

Abstract
In neurons, local translation in dendritic and axonal compartments allows for the fast and on-demand modification of the local proteome. As the last few years have witnessed dramatic advancements in our appreciation of the brain’s neuronal diversity, it is increasingly relevant to understand how local translation is regulated according to cell type. To this end, both sequencing-based and imaging-based techniques have recently been reported. Here, we present a subcellular single cell RNA sequencing protocol that allows molecular quantification from the soma and dendrites of single neurons, and which can be scaled up for the characterization of several hundreds to thousands of neurons. Somata and dendrites of cultured neurons are dissected using laser capture microdissection, followed by cell lysis to release mRNA content. Reverse transcription is then conducted using an indexed primer that allows the downstream pooling of samples. The pooled cDNA library is prepared for and sequenced in an Illumina platform. Finally, the data generated are processed and converted into a gene vs. cells digital expression table. This protocol provides detailed instructions for both wet lab and bioinformatic steps, as well as insights into controls, data analysis, interpretations, and ways to achieve robust and reproducible results.
Graphic abstract:
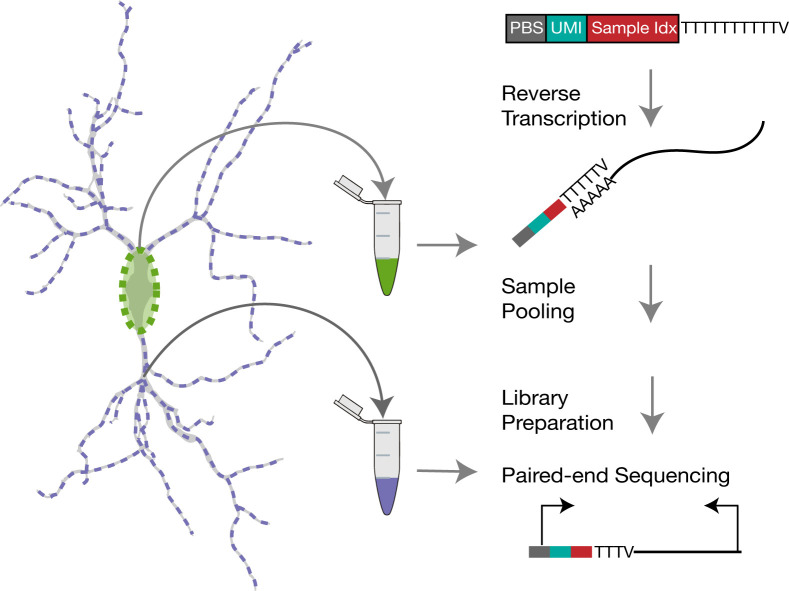
Subcellular Single Cell RNA-seq in Neurons.
Keywords: Local translation, Local protein synthesis, Local transcriptome, Subcellular transcriptomics, Single cell RNA-seq, Neuron, Dendrites, Neuronal compartments
Background
Within their highly polarized and complex structure, neurons create subcellular compartments that optimize operations requiring spatial and temporal isolation. The functional specialization of these compartments is, in part, accomplished via the selective transport and local translation of mRNAs ( Holt et al., 2019 ). To characterize the neuronal local transcriptome, many studies have performed bulk RNA profiles in brain regions enriched in dendrites and axons ( Zhong et al., 2006 ; Cajigas et al., 2012 ; Glock et al., 2020 ), on synaptic particles isolated from tissue ( Hafner et al., 2019 ), or from neuronal cultures in chambers that separate cell bodies and neurites ( Gumy et al., 2011 ; Poon et al., 2006 ). These studies have revealed that protein functions such as synaptic transmission, cytoskeletal regulation, and translation itself (among others) are encoded in the local transcriptome ( Holt et al., 2019 ). However, as recent advancements in single cell transcriptomics have revealed, the brain contains a complex array of neuronal types, raising the question of how variable the local transcriptome is across diverse cell types ( Wang et al., 2020 ; Perez et al., 2021 ).
