

ABSTRACT
Whole-genome sequencing (WGS) has recently been used to investigate acquisition of Mycobacterium abscessus. Investigators have reached conflicting conclusions about the meaning of genetic distances for interpretation of person-to-person transmission. Existing genomic studies were limited by a lack of WGS from environmental M. abscessus isolates. In this study, we retrospectively analyzed the core and accessory genomes of 26 M. abscessus subsp. abscessus isolates collected over 7 years. Clinical isolates (n = 22) were obtained from a large hospital-associated outbreak of M. abscessus subsp. abscessus, the outbreak hospital before or after the outbreak, a neighboring hospital, and two outside laboratories. Environmental M. abscessus subsp. abscessus isolates (n = 4) were obtained from outbreak hospital water outlets. Phylogenomic analysis of study isolates revealed three clades with pairwise genetic distances ranging from 0 to 135 single-nucleotide polymorphisms (SNPs). Compared to a reference environmental outbreak isolate, all seven clinical outbreak isolates and the remaining three environmental isolates had highly similar core and accessory genomes, differing by up to 7 SNPs and a median of 1.6% accessory genes, respectively. Although genomic comparisons of 15 nonoutbreak clinical isolates revealed greater heterogeneity, five (33%) isolates had fewer than 20 SNPs compared to the reference environmental isolate, including two unrelated outside laboratory isolates with less than 4% accessory genome variation. Detailed genomic comparisons confirmed environmental acquisition of outbreak isolates of M. abscessus subsp. abscessus. SNP distances alone, however, did not clearly differentiate the mechanism of acquisition of outbreak versus nonoutbreak isolates. We conclude that successful investigation of M. abscessus subsp. abscessus clusters requires molecular and epidemiologic components, ideally complemented by environmental sampling.
KEYWORDS: Mycobacterium abscessus, nontuberculous mycobacteria, whole-genome sequencing, hospital outbreak, infection prevention
INTRODUCTION
Mycobacterium abscessus includes three subspecies (subsp. abscessus, massiliense, and bolletii) of nontuberculous mycobacteria (NTM) that typically cause subacute infections of lungs, skin, and soft tissue (1). Despite long courses of combination antimicrobial therapy and aggressive surgical debridement, many patients do not achieve eradication (2, 3).
The worldwide prevalence of M. abscessus disease is increasing (4–7), which has sparked interest in better understanding how patients acquire this emerging pathogen and how infections might be prevented. Acquisition of M. abscessus and other NTM historically has been thought to occur through independent exposure to soil, air, or water harboring NTM, including colonized health care facility water systems (1, 8). However, recent studies have utilized whole-genome sequencing (WGS) to examine global collections of M. abscessus isolates (9–19), and some analyses have challenged traditional hypotheses on NTM transmission. For example, one large genomic study of M. abscessus isolates obtained from people with cystic fibrosis (CF) revealed that dominant M. abscessus clones found across Europe, Australia, and one U.S. site had isolates that differed by fewer than 20 single-nucleotide polymorphisms (SNPs) in the core genomes (10). Based on these genomic data, investigators concluded that acquisition of M. abscessus likely occurs through person-to-person transmission, perhaps indirectly via fomites and aerosols. A population genomics study of M. abscessus isolates from U.S. CF centers also identified dominant M. abscessus clones and clusters of genetically similar isolates (<20 SNPs) that originated from the same states as well as geographically distant regions (19). Other studies that included both WGS and epidemiologic analyses of M. abscessus clinical isolates concluded that SNP thresholds do not accurately predict transmission and that interhuman transmission of M. abscessus is quite uncommon (15, 17, 18). Each of the preceding studies was limited by the absence of corresponding genomic analysis of environmental M. abscessus isolates.
We previously investigated a large, biphasic outbreak of M. abscessus that occurred from 2013 to 2015 at Duke University Hospital (DUH) in North Carolina (NC) and affected over 100 patients, including more than 50 lung transplant recipients, and greater than 20 cardiac surgery patients (20). The outbreak was epidemiologically linked to a new-hospital-addition water system that was colonized with M. abscessus subsp. abscessus. During the investigation, we performed molecular fingerprinting on clinical isolates and environmental isolates obtained from biofilms of water outlets in the new hospital addition that included multilocus gene sequencing of erm(41) and rpoβ genes (21, 22), variable-number tandem repeat (VNTR) analysis (23), and pulsed-field gel electrophoresis (PFGE) (20). Genetic analyses with non-WGS methods revealed that over 75% of patient isolates and all environmental isolates represented an unusual clone of M. abscessus subsp. abscessus. A small proportion of patient isolates were identified as a clone of M. abscessus subsp. massiliense, but environmental isolates of M. abscessus subsp. massiliense were not recovered. Interventions designed to prevent hospital tap water contact among vulnerable patients mitigated the outbreak, and incidence rates of hospital-associated M. abscessus acquisition returned to baseline (20).
In this follow-up study, we performed WGS of DUH clinical and environmental outbreak isolates of M. abscessus subsp. abscessus that represented the primary outbreak clone. We also analyzed WGS from nonoutbreak clinical isolates of M. abscessus subsp. abscessus obtained at DUH and external hospitals. Nonoutbreak isolates were required to have molecular fingerprinting signatures that matched the outbreak clone. To confirm the molecular fingerprinting results from the original outbreak investigation (20), we first assessed if outbreak hospital patients within the outbreak time period acquired isolates from the same source as environmental isolates. Next, to test whether the outbreak strains predated or remained in the geographic region after the outbreak period, we compared isolates from outbreak hospital patients outside the outbreak time period to isolates from patients within the outbreak period. Finally, to compare isolate genotyping by multilocus gene sequencing versus WGS, we tested whether control isolates from a neighboring hospital and remote outside laboratories (OLs) had genetic distances similar to those of outbreak isolates, as predicted by shared erm(41) and rpoβ gene sequences. We hypothesized that core and accessory genome analyses of this isolate cohort would improve interpretation of genetic distances between M. abscessus subsp. abscessus isolates, understanding of M. abscessus subsp. abscessus acquisition, and strategies for prevention of health care facility-associated (HCFA) NTM infection.
MATERIALS AND METHODS
Study setting.
