

Abstract
Feature selection is crucial for the analysis of high-dimensional data, but benchmark studies for data with a survival outcome are rare. We compare 14 filter methods for feature selection based on 11 high-dimensional gene expression survival data sets. The aim is to provide guidance on the choice of filter methods for other researchers and practitioners. We analyze the accuracy of predictive models that employ the features selected by the filter methods. Also, we consider the run time, the number of selected features for fitting models with high predictive accuracy as well as the feature selection stability. We conclude that the simple variance filter outperforms all other considered filter methods. This filter selects the features with the largest variance and does not take into account the survival outcome. Also, we identify the correlation-adjusted regression scores filter as a more elaborate alternative that allows fitting models with similar predictive accuracy. Additionally, we investigate the filter methods based on feature rankings, finding groups of similar filters.
Keywords: benchmark, feature selection, filter methods, high-dimensional data, survival analysis
1 Introduction
Feature selection is one of the most fundamental problems in the analysis of high-dimensional data. Especially for high-dimensional data sets, it is often advantageous with respect to predictive performance, run time and interpretability to disregard the irrelevant and redundant features. This can be achieved by choosing a suitable subset of features that are relevant for target prediction. In bioinformatics, feature selection often allows identifying the features that are important for biological processes of interest. Due to the enormous amount of existing feature selection methods, benchmark studies are of great importance for identifying the best methods to use in data analyses.
In the past decades, many feature selection methods have been proposed. The methods can be categorized into three classes: filter methods, wrapper methods and embedded methods [1]. Filter methods rank features by calculating a score for each feature independent of a model. Either the 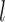 features with the highest scores or all features whose scores exceed a threshold
features with the highest scores or all features whose scores exceed a threshold 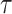 are selected (with
are selected (with 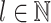 or
or 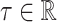 being pre-specified). For many filter methods, the score calculation can be done in parallel, thus resulting in increased computational efficiency. An extensive overview of existing filter methods is given in [2]. Wrapper methods [3] consider subsets of the set of all features. For each of the subsets, a predictive model is fitted and the subsets are evaluated by a performance measure calculated from the resulting model. Wrapper methods include not only simple approaches like greedy sequential searches [4], but also more elaborate algorithms like recursive feature elimination [5] as well as evolutionary and swarm intelligence algorithms for feature selection [6–8]. Embedded methods include the feature selection in the model fitting process. Examples of predictive methods that perform embedded feature selection are lasso regression [9] and tree-based methods like random forests [10] or gradient boosting [11, 12]. There are many overview papers that describe in detail, categorize and suggest how to evaluate existing feature selection methods, for example [1, 13–21].
being pre-specified). For many filter methods, the score calculation can be done in parallel, thus resulting in increased computational efficiency. An extensive overview of existing filter methods is given in [2]. Wrapper methods [3] consider subsets of the set of all features. For each of the subsets, a predictive model is fitted and the subsets are evaluated by a performance measure calculated from the resulting model. Wrapper methods include not only simple approaches like greedy sequential searches [4], but also more elaborate algorithms like recursive feature elimination [5] as well as evolutionary and swarm intelligence algorithms for feature selection [6–8]. Embedded methods include the feature selection in the model fitting process. Examples of predictive methods that perform embedded feature selection are lasso regression [9] and tree-based methods like random forests [10] or gradient boosting [11, 12]. There are many overview papers that describe in detail, categorize and suggest how to evaluate existing feature selection methods, for example [1, 13–21].
Regarding the comparison of different methods, benchmark studies have gained increasing attention. The majority of these studies are based on classification data. For these, the feature selection methods are combined with classification methods in order to assess the predictive performance of the selected features. In [22], filter methods are compared based on two gene expression data sets, counting the number of misclassified samples. In [23], the classification accuracy of different filter, wrapper, and embedded methods on several artificial data sets is analyzed. In [24, 25], filter methods are compared with respect to classification accuracy based on microarray data sets. In [26, 27], extensive comparisons based on text classification data sets are conducted. The authors analyze filter and wrapper methods, respectively. In [28], filter methods are compared with respect to classification accuracy on malware detection data. In [29, 30], filter methods are studied on large data sets, analyzing the predictive accuracy with respect to the number of features to select. In [31, 32], small artificial data sets are used to assess whether the correct features are selected. In [31], different feature selection methods are compared while in [32], only filter methods are considered. In [33], filter and wrapper methods are compared on large simulated data sets with respect to the correctness of the selected features. Additionally, the authors conduct comparisons with respect to classification accuracy on real data sets. In [34], filter and wrapper methods are compared comprehensively with respect to classification accuracy and run time, considering each of the two objectives separately. Most of the data sets on which the comparison is based contain a small or medium number of features. In [35, 36], several filter methods that are based on mutual information are compared. In [35], the accuracy and the run time of the methods are analyzed separately. Additionally, the authors take into account theoretical properties and look at the proportions of correctly identified features on artificial data. In [36], the authors analyze the classification accuracy with respect to the number of selected features and search for Pareto optimal methods considering the accuracy and feature selection stability. In [37], an extensive study of correlation-based feature selection is conducted. The author analyzes the classification accuracy based on real data sets as well as the choice of relevant or irrelevant features based on artificial data sets. In [38], an extensive comparison of 22 filter methods on 16 large or high-dimensional data sets is conducted with respect to both classification accuracy and run time jointly. Also, the empirical similarity of the filter methods based on the rankings of all features of all considered data sets is analyzed. In [39], the authors perform hyper parameter tuning of predictive models on survival data sets. They use combined methods consisting of a filter and a survival prediction method and consider the choice of filter method as a hyper parameter. This way, they find out which filter methods yield good results on many data sets.
In this article, we analyze feature selection methods on gene expression survival data sets. The features of gene expression data contain information about the activity of genes, for example in cancerous tissues. Gene expression data are a typical example of high-dimensional data used in bioinformatics. The outcome of survival data consists of two variables: one variable indicating the observed outcome time and one variable indicating if an event occurred or if the observation is censored. For right-censored data, a censored observation means that no event has occurred up to this point in time, but no information about events occurring afterward is available. For survival data sets, commonly used regression or classification techniques are not suitable because they cannot take into account the censoring information. Since censored survival data are frequently used in bioinformatics and since feature selection methods for survival data have not been thoroughly benchmarked before, it is necessary to conduct such a benchmark for survival data.
This article focuses on the comparison of filter methods for feature selection. Our focus on filter methods is motivated by the following considerations: Most wrapper methods are computationally infeasible for high-dimensional data sets. Embedded methods require the use of a certain predictive model. Most filter methods, however, are fast to calculate and can be combined with any kind of predictive method, even methods with embedded feature selection, see [40, 41]. Also, they can heavily reduce the run time for fitting the subsequent model. So, for data sets with really large numbers of features, it can be necessary to pre-filter the data set in order to make further analyses possible. To the best of our knowledge, a thorough and extensive filter comparison study has not yet been conducted for survival data.
In this article, 14 filter methods from different toolboxes are benchmarked based on 11 gene expression survival data sets. The filter methods considered here are representatives of the most prominent general concepts for filter methods. These classes of filter methods are univariate filters, feature importance filters based on multivariate models and information theoretic measures. Most of the compared filter methods have been integrated into the machine learning R package mlr3 [42] and are ready to use. mlr3 is a comprehensive package for machine learning and a standard in the R community.
The aim of benchmarking the filter methods is to identify the best filter methods, so that these methods can be employed in future data analyses. The best filter methods are assessed with respect to predictive performance when combined with a predictive model and with respect to run time. Also, the number of selected features required for obtaining a model with high predictive accuracy and the feature selection stability is assessed. Feature selection stability is defined as the robustness of the set of selected features with respect to different data sets from the same data generating distribution and is crucial for the reliability of the results [43]. Additionally, we analyze the empirical similarity of the filter methods. For finding groups of similar filter methods, we investigate which methods select the top features in a similar order. Our analysis identifies three groups of filter methods with a similar behavior, as well as several filter methods that are not very similar to any other filter method. There is one simple filter method that performs best with respect to predictive accuracy, run time and feature selection stability. We also identify a more elaborate filter method that allows fitting models with similar predictive performance.
The remainder of this article is organized as follows: In the Methods section, basic concepts of survival data are briefly introduced, the filter methods considered in this article are described and a feature selection stability measure is defined. In the Experiments section, the considered data sets are given. For both the similarity analysis and the comparison of the filters’ performances, the setup of the experiments is explained and the results are analyzed in detail. The Conclusions section contains a summary of the findings and concluding remarks.
2 Methods
2.1 Survival data basics
2.1.1 Notation
A data set with 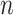 observations of the
observations of the 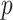 features
features 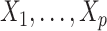 is considered. Furthermore,
is considered. Furthermore, 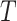 denotes the observed outcome time and is defined as
denotes the observed outcome time and is defined as 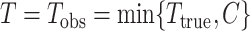 with
with 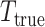 denoting the true survival time and
denoting the true survival time and 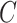 denoting the censoring time.
denoting the censoring time. 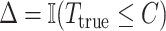 is the event indicator with ‘0’ corresponding to a right-censored and ‘1’ corresponding to an uncensored observation.
is the event indicator with ‘0’ corresponding to a right-censored and ‘1’ corresponding to an uncensored observation.
2.1.2 Cox proportional hazards regression model
The Cox proportional hazards model is a regression technique for modeling survival outcomes. It is defined as
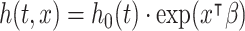 |
(1) |
with 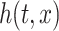 denoting the hazard at time
denoting the hazard at time 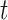 for an individual with covariate vector
for an individual with covariate vector 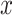 representing the features. The term
representing the features. The term 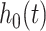 denotes the baseline hazard at time
denotes the baseline hazard at time 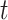 and
and 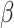 is the vector of regression parameters. When fitting a Cox proportional hazards model to a data set, the baseline hazard and the regression parameters are estimated by maximizing the partial log-likelihood. For more details about Cox proportional hazards models see [44].
is the vector of regression parameters. When fitting a Cox proportional hazards model to a data set, the baseline hazard and the regression parameters are estimated by maximizing the partial log-likelihood. For more details about Cox proportional hazards models see [44].
