply filtering procedures to remove FPs, but that may also reject true circRNAs [21–24], increasing the number of FNs and the risk of overlooking circRNAs of interest, as suggested by our analysis. Moreover, the frequent practice of filtering out low-count circRNAs to improve precision may result in the loss of differentially expressed elements in comparative studies, as demonstrated by systematic studies on gene expression [52, 53]. Notably, unlike FPs, experimental validations cannot amend the bias derived by FN errors. These considerations prompted us to consider a method evaluation metric that equally weighted precision and recall, such as the F1-score.
Interestingly, the evaluation by F1-score in our simulated data analysis highlighted that some methods with diverging precision and recall, such as CIRI2 and Segemehl (Figure 3A), were equally ranked. Moreover, the real data set analysis showed an ample recall variation of each method across the samples, while precision scores were tighter both between and within methods. These results confirmed that choosing a single method for circRNA discovery is problematic since no single best performing method exists. We showed that an approach leveraging the advantages brought by each of the integrated methods allows CirComPara2 to perform better than the single methods consistently in different experimental contexts. Besides, we showed that CirComPara2 predictions are more inclusive and robust than single-method ones, even upon low-count filtering, indicating that the recovered circRNAs missed by other methods are not merely of low abundance.
To our knowledge, CirComPara has been the first bioinformatics tool to combine multiple circRNA detection methods in an automated software pipeline. Since its former implementation, the continuous upgrade of CirComPara evolved into a substantially improved new tool, CirComPara2, with an efficacious method combination strategy. Other computational pipelines that employed multiple circRNA detection tools, such as RAISE [54] and circRNAwrap [55], considered the circRNAs predicted by the union or the intersection of all methods, respectively, without validating which method combination was best. Our data show that those two approaches could be suboptimal as they suffer either low precision or sensitivity, whereas the best trade-off between precision and recall is achieved with predictions of methods taken pairwise.
Recently, large circRNA databases have been compiled using method combination strategies [56]. For instance, circAtlas2.0 [57] retained circRNAs identified by at least two methods among CIRI2, CIRCexplorer2, Findcirc and DCC; the same combination but replacing DCC with circRNA_finder has been used in CircRic [58]. Applying these two approaches to our real data sets analysis, we observed that they performed better than most of the algorithms except CirComPara2 (Supplementary Table S4 available online at http://bib.oxfordjournals.org/), which achieved similar precision but slightly more than 0.2 better recall. Such a result suggests that CirComPara2 could be employed to compile comprehensive and reliable circRNA databases in future works.
Importantly, the computational pipelines used to compile circAtlas2.0 and CircRic were not implemented as software tools available to the scientific community. Instead, we made the automated and computationally efficient CirComPara2 pipeline ready-to-use and portable through a Docker container, freeing bioinformaticians from the several difficulties posed by implementing a computational pipeline, such as installation of multiple tools, software portability, code maintenance and documentation [59].
CirComPara2 is the only tool that aggregates various method expression estimates into unified values that eliminate redundant counts of BJR identified by multiple tools without relying on additional re-alignment of the reads. CircRNAwrap, RAISE and CircAtlas2.0 applied a quantification step downstream of circRNA detection to estimate the circRNA expression. They considered the re-alignment of the reads onto pseudo-reference circRNA sequences through Sailfish-cir [60], RAISE itself and CIRIquant [46], respectively, thus increasing the computational requirements of the pipeline. We applied CircAST, Sailfish-cir and CIRIquant to the CirComPara2 predictions. The additional computational load was modest for Sailfish-cir and CIRIquant, but we did not obtain a better quantification of the expression than the BJR counts reported by CirComPara2. Moreover, CircAST and Sailfish-cir failed to quantify some circRNAs, which eventually reduced the accuracy of CirComPara2 predictions. Notably, in our test, CirComPara2 reported the best correlation with the true BJR counts with no need for additional re-alignment steps.
The various tools integrated into CirComPara2 determine its computational requirements. As pointed out by Jakobi and Dieterich [19], the read mapping phase is the limiting factor in terms of memory requirements, and the genome size of the target organism determines the amount of memory requested. For mammalian genomes, Segemehl bounds CirComPara2 to require at least 50 gigabytes (GB) of random access memory (RAM). Future development of CirComPara2 could reduce the peak memory by splitting the Segemehl and STAR indexes into chromosomes and allow CirComPara2 to run also on lower-end hardware. On the other hand, the task parallelization of CirComPara2 allows modulating the computational requirements to exploit higher-end hardware fully. Nevertheless, we think that the computational needs of CirComPara2 are compensated by its asset of detecting and quantifying circRNA expression.