DUH (“outbreak hospital”) is a 957-bed academic medical center in central North Carolina that utilizes the municipal water supply (surface water). A new hospital addition that included intensive care units, intermediate beds, and operating suites opened for patient care in late July 2013.
The institutional review boards at Duke University, the University of Texas Health Science Center at Tyler (UTHSCT), and National Jewish Health approved this research.
Mycobacterium abscessus subsp. abscessus isolates.
A total of 26 M. abscessus subsp. abscessus isolates meeting specific genetic inclusion criteria were analyzed in the study cohort, including 19 isolates sequenced for this study (Table 1). Inclusion criteria were identification as M. abscessus subsp. abscessus, type VI erm(41) gene (21), and a C→T mutation at base pair 207 in region V of the rpoβ gene (22). A subset of 15 outbreak hospital isolates meeting these criteria were selected for WGS, including clinical (n = 7) and environmental (n = 4) isolates obtained from unique patients and water outlets, respectively, during phase 1 (August 2013 to May 2014) or phase 2 (December 2014 to June 2015) of the outbreak. All outbreak hospital clinical isolates were suspected to have been hospital acquired, and all environmental isolates were obtained from biofilms of water outlets in the hospital addition. Outbreak hospital patient isolates collected before (n = 3) and after (n = 1) the outbreak and presumed to be hospital acquired were also sequenced. Regional controls (n = 2) were clinical isolates obtained from patients who received care at a neighboring hospital in NC with a shared municipal water supply; these patients did not have a history of outbreak hospital contact. Other controls included pulmonary isolates that also met genotypic inclusion criteria and were obtained from two outside laboratories (OLs): the Mycobacteria/Nocardia Research Laboratory at the UTHSCT (n = 2) and previously sequenced isolates from the Colorado CF Research & Development Program (19) (CO-RDP; n = 7). OL isolates were collected from 1 month before to 2 years after the outbreak.
TABLE 1.
Characteristics of clinical (n = 22) and environmental (n = 4) study isolatesa
| Label | Date collected (mo/yr) | Source | Patient underlying condition(s) | Suspected U.S. state of acquisition | Suspected site of acquisition |
|---|---|---|---|---|---|
| P1-E1 | 4/2014 | Utility room water basin | NA | NC | NA |
| P1-E2 | 4/2014 | Patient room faucet | NA | NC | NA |
| P2-E1 | 5/2015 | OR scrub sink | NA | NC | NA |
| P2-E2 | 5/2015 | Patient room faucet | NA | NC | NA |
| P1-C1 | 12/2013 | Sputum | Lung transplant | NC | Outbreak hospital |
| P1-C2 | 12/2013 | Pleural fluid | Esophageal perforation | NC | Outbreak hospital |
| P1-C3 | 2/2014 | BAL fluid | Lung transplant | NC | Outbreak hospital |
| P1-C4 | 4/2014 | Blood | Heart transplant | NC | Outbreak hospital |
| P2-C1 | 2/2015 | RVAD driveline site | RVAD, heart transplant | NC | Outbreak hospital |
| P2-C2 | 3/2015 | Sternal wound | Tricuspid valve replacement | NC | Outbreak hospital |
| P2-C3 | 3/2015 | Pleural fluid | LVAD | NC | Outbreak hospital |
| NH-C1 | 12/2013 | Sputum | COPD, GPA | NC | Community or NH |
| NH-C2 | 1/2016 | Abdominal abscess | PEG tube placement | NC | Community or NH |
| BO-C1 | 8/2010 | BAL fluid | Lung transplant | NC | Outbreak hospital |
| BO-C2 | 6/2012 | BAL fluid | Lung transplant | NC | Outbreak hospital |
| BO-C3 | 5/2013 | BAL fluid | Lung transplant | NC | Outbreak hospital |
| AO-C1 | 3/2017 | BAL fluid | Lung transplant | NC | Outbreak hospital |
| OL1-C1 | 7/2013 | BAL fluid | Bronchiectasis | NC | Community |
| OL1-C2 | 5/2014 | Sputum | Bronchiectasis | LA | Community |
| OL2-C1 | 2015 | Pulmonary | Cystic fibrosis | NCb | Community |
| OL2-C2 | 2016 | Pulmonary | Cystic fibrosis | OHb | Community |
| OL2-C3 | 2016 | Pulmonary | Cystic fibrosis | UTb | Community |
| OL2-C4 | 2017 | Pulmonary | Cystic fibrosis | LAb | Community |
| OL2-C5 | 2017 | Pulmonary | Cystic fibrosis | IAb | Community |
| OL2-C6 | 2017 | Pulmonary | Cystic fibrosis | LAb | Community |
| OL2-C7 | 2017 | Pulmonary | Cystic fibrosis | OHb | Community |
Abbreviations: AO, after outbreak; BAL, bronchoalveolar lavage; BO, before outbreak; C, clinical; COPD, chronic obstructive pulmonary disease; E, environmental; GPA, granulomatosis with polyangiitis; NA, not applicable; NH, neighboring hospital; OL, outside laboratory; P, phase of outbreak; PEG, percutaneous endoscopic gastrostomy; OR, operating room; RVAD, right ventricular assist device.
Patient home state not known and state of facility performing culture given for state of origin (19).
Whole-genome sequencing and SNP analysis.
DNA was extracted from study isolates as previously described (24), and samples were sequenced on the Illumina MiSeq. A selection of publicly available M. abscessus genomes that did not meet genetic inclusion criteria were downloaded from the National Center for Biotechnology Information (NCBI) to include in genomic comparisons. Core genomes of M. abscessus subsp. abscessus isolates in the study cohort (n = 26) (Table 1) and publicly available M. abscessus genomes (n = 25) were analyzed for SNPs using a reference-based mapping approach (13). All isolates in the study cohort were confirmed to have a type VI erm(41) gene and C→T mutation at base pair 207 in region V of the rpoβ gene in the WGS data (Fig. S1). Details are provided in the supplemental material.
Phylogenomic analysis.