A variant of the Cox proportional hazards model is the 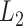 -regularized Cox proportional hazards model [45]. For the parameter estimation of this model, the partial log-likelihood is modified with a
-regularized Cox proportional hazards model [45]. For the parameter estimation of this model, the partial log-likelihood is modified with a 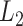 -penalty. The
-penalty. The 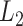 -penalty shrinks all regression coefficients toward zero. In contrast to an unregularized Cox proportional hazards model, a
-penalty shrinks all regression coefficients toward zero. In contrast to an unregularized Cox proportional hazards model, a 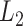 -regularized Cox proportional hazards model can be fitted on data sets that contain more features than observations.
-regularized Cox proportional hazards model can be fitted on data sets that contain more features than observations.
2.1.3 Integrated Brier score
The integrated Brier score [46] is a measure of prediction error that reflects both discrimination and calibration aspects. It is estimated using inverse probability weighting. The integrated Brier score measures the quadratic difference between the individual estimated survival function of each observation and its observed indicator function of individual survival and then sums these values for all observations. The range of the integrated Brier score is 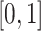 with 0 being the best value.
with 0 being the best value.
2.1.4 Transforming survival data into regression data using martingale residuals
Some of the filter methods described below require the transformation of the survival outcome to an uncensored continuous outcome. This can be accomplished by calculating martingale residuals [47]. For a survival data set with right-censored data, first, a Cox proportional hazards regression model is fitted without covariates. This model incorporates the information given by the observed outcome time  and the event indicator
and the event indicator 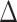 . Then, for each observation, the martingale residual is calculated based on the Cox model. Martingale residuals are real-valued and can be used as uncensored continuous outcomes. For more details see [47]. In the following subsection, we will refer to the transformed target variable containing the martingale residuals as
. Then, for each observation, the martingale residual is calculated based on the Cox model. Martingale residuals are real-valued and can be used as uncensored continuous outcomes. For more details see [47]. In the following subsection, we will refer to the transformed target variable containing the martingale residuals as 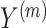 .
.
2.2 Filter methods
In this subsection, the 14 filter methods analyzed in this article are defined, using the notation of the previous subsection. All filter methods in this subsection are described for survival data sets with continuous features, because gene expression data sets usually only contain continuous features. Two kinds of filter methods are presented: Most filter methods calculate a score for all features and then select the features with the highest scores. Some filter methods, however, select features iteratively in a greedy forward fashion. For these filters, in each iteration, the feature with the maximal score is selected but the scores of different iterations are not comparable. We first describe univariate filter methods, which do not consider interactions between the 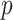 features. Subsequently, we discuss multivariate filter methods. All filter methods are described in the way they are implemented in the software used for the comparative experiments, which is indicated at the end of this subsection.
features. Subsequently, we discuss multivariate filter methods. All filter methods are described in the way they are implemented in the software used for the comparative experiments, which is indicated at the end of this subsection.
2.2.1 Univariate filter methods
The variance filter uses the variance of a feature as its score
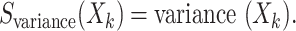 |
(2) |
The idea of this filter is to remove features that only consist of noise and therefore have very little variation. This filter only makes sense for data sets where the features are measured on the same scale and have not been scaled to unit variance.
For the correlation filter, the survival targets are transformed into the continuous target variable 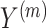 as described above. Then, for each feature, the absolute Pearson correlation between this feature and
as described above. Then, for each feature, the absolute Pearson correlation between this feature and 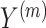 is computed and used as filter score
is computed and used as filter score
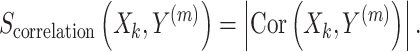 |
(3) |
The idea of this filter is to keep features that have a strong linear association with the target variable.
The cox.score filter assesses how well each feature can explain the survival outcome univariately. For feature 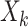 , a Cox proportional hazards regression model with only
, a Cox proportional hazards regression model with only 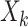 as independent variable is fitted. Then, a score test [44] is calculated for this model. The test statistic of the score test is used as filter score
as independent variable is fitted. Then, a score test [44] is calculated for this model. The test statistic of the score test is used as filter score
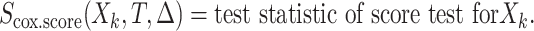 |
(4) |
The idea of this filter is that, the more important a feature is for explaining the survival outcome, the higher is the test statistic of the respective score test.
The carss filter calculates absolute correlation-adjusted regression survival scores 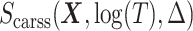 , in which
, in which 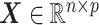 is defined as the matrix containing all covariates and
is defined as the matrix containing all covariates and 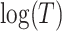 corresponds to the logarithmic observed outcome time. To estimate these scores, first, the correlation between
corresponds to the logarithmic observed outcome time. To estimate these scores, first, the correlation between 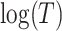 and each covariate
and each covariate 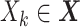 is adjusted by inverse probability of censoring weighting [48] in the formula
is adjusted by inverse probability of censoring weighting [48] in the formula
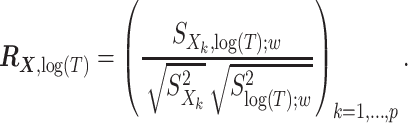 |
(5) |
This equation yields the usual correlation definition with the weighted covariance 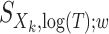 , weighted variance
, weighted variance 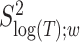 and variance
and variance 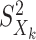 . The weights
. The weights 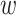 are calculated based on an estimate of the Kaplan–Meier survival function of the logarithmic censoring process. Then, the estimated correlations
are calculated based on an estimate of the Kaplan–Meier survival function of the logarithmic censoring process. Then, the estimated correlations 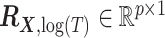 are decorrelated with the estimated covariate correlation matrix
are decorrelated with the estimated covariate correlation matrix 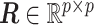 by
by
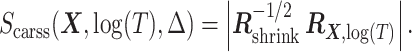 |
(6) |
The matrix 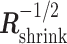 is estimated by shrinkage toward the identity matrix following the approach of [49, 50]. No additional tuning parameter is necessary, because the amount of shrinkage can be estimated directly from the data [51]. For a more detailed description of this method, we refer to [52].
is estimated by shrinkage toward the identity matrix following the approach of [49, 50]. No additional tuning parameter is necessary, because the amount of shrinkage can be estimated directly from the data [51]. For a more detailed description of this method, we refer to [52].
2.2.2 Filter methods based on feature importance measures
Three feature importance filters based on multivariate models are considered: random forest permutation importance, random forest impurity importance and gradient boosting importance. Random survival forests are bagging ensembles with survival trees as base learners [10]. To calculate the permutation importance, the out of bag (oob) observations for each tree, that is, the observations that were not used for fitting this tree, are considered. For the oob observations of each tree, feature 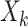 is permuted. Then, for the permuted observations, the cumulative hazards are predicted by the corresponding trees. The resulting predictive accuracy is compared to the predictive accuracy without permuting feature
is permuted. Then, for the permuted observations, the cumulative hazards are predicted by the corresponding trees. The resulting predictive accuracy is compared to the predictive accuracy without permuting feature 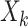 . To measure the predictive accuracy, the concordance index (C-index) by Harrell [53] is employed, see [10] for details on computing the C-index based on cumulative hazards. The score of the permutation importance filter is the decrease in predictive accuracy of the random forest from original oob observations to permuted observations. Features that are important for survival prediction cause a large decrease in accuracy because their relevant information is not available when the feature is permuted.
. To measure the predictive accuracy, the concordance index (C-index) by Harrell [53] is employed, see [10] for details on computing the C-index based on cumulative hazards. The score of the permutation importance filter is the decrease in predictive accuracy of the random forest from original oob observations to permuted observations. Features that are important for survival prediction cause a large decrease in accuracy because their relevant information is not available when the feature is permuted.
 |
(7) |
The impurity filter considers the node impurities of the trees. Nodes containing observations with similar survival are called pure, nodes with many dissimilar cases are called impure. When constructing a survival tree, the split variables and split points are chosen based on maximal difference in survival measured by the statistic of the log-rank test. To assess the gains in purity due to feature 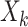 , the sum of all log-rank test statistics for all splits based on
, the sum of all log-rank test statistics for all splits based on 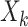 is calculated and used as filter score. A feature that is important for survival prediction causes on average a large gain in purity.
is calculated and used as filter score. A feature that is important for survival prediction causes on average a large gain in purity.
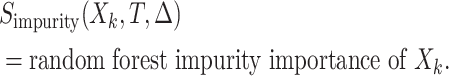 |
(8) |
The boosting filter uses the gradient boosting feature importance as filter score. Gradient boosting is an ensemble method that additively combines many weak learners into one strong prediction method [11, 12]. Here, survival trees (actual trees, not just stumps) are used as weak learners and the negative partial log-likelihood is employed as loss function. To assess the importance of the features in the boosting model, first, for each split in each tree, the improvement caused by this split is assessed. Then, for each feature, the sum of the corresponding improvement values is calculated [54]. A feature that is important for survival prediction causes on average large improvements.
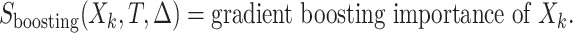 |
(9) |
2.2.3 Mutual information-based filter methods
For applying the following mutual information-based filter methods, a data set with categorical features and categorical target variable is required. To transform a survival data set accordingly, first, the continuous variable  is created as described above. Then,
is created as described above. Then,  is transformed into the categorical target variable
is transformed into the categorical target variable 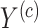 by cutting its range into
by cutting its range into 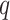 equally spaced intervals and using these intervals as categories. The number of intervals is determined as
equally spaced intervals and using these intervals as categories. The number of intervals is determined as 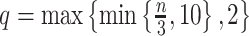 where
where 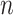 is the number of observations in the data set [55]. Continuous features are categorized analogously. The categorized version of feature
is the number of observations in the data set [55]. Continuous features are categorized analogously. The categorized version of feature 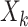 is denoted as
is denoted as  .