CircRNA studies are likely to grow in several branches of biology, both on model [61] and non-model organisms and beyond the biomedical field [62–64], prompting the development of improved tools allowing more extensive circRNA investigation to unravel circRNA-related condition peculiarities, such as differential circRNA expression [65–67], imbalances of the CLP [53] and prevailing circular transcript isoform expression [68]. CirComPara2 is a resource tool meeting these needs, as already proved by the successful application of its embryonic implementations in several studies of human diseases and of other species, including plants [53, 65, 69, 70].
In this work, we described the main features of CirComPara2, our automated and computationally efficient software pipeline for circRNA expression characterization that also allows traditional gene expression analysis and the computation of circRNA to host–gene linear transcript abundance. Importantly, we demonstrated the asset given by the CirComPara2 method combination strategy for circRNA discovery, which provides robust and inclusive predictions in diverse biological contexts. With CirComPara2, we aim to provide a helpful bioinformatics tool to obtain a more comprehensive picture of transcriptomes and boost the understanding of circRNA features, biological and pathogenetic roles.
Methods
Simulated data set
CircRNA reads were simulated with the CIRI_simulator from the CIRI2 tool suite using the whole GRCh38 human genome and Gencode v29 gene annotation.
The parameters used in CIRI_simulator were: -C 20 -LC 0 -R 1 -LR 1 -L 101 -E 1 -CHR1 0 -M 250 -M2 450 -PM 0 -S 70 -S2 0 -SE 0 -PSI 10.
A total of 10% (30% for the highly pruned annotation simulation) of the gene annotation for the simulated circRNA parent genes was removed from the annotation file and used as input to circRNA detection methods and to simulate linear transcript reads with Polyester (the reads_per_transcript parameter was set to 300). The linear transcript read files were then concatenated to the circRNA read files.
Code for generating the simulated data is available as a software tool CCP_simulator at https://github.com/egaffo/CCP_simulator. The parameter ANNOPARTS used in CCP_simulator for the two filters on gene annotation (standard and high pruning) was set to ‘85,4,5,1,0,0,0’ and ‘60,15,10,2.5,6,3.5,3’, respectively.
Real data sets
Overall, 71 samples with matched rRNA depletion and rRNA depletion followed by RNase R treatment libraries from six studies (Table 1), for a total of 142 samples processed, were retrieved from Gene Expression Omnibus (GEO) or the National Genomics Data Center (NGDC) databases.
Reads from PRJCA000751 were trimmed to 150 bp in the pre-processing phase, as reported in the original work.
Method predictions in genomic scaffolds or from the mitochondrial genome were not considered. CircRNAs predicted with a length shorter than the library read length or longer than the longest gene expressed in the sample (computed as genes with TPM ≥ 1 computed by StringTie v2.1.4) were filtered out.
CircRNA detection methods’ parameters
The following genome and gene annotations from the Ensembl database were used in the analyses: GRCh38 human genome and v97 gene annotation, Mmul 10 Macaca mulatta genome and v101 gene annotation, and GRCm38 Mus musculus genome and v101 gene annotation.
CirComPara2 default parameters were set for the analyses, which are as follows: adaptors from the Trimmomatic v0.39 TruSeq3-PE-2.fa file; PREPROCESSOR = ‘trimmomatic’; PREPROCESSOR_PARAMS = ‘MAXINFO:40:0.5 LEADING:20 TRAILING:20 SLIDINGWINDOW:4:30 MINLEN:50 AVGQUAL:26’. For the PRJCA000751 data sets, the CROP:150 option was appended to the parameter. STAR_PARAMS = ‘—runRNGseed 123 —outSJfilterOverhangMin 15 15 15 15 —alignSJoverhangMin 15 —alignSJDBoverhangMin 15 —seedSearchStartLmax 30 —outFilterScoreMin 1 —outFilterMatchNmin 1 —outFilterMismatchNmax 2 —chimSegmentMin 15 —chimScoreMin 15 —chimScoreSeparation 10 —chimJunctionOverhangMin 15’. CIRCRNA_METHODS = ‘circexplorer2_bwa, circexplorer2_segemehl, circexplorer2_star, circexplorer2_tophat, ciri, dcc, findcirc’ (‘circrna_finder’ and ‘testrealign’ values were used in additional runs to obtain CircRNA_finder and Segemehl predictions); CPUS = 12; BWA_PARAMS =’-T 19′; SEGEMEHL_PARAMS =’-D 0′; BOWTIE2_PARAMS =’—reorder —score-min = C,-15,0 -q —seed 123′; DCC_EXTRA_PARAMS =’-fg -M -F -Nr 1 1 -N’; TESTREALIGN_PARAMS = ‘q median_1’; FINDCIRC_EXTRA_PARAMS =’—best-qual 40 —filter-tags UNAMBIGUOUS_BP —filter-tags ANCHOR_UNIQUE’ (this setting implements the optimization suggested by Hansen [24]). MIN_METHODS = 2; MIN_READS = 2; CIRC_MAPPING = “{'SE':['STAR','TOPHAT','BOWTIE2'],'PE':['BWA','SEGEMEHL']}”; HISAT2_PARAMS = ‘—seed 123’.