Sequence alignments were created from concatenated core genome base calls, and phylogenetic trees were generated with Seaview v4.5.4 (25) using the neighbor-joining method and 100 bootstrap replicates. Phylogenetic tree visualizations were created with ggtree (26). Pairwise distances of core genome SNPs were estimated between all isolate pairs using MEGA7 (27). Statistical comparisons of SNP distances and accessory genes were performed with Mann-Whitney tests in R v3.4.3 (28).
Pangenome analysis.
Draft genomes of study isolates were assembled with Unicycler (29), and pangenome analyses were performed using Prokka (30) and Roary (31) (see Table S1 and details in the methods section of the supplemental material). Core genes are defined as homologous genes shared among all isolates, and accessory genes are present in one or more, but not all, isolates analyzed. Accessory genome comparisons reveal gene content variation, which includes the gain or loss of single genes, genomic islands, prophages, or plasmids (14, 32–34). Accessory genome content between isolate pairs was calculated as percent accessory genes (accessory genes from isolate 1 + accessory genes from isolate 2/sum of total genes from isolate 1 + isolate 2) × 100% (35). Accessory genes for the entire sample set were converted to binary data (1 or 0 for present or absent) and clustered by genes (columns) and samples (rows) using a hierarchical clustering algorithm (hclust) and a Euclidean distance metric in R v3.4.3 (28).
Data availability.
WGS data generated through this study are available at NCBI (PRJNA734909).
RESULTS
Phylogenetic relationships of M. abscessus subsp. abscessus isolates in the study cohort.
Phylogenomic comparisons of isolates in the study cohort versus publicly available M. abscessus genomes without genetic inclusion criteria, including representative isolates of M. abscessus subsp. massiliense and M. abscessus subsp. bolletii (Fig. 1A), confirmed the study isolates as M. abscessus subsp. abscessus. The study isolates clustered into a monophyletic clade that is genetically distinct (>1,800 SNPs) from the previously described dominant circulating clone of M. abscessus subsp. abscessus and the type strain ATCC 19977T (Fig. 1A, yellow box) (10, 14, 19).
FIG 1.
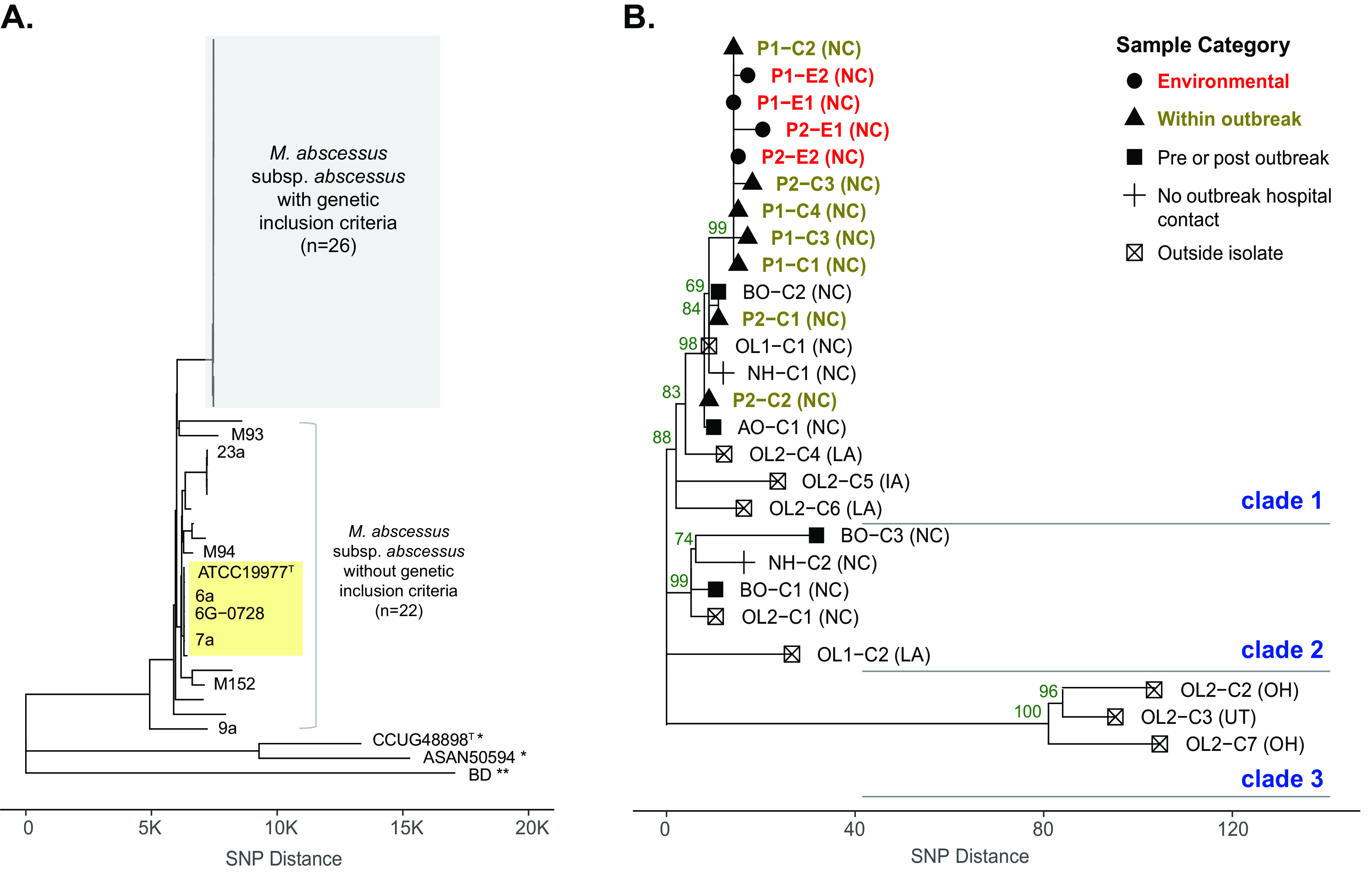
Phylogenomic analysis of study cohort including outbreak and control M. abscessus subsp. abscessus isolates. (A) Genome-wide SNP data from study isolates with genetic inclusion criteria (n = 26) and isolates without genetic inclusion criteria (n = 22) were analyzed using the neighbor-joining (NJ) method. The x axis represents SNP distance. Outgroup isolates of M. abscessus subsp. massiliense (*) and M. abscessus subsp. bolletii (**) were included. The clade with the previously described “dominant circulating clone,” including the M. abscessus subsp. abscessus type strain ATCC 19977T, is labeled in yellow. (B) Study isolates with genetic inclusion criteria were analyzed in detail. Sample categories are designated shapes, and color-coded labels are indicated for environmental (red) and within outbreak (brown) isolates. Bootstrap support values from 100 replicate searches are shown on selected nodes (values of >50), and three genetic clades are labeled. Suspected U.S. state of acquisition is given in parentheses. Abbreviations: AO, after outbreak; BO, before outbreak; C, clinical; E, environmental; NH, neighboring hospital; OL, outside laboratory; P, phase of outbreak.