.
Let 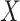 and
and 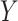 be two discrete variables with respective (empirical) probability mass function
be two discrete variables with respective (empirical) probability mass function 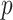 . Then, the entropy of
. Then, the entropy of 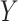 is defined as
is defined as
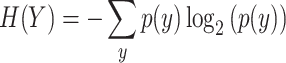 |
(10) |
and the conditional entropy of 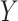 given
given 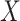 is given by
is given by
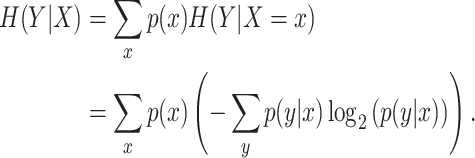 |
(11) |
The entropy measures the uncertainty of a variable. When all possible values occur with roughly the same probability, the entropy is high. If the probabilities of occurrence are very different from each other, the entropy is low. The mutual information of two variables is defined as
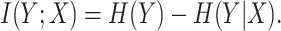 |
(12) |
It can be interpreted as the decrease in uncertainty about 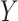 conditional on knowing
conditional on knowing 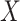 . Considering the symmetry property
. Considering the symmetry property 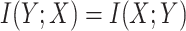 , it can also be seen as the amount of information shared by
, it can also be seen as the amount of information shared by 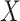 and
and 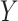 .
.
The mim filter [36] ranks all features according to the information they share with the target variable 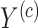
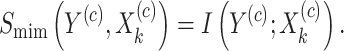 |
(13) |
The following filter methods calculate the scores of all features iteratively, implying that the features are selected in a greedy forward manner. Let 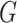 denote the set of features that are already selected.
denote the set of features that are already selected. 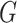 is initialized as
is initialized as  with
with 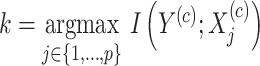 . In each iteration, the feature that maximizes the respective score is added to
. In each iteration, the feature that maximizes the respective score is added to  .
.
The mrmr filter [30] uses the score
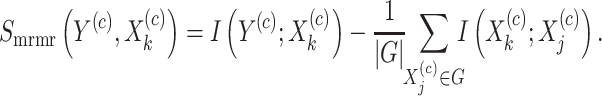 |
(14) |
The term 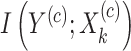 measures the relevance of the feature based on the information this feature has about
measures the relevance of the feature based on the information this feature has about 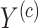 . The term
. The term 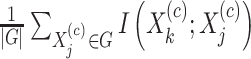 judges the redundancy of
judges the redundancy of  by assessing the mean information that the feature shares with the features in
by assessing the mean information that the feature shares with the features in  . The idea is to find maximally relevant and minimally redundant (mrmr) features.
. The idea is to find maximally relevant and minimally redundant (mrmr) features.
For the jmi filter [56], the score
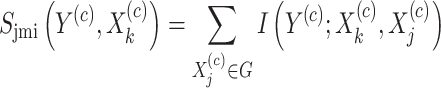 |
(15) |
is employed. 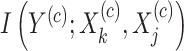 is the amount of information about
is the amount of information about  that
that  and
and 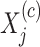 provide jointly. This quantity can be calculated by using the variable
provide jointly. This quantity can be calculated by using the variable 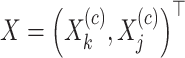 and its multivariate probability mass function in the definition of mutual information. The idea of this score is to include features that are complementary to the already selected features.
and its multivariate probability mass function in the definition of mutual information. The idea of this score is to include features that are complementary to the already selected features.
The jmim filter [57] is a modification of the jmi filter. The score
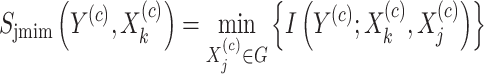 |
(16) |
considers the minimal joint information over all already selected features instead of the sum.
For the disr filter [58], the score
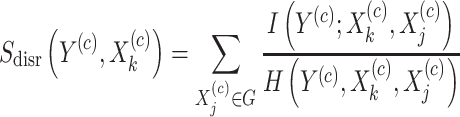 |
(17) |
is used. Like the jmi filter, it uses the information about 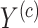 provided jointly by
provided jointly by 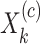 and
and 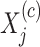 . But additionally, this information is divided by the joint entropy of
. But additionally, this information is divided by the joint entropy of 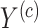 ,
, 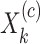 , and
, and 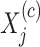 . To obtain this entropy, consider the variable
. To obtain this entropy, consider the variable 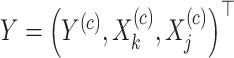 and plug it into the above definition of the entropy.
and plug it into the above definition of the entropy.
The njmim filter [57] is a modification of the disr filter. Its score
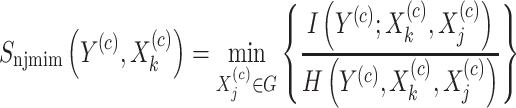 |
(18) |
considers the minimal relative joint information over all already selected features instead of the sum.
The cmim filter [59] has the score
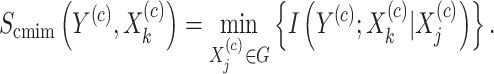 |
(19) |
It uses the conditional mutual information
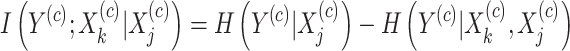 |
(20) |
that can be interpreted as the difference in uncertainty about  before and after
before and after  is known, while
is known, while 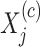 is known anyway. The idea is to select features that provide much information about the class variable, given the information of the already selected features.
is known anyway. The idea is to select features that provide much information about the class variable, given the information of the already selected features.
2.2.4 Implementation of filter methods
Table 1 provides an overview of the filter methods and the implementations used for the benchmark experiments in this article.
Table 1.
Overview of the filter methods: Name of the filter method (filter), short description of filter (description), information if filter is multivariate (multivariate), information if filter uses the survival outcome or a transformed target variable (target), information if filter requires categorization of numeric features (features category), and information about R package from which the implementation is taken
| Filter | Description | Multivariate | Target | Features category | Implementation |
|---|---|---|---|---|---|
| variance | Feature variance | No |
|
No | mlr3filters [60] |
| correlation | Pearson correlation | No |
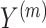
|
No | mlr3filters [60] |
| cox.score | Score test | No |
|
No | survival [61] |
| carss | Correlation-adjusted regression survival scores | No |
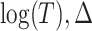
|
No | carSurv [62] |
| permutation | Random forest permutation importance | Yes |
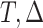
|
No | ranger [63] with default hyper parameter settings |
| impurity | Random forest impurity importance | Yes |
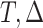
|
No | ranger [63] with default hyper parameter settings |
| boosting | Boosting importance | Yes |
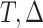
|
No | xgboost [64] with 2 000 boosting iterations, step size 0.05 and maximum tree depth 10 |
| mim | Mutual information | No |
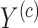
|
Yes | praznik [55] |
| mrmr | Mutual information | Yes |
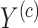
|
Yes | praznik [55] |
| jmi | Mutual information | Yes |
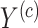
|
Yes | praznik [55] |
| jmim | Mutual information | Yes |
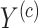
|
Yes | praznik [55] |
| disr | Mutual information | Yes |
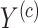
|
Yes | praznik [55] |
| njmim | Mutual information | Yes |
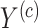
|
Yes | praznik [55] |
| cmim | Mutual information | Yes |
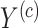
|
Yes | praznik [55] |
2.3 Feature selection stability measure
Let 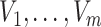 denote
denote 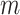 sets of selected features and
sets of selected features and 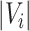 the cardinality of set
the cardinality of set 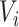 . Let
. Let 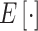 denote the expected value for a random selection of two feature sets that have the same cardinality as
denote the expected value for a random selection of two feature sets that have the same cardinality as 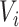 and
and 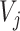 , respectively (with equal selection probabilities for all sets that have the respective cardinality). Let
, respectively (with equal selection probabilities for all sets that have the respective cardinality). Let 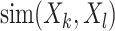 be the similarity of two features
be the similarity of two features 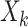 and
and 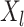 , assessed with a similarity measure that attains values in the interval
, assessed with a similarity measure that attains values in the interval 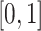 , for example the absolute Pearson correlation, and let
, for example the absolute Pearson correlation, and let 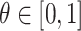 be a threshold. The stability measure SMA-Count [65] is defined as
be a threshold. The stability measure SMA-Count [65] is defined as
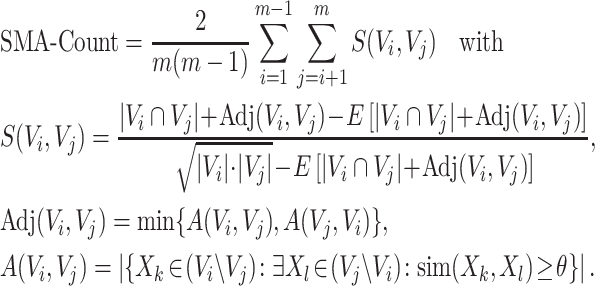 |
(21) |
The term 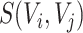 measures the similarity of the two sets
measures the similarity of the two sets  and
and  . It takes into account features that are included in both sets (
. It takes into account features that are included in both sets (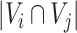 ) as well as features that are not included in both sets but that are very similar to at least one feature in the other set (
) as well as features that are not included in both sets but that are very similar to at least one feature in the other set (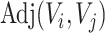 ). The maximum value of SMA-Count is 1 and indicates a perfectly stable feature selection. The stability measure SMA-Count is suitable for data sets that contain highly correlated features such as gene expression data [41].
). The maximum value of SMA-Count is 1 and indicates a perfectly stable feature selection. The stability measure SMA-Count is suitable for data sets that contain highly correlated features such as gene expression data [41].
3 Experiments
3.1 Data sets and software
For our benchmark experiments, we use 11 high-dimensional survival data sets. All of them contain gene expression data from cancer patients and right-censored survival outcomes. The data sets are taken from [66]. We select data sets that have at least 50 events and we only use the features that provide RNA and miRNA data. The data sets thus do not contain clinical features. An overview of the considered data sets is displayed in Table 2. The variance filter is applicable to all of these data sets because the features are measured on the same scale and have not been scaled to unit variance.