To address this question, single cell resolution of the local transcriptome is needed. There are three main challenges to implementing such an approach: (1) the isolation of mRNAs from distinct subcellular compartments of a single neuron, (2) the unbiased characterization of the local mRNAs, and (3) the characterization of hundreds to thousands of samples for robust cell type classification. Two pioneering studies used single-cell nanobiopsies or micropipettes to isolate material from the soma and dendrites of single neurons in culture, and RNA-seq to profile hundreds and thousands local mRNAs, respectively ( Tóth et al., 2018 ; Middleton et al., 2019 ). However, both studies contained a relatively low number of samples (a few dozen), and neither investigated cell type effects on the local transcriptome. Recent breakthroughs in spatial transcriptomics have provided unprecedented in situ single molecule resolution of the local transcriptome, allowing for the characterization of mRNAs across and within subdomains of dendritic and axonal compartments. Using multiplexed error-robust fluorescence in situ hybridization, Wang et al. (2020) profiled the spatial location of hundreds of mRNAs in hundreds of cultured neurons, resulting in the identification of dendritic and axonal transcripts in glutamatergic and GABAergic neurons. However, this technique requires the experimenter to select a priori which mRNAs to target, and therefore, cannot provide an unbiased view of the local transcriptome. To circumvent this limitation, Alon et al. (2021) recently developed expansion sequencing (ExSeq), which combines expansion microscopy with fluorescent in situ sequencing. This technique allows the unbiased characterization of mRNAs anywhere within a neuron, in either cultured cells or tissue samples. However, so far untargeted ExSeq resolves only dozens of mRNA species per cell, and thus has not been used to profile cell type-specific variation in the local transcriptome.
Recently, we developed a method that allows for the separate isolation of dendritic and somatic mRNAs, as well as the unbiased characterization and molecular tallying of local mRNAs, in hundreds to thousands of single neurons ( Perez et al., 2021 ). Using this method, we profiled the dendritic transcriptome of glutamatergic and various GABAergic interneurons in culture, and identified dozens of mRNAs whose dendritic localization is regulated according to cell-type. Cell-type specific differences are substantially more common among somata as expected, since besides its own transcriptome, the soma also harbors the transcriptome of dendritic and axonal compartments. Similar observations were made by Wang et al. (2020) using an alternative approach. Our method combines laser capture microdissection (LCM) for the isolation of dendritic and somatic compartments (micron resolution; Figure 1A), with a sensitive scRNA-seq protocol (adapted from Picelli et al., 2014 ; Macosko et al., 2015 ), which tags mRNAs with a unique molecular identifier (UMI) and an index (Figure 1B). This index allows iterative pooling steps during library preparation, enabling the sequencing of 384 samples per run. The protocol can be executed from beginning-to-end in two weeks, and, if repeated 3 times or more, thousands of subcellular samples can be accumulated. Since every cell is imaged before collection, this method can also be used to investigate correlations between the transcriptome and neuronal morphology. Nonetheless, several drawbacks of the protocol should be considered before starting. First, the method (as calculated using ERCC RNA standards) captures one of every 4 molecules present after LCM collection. This, however, likely overestimates the sensitivity to the actual number of molecules present inside the cell, as not all LCM catapulted pieces of cellular material land in the collection cap, an issue that appears more severe for dendrites. Thus, the more abundant an mRNA is, the more likely it is to be detected, and lower abundance mRNAs are more frequently missed. To compensate for this, we recommend increasing the number of samples until the number of detected mRNAs in dendrites reaches saturation. Second, the protocol requires the selection of neurons with little-to-no overlapping cellular processes in most of its dendritic arbor. This selection can introduce biases for some cell types over others. Indeed, we observed that, on average, GABAergic neurons have more accessible somata and processes, while glutamatergic processes are often heavily entangled with those of other cells. To estimate this potential bias, we suggest collecting somata-only samples from less accessible neurons in the same dish. The inclusion of such samples also improves unsupervised clustering, and thus enables more accurate cell-type determination of those neurons for which both soma and dendrites are collected.
Figure 1. Subcellular scRNA-seq method.

A. Images showing the dissection of the soma and dendrites of a neuron using LCM. B. Library preparation workflow, showing the sequences, primers, and key enzymes used at every step.