A detailed phylogenetic analysis of the M. abscessus subsp. abscessus study cohort revealed three clades with pairwise genetic distances ranging from 0 to 135 SNPs (Fig. 1B) out of 4,602,349 core genome positions analyzed. In clade 1, environmental isolates clustered together and were closely related to patient isolates collected during phase 1 and phase 2 of the outbreak. Clade 1 also contained seven more distantly related isolates, including two isolates collected before and after the outbreak period (BO-C2 and AO-C1), one isolate from a neighboring hospital (NH-C1), and four OL isolates that originated in NC (OL1-C1) and outside NC (OL2-C4, OL2-C5, and OL2-C6). Clade 2 included two isolates from before the outbreak period (BO-C1 and BO-C3), one isolate from a neighboring hospital (NH-C2), and two isolates from OLs (OL1-C2 and OL2-C1). Three control isolates from the CO-RDP (OL2-C2, OL2-C3, and OL2-C7) clustered distantly from environmental isolates at >100 SNPs (clade 3).
Clinical isolates in clade 1 that clustered most closely with the environmental isolates had suspected acquisition from NC, including outbreak and control isolates. Two other closely-related OL isolates in clade 1 originated in Louisiana (LA) and Iowa (IA). More distantly related isolates in clades 2 and 3 originated from NC, LA, Ohio (OH), and Utah (UT).
Genomic variation among isolate categories.
To test our study hypotheses, we chose an environmental isolate from phase 1 of the outbreak (P1-E1) as a reference strain to compare the genetic distances of all other isolates in the study cohort. We analyzed genetic distances in terms of SNPs (core genome) and percent accessory genes (accessory genome), and isolates were categorized into five comparison groups: environmental, within outbreak period, pre/postoutbreak period, no outbreak hospital contact (neighboring hospital isolates), and OL isolates (Fig. 2).
FIG 2.
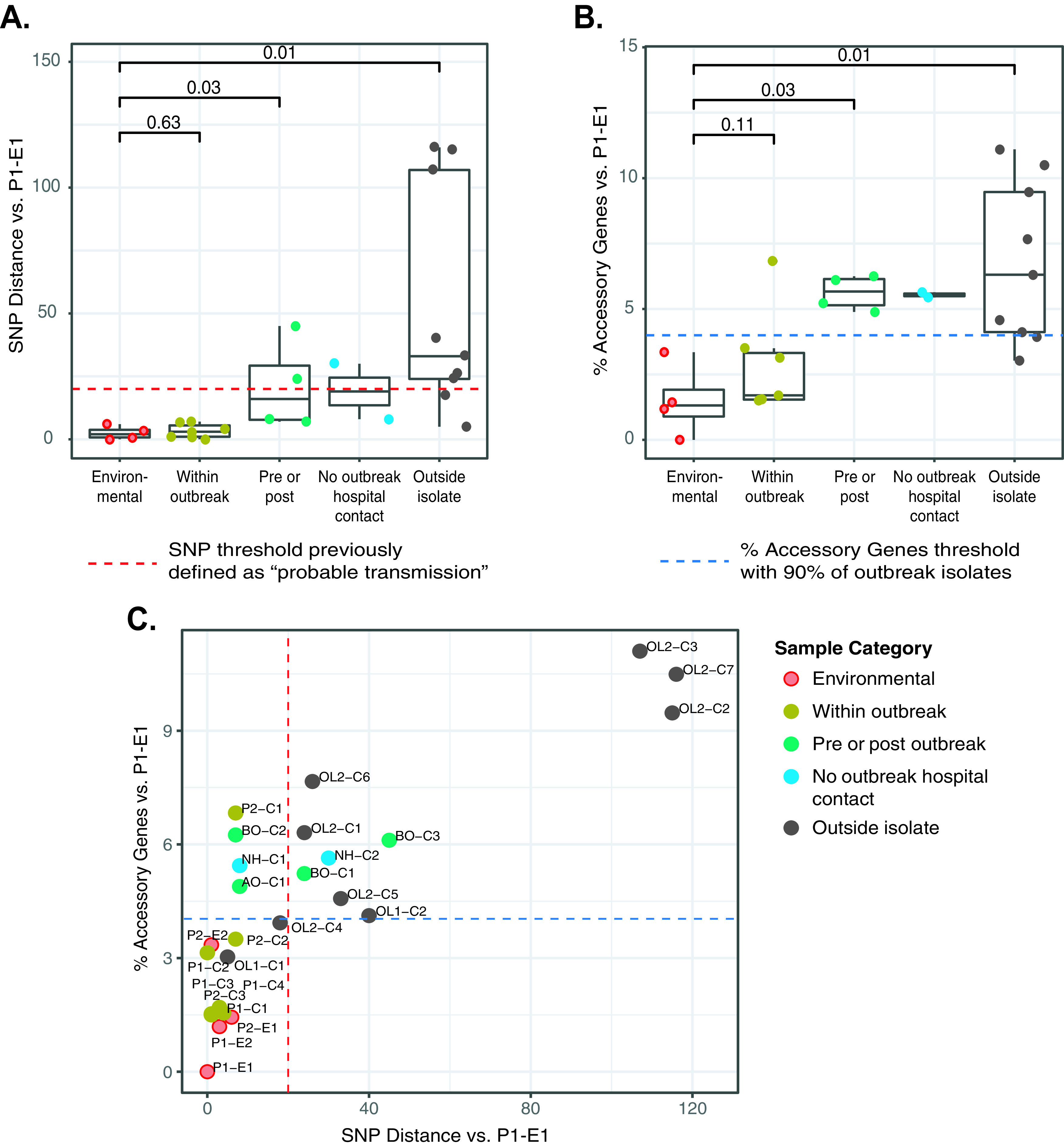
Genomic similarities of M. abscessus subsp. abscessus isolates in the study cohort by isolate categories. (A) Genome-wide SNP distances relative to a reference environmental strain collected during phase 1 of the outbreak (P1-E1) are shown on the y axis by sample categories (x axis). The previously described 20-bp SNP threshold is shown as a red dashed line. (B) Accessory genome variation for all isolates compared to P1-E1 is shown on the y axis as percent accessory genes. Sample categories are indicated on the x axis. The percent accessory gene threshold (4%) that includes 90% of clinical outbreak isolates is shown as a blue dashed line. (C) Integrated scatterplot of core genome SNPs versus percent accessory genes relative to P1-E1. Samples are color coded by sample category, and threshold lines are indicated as for panels A and B. Abbreviations: AO, after outbreak; BO, before outbreak; C, clinical; E, environmental; NH, neighboring hospital; OL, outside laboratory; P, phase of outbreak.