Table 2.
Information about the data sets: number of observations ( ), number of features (
), number of features (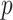 ), number of events (
), number of events (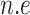 ), and relative number of events (
), and relative number of events (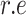 )
)
| Data set |
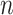
|
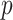
|
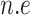
|
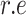
|
|---|---|---|---|---|
| BLCA | 382 | 23 906 | 103 | 0.27 |
| BRCA | 735 | 23 531 | 72 | 0.10 |
| HNSC | 443 | 22 313 | 152 | 0.34 |
| KIRC | 249 | 23 697 | 62 | 0.25 |
| LGG | 419 | 22 942 | 77 | 0.18 |
| LUAD | 426 | 24 480 | 101 | 0.24 |
| LUSC | 418 | 24 419 | 132 | 0.32 |
| OV | 219 | 25 483 | 109 | 0.50 |
| PAAD | 124 | 22 960 | 52 | 0.42 |
| SKCM | 249 | 23 250 | 87 | 0.35 |
| STAD | 295 | 26 814 | 62 | 0.21 |
The benchmark is conducted using the software R [67] and the machine learning packages mlr3 [42], mlr3filters [60], mlr3proba [68], mlr3learners [69] and mlr3pipelines [70]. Moreover, the filter implementations are based on the R packages carSurv [62], ranger [63], xgboost [64] and praznik [55], see Table 1. The R package survival [61] is employed for fitting unregularized Cox regression models and computing martingale residuals while glmnet [71] is used for fitting 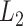 -regularized Cox regression models. The experiments are rolled out on a high-performance compute cluster using the R package batchtools [72]. The feature selection stability is computed with the R package batchtools [73]. For analyzing the results, the R package ggplot2 [74] is used. The R source code for all analyses presented in this article is publicly available at https://github.com/bommert/survival-filter-benchmark.
-regularized Cox regression models. The experiments are rolled out on a high-performance compute cluster using the R package batchtools [72]. The feature selection stability is computed with the R package batchtools [73]. For analyzing the results, the R package ggplot2 [74] is used. The R source code for all analyses presented in this article is publicly available at https://github.com/bommert/survival-filter-benchmark.
3.2 Similarity of feature rankings
In this first part of the analyses, the similarity of the filter methods is assessed. The aim is to identify groups of filter methods that rank the features in a similar way.
3.2.1 Experimental setup
The 14 filter methods described in the Methods section are applied to the 11 data sets presented in the previous subsection. Each filter is used to rank all features of each data set. Then, for each data set, the rankings of all filter methods are compared. This is done in the following way: Let 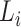 and
and 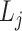 denote the lists of features ordered by the rankings of filters
denote the lists of features ordered by the rankings of filters 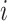 and
and 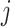 , respectively. That is, the first entry of list
, respectively. That is, the first entry of list 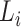 is the most important feature according to filter method
is the most important feature according to filter method 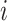 and the last entry of
and the last entry of 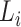 is the least important feature according to filter method
is the least important feature according to filter method 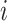 . Let
. Let 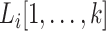 denote the sublist that contains the first
denote the sublist that contains the first 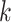 entries of
entries of 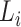 . Then,
. Then, 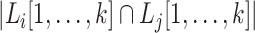 is the number of features that are among the first
is the number of features that are among the first 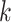 features in both lists. Based on theses numbers, the ordered list (OL) score is calculated [75]:
features in both lists. Based on theses numbers, the ordered list (OL) score is calculated [75]:
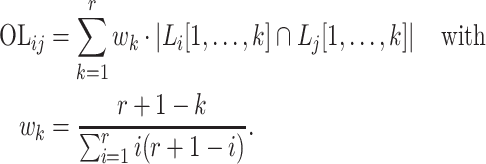 |
(22) |
This score is a weighted sum of the numbers of features included in both lists, giving more importance to features placed in the first positions. The weights are linearly decreasing with the lengths of the considered sublists and scaled such that the maximum value of the OL score is 1. The parameter 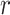 determines the maximal lengths of the sublists. The OL score is taken from [75], but we use linearly decreasing weights instead of exponentially decreasing weights. The reason is that with exponentially decreasing weights, the OL scores are mostly determined by the first few positions of the sublists, giving almost no relevance to the rest of the sublists.
determines the maximal lengths of the sublists. The OL score is taken from [75], but we use linearly decreasing weights instead of exponentially decreasing weights. The reason is that with exponentially decreasing weights, the OL scores are mostly determined by the first few positions of the sublists, giving almost no relevance to the rest of the sublists.
3.2.2 Results
In order to assess the similarity of the filter methods, for each data set, OL scores are computed for all pairs of filter methods. Then, the scores are aggregated across data sets with the arithmetic mean. For the computation of the OL scores, only the 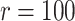 top ranked features are considered. Figure 1 displays the mean OL scores. Three groups of similar filter methods can be identified. The first group consists of the mutual information filters mim, cmim and mrmr. The second ground is formed by the remaining mutual information filters njmim, jmim, jmi and disr. The third group consists of the filters cox.score and correlation. There exist weak similarities between the two groups of mutual information filters as well as between the two random forest feature importance filters permutation and impurity. The other filter methods are not similar in the way they rank the top features.
top ranked features are considered. Figure 1 displays the mean OL scores. Three groups of similar filter methods can be identified. The first group consists of the mutual information filters mim, cmim and mrmr. The second ground is formed by the remaining mutual information filters njmim, jmim, jmi and disr. The third group consists of the filters cox.score and correlation. There exist weak similarities between the two groups of mutual information filters as well as between the two random forest feature importance filters permutation and impurity. The other filter methods are not similar in the way they rank the top features.
Figure 1 .
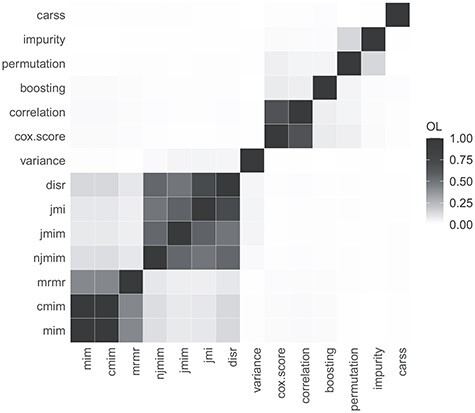
Mean OL scores between the filter methods averaged across data sets. The filter methods are ordered based on single linkage clustering.
It is plausible that the mutual information filters yield similar feature rankings because they use similar concepts for score calculation and they categorize the features in the same way. Especially for the second group of mutual information filters, the high similarity values make sense because all of these filters rank the features iteratively with respect to joint mutual information terms. Considering the filters mim and cmim, it is rather surprising that they are so highly similar, because mim is a univariate filter method and cmim iteratively assesses conditional mutual information terms based on the already selected features. A possible reason for this similarity could be that the features do not provide much information about the target variable, making the conditional mutual information term in 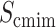 take on similar values as the mutual information term in
take on similar values as the mutual information term in 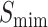 . Regarding the filter methods cox.score and correlation, their similarity can be explained by both filters being indicators of univariate associations between the features and the target.
. Regarding the filter methods cox.score and correlation, their similarity can be explained by both filters being indicators of univariate associations between the features and the target.
3.3 Comparison of the performance
3.3.1 Experimental setup
The aim of this analysis is to identify the best filter methods with respect to predictive performance and to run time. To assess the predictive performance of the filter method, each filter method is combined with  -regularized Cox regression [45] such that the filter is applied first and the Cox model is trained only on the features selected by the filter. We choose
-regularized Cox regression [45] such that the filter is applied first and the Cox model is trained only on the features selected by the filter. We choose 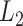 -regularized Cox regression because it is a frequently used regression technique that is known to perform well [76] and it does not perform embedded feature selection. The latter aspect is important for judging the predictive quality of the entire set of selected features.
-regularized Cox regression because it is a frequently used regression technique that is known to perform well [76] and it does not perform embedded feature selection. The latter aspect is important for judging the predictive quality of the entire set of selected features.
Each filter method has one hyper parameter, prop, indicating the proportion of features to be selected by the filter. To tune this hyper parameter, we perform a grid search. We consider the 100 equidistant values 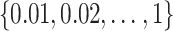 and transform them with
and transform them with 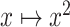 , focusing more on small proportions of selected features. We conduct nested cross-validation [77] with 10 inner and 10 outer iterations (see Figure 2 for a visualization). Both the inner and the outer cross-validation splits are stratified based on the event indicator
, focusing more on small proportions of selected features. We conduct nested cross-validation [77] with 10 inner and 10 outer iterations (see Figure 2 for a visualization). Both the inner and the outer cross-validation splits are stratified based on the event indicator 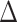 . For each filter method and data set, in each outer iteration, we consider the best prop-value based on the inner cross-validation. For choosing the best prop-value, the filter method ranks the features on the inner training data sets. For each of the 100 prop-values, the best
. For each filter method and data set, in each outer iteration, we consider the best prop-value based on the inner cross-validation. For choosing the best prop-value, the filter method ranks the features on the inner training data sets. For each of the 100 prop-values, the best 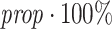 of the features are used to train a
of the features are used to train a 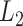 -regularized Cox regression model on the inner training data sets. Then, the 10 Cox models are evaluated with respect to predictive performance on the respective test data sets. Based on the mean predictive performance, the best of the 100 prop-values is chosen. The next step is to evaluate the performance of the filter method on the validation data set. For this, the filter is applied on the outer training data set, selecting
-regularized Cox regression model on the inner training data sets. Then, the 10 Cox models are evaluated with respect to predictive performance on the respective test data sets. Based on the mean predictive performance, the best of the 100 prop-values is chosen. The next step is to evaluate the performance of the filter method on the validation data set. For this, the filter is applied on the outer training data set, selecting 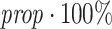 of the features (with prop set to the chosen value). Then, a
of the features (with prop set to the chosen value). Then, a  -regularized Cox model is fitted on the selected features of the outer training data set. The time for filtering and model fitting on the outer training data set as well as the time for predicting on the validation data set is recorded. Also, the predictive performance on the validation data set is assessed. This procedure is repeated for all 10 outer cross-validation iterations, resulting in 10 values per filter method, data set, and performance criterion.