Segemehl predictions reported in the sngl.bed and trns.txt files were merged to include spliced reads spanning >20 000 bps. The same approach has been implemented in CirComPara2.
Read alignment and circRNA detection methods’ parameters were set as the value in the corresponding CirComPara2 parameters. All other parameters not mentioned were left with default values.
Details of the CirComPara2 method
CircRNA expression estimates in CirComPara2 are represented as the sum of all unique BJRs identified by the circRNA methods. Then, the number of BJR fragments is counted while keeping track of the number of methods detecting each circRNAs. Finally, circRNAs with at least two reads identified by two or more methods are reported. The MIN_METHODS and MIN_READS options can be used to modify the required minimum reads and methods.
The CirComPara2 pipeline considers a preliminary alignment step that maps the reads linearly on the genome. Linearly aligned reads are then used to characterize canonical gene and transcript expression and count the linearly spliced reads spanning the back-splice junctions. Instead, linearly unmapped reads are used as input to the circRNA detection methods.
Evaluation metrics and statistical tests
In evaluating method predictions, to compare samples with their matched control libraries, the different sequencing depths of the control library and the biochemical variability of the exoribonuclease in RNase R treatment have been adjusted by considering the proportion between the expression of the predicted circRNAs and the linear transcripts sharing the back-spliced exons. In each sample and for each method, we computed the circular-to-linear expression proportion (CLP) of each predicted circRNA by counting the number of reads back-spliced (BSreads) and linearly-spliced (LSreads) on the circRNA back-splice junctions, respectively. Then, the CLPs were calculated as BSreads/(BSreads + LSreads). Szabo and Salzman [43] also suggested using the ratio between the expression of circRNAs and their linear counterparts to overcome the evaluation issues deriving by RNase R-treated control samples.
Assuming that RNase R should degrade linear transcripts more than circRNAs regardless of its efficiency in different samples, circRNAs with an equal or increased CLP in the control samples were deemed true-positives (TPs); false-positives (FPs) otherwise. Only circRNAs detected in the control libraries were considered to limit incorrect FP calls due to a lower sequencing depth of the control sample and to RNase R-sensitive circRNAs [47].
The precision was defined as TP/(TP + FP). The recall was computed as TP/(TP + FN) and F1-score as (2 × Precision × Recall)/(Precision + Recall), where TP and FN denote the true-positive and false-negative numbers.
Wilcoxon one-tailed paired tests were used to compare CirComPara2 greater recall and F1-score or lower precision with each method. Bonferroni multiple test correction was applied to compute adjusted P-values (q-values). The q-values reported in the main text refer to the highest value among the pairwise comparisons for recall and F1-score, whereas to the lowest value for precision comparisons.
Software versions
The following software versions of the circRNA detection methods and chimeric read aligners were used in this study: CIRCexplorer2 [31] v2.3.8, CircRNA_finder [29] v1.1, CIRI2 [27] v2.0.6, DCC v0.4.8 [30], Findcirc [28] v1.2, Segemehl [17] v0.3.4 (used also by CIRCexplorer2), BWA MEM [16] v0.7.15 (used by CIRI2 and CIRCexplorer2), STAR [15] v2.6.1e (used by circRNA_finder, DCC and CIRCexplorer2) and TopHat2 [14] v2.1.0 (used for the TopHat-Fusion algorithm by CIRCexplorer2). CirComPara2 v0.1.2.1 was used to run the analysis. Other method versions used were as follows: CirComPara v0.1.3, CIRI-full v2.0 (the embedded CIRI-vis had to be replaced with an updated version CIRI-vis v1.4), CIRIquant v1.1.2, CircAST v1.0.2 [71] and Sailfish-cir v0.11a.