Core genome comparisons demonstrated that the four environmental isolates were highly similar to each other and ranged from 0 to 6 SNPs relative to P1-E1 (Fig. 2A). All seven clinical isolates within the outbreak period, obtained from a variety of pulmonary and extrapulmonary specimen sources (Table 1), were highly similar to the reference environmental isolate and ranged from 0 to 7 SNPs. The three clinical isolates collected before the outbreak period ranged from 7 to 45 SNPs, and a single postoutbreak clinical isolate had only 8 SNP differences compared to P1-E1. Clinical isolates from neighboring hospital patients without outbreak hospital contact had SNP distances of 8 and 30 SNPs. Of the nine OL control isolates that met the genotyping inclusion criteria, six isolates had SNP distances from 5 to 40 SNPs compared to P1-E1, and three outlier isolates had 107 to 116 SNP differences. Statistically significant SNP differences compared to the environmental isolate group were observed for the pre/postoutbreak isolates (P = 0.03) and OL isolates (P = 0.01). In total, 15 isolates in the study cohort were <20 SNPs from P1-E1 (Fig. 2A, red line), including three environmental isolates, seven clinical outbreak isolates, isolates collected at the outbreak hospital before and after the outbreak period, an isolate from a neighboring hospital, and two OL isolates.
Accessory genome comparisons revealed that environmental isolates had only 0 to 3.4% accessory gene differences relative to P1-E1, while clinical isolates from outbreak patients had a median of 1.7% accessory genome differences (range, 1.5 to 6.8%) (Fig. 2B). Clinical isolates from before and after the outbreak period ranged from 4.9 to 6.3%, similar to isolates from local NC patients with no outbreak hospital contact, which had 5.4 and 5.6% accessory genes. Finally, OL isolates had a median of 6.3% accessory genes compared to P1-E1 and had the widest range of all isolate groups (3.0 to 11.1%). Significantly higher proportions of accessory genes were observed for the pre/postoutbreak isolates (P = 0.03) and OL isolates (P = 0.01) compared to the environmental isolate group.
We also compared genetic distances using an integrated plot of core genome SNPs versus percent accessory genes relative to P1-E1 (Fig. 2C). Of the 10 environmental and clinical outbreak isolates, nine (90%) had fewer than 20 SNPs and less than 4% accessory genome variation (Fig. 2C, red and blue lines). Among 15 nonoutbreak clinical isolates, only two (13%) isolates met this combined threshold (OL1-C1 and OL2-C4). Three additional isolates (20%) had fewer than 20 SNPs but exceeded 4% accessory genome variation (AO-C1, BO-C2, and NH-C1).
Accessory genome clustering analysis.
To visualize the accessory genome, we performed hierarchical clustering analysis on a presence-absence matrix of 1,766 accessory genes identified in the study cohort with a corresponding heatmap (Fig. 3). Of the 1,776 accessory genes, 230 genes (13%) were present in 25 of 26 isolates; 1,125 genes (64%) were present in 2 to 24 isolates; and 411 genes (23%) were strain specific.
FIG 3.

Clustering of M. abscessus subsp. abscessus study isolates by accessory genes. Accessory genes (n = 1,766) identified among study isolates (n = 26) were analyzed by hierarchical cluster analysis using a Euclidean distance metric. The dendrogram on the left shows the clustering pattern of isolates (rows) based on gene content, and the heat map illustrates the presence (blue) or absence (light yellow) of accessory genes (columns). Suspected U.S. state of acquisition is given in parentheses. Abbreviations: AO, after outbreak; BO, before outbreak; C, clinical; E, environmental; NH, neighboring hospital; OL, outside laboratory; P, phase of outbreak.
Hierarchical clustering analysis (Fig. 3) showed clustering patterns similar to those of the core genome phylogeny (Fig. 1B), with a few exceptions. Three of the four environmental outbreak isolates showed highly similar accessory genomes, while P2-E2 had a unique pattern shared with one clinical outbreak isolate (P1-C2). One notable difference from the core genome phylogeny was that BO-C2, P2-C1, and NH-C1 clustered away from clinical outbreak isolates due to a large set of shared genes that were absent from all other isolates. These shared genes include a Tn916 transposase and DNA-invertase hin, suggesting acquisition via horizontal gene transfer (36, 37). Multiple OL isolates, including OL2-C1 and OL2-C6, also showed strain-specific sets of genes and unique accessory genomes compared to outbreak hospital isolates. Finally, the three OL isolates that were outliers by core genome SNPs (OL2-C2, OL2-C3, and OL2-C7) had clade-specific genes not present in other isolates and were also missing genes that were present in the rest of the study cohort.
DISCUSSION
We performed WGS of clinical and environment isolates of a unique clone of M. abscessus subsp. abscessus obtained at an NC hospital during an outbreak of M. abscessus. We also analyzed genomes of nonoutbreak clinical M. abscessus subsp. abscessus isolates obtained over 7 years from a variety of U.S. locations that contained the same unique erm(41) gene and rpoβ gene sequences exhibited by the outbreak strains.