-regularized Cox model is fitted on the selected features of the outer training data set. The time for filtering and model fitting on the outer training data set as well as the time for predicting on the validation data set is recorded. Also, the predictive performance on the validation data set is assessed. This procedure is repeated for all 10 outer cross-validation iterations, resulting in 10 values per filter method, data set, and performance criterion.
Figure 2 .

Visualization of nested cross-validation.
In addition to the 14 filter methods, we also consider the approach of not applying any filter before fitting the 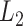 -regularized Cox model, which serves as a baseline. For this approach, there exists no hyper parameter prop that would require tuning. Therefore the
-regularized Cox model, which serves as a baseline. For this approach, there exists no hyper parameter prop that would require tuning. Therefore the 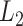 -regularized Cox model is directly fitted on the outer training data sets, using all features, and it is evaluated as described above. Moreover, we also include the simple Kaplan–Meier estimator of the survival function [44] as a baseline. The Kaplan–Meier estimator does not consider any of the features.
-regularized Cox model is directly fitted on the outer training data sets, using all features, and it is evaluated as described above. Moreover, we also include the simple Kaplan–Meier estimator of the survival function [44] as a baseline. The Kaplan–Meier estimator does not consider any of the features.
The hyper parameter of 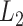 -regularized Cox regression that balances the goodness of the fit and the size of the regression parameters is chosen automatically by [71] using 10-fold cross-validation. The predictive performance is evaluated with the integrated Brier score. To ensure a fair comparison of the filter methods, the same inner and outer cross-validation splits are used for all filter methods.
-regularized Cox regression that balances the goodness of the fit and the size of the regression parameters is chosen automatically by [71] using 10-fold cross-validation. The predictive performance is evaluated with the integrated Brier score. To ensure a fair comparison of the filter methods, the same inner and outer cross-validation splits are used for all filter methods.
3.3.2 Results
First, the filter methods are compared with respect to predictive performance. Figure 3 shows the integrated Brier score values of the best configurations of the 10 outer cross-validation iterations separately for each data set. It can be observed that for all data sets, there are some filter methods that lead to considerably better results than other filter methods. However, many of the boxplots overlap. For most data sets, applying the filter methods variance or carss before fitting the 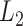 -regularized Cox model, results in models with high predictive accuracy. Applying no filter before fitting the
-regularized Cox model, results in models with high predictive accuracy. Applying no filter before fitting the 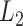 -regularized Cox model leads to comparably bad results on most data sets. The variation of the results between the 10 outer cross-validation iterations is rather small for the filter methods that achieve good results. In comparison to the simple Kaplan–Meier estimator, no gain in predictive performance is achieved with filtering and
-regularized Cox model leads to comparably bad results on most data sets. The variation of the results between the 10 outer cross-validation iterations is rather small for the filter methods that achieve good results. In comparison to the simple Kaplan–Meier estimator, no gain in predictive performance is achieved with filtering and 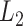 -regularized Cox regression.
-regularized Cox regression.
Figure 3 .
Boxplots of the integrated Brier score of the best configurations of the 10 outer cross-validation iterations per data set. Each boxplot represents 10 performance values. Small values indicate a good predictive performance.
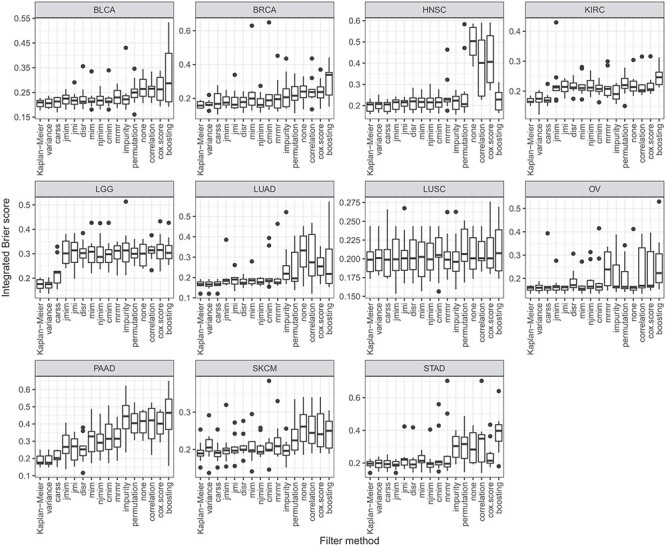
Figure 4 provides an aggregation of the predictive performances of the filter methods over all data sets. In order to obtain one performance value per filter method and data set, the mean integrated Brier score of the 10 outer cross-validation iterations is considered. Then, for each pair of filter methods, it is investigated, on how many data sets one filter outperforms the other. In Figure 4, the number displayed in the row of filter A and in the column of filter B indicates the number of data sets on which filter A achieves a lower mean integrated Brier score than filter B. Filters carss and variance achieve lower mean integrated Brier scores than most other filter methods on all data sets. They perform better than boosting, cox.score, correlation, permutation, mrmr and cmim as well as applying no filter on all of the 11 considered data sets. In comparison to the filters impurity, mim, njmim, disr, jmim and jmi, the carss and variance filters resulted in a better performance on at least 8 of the data sets. Filter carss wins against filter variance on 5 data sets and loses on 6 data sets. The simple Kaplan–Meier estimator outperforms all other approaches on at least 9 of the 11 data sets, however, compared to the filters variance and carss, almost always only by a very small margin.
Figure 4 .
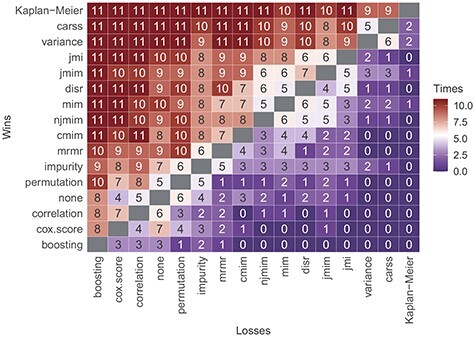
Number of data sets on which the filter method displayed in the row achieves a lower mean integrated Brier score (corresponding to a better performance) than the filter method displayed in the column. The filter methods are ordered by the sums of the rows, that is, the total number of wins against any of the other filter methods on any of the 11 data sets.
In the next step, we jointly consider the run time and the predictive performance of the filters when combined with a 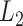 -regularized Cox regression model. Ideally, a filter method that provides low prediction errors and is fast to compute is desired. Figure 5 presents an aggregation of both performance criteria over the 11 data sets. The sizes of the considered data sets are different (see Table 2) and also the difficulties of the survival prediction tasks differ between the data sets (see Figure 3). This makes it necessary to scale both performance criteria before they can be aggregated across data sets. Regarding the prediction error, for each data set, we subtract the best observed mean integrated Brier score from all mean integrated Brier scores. The best filter method per data set therefore has ‘relative mean integrated Brier score’ 0. A relative mean integrated Brier score of
-regularized Cox regression model. Ideally, a filter method that provides low prediction errors and is fast to compute is desired. Figure 5 presents an aggregation of both performance criteria over the 11 data sets. The sizes of the considered data sets are different (see Table 2) and also the difficulties of the survival prediction tasks differ between the data sets (see Figure 3). This makes it necessary to scale both performance criteria before they can be aggregated across data sets. Regarding the prediction error, for each data set, we subtract the best observed mean integrated Brier score from all mean integrated Brier scores. The best filter method per data set therefore has ‘relative mean integrated Brier score’ 0. A relative mean integrated Brier score of 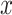 means that the predictive performance of a filter method is worse by the additive factor
means that the predictive performance of a filter method is worse by the additive factor 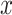 compared to the best filter method on the same data set. Regarding the run times, we consider the (base 10) logarithmic median run time of the 10 outer cross-validation iterations for each filter and data set. We subtract the fastest logarithmic median run time per data set from all logarithmic median run times measured on the same data set. A ‘relative logarithmic median run time’ of
compared to the best filter method on the same data set. Regarding the run times, we consider the (base 10) logarithmic median run time of the 10 outer cross-validation iterations for each filter and data set. We subtract the fastest logarithmic median run time per data set from all logarithmic median run times measured on the same data set. A ‘relative logarithmic median run time’ of 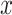 means that the median run time of a filter equals the median run time for the fastest filter on the same data set multiplied with
means that the median run time of a filter equals the median run time for the fastest filter on the same data set multiplied with 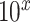 . The scaled performance criteria are displayed in Figure 5 in the following way: The median of both criteria across the 11 data sets is displayed by a symbol. Horizontal and vertical lines reaching from the minimum to the maximum value of the respective performance criterion observed on any of the 11 data sets are added. The symbol represents the central location of the performance measures while the lines indicate the spread across data sets. The Kaplan–Meier estimator requires the least run time among all considered approaches as it does not take into account the features. Also, it provides the highest predictive accuracy on most of the data sets. Among the filter methods, looking at the symbols, it can be seen that filter variance outperforms all other filter methods as well as applying no filter. It obtains a better median relative mean integrated Brier score and a better median relative logarithmic median run time than all other filter methods. Filter carss also provides a very small median relative mean integrated Brier score but requires more time for calculation. The filters jmim, njmim, mim, cmim and mrmr are comparably fast to compute but achieve a noticeably lower predictive performance.