Altogether, this method can serve as a powerful tool to achieve an unbiased investigation of cell type effects in the local transcriptome, and can be implemented across cells derived from different brain regions, developmental stages, or species. Additionally, it may be used to study single cell responses to pharmacological treatments, or other manipulations that induce changes in cell states (e.g., paradigms of synaptic plasticity). It may also be useful to study the local transcriptome of other polarized cell types, such as astrocytes ( Sakers et al., 2017 ; Mazaré et al., 2021 ), or epithelial cells ( Moor et al., 2017 ). Finally, it should be possible to adjust this protocol to profile non-coding RNAs, such as small RNAs (Hagemann- Jensen et al., 2018 ).
Materials and Reagents
Glass-bottom culture 35 mm dishes, 14 mm glass diameter (MatTek, catalog number: P35G-1.5-14-C)
Qubit Assay Tubes (ThermoFisher, catalog number: Q32856)
Reagent Reservoir 25 mL (VWR, catalog number: 89094-662)
96-well DNA LoBind Plate (Eppendorf, catalog number: 30129504)
AdhesiveCap-200 Clear (Zeiss, catalog number: 415190-9191-000)
Agencourt AMPure XP Magnetic Beads (Beckman Coulter, catalog number: A63881)
Betaine (ThermoFisher, catalog number: J77507AE)
Bioanalyzer HS-DNA Kit (Agilent, catalog number: 5067-4626)
Cultured neurons. We use Rat hippocampus primary neuronal cultures (prepared as described in Aakalu et al., 2001 ).
CustomSeqB Primer (IDT, see Supplementary File 1 with oligo information)
Dithiothreitol (DTT) (Bio-Rad, catalog number: 1610611)
dNTP mix (ThermoFisher, catalog number: R0192)
Elution Buffer (Qiagen, catalog number: 1014609)
ERCC RNA Spike-Ins (ThermoFisher, catalog number: 4456740)
ISPCR Primer (IDT, see Supplementary File 1 with oligo information)
KAPA HiFi HotStart Mix (Roche, catalog number: KK2602)
Magnesium Chloride (ThermoFisher, catalog number: AM9530G)
Microseal B adhesive film (Bio-Rad, catalog number: MSB1001)
Molecular Biology Grade Ethanol (Sigma, catalog number: BP2818-100)
Nextera XT DNA Library Prep Kit (96 samples) (Illumina, catalog number: FC-131-1096)
Nextera XT Index Kit v2 Set A (Illumina, catalog number: FC-131-2001)
Nextera XT Index Kit v2 Set B (Illumina, catalog number: FC-131-2002)
NextSeq 1000/2000 P2 reagents (100 cycles) (Illumina, catalog number: 20046811)
Nuclease-free H2O (ThermoFisher, catalog number: 10977-035)
P5-ISPCR Primer (IDT, see Supplementary File 1 with oligo information)
Parafilm (Sigma, catalog number: PM-992)
Phosphate Buffer Saline (Sigma, catalog number: P5493-1L)
Poly-D-lysine (Corning, catalog number: 354210)
Qiagen Protease (Qiagen, catalog number: 19155)
Qubit dsDNA BR kit (ThermoFisher, catalog number: Q32853)
Qubit dsDNA HS kit (ThermoFisher, catalog number: Q32851)
RNase Inhibitor (Takara, catalog number: 2313A)
RNaseZap (ThermoFisher, catalog number: AM9780)
RT-UMI-Index Primer (16 variants) (IDT, see Supplementary File 1 with oligo information)
SingleShot Cell Lysis Kit (Bio-Rad, catalog number: 1725080)
SuperScript IV (ThermoFisher, catalog number: 18090200)
Template Switch Oligo (IDT, see Supplementary File 1 with oligo information)
Cell Lysis Mix (see Recipes)
RT Mix (see Recipes)
PreAmp PCR Mix (see Recipes)
Final PCR Amplification Mix (see Recipes)
Equipment
2-20, µL 12-channel, Multichannel pipette (e.g., Ranin, catalog number: 17013808)
20-200 µL, 12-channel, Multichannel pipette (e.g., Ranin, catalog number: 17013810)
2100 Bioanalyzer Instrument (Agilent, catalog number: G2939BA)
Cell culture incubator (e.g., ThermoFisher)
Curved, cover glass forceps (VWR, catalog number: HAMMHSC817-13)
DISH 35 CC Adapter (Zeiss, catalog number: 415101-2000-835)