Outbreak isolates obtained from hospital water outlets and patients had remarkably similar core and accessory genomes. In fact, compared to the reference outbreak environmental isolate, the other three environmental and seven clinical outbreak isolates had 7 or fewer SNP differences and, except for a single clinical isolate, accessory genome variation of less than 4%. These results confirm the findings of the initial outbreak investigation using non-WGS methods that revealed acquisition of M. abscessus subsp. abscessus from a colonized hospital water supply and led to successful outbreak mitigation via water avoidance interventions (20). Furthermore, clinical isolates with suspected acquisition at the outbreak hospital as many as 3 years before outbreak onset and 1 year after outbreak resolution also had few SNP differences (<10 SNPs) compared to environmental isolates. These longitudinal data strongly support the hypothesis that hospital plumbing system colonization was the result of municipal water colonization and preceded construction of the hospital addition associated with the outbreak.
Some nonoutbreak isolates also had genomes highly similar to those of outbreak isolates. Not surprisingly, a clinically unrelated M. abscessus subsp. abscessus isolate that was obtained from a neighboring hospital with the same municipal water supply as the outbreak hospital had a core genome distance similar to that of outbreak isolates, although it showed genetic diversification in the accessory genome. Interestingly, two unrelated OL study isolates (CF and non-CF pulmonary) that originated elsewhere in the southeastern United States showed core and accessory genomes similar to those of the reference outbreak environmental isolate by fewer than 20 SNPs and 4% accessory genome variation. Conversely, three OL isolates originating from OH and UT had genomes that proved quite dissimilar to the reference isolate with SNP distances of >100 and accessory genome variation of >9%. These results highlight the potential for clinically and geographically distinct M. abscessus subsp. abscessus isolates to have nearly identical genomes while also demonstrating that shared erm(41) and rpoβ sequencing alone, even for unusual gene combinations, does not unequivocally imply similar core and accessory genomes.
This study was unique compared to prior WGS analyses of M. abscessus isolates (9–19), because we included clinical isolates obtained from a well-characterized outbreak with a known environmental source and timing of M. abscessus subsp. abscessus acquisition. Therefore, we were able to compare the genomic diversity of clinical isolates of M. abscessus subsp. abscessus acquired over 7 years from a single outbreak hospital, environmental outbreak isolates, and clinical isolates obtained from external facilities. Importantly, to our knowledge, this study is the first to compare WGS of clinical and environmental M. abscessus subsp. abscessus isolates. Our study also evaluates the usefulness of the accessory genome for examining genetic relationships beyond core genome SNP distances.
Results from this study support the conclusions of other recent studies that caution against using SNP distance thresholds alone to indicate suspected interhuman M. abscessus transmission (17, 18). While potential exists for interhuman transmission of M. abscessus among CF patients (9, 38–40), this analysis suggests that independent environmental acquisition of certain highly similar M. abscessus strains is common. Furthermore, these strains may be prevalent in geographically distinct environments in both health care and community settings. Therefore, while adherence to CF Foundation guidelines designed to prevent health care transmission of a variety of resistant pathogens, including M. abscessus, remains paramount (41), current data do not support routine use of specialized infection prevention precautions for other patient populations on account of NTM colonization or infection alone.
These findings also emphasize the importance of a multifaceted approach when investigating HCFA clusters of M. abscessus and other NTM (40). In addition to genetic screening and genomic sequencing of clinical isolates, NTM investigations should include careful clinical and epidemiologic components, including environmental sampling. For example, genomic data from a cluster of NTM isolates may suggest a clonal outbreak propagated by interhuman transmission within a health care facility. However, epidemiologic investigation could instead reveal an environmental outbreak source intrinsic to the health care facility or reveal community acquisition of an endemic NTM clone with environmental reservoirs.
This study had two primary limitations. First, we cannot exclude the possibility that person-to-person transmission contributed to the M. abscessus subsp. abscessus outbreak linked to the colonized plumbing system at the outbreak hospital. However, we suspect any transmission events were quite uncommon because targeted interventions that prevented tap water exposure to high-risk patients promptly mitigated both phases of the outbreak (20, 42). Furthermore, outbreak mitigation occurred without placing patients with NTM colonization on contact or other specialized isolation precautions. Second, the number of outbreak isolates in this study was limited, so it is unclear whether the genomic similarity thresholds observed here would extend to other environmental-clinical isolate pairs.
Prompt and thorough investigation of HCFA M. abscessus clusters is essential for mitigation of potential outbreaks and prevention of new infections. Molecular techniques, including WGS, have become critical components of such investigations; however, despite its power, WGS of NTM isolates has limitations. In this study, genomic distances alone were unable to differentiate hospital plumbing M. abscessus subsp. abscessus acquisition from interhuman transmission, neighboring hospital acquisition, or remote community acquisition. Use of core and accessory genome comparisons combined with epidemiologic analyses provides the best opportunity for timely investigation and successful interventions.
ACKNOWLEDGMENTS
R.M.D. was supported by NIH-NIAID (K01-AI125726). R.M.D., N.W., N.A.H., L.E.E., M.S., and J.A.N. were supported by the U.S. Cystic Fibrosis Foundation (NICK20Y2-SVC, NICK20Y2-OUT). A.W.B. was supported by NIH-NIAID (5T32AI100851 and K08-AI163462).
We thank Jane E. Gross for reviewing the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Rebecca M. Davidson, Email: DavidsonR@NJHealth.org.
Alexander Mellmann, University Hospital Münster.