. The scaled performance criteria are displayed in Figure 5 in the following way: The median of both criteria across the 11 data sets is displayed by a symbol. Horizontal and vertical lines reaching from the minimum to the maximum value of the respective performance criterion observed on any of the 11 data sets are added. The symbol represents the central location of the performance measures while the lines indicate the spread across data sets. The Kaplan–Meier estimator requires the least run time among all considered approaches as it does not take into account the features. Also, it provides the highest predictive accuracy on most of the data sets. Among the filter methods, looking at the symbols, it can be seen that filter variance outperforms all other filter methods as well as applying no filter. It obtains a better median relative mean integrated Brier score and a better median relative logarithmic median run time than all other filter methods. Filter carss also provides a very small median relative mean integrated Brier score but requires more time for calculation. The filters jmim, njmim, mim, cmim and mrmr are comparably fast to compute but achieve a noticeably lower predictive performance.
Figure 5 .
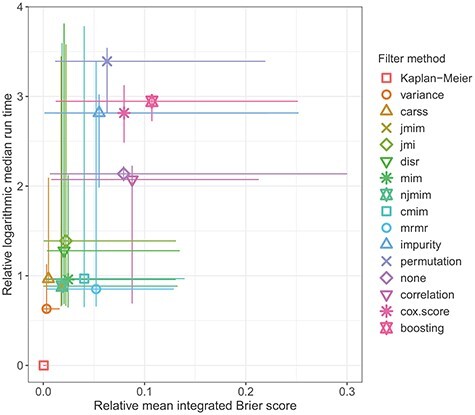
Relative mean integrated Brier score and relative logarithmic median run time. A symbol indicates the median across the 11 data sets. The lines span from the minimum to the maximum value observed for any of the data sets. Small values are desirable for both criteria.
In the next step, the feature selection of the filter methods is analyzed in more detail. Figure 6 displays the proportion of selected features per filter method for the best configurations. Remember that for each filter method, the proportion of features to select is optimized based on the predictive performance of the subsequent 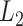 -regularized Cox model. The boxplots in Figure 6 represent the proportions of selected features in the 10 outer cross-validation iterations for each of the 11 data sets. The filter methods are sorted by predictive performance. For the filter methods that lead to a good predictive performance, comparably few features are selected for all data sets. This means that it is sufficient to include only a small number of features in the survival models for obtaining high predictive accuracy. Fitting models based on only a small number of features has the advantage of a faster run time and lower memory consumption as well as an easier interpretation of the
-regularized Cox model. The boxplots in Figure 6 represent the proportions of selected features in the 10 outer cross-validation iterations for each of the 11 data sets. The filter methods are sorted by predictive performance. For the filter methods that lead to a good predictive performance, comparably few features are selected for all data sets. This means that it is sufficient to include only a small number of features in the survival models for obtaining high predictive accuracy. Fitting models based on only a small number of features has the advantage of a faster run time and lower memory consumption as well as an easier interpretation of the 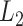 -regularized Cox model. The comparably bad predictive performance of applying no filter before fitting the
-regularized Cox model. The comparably bad predictive performance of applying no filter before fitting the 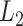 -regularized Cox model shows that it is not only sufficient but also necessary to filter out many of the features in the considered data sets. The filter methods with low predictive accuracy select comparably many features. This means that they fail at selecting only the few important features for target prediction.
-regularized Cox model shows that it is not only sufficient but also necessary to filter out many of the features in the considered data sets. The filter methods with low predictive accuracy select comparably many features. This means that they fail at selecting only the few important features for target prediction.
Figure 6 .
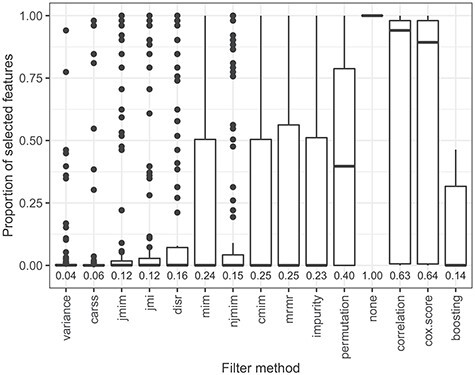
Boxplots of the proportion of selected features of the best configurations of the 10 outer cross-validation iterations for all data sets. Each boxplot represents 110 proportion values. The mean proportions of selected features are indicated by numbers.
In Figure 7, the feature selection stability of the filter methods is assessed. For each filter method and each outer iteration of each data set, the following is done: the 10 sets of features that were selected by the filter method during the inner cross-validation iterations with the best prop-value are considered. The value of the stability measure SMA-Count (see Equation 21) is calculated, using the absolute Pearson correlation between the features as a measure of feature similarity and the similarity threshold 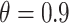 , which implies a strong association. Large stability values are desirable, as they indicate a consistent choice of features irrespective of some variation in the data set. Figure 7 shows that the feature selection conducted by filter variance is by far the most stable among all considered filter methods. The other filter methods provide a much less stable feature selection. Filter carss is the second best filter method with respect to feature selection stability.
, which implies a strong association. Large stability values are desirable, as they indicate a consistent choice of features irrespective of some variation in the data set. Figure 7 shows that the feature selection conducted by filter variance is by far the most stable among all considered filter methods. The other filter methods provide a much less stable feature selection. Filter carss is the second best filter method with respect to feature selection stability.
Figure 7 .
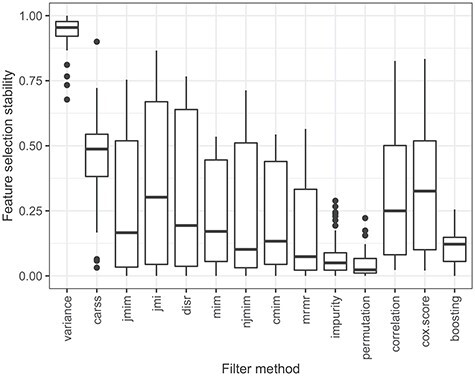
Boxplots of the feature selection stability of the best configurations of the 10 outer cross-validation iterations for all data sets. Each boxplot represents 110 stability values assessed with the stability measure SMA-Count. Large values are desirable.
Finally, we compare the results of the similarity analyses and the performance analyses. The groups of similar filter methods identified on the basis of Figure 1 also provides a similar predictive accuracy. They are positioned next to each other in the ranking in Figure 4 and they obtain similar scaled predictive performance values in Figure 5. Furthermore, the groups of similar filters also select similar proportions of features (see Figure 6) with a similar feature selection stability (see Figure 7) for the respective optimal prop-values. All in all, the results of the two analyses are consistent.
4 Conclusion
Feature selection is a fundamental problem in statistical research, especially for the analysis of high-dimensional biomedical data sets. It is often advantageous with respect to predictive performance, run time and interpretability to disregard the irrelevant and redundant features. Filter methods are a popular class of feature selection methods because they are fast to compute and can be combined with any subsequent predictive model.
We considered gene expression survival data, which is a typical example of high-dimensional, censored data used in bioinformatics. Existing benchmark studies of filter methods are conducted on the basis of classification or regression data sets. But commonly used regression or classification techniques are not suitable for survival data sets, because they cannot take into account the censoring information. Since censored survival data are frequently used in bioinformatics and since filter methods for survival data had not been thoroughly benchmarked before, we conducted such a benchmark for survival data.
We compared 14 filter methods based on 11 high-dimensional survival data sets containing gene expression data. The data sets were chosen as a subset of the data sets used in [66] based on the number of events. This choice of data sets can be seen as unbiased. First, we analyzed the orders in which the filter methods rank the top 100 features, identifying groups of similar filter methods. Filter methods that were classified as similar often used similar concepts such as mutual information.
Next, we compared the filter methods with respect to predictive performance when combined with a 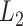 -regularized Cox proportional hazards model. We also included the approach of not applying a filter method before fitting the
-regularized Cox proportional hazards model. We also included the approach of not applying a filter method before fitting the 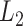 -regularized Cox model in our analyses. We could conclude that the filter methods variance and carss perform best with respect to the integrated Brier score on all considered data sets. Filter variance ranks the features based on their variance, independent of the survival outcome, while filter carss computes correlation-adjusted regression survival scores. In comparison to fitting
-regularized Cox model in our analyses. We could conclude that the filter methods variance and carss perform best with respect to the integrated Brier score on all considered data sets. Filter variance ranks the features based on their variance, independent of the survival outcome, while filter carss computes correlation-adjusted regression survival scores. In comparison to fitting 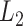 -reqularized Cox models with all features, models with much better predictive accuracy were obtained when first applying one of these filters. When considering both the predictive performance and the run time for applying the filter, fitting the model and prediction and then aggregating these performance criteria across data sets, filter variance outperformed all other approaches.
-reqularized Cox models with all features, models with much better predictive accuracy were obtained when first applying one of these filters. When considering both the predictive performance and the run time for applying the filter, fitting the model and prediction and then aggregating these performance criteria across data sets, filter variance outperformed all other approaches.
Next, we analyzed the feature selection of the filter methods. We observed that all filter methods that lead to models with high predictive accuracy only select a small number of features on all data sets. An analysis of the feature selection stability of the filter methods showed that filter variance provides by far the most consistent sets of selected features.
Comparing the results of the similarity and the performance analysis, we found them to be in accordance. We observed that the filter methods that were categorized as similar based on their feature rankings also achieved a similar predictive performance and selected a similar number of features with a similar feature selection stability.
Based on our extensive analyses, we recommend using the simple variance filter before fitting a Cox regression model on a high-dimensional gene expression survival data set. This filter method allowed fitting models with the best predictive performance, required the least run time for filter score calculation, model fitting and prediction and at the same time produced the most stable results. When a more elaborate filter method is desired, we recommend the carss filter. This filter achieved a comparable predictive accuracy, required a bit more run time and ranked second with respect to feature selection stability.
Both the variance and the carss filter do not have any hyper parameters (other than the proportion of features to select) that require tuning or a robust choice. This is an advantage over filter methods that are based on feature importance values calculated from models like random forest or boosting. In our benchmark study, we did not tune the hyper parameters of random forest or boosting. Tuning them could have led to an increase in predictive accuracy for the respective filter methods. This, however, would have come at the expense of a large increase in run time and the run time of these filters already was the longest among all considered filter methods.