Key Points
Current circRNA detection methods achieve either high precision or high recall, possibly overlooking circRNAs of interest.
Extensive tests on simulated RNA-seq expression data determined the optimal method integration strategy for circRNA detection.
CirComPara2 achieves high detection recall with no loss of precision by combining seven circRNA detection methods.
At benchmarking on 142 real data sets, CirComPara2 consistently outperforms other methods regardless of the biological context and the genome annotation quality.
Supplementary Material
Acknowledgements
We thank Dr Geertruij te Kronnie for insightful suggestions and critical revision of the manuscript.
Enrico Gaffo is post-doc at the Computational Genomics Laboratory at the Department of Molecular Medicine, University of Padova. His research interests include circular RNA, microRNA, advanced methods for RNA-seq data analysis, and bioinformatics applied to cancer research.
Alessia Buratin is PhD student in Biosciences (curriculum Genetics, Genomics and Bioinformatics) of the University of Padova. Her main interests are biostatistics and bioinformatics, transcriptomics of hematologic malignancies, circular RNA function prediction and biogenesis.
Anna Dal Molin is post-doc at the Computational Genomics Laboratory at the Department of Molecular Medicine, working on circular RNAs in leukemias, and developing computational methods for circular RNA function prediction.
Stefania Bortoluzzi is associate professor of Applied Biology at the Department of Molecular Medicine of the University of Padova, where she leads the Computational Genomics Laboratory. Her research interests include cancer genomics and transcriptomics, bioinformatics, systems biology, noncoding RNAs, circular RNAs, exosomal RNAs, and hematologic malignancies.
Contributor Information
Enrico Gaffo, Department of Molecular Medicine, University of Padova, Padova, Italy.
Alessia Buratin, Department of Molecular Medicine, University of Padova, Padova, Italy; Department of Biology, University of Padova, Padova, Italy.
Anna Dal Molin, Department of Molecular Medicine, University of Padova, Padova, Italy.
Stefania Bortoluzzi, Department of Molecular Medicine, University of Padova, Padova, Italy; Interdepartmental Center For Innovative Biotechnologies, University of Padova, Padova, Italy.
Funding
Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN 2017 2017PPS2X4_003 to S.B.); Associazione Italiana per la Ricerca sul Cancro, Milan, Italy (Investigator Grant 2017 20052 to S.B.); and Fondazione Umberto Veronesi, Milan, Italy (Fellowship 2020 to E.G.).
Data Availability
The data underlying this article are available in the Gene Expression Omnibus (at https://www.ncbi.nlm.nih.gov/geo/) and the National Genomics Data Center (at https://bigd.big.ac.cn/) and can be accessed with SRR3476956, SRR1636985, SRR1636986, SRR3476958, SRR1637089, SRR1637090, SRR3479244, SRR3479243, GSE130905, SRR444655, SRR444975, SRR445016, SRR444974, GSE113120 and CRA001838 and PRJCA000751 accession numbers. Further details are referenced in Table 1 of this manuscript.
The code of CirComPara2 is available on GitHub at https://github.com/egaffo/circompara2.
Author Contributions
Conceptualization, E.G.; Data Curation, E.G.; Formal Analysis, E.G. and A.B.; Funding Acquisition, E.G. and S.B.; Methodology, E.G. and A.B.; Project Administration, S.B.; Resources, S.B.; Software, E.G. and A.B.; Supervision, E.G. and S.B.; Visualization, E.G.; Writing-Original Draft, E.G., A.B. and S.B.; Writing-Review & Editing, E.G., A.B., A.D.M. and S.B. All authors read and approved the final manuscript.