REFERENCES
- 1.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ, Jr, Winthrop K, Infectious Disease Society of America. 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 175:367–416. 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 2.Pasipanodya JG, Ogbonna D, Ferro BE, Magombedze G, Srivastava S, Deshpande D, Gumbo T. 2017. Systematic review and meta-analyses of the effect of chemotherapy on pulmonary Mycobacterium abscessus outcomes and disease recurrence. Antimicrob Agents Chemother 61:e01206-17. 10.1128/AAC.01206-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee MR, Sheng WH, Hung CC, Yu CJ, Lee LN, Hsueh PR. 2015. Mycobacterium abscessus complex infections in humans. Emerg Infect Dis 21:1638–1646. 10.3201/2109.141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Floto RA, Olivier KN, Saiman L, Daley CL, Herrmann JL, Nick JA, Noone PG, Bilton D, Corris P, Gibson RL, Hempstead SE, Koetz K, Sabadosa KA, Sermet-Gaudelus I, Smyth AR, van Ingen J, Wallace RJ, Winthrop KL, Marshall BC, Haworth CS, US Cystic Fibrosis Foundation and European Cystic Fibrosis Society. 2016. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax 71(Suppl 1):i1–i22. 10.1136/thoraxjnl-2015-207360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alcaide F, Pena MJ, Perez-Risco D, Camprubi D, Gonzalez-Luquero L, Grijota-Camino MD, Dorca J, Santin M. 2017. Increasing isolation of rapidly growing mycobacteria in a low-incidence setting of environmental mycobacteria, 1994-2015. Eur J Clin Microbiol Infect Dis 36:1425–1432. 10.1007/s10096-017-2949-0. [DOI] [PubMed] [Google Scholar]
- 6.Velayati AA, Rahideh S, Nezhad ZD, Farnia P, Mirsaeidi M. 2015. Nontuberculous mycobacteria in Middle East: current situation and future challenges. Int J Mycobacteriol 4:7–17. 10.1016/j.ijmyco.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Prevots DR, Shaw PA, Strickland D, Jackson LA, Raebel MA, Blosky MA, Montes de Oca R, Shea YR, Seitz AE, Holland SM, Olivier KN. 2010. Nontuberculous mycobacterial lung disease prevalence at four integrated health care delivery systems. Am J Respir Crit Care Med 182:970–976. 10.1164/rccm.201002-0310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanamori H, Weber DJ, Rutala WA. 2016. Healthcare outbreaks associated with a water reservoir and infection prevention strategies. Clin Infect Dis 62:1423–1435. 10.1093/cid/ciw122. [DOI] [PubMed] [Google Scholar]
- 9.Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, Reacher M, Haworth CS, Curran MD, Harris SR, Peacock SJ, Parkhill J, Floto RA. 2013. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 381:1551–1560. 10.1016/S0140-6736(13)60632-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bryant JM, Grogono DM, Rodriguez-Rincon D, Everall I, Brown KP, Moreno P, Verma D, Hill E, Drijkoningen J, Gilligan P, Esther CR, Noone PG, Giddings O, Bell SC, Thomson R, Wainwright CE, Coulter C, Pandey S, Wood ME, Stockwell RE, Ramsay KA, Sherrard LJ, Kidd TJ, Jabbour N, Johnson GR, Knibbs LD, Morawska L, Sly PD, Jones A, Bilton D, Laurenson I, Ruddy M, Bourke S, Bowler IC, Chapman SJ, Clayton A, Cullen M, Daniels T, Dempsey O, Denton M, Desai M, Drew RJ, Edenborough F, Evans J, Folb J, Humphrey H, Isalska B, Jensen-Fangel S, Jonsson B, Jones AM, et al. 2016. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 354:751–757. 10.1126/science.aaf8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davidson RM, Hasan NA, de Moura VC, Duarte RS, Jackson M, Strong M. 2013. Phylogenomics of Brazilian epidemic isolates of Mycobacterium abscessus subsp. bolletii reveals relationships of global outbreak strains. Infect Genet Evol 20:292–297. 10.1016/j.meegid.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tettelin H, Davidson RM, Agrawal S, Aitken ML, Shallom S, Hasan NA, Strong M, de Moura VC, De Groote MA, Duarte RS, Hine E, Parankush S, Su Q, Daugherty SC, Fraser CM, Brown-Elliott BA, Wallace RJ, Jr, Holland SM, Sampaio EP, Olivier KN, Jackson M, Zelazny AM. 2014. High-level relatedness among Mycobacterium abscessus subsp. massiliense strains from widely separated outbreaks. Emerg Infect Dis 20:364–371. 10.3201/eid2003.131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davidson RM, Hasan NA, Reynolds PR, Totten S, Garcia B, Levin A, Ramamoorthy P, Heifets L, Daley CL, Strong M. 2014. Genome sequencing of Mycobacterium abscessus isolates from patients in the united states and comparisons to globally diverse clinical strains. J Clin Microbiol 52:3573–3582. 10.1128/JCM.01144-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson RM. 2018. A closer look at the genomic variation of geographically diverse Mycobacterium abscessus clones that cause human infection and disease. Front Microbiol 9:2988. 10.3389/fmicb.2018.02988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris KA, Underwood A, Kenna DT, Brooks A, Kavaliunaite E, Kapatai G, Tewolde R, Aurora P, Dixon G. 2015. Whole-genome sequencing and epidemiological analysis do not provide evidence for cross-transmission of Mycobacterium abscessus in a cohort of pediatric cystic fibrosis patients. Clin Infect Dis 60:1007–1016. 10.1093/cid/ciu967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redondo N, Mok S, Montgomery L, Flanagan PR, McNamara E, Smyth EG, O'Sullivan N, Schaffer K, Rogers TR, Fitzgibbon MM. 2020. Genomic analysis of Mycobacterium abscessus complex isolates collected in Ireland between 2006 and 2017. J Clin Microbiol 58:e00295-20. 10.1128/JCM.00295-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doyle RM, Rubio M, Dixon G, Hartley J, Klein N, Coll P, Harris KA. 2019. Cross-transmission is not the source of new Mycobacterium abscessus infections in a multi-centre cohort of cystic fibrosis patients. Clin Infect Dis 10.1093/cid/ciz526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tortoli E, Kohl TA, Trovato A, Baldan R, Campana S, Cariani L, Colombo C, Costa D, Cristadoro S, Di Serio MC, Manca A, Pizzamiglio G, Rancoita PMV, Rossolini GM, Taccetti G, Teri A, Niemann S, Cirillo DM. 2017. Mycobacterium abscessus in patients with cystic fibrosis: low impact of inter-human transmission in Italy. Eur Respir J 50:1602525. 10.1183/13993003.02525-2016. [DOI] [PubMed] [Google Scholar]