In comparison to the considered mutual information filters, for both the variance and the carss filter, no categorization of the survival outcome and features, resulting in a loss of information, is required. An advantage of the variance filter over the carss filter is that it allows unbiased testing in a subsequent unregularized model, if the proportion of features to select is prespecified. This is because the variance filter does not take into account the survival outcome for feature selection, see also [78]. A disadvantage of the variance filter is that it requires that all features are measured on the same scale, because the feature variance is not scale invariant. On data sets with features that are measured on strongly differing scales, the variance filter will likely be misguided. Also, the features must not have been scaled to unit variance during preprocessing. The carss filter does not have these limitations.
In conclusion, our results suggest that the filters variance and carss are a well-performing and convenient tool to eliminate overfitting caused by the high dimensionality of the gene expression data. However, when comparing the integrated Brier score values of the 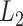 -regularized Cox regression models based on the features selected by the best-performing filters to the respective integrated Brier score values of the non-informative Kaplan–Meier estimator, it must be pointed out that the former did not show a clear tendency to outperform the latter. This result is in line with an earlier comparison study by Herrmann et al. [66] based on the same data sets, who noted that ‘in general, conclusions about the superiority of one method over the other with respect to the prediction performance must be drawn with caution, as the differences in performance can be very small and the confidence intervals often show a remarkable overlap’ [66]. Furthermore, this finding highlights the importance of the inclusion of clinical (non-genetic) features such as age, sex and disease stage in survival prediction models. Adding clinical covariates to the genetic features could improve the survival predictive performance over the covariate-free Kaplan–Meier estimator. In our study, we only included genetic features in the covariate sets, since these are high-dimensional and subject to filtering, and since we aimed at investigating the performance of filter methods.
-regularized Cox regression models based on the features selected by the best-performing filters to the respective integrated Brier score values of the non-informative Kaplan–Meier estimator, it must be pointed out that the former did not show a clear tendency to outperform the latter. This result is in line with an earlier comparison study by Herrmann et al. [66] based on the same data sets, who noted that ‘in general, conclusions about the superiority of one method over the other with respect to the prediction performance must be drawn with caution, as the differences in performance can be very small and the confidence intervals often show a remarkable overlap’ [66]. Furthermore, this finding highlights the importance of the inclusion of clinical (non-genetic) features such as age, sex and disease stage in survival prediction models. Adding clinical covariates to the genetic features could improve the survival predictive performance over the covariate-free Kaplan–Meier estimator. In our study, we only included genetic features in the covariate sets, since these are high-dimensional and subject to filtering, and since we aimed at investigating the performance of filter methods.
Many of the filter methods investigated in this benchmark study are analyzed in [38], too. But there, classification data sets from various domains were used instead of gene expression survival data sets. In [38], especially random forest feature importance filters and mutual information filters performed well with respect to predictive accuracy and run time. The carss filter was not analyzed, since it is not suitable for classification data. With the variance filter, comparably poor results were obtained. Comparing the findings of this benchmark study with the results of [38], it becomes obvious that the best filter methods differ for different types of data. This stresses the importance of having conducted this benchmark study.
For future analyses one could consider additional data sets from different domains. Also, one could compare the filter methods in simulation studies. In contrast to real data, this would allow assessing whether the filter methods select the features that were used for generating the target. Moreover, one could consider further survival prediction models.
Key Points
On the considered high-dimensional gene expression survival data sets, it is beneficial to first apply a filter method before fitting a
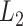 -regularized Cox proportional hazards model in order to achieve high predictive accuracy.
-regularized Cox proportional hazards model in order to achieve high predictive accuracy.The simple variance filter outperforms all other considered filter methods with respect to the predictive accuracy of a subsequent
 -regularized Cox proportional hazards model, the run time and the feature selection stability. This filter selects the features with the largest variance and does not take into account the survival outcome.
-regularized Cox proportional hazards model, the run time and the feature selection stability. This filter selects the features with the largest variance and does not take into account the survival outcome.The correlation-adjusted regression scores filter is a more elaborate alternative to the variance filter that allows fitting models with similar predictive accuracy.
Groups of similar filter methods can be identified based on feature rankings. Filter methods of the same group achieve a similar predictive performance and select a similar number of features with a similar feature selection stability.
Acknowledgments
This work was supported by German Research Foundation (Deutsche Forschungsgemeinschaft, DFG), Collaborative Research Center SFB 876, A3 and German Research Foundation (Deutsche Forschungsgemeinschaft, DFG), grant number SCHM 2966/2-1. We acknowledge the computing time provided on the Linux HPC cluster at TU Dortmund University (LiDO3), partially funded in the course of the Large-Scale Equipment Initiative by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) as Project 271512359.
Andrea Bommert is a postdoctoral fellow at the Department of Statistics, TU Dortmund University, Germany.
Thomas Welchowski is a postdoctoral fellow at the Institute of Medical Biometry, Informatics and Epidemiology (IMBIE), Medical Faculty, University of Bonn, Germany.
Matthias Schmid is a full professor at the Institute of Medical Biometry, Informatics and Epidemiology (IMBIE), Medical Faculty, University of Bonn, Germany.
Jörg Rahnenführer is a full professor at the Department of Statistics, TU Dortmund University, Germany.
Contributor Information
Andrea Bommert, Department of Statistics, TU Dortmund University, Vogelpothsweg 87, 44227, Dortmund, Germany.
Thomas Welchowski, Institute of Medical Biometry, Informatics and Epidemiology (IMBIE), Medical Faculty, University of Bonn, Venusberg-Campus 1, 53127, Bonn, Germany.
Matthias Schmid, Institute of Medical Biometry, Informatics and Epidemiology (IMBIE), Medical Faculty, University of Bonn, Venusberg-Campus 1, 53127, Bonn, Germany.
Jörg Rahnenführer, Department of Statistics, TU Dortmund University, Vogelpothsweg 87, 44227, Dortmund, Germany.
Data availability
All analyses are reproducible using the R code publicly available on Github. The data sets were derived from OpenML.
Author contributions statement
A.B. conceived the experiments with input from J.R., T.W. and M.S.; A.B. conducted the experiments; A.B. analyzed the results with input from J.R., T.W. and M.S.; A.B. wrote the manuscript; and J.R., T.W. and M.S. reviewed the manuscript.
References
- 1. Guyon I, Elisseeff A. An introduction to variable and feature selection. Journal of Machine Learning Research 2003;3:1157–82. [Google Scholar]
- 2. Lazar C, Taminau J, Meganck S, et al. A survey on filter techniques for feature selection in gene eexpression microarray analysis. IEEE/ACM Trans Comput Biol Bioinform 2012;9(4):1106–19. [DOI] [PubMed] [Google Scholar]
- 3. Kohavi R, John GH. Wrappers for feature subset selection. Artificial Intelligence 1997;97(1–2):273–324. [Google Scholar]
- 4. Kittler J. Feature Set Search Algorithms. Alphen aan den Rijn, Netherlands: Sijthoff and Noordhoff, 1978, 41–60. [Google Scholar]
- 5. Huang X, Zhang L, Wang B, et al. Feature clustering based support vector machine recursive feature elimination for gene selection. Applied Intelligence 2018;48(3):594–607. [Google Scholar]
- 6. Yang J, Honavar V. Feature Subset Selection Using a Genetic Algorithm. New York, USA: Springer, 1998, 117–36. [Google Scholar]
- 7. Xue B, Zhang M. Will N Browne, and Xin Yao. A survey on evolutionary computation approaches to feature selection. IEEE Transactions on Evolutionary Computation 2016;20(4):606–26. [Google Scholar]
- 8. Brezočnik L, Fister I, Podgorelec V. Swarm intelligence algorithms for feature selection: A review. Applied Sciences 2018;8(9). [Google Scholar]
- 9. Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med 1997;16(4):385–95. [DOI] [PubMed] [Google Scholar]
- 10. Ishwaran H, Kogalur UB, Blackstone EH, et al. Random survival forests. The Annals of Applied Statistics 2008;2(3). [Google Scholar]
- 11. Friedman JH. Greedy function approximation: A gradient boosting machine. Annals of Statistics 2001;29(5):1189–232. [Google Scholar]
- 12. Bühlmann P, Hothorn T. Boosting algorithms: Regularization, prediction and model fitting. Statistical Science 2007;22(4):477–505. [Google Scholar]
- 13. Liu H, Yu L. Toward integrating feature selection algorithms for classification and clustering. IEEE Transactions on Knowledge and Data Engineering 2005;17(4):491–502. [Google Scholar]
- 14. Saeys Y, Inza I, Larrañaga P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007;23(19):2507–17. [DOI] [PubMed] [Google Scholar]
- 15. Chandrashekar G, Sahin F. A survey on feature selection methods. Computers & Electrical Engineering 2014;40(1):16–28. [Google Scholar]
- 16. Tang J, Alelyani S, Liu H. Feature Selection for Classification: A Review, pages 37–64. Boca Raton, FL, USA: CRC Press, 2014. [Google Scholar]
- 17. Hira ZM, Gillies DF. A review of feature selection and feature extraction methods applied on microarray data. Advances in Bioinformatics 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jović A, Brkić K, Bogunović N. A review of feature selection methods with applications. In: 38th International Convention on Information and Communication Technology, Electronics and Microelectronics, 2015, 1200–5.