References
- 1. Xiao M-S, Ai Y, Wilusz JE. Biogenesis and functions of circular RNAs come into focus. Trends Cell Biol 2020;30:226–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonizzato A, Gaffo E, Te Kronnie G, et al. CircRNAs in hematopoiesis and hematological malignancies. Blood Cancer J 2016;6:e483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature 2013;495:384–8. [DOI] [PubMed] [Google Scholar]
- 4. Du WW, Yang W, Liu E, et al. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 2016;44:2846–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu X, Xiao S, Zhang M, et al. A novel protein encoded by circular SMO RNA is essential for Hedgehog signaling activation and glioblastoma tumorigenicity. Genome Biol 2021;22:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li Z, Huang C, Bao C, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 2015;22:256–64. [DOI] [PubMed] [Google Scholar]
- 7. Hanniford D, Ulloa-Morales A, Karz A, et al. Epigenetic silencing of CDR1as drives IGF2BP3-mediated melanoma invasion and metastasis. Cancer Cell 2020;37:55–70.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell 2019;179:1033–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rajappa A, Banerjee S, Sharma V, et al. Circular RNAs: emerging role in cancer diagnostics and therapeutics. Front Mol Biosci 2020;7:577938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Santer L, Bär C, Thum T. Circular RNAs: a novel class of functional RNA molecules with a therapeutic perspective. Mol Ther 2019;27:1350–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hua JT, Chen S, He HH. Landscape of noncoding RNA in prostate cancer. Trends Genet 2019;35:840–51. [DOI] [PubMed] [Google Scholar]
- 12. Awan FM, Yang BB, Naz A, et al. The emerging role and significance of circular RNAs in viral infections and antiviral immune responses: possible implication as theranostic agents. RNA Biol 2021;18:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L, Wang C, Sun H, et al. The bioinformatics toolbox for circRNA discovery and analysis. Brief Bioinform 2021;22(2):1706–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D, Salzberg SL. TopHat-fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 2011;12:R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM, 2013. arXiv.org, arXiv:1303.3997v2.
- 17. Hoffmann S, Otto C, Doose G, et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol 2014;15:R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang K, Singh D, Zeng Z, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 2010;38:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jakobi T, Dieterich C. Computational approaches for circular RNA analysis. Wiley Interdiscip Rev RNA 2019;10:e1528. [DOI] [PubMed] [Google Scholar]
- 20. Jiao S, Wu S, Huang S, et al. Advances in the identification of circular RNAs and research into circRNAs in human diseases. Front Genet 2021;12:665233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen I, Chen C-Y, Chuang T-J. Biogenesis, identification, and function of exonic circular RNAs. Wiley Interdiscip Rev RNA 2015;6:563–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hansen TB, Venø MT, Damgaard CK, et al. Comparison of circular RNA prediction tools. Nucleic Acids Res 2016;44:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeng X, Lin W, Guo M, et al. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol 2017;13:e1005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hansen TB. Improved circRNA identification by combining prediction algorithms. Front Cell Dev Biol 2018;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaffo E, Bonizzato A, Kronnie GT, et al. CirComPara: a multi-method comparative bioinformatics pipeline to detect and study circRNAs from RNA-seq data. Noncoding RNA 2017;3(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang X-O, Wang H-B, Zhang Y, et al. Complementary sequence-mediated exon circularization. Cell 2014;159:134–47. [DOI] [PubMed] [Google Scholar]
- 27. Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform 2018;19:803–10. [DOI] [PubMed] [Google Scholar]
- 28. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495:333–8. [DOI] [PubMed] [Google Scholar]
- 29. Westholm JO, Miura P, Olson S, et al. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 2014;9:1966–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics 2016;32:1094–6. [DOI] [PubMed] [Google Scholar]
- 31. Zhang X-O, Dong R, Zhang Y, et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res 2016;26:1277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Andrews S. FastQC: a quality control tool for high throughput sequence data. FastQC 2010. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- 34. Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015;33:290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 2015;4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Love MI, Soneson C, Hickey PF, et al. Tximeta: reference sequence checksums for provenance identification in RNA-seq. PLoS Comput Biol 2020;16:e1007664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tange O. GNU Parallel 20200922 (‘Ginsburg'). 2020:10.