- 19.Davidson RM, Hasan NA, Epperson LE, Benoit JB, Kammlade SM, Levin AR, Calado de Moura V, Hunkins J, Weakly N, Beagle S, Sagel SD, Martiniano SL, Salfinger M, Daley CL, Nick JA, Strong M. 2021. Population genomics of Mycobacterium abscessus from United States cystic fibrosis care centers. Ann Am Thorac Soc 10.1513/AnnalsATS.202009-1214OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker AW, Lewis SS, Alexander BD, Chen LF, Wallace RJ, Jr, Brown-Elliott BA, Isaacs PJ, Pickett LC, Patel CB, Smith PK, Reynolds JM, Engel J, Wolfe CR, Milano CA, Schroder JN, Davis RD, Hartwig MG, Stout JE, Strittholt N, Maziarz EK, Saullo JH, Hazen KC, Walczak RJ, Jr, Vasireddy R, Vasireddy S, McKnight CM, Anderson DJ, Sexton DJ. 2017. Two-phase hospital-associated outbreak of Mycobacterium abscessus: investigation and mitigation. Clin Infect Dis 64:902–911. 10.1093/cid/ciw877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown-Elliott BA, Vasireddy S, Vasireddy R, Iakhiaeva E, Howard ST, Nash K, Parodi N, Strong A, Gee M, Smith T, Wallace RJ, Jr.. 2015. Utility of sequencing the erm(41) gene in isolates of Mycobacterium abscessus subsp. abscessus with low and intermediate clarithromycin MICs. J Clin Microbiol 53:1211–1215. 10.1128/JCM.02950-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adékambi T, Colson P, Drancourt M. 2003. rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J Clin Microbiol 41:5699–5708. 10.1128/JCM.41.12.5699-5708.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong YL, Ong CS, Ngeow YF. 2012. Molecular typing of Mycobacterium abscessus based on tandem-repeat polymorphism. J Clin Microbiol 50:3084–3088. 10.1128/JCM.00753-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Epperson LE, Strong M. 2020. A scalable, efficient, and safe method to prepare high quality DNA from mycobacteria and other challenging cells. J Clin Tuberc Other Mycobact Dis 19:100150. 10.1016/j.jctube.2020.100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224. 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 26.Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y. 2017. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol 8:28–36. 10.1111/2041-210X.12628. [DOI] [Google Scholar]
- 27.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. [Google Scholar]
- 29.Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13:e1005595. 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 31.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segerman B. 2012. The genetic integrity of bacterial species: the core genome and the accessory genome, two different stories. Front Cell Infect Microbiol 2:116. 10.3389/fcimb.2012.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O'Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc Natl Acad Sci USA 102:13950–13955. 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dedrick RM, Aull HG, Jacobs-Sera D, Garlena RA, Russell DA, Smith BE, Mahalingam V, Abad L, Gauthier CH, Hatfull GF. 2021. The prophage and plasmid mobilome as a likely driver of Mycobacterium abscessus diversity. mBio 12:e03441-20. 10.1128/mBio.03441-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davidson RM, Benoit JB, Kammlade SM, Hasan NA, Epperson LE, Smith T, Vasireddy S, Brown-Elliott BA, Nick JA, Olivier KN, Zelazny AM, Daley CL, Strong M, Wallace RJ. 2021. Genomic characterization of sporadic isolates of the dominant clone of Mycobacterium abscessus subspecies massiliense. Sci Rep 11:15336. 10.1038/s41598-021-94789-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson RC. 2015. Site-specific DNA inversion by serine recombinases. Microbiol Spectr 3:MDNA3-0047-2014. 10.1128/microbiolspec.MDNA3-0047-2014. [DOI] [PubMed] [Google Scholar]
- 37.Roberts AP, Mullany P. 2009. A modular master on the move: the Tn916 family of mobile genetic elements. Trends Microbiol 17:251–258. 10.1016/j.tim.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Aitken ML, Limaye A, Pottinger P, Whimbey E, Goss CH, Tonelli MR, Cangelosi GA, Dirac MA, Olivier KN, Brown-Elliott BA, McNulty S, Wallace RJ, Jr.. 2012. Respiratory outbreak of Mycobacterium abscessus subspecies massiliense in a lung transplant and cystic fibrosis center. Am J Respir Crit Care Med 185:231–232. 10.1164/ajrccm.185.2.231. [DOI] [PubMed] [Google Scholar]
- 39.Yan J, Kevat A, Martinez E, Teese N, Johnson K, Ranganathan S, Harrison J, Massie J, Daley A. 2020. Investigating transmission of Mycobacterium abscessus amongst children in an Australian cystic fibrosis centre. J Cyst Fibros 19:219–224. 10.1016/j.jcf.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 40.Gross JE, Martiniano SL, Nick JA. 2019. Prevention of transmission of Mycobacterium abscessus among patients with cystic fibrosis. Curr Opin Pulm Med 25:646–653. 10.1097/MCP.0000000000000621. [DOI] [PubMed] [Google Scholar]
- 41.Saiman L, Siegel JD, LiPuma JJ, Brown RF, Bryson EA, Chambers MJ, Downer VS, Fliege J, Hazle LA, Jain M, Marshall BC, O'Malley C, Pattee SR, Potter-Bynoe G, Reid S, Robinson KA, Sabadosa KA, Schmidt HJ, Tullis E, Webber J, Weber DJ, Cystic Fibrous Foundation, Society for Healthcare Epidemiology of America. 2014. Infection prevention and control guideline for cystic fibrosis: 2013 update. Infect Control Hosp Epidemiol 35(Suppl 1):S1–S67. 10.1086/676882. [DOI] [PubMed] [Google Scholar]
- 42.Baker AW, Stout JE, Anderson DJ, Sexton DJ, Smith B, Moehring RW, Huslage K, Hostler CJ, Lewis SS. 2020. Tap water avoidance decreases rates of hospital-onset pulmonary nontuberculous mycobacteria. Clin Infect Dis 10.1093/cid/ciaa1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods, Fig. S1, and Table S1. Download JCM.01547-21-s0001.pdf, PDF file, 0.7 MB (752.9KB, pdf)
Data Availability Statement
WGS data generated through this study are available at NCBI (PRJNA734909).