- 19. Cai J, Luo J, Wang S, et al. Feature selection in machine learning: A new perspective. Neurocomputing 2018;300:70–9. [Google Scholar]
- 20. Li J, Cheng K, Wang S, et al. Feature selection: A data perspective. ACM Computing Surveys 2018;50(6). [Google Scholar]
- 21. Venkatesh B, Anuradha J. A review of feature selection and its methods. Cybernetics and Information Technologies 2019;19(1):3–26. [Google Scholar]
- 22. Liu H, Li J, Wong L. A comparative study on feature selection and classification methods using gene expression profiles and proteomic patterns. Genome Inform 2002;13:51–60. [PubMed] [Google Scholar]
- 23. Bolón-Canedo V, Sánchez-Maroño N, Alonso-Betanzos A. A review of feature selection methods on synthetic data. Knowledge and Information Systems 2013;34(3):483–519. [Google Scholar]
- 24. Bolón-Canedo V, Sánchez-Marono N, Alonso-Betanzos A, et al. A review of microarray datasets and applied feature selection methods. Inform Sci 2014;282:111–35. [Google Scholar]
- 25. Inza I, Larrañaga P, Blanco R, et al. Filter versus wrapper gene selection approaches in dna microarray domains. Artif Intell Med 2004;31(2):91–103. [DOI] [PubMed] [Google Scholar]
- 26. Forman G. An extensive empirical study of feature selection metrics for text classification. Journal of Machine Learning Research 2003;3:1289–305. [Google Scholar]
- 27. Aphinyanaphongs Y, Fu LD, Li Z, et al. A comprehensive empirical comparison of modern supervised classification and feature selection methods for text categorization. J Assoc Inf Sci Technol 2014;65(10):1964–87. [Google Scholar]
- 28. Darshan SLS, Jaidhar CD. Performance evaluation of filter-based feature selection techniques in classifying portable executable files. Procedia Computer Science 2018;125:346–56. [Google Scholar]
- 29. Liu Y. A comparative study on feature selection methods for drug discovery. J Chem Inf Comput Sci 2004;44(5):1823–8. [DOI] [PubMed] [Google Scholar]
- 30. Peng H, Long F, Ding C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell 2005;27(8):1226–38. [DOI] [PubMed] [Google Scholar]
- 31. Dash M, Liu H. Feature selection for classification. Intelligent Data Analysis 1997;1:131–56. [Google Scholar]
- 32. Sánchez-Maroño N, Alonso-Betanzos A, Tombilla-Sanromán M. Filter methods for feature selection – a comparative study. In International Conference on Intelligent Data Engineering and Automated Learning 2007;178–87. [Google Scholar]
- 33. Wah YB, Ibrahim N, Hamid HA, et al. Feature selection methods: Case of filter and wrapper approaches for maximising classification accuracy. Pertanika Journal of Science & Technology 2018;26(1):329–40. [Google Scholar]
- 34. Xue B, Zhang M, Browne WN. A comprehensive comparison on evolutionary feature selection approaches to classification. International Journal of Computational Intelligence and Applications 2015;14(2). [Google Scholar]
- 35. Meyer PE, Schretter C, Bontempi G. Information-theoretic feature selection in microarray data using variable complementarity. IEEE Journal of Selected Topics in Signal Processing 2008;2(3):261–74. [Google Scholar]
- 36. Brown G, Pocock A, Zhao M-J, et al. Conditional likelihood maximisation: A unifying framework for information theoretic feature selection. Journal of Machine Learning Research 2012;13:27–66. [Google Scholar]
- 37. Hall MA. Correlation-Based Feature Selection for Machine Learning. PhD thesis, University of Waikato, Hamilton, New Zealand, 1999. [Google Scholar]
- 38. Bommert A, Sun X, Bischl B, et al. Benchmark for filter methods for feature selection in high-dimensional classification data. Computational Statistics & Data Analysis 2020;143. [Google Scholar]
- 39. Lang M, Kotthaus H, Marwedel P, et al. Automatic model selection for high-dimensional survival analysis. Journal of Statistical Computation and Simulation 2015;85(1):62–76. [Google Scholar]
- 40. Bommert A, Rahnenführer J, Lang M. A multicriteria approach to find predictive and sparse models with stable feature selection for high-dimensional data. Comput Math Methods Med 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bommert A. Integration of Feature Selection Stability in Model Fitting. PhD thesis, TU Dortmund University, Germany, 2020. [Google Scholar]
- 42. Lang M, Binder M, Richter J, et al. ref42: A modern object-oriented machine learning framework in ref67. Journal of Open Source Software 2019. [Google Scholar]
- 43. Kalousis A, Prados J, Hilario M. Stability of feature selection algorithms: A study on high-dimensional spaces. Knowledge and Information Systems 2007;12(1):95–116. [Google Scholar]
- 44. Klein JP, Moeschberger ML. Survival Analysis: Techniques for Censored and Truncated Data. New York, USA: Springer, 2003. [Google Scholar]
- 45. Simon N, Friedman J, Hastie T, et al. Regularization paths for Cox’s proportional hazards model via coordinate descent. J Stat Softw 2011;39(5):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gerds TA, Schumacher M. Consistent estimation of the expected brier score in general survival models with right-censored event times. Biom J 2006;48(6):1029–40. [DOI] [PubMed] [Google Scholar]
- 47. Therneau TM, Grambsch PM, Fleming TR. Martingale-based residuals for survival models. Biometrika 1990;77(1):147–60. [Google Scholar]
- 48. Van der Laan MJ, Robins JM. Unified Methods for Censored Longitudinal Data and Causality. New York, USA: Springer, 2003. [Google Scholar]
- 49. Zuber V, Strimmer K. High-dimensional regression and variable selection using CAR scores. Stat Appl Genet Mol Biol 2011;10(34):2194–6302. [Google Scholar]
- 50. Kessy A, Lewin A, Strimmer K. Optimal whitening and decorrelation. The American Statistician 2018;72(4):309–14. [Google Scholar]
- 51. Schäfer J, Strimmer K. A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat Appl Genet Mol Biol 2005;4(1):1–30. [DOI] [PubMed] [Google Scholar]
- 52. Welchowski T, Zuber V, Schmid M. Correlation-adjusted regression survival scores for high-dimensional variable selection. Stat Med 2019;38(13):2413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harrell FE, Jr, Lee KL, Mark DB. Multivariable prognostic models: Issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med 1996;15(4):361–87. [DOI] [PubMed] [Google Scholar]
- 54. Hastie T, Tibshirani R, Friedman J. The elements of Statistical Learning: Data Mining, Inference, and Prediction. New York, USA: 2 edition: Springer, 2009. [Google Scholar]
- 55. Kursa MB. praznik: Tools for Information-Based Feature Selection, R package version 8.0.0, 2020.
- 56. Yang HH, Moody JE. Data visualization and feature selection: New algorithms for nongaussian data. In: Advances in Neural Information Processing Systems 12 (NIPS 1999), 1999, 687–93.
- 57. Bennasar M, Hicks Y, Setchi R. Feature selection using joint mutual information maximisation. Expert Systems with Applications 2015;42(22):8520–32. [Google Scholar]
- 58. Meyer PE, Bontempi G. On the use of variable complementarity for feature selection in cancer classification. In: EvoWorkshops 2006: Applications of Evolutionary Computing, 2006, 91–102.
- 59. Fleuret F. Fast binary feature selection with conditional mutual information. Journal of Machine Learning Research 2004;5:1531–55. [Google Scholar]
- 60. Schratz P, Lang M, Bischl B, et al. mlr3filters: Filter Based Feature Selection for ‘mlr3’, 2020.
- 61. Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. New York, USA: Springer, 2000. [Google Scholar]
- 62. Welchowski T. carSurv: Correlation-Adjusted Regression Survival (CARS) Scores, 2018, R package version 1.0.0. [DOI] [PMC free article] [PubMed]
- 63. Wright MN, Ziegler A. ref63: A fast implementation of random forests for high dimensional data in C++ and ref67. J Stat Softw 2017;77(1):1–17. [Google Scholar]
- 64. Chen T, He T, Benesty M, et al. xgboost: Extreme Gradient Boosting, 2020, R package version 1.1.1.1.
- 65. Bommert A and Rahnenführer J. Adjusted measures for feature selection stability for data sets with similar features. In Nicosia G, Ojha V, Malfa EL, Jansen G, Sciacca V, Pardalos Panos, Giuffrida G, and Umeton R, editors, Machine Learning, Optimization, and Data Science, pages 203–14, 2020.
- 66. Herrmann M, Probst P, Hornung R, et al. Large-scale benchmark study of survival prediction methods using multi-omics data. Brief Bioinform 2021;22(3):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2020. [Google Scholar]
- 68. Sonabend R, Kiraly F, Lang M. mlr3proba: Probabilistic Supervised Learning for ‘mlr3’, 2020, R package version 0.2.6.
- 69. Lang M, Quay A, Coors S, et al. mlr3learners: Recommended Learners for ‘mlr3’, 2020, R package version 0.4.3.
- 70. Binder M, Pfisterer F, Schneider L, et al. mlr3pipelines: Preprocessing Operators and Pipelines for ‘mlr3’, 2020, R package version 0.3.1.
- 71. Simon N, Friedman J, Hastie T, et al. Regularization paths for Cox’s proportional hazards model via coordinate descent. J Stat Softw 2011;39(5):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bischl B, Lang M, Mersmann O, et al. BatchJobs and BatchExperiments: Abstraction mechanisms for using ref67 in batch environments. J Stat Softw 2015;64(11):1–25. [Google Scholar]
- 73. Bommert A, Lang M. stabm: Stability measures for feature selection. Journal of Open Source Software 2021;6(59):3010. [Google Scholar]
- 74. Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York, USA: Springer, 2016. [Google Scholar]
- 75. Yang X, Scheid S, Lottaz C. OrderedList: Similarities of Ordered Gene Lists, 2020, R package version 1.62.0.
- 76. Bøvelstad HM, Nygärd S, Størvold HL, et al. Predicting survival from microarray data – a comparative study. Bioinformatics 2007;23(16):2080–7. [DOI] [PubMed] [Google Scholar]
- 77. Bischl B, Mersmann O, Trautmann H, et al. Resampling methods for meta-model validation with recommendations for evolutionary computation. Evol Comput 2012;20(2):249–75. [DOI] [PubMed] [Google Scholar]
- 78. Hackstadt AJ, Hess AM. Filtering for increased power for microarray data analysis. BMC Bioinformatics 2009;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All analyses are reproducible using the R code publicly available on Github. The data sets were derived from OpenML.