Magnetic Stand for 96-well plates (ThermoFisher, catalog number: AM10027)
Minicentrifuge (e.g., ThermoFisher, catalog number: 75004061)
lllumina DNA Sequencer (e.g., NextSeq 2000)
PALM MicroBeam Axio Observer Laser Capture Microdissection Microscope (Zeiss)
PCR Plate Spinner (VWR, catalog number: 89184-610) or 96-well plate-compatible centrifuge
Qubit Fluorometer (ThermoFisher, catalog number: Q33238)
SingleCap Collector II 200 RM (Zeiss, catalog number: 415101-2000-951)
Thermal cycler (e.g., Bio-Rad, C1000, catalog number: 1851197)
Tissue culture hood (e.g., ThermoFisher)
Vortex (e.g., VWR, catalog number: 10153-840)
Software
PALM RoboSoftware v3 or above (Zeiss, https://www.zeiss.com/microscopy/us/products/microscope-software/palm-robosoftware.html/palm46)
bcl2fastq v2.20.0.422 (Illumina, https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html)
FastQC v.0.11.9 (Babraham Bioinformatics/FastQC, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
Picard Tools v.2.20.2 or above (broadinstitute/picard, https://github.com/broadinstitute/picard/releases/tag/2.20.2)
Drop-seq Tools v.2.3.0 or above (broadinstitute/Drop-seq, https://github.com/broadinstitute/Drop-seq/releases/tag/v2.3.0)
STAR v.2.7.2b or above (alexdobin/STAR, https://github.com/alexdobin/STAR/releases/tag/2.7.2b)
Fasta file containing the genome of the species being used. Genome fasta files of various species can be found in https://hgdownload.soe.ucsc.edu/downloads.html or http://ftp.ensembl.org/pub/release-99/fasta/
GTF file containing transcript coordinates in the genome of the species being used. GTF files of various species can be found in https://hgdownload.soe.ucsc.edu/downloads.html or http://ftp.ensembl.org/pub/release-99/gtf/
Seurat 3 or above (satijalab/seurat, https://satijalab.org/seurat/articles/install.html)
Procedure
Determine the desired number of samples per sequencing run (see Note 1 for limitations and considerations). This procedure is designed for the sequencing analysis of 384 samples, including both positive and negative controls (see Note 2), which are indexed, and subsequently pooled in three separate steps (see Note 3).
Before starting, clean pipettes, racks, centrifuges, and surfaces in the LCM microscope and bench space with RNaseZap, to avoid RNA degradation.
-
Primary neuronal cultures
Prepare mammalian neuronal cultures according to protocol of choice. In our case, we culture neurons from the hippocampus or cortex of newborn rats (P1), as previously described ( Aakalu et al., 2001 ).
Plate cells on coverslips of MatTek 14mm diameter glass bottom dishes coated with a 0.1 mg/mL solution of poly-D-lysine, at a density of 20,000 cells/coverslip.
Maintain plated cells in the cell medium of choice for at least 2 weeks and no longer than 5 weeks in culture for best confluency of neuronal processes. Unless cell age is a variable of interest, we strongly recommend that all neurons in an experiment share the same age, as the transcriptome may vary significantly over the lifespan of cultured neurons.
-
Ethanol fixation
Discard cell medium.
Wash cultures by adding 5 mL of 1× PBS and immediately discard it. Repeat once, for a total of 2 washes.
Add 5 mL of cold 70% ethanol (kept at -20°C). Wait 5 min and discard ethanol.
Seal the dish with parafilm and store at -80°C. RNA in cells will remain stable for at least 3 months.
-
Laser capture microdissection
Make 4 separate cell lysis master mixes, differing in which of the 16 RT-UMI-Index (1-16) Primer is used (see Recipe 1). Keep on ice.
Remove parafilm and cap from dish and place it on the DISH 35 CC adapter of a Zeiss PALM LCM microscope. Cells should be allowed to thaw for 5-10 min before dissection. Proceed to the next step in the meantime.