- 41. Zheng Y, Ji P, Chen S, et al. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med 2019;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y, Lun ATL, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res 2016;5:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Szabo L, Salzman J. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 2016;17:679–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gao Y, Wang J, Zheng Y, et al. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun 2016;7:12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiao M-S, Wilusz JE. An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3′ ends. Nucleic Acids Res 2019;47:8755–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang J, Chen S, Yang J, et al. Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat Commun 2020;11:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013;19:141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen S, Huang V, Xu X, et al. Widespread and functional RNA circularization in localized prostate cancer. Cell 2019;176:831–843.e22. [DOI] [PubMed] [Google Scholar]
- 49. Ji P, Wu W, Chen S, et al. Expanded expression landscape and prioritization of circular RNAs in mammals. Cell Rep 2019;26:3444–3460.e5. [DOI] [PubMed] [Google Scholar]
- 50. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 2013;41:D991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. National Genomics Data Center Members and Partners . Database resources of the National Genomics Data Center in 2020. Nucleic Acids Res 2020;48:D24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu A, Ibrahim JG, Love MI. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 2019;35:2084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Buratin A, Paganin M, Gaffo E, et al. Large-scale circular RNA deregulation in T-ALL: unlocking unique ectopic expression of molecular subtypes. Blood Adv 2020;4:5902–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li L, Zheng Y-C, Kayani MUR, et al. Comprehensive analysis of circRNA expression profiles in humans by RAISE. Int J Oncol 2017;51:1625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li L, Bu D, Zhao Y. CircRNAwrap—a flexible pipeline for circRNA identification, transcript prediction, and abundance estimation. FEBS Lett 2019;593:1179–89. [DOI] [PubMed] [Google Scholar]
- 56. Vromman M, Vandesompele J, Volders P-J. Closing the circle: current state and perspectives of circular RNA databases. Brief Bioinform 2021;22(1):288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu W, Ji P, Zhao F. CircAtlas: an integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol 2020;21:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ruan H, Xiang Y, Ko J, et al. Comprehensive characterization of circular RNAs in ~1000 human cancer cell lines. Genome Med 2019;11:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Menegidio FB, Jabes DL, Costa de Oliveira R, et al. Dugong: a Docker image, based on Ubuntu Linux, focused on reproducibility and replicability for bioinformatics analyses. Bioinformatics 2018;34:514–5. [DOI] [PubMed] [Google Scholar]
- 60. Li M, Xie X, Zhou J, et al. Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics 2017;33:2131–9. [DOI] [PubMed] [Google Scholar]
- 61. Weigelt CM, Sehgal R, Tain LS, et al. An insulin-sensitive circular RNA that regulates lifespan in Drosophila. Mol Cell 2020;79:268–279.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu Q, Ning X, Sun L. Megalocytivirus induces complicated fish immune response at multiple RNA levels involving mRNA, miRNA, and circRNA. Int J Mol Sci 2021;22(6):3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chu Q, Zheng W, Su H, et al. A highly conserved circular RNA circRasGEF1B enhances antiviral immunity by regulating miR-21-3p/MITA pathway in lower vertebrates. J Virol 2021;95(7):e02145–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liang Y, Zhang Y, Xu L, et al. CircRNA expression pattern and ceRNA and miRNA–mRNA networks involved in anther development in the CMS line of Brassica campestris. Int J Mol Sci 2019;20:4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gaffo E, Boldrin E, Dal Molin A, et al. Circular RNA differential expression in blood cell populations and exploration of circRNA deregulation in pediatric acute lymphoblastic leukemia. Sci Rep 2019;9:14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu Y, Zhao T, Deng R, et al. A study of differential circRNA and lncRNA expressions in COVID-19-infected peripheral blood. Sci Rep 2021;11:7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tian J, Fu Y, Li Q, et al. Differential expression and bioinformatics analysis of CircRNA in PDGF-BB-induced vascular smooth muscle cells. Front Genet 2020;11:530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Izuogu OG, Alhasan AA, Mellough C, et al. Analysis of human ES cell differentiation establishes that the dominant isoforms of the lncRNAs RMST and FIRRE are circular. BMC Genomics 2018;19:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Frydrych Capelari É, Fonseca GC, Guzman F, et al. Circular and micro RNAs from Arabidopsis thaliana flowers are simultaneously isolated from AGO-IP libraries. Plan Theory 2019;8(9):302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dal Molin A, Hofmans M, Gaffo E, et al. CircRNAs dysregulated in juvenile Myelomonocytic Leukemia: CircMCTP1 stands out. Front Cell Dev Biol 2020;8:613540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jing Wu, Yan Li, Cheng Wang, et al. CircAST: Full-length Assembly and Quantification of Alternatively Spliced Isoforms in Circular RNAs. Genomics Proteomics Bioinformatics 2019;17(5):522–534. doi: 10.1016/j.gpb.2019.03.004. PMID: 32007626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the Gene Expression Omnibus (at https://www.ncbi.nlm.nih.gov/geo/) and the National Genomics Data Center (at https://bigd.big.ac.cn/) and can be accessed with SRR3476956, SRR1636985, SRR1636986, SRR3476958, SRR1637089, SRR1637090, SRR3479244, SRR3479243, GSE130905, SRR444655, SRR444975, SRR445016, SRR444974, GSE113120 and CRA001838 and PRJCA000751 accession numbers. Further details are referenced in Table 1 of this manuscript.
The code of CirComPara2 is available on GitHub at https://github.com/egaffo/circompara2.