Open the PALMRobo software. Using the 20× objective, identify and register locations for microdissection. We recommend the collection of no more than 48 samples per plate (see Note 1): the somata and dendritic arbors of 16 neurons amenable for dissection, 12 somata whose dendritic arbors are not amenable for collection, and 4 empty cuts (see Note 2). Thus, at this point the location of 28 neurons and 4 empty regions should be saved. Neurons amenable for collection have isolated somata free of processes, AND isolated dendritic arbors in which most processes can be unambiguously assigned to the same neuron (See example in Figure 1A and Supplementary File 2). Avoid neurons at the edges of the coverslip, as these are inefficiently catapulted.
Switch to the 40× objective, go to the first registered location, and take a picture.
For somata or empty regions, select AutoLPC from the Cut Tools menu, and the circle drawing tool to delineate the soma or region of interest. For dendrites, select LineAutoLPC from the Cut Tools menu, and the free-hand drawing tool to delineate processes. Because of the continuous nature of neuronal compartments, there is no obvious point where the soma ends and dendrites begin, and thus what we consider the border between the two compartments is ultimately arbitrary. In our experiments, we delineate the soma as the cellular area containing and surrounding the nucleus, which dilates out until drastic decrements in width suddenly occur. The processes that continue are considered dendrites. However, processes that are qualitatively thinner than the rest are excluded as these might be axons (see example in Figure 1A, Supplementary File 2, and Video 1).
Place cap of an AdhesiveCap-200 clear tube in the microscope’s RoboMover containing the SingleCap Collector II 200 RM. Cut the stretch of the tube linking the cap to the tube, and discard the tube.
Click on Capture Device icon, go to the Adjust tab, and set working height to -12,500. Go to Operation, and double-click on top of the cap icon. This will bring the cap in the collection position above the culture dish. Adjust focus if necessary, to obtain a clear image of the neuron.
Set the following parameters for Laser Pressure Catapulting (LPC): Energy = 40, focus = 70%, and speed = 15%.
Go to the Colors icon on the Graphic toolbar, and on the LPC Distances tab set the Distance of AutoLPC shots to 2. This determines the density of the LPC punches.
Go to Element List, and select only the element delineating the soma. Click on the Start Cutting Laser icon. The area delineating the soma will be catapulted to the collection cap in many individual punches.
Click on Capture Device icon, go to the Operation tab, and click on the Home icon. This will bring the cap back to the loading position.
Using curved forceps, carefully remove the cap from the collection arm and place it on a surface, with the side where the material was catapulted into facing up.
Add 3 µL of the respective Cell lysis master mix to the cap containing the catapulted material.
Using curved forceps, carefully place the cap in a well of a 96-well DNA LoBind plate, with the side where the material was catapulted into facing down.
Repeat steps 5 to 7.
Go to Element List and select all of the elements delineating the dendrites of that particular neuron. Click on the Start Cutting Laser icon. The area delineating each dendrite will be catapulted to the collection cap in many individual punches.
Repeat steps 11 to 14.
Repeat steps 5 to 17 for each location registered in step 3. We recommend organizing samples shown in Figure 4A in each 96-well plate.
Proceed immediately to the next step.
-
Cell lysis
Seal the plate containing collection caps tightly with Microseal B adhesive film, turn it upside down, and vortex the side containing caps for 15 s.
Centrifuge plate for 1 min in plate spinner, or centrifuge for 1min at 1,000 × g, to bring volumes from the cap to the bottom of the well. Discard caps and reseal plate with Microseal B adhesive film.
-
Place plate in thermal cycler and run the following program (lid set to 105°C):
Step 1 (Protein Digestion): 50°C for 10 min
Step 2 (Protease Inactivation): 75°C for 10 min
Step 3: 4°C hold
Proceed immediately to the next step.
-
Reverse Transcription
Prepare RT master mix (see Recipe 2).
Split RT master mix into 12 PCR tubes, each containing 15.3 µL.
Using a multichannel pipette, pipette 3.4 µL of RT master mix out of the 12 PCR tubes prepared in the previous step, and add it to each reaction.
Seal plate with Microseal B adhesive film.
Mix, by quick vortex and 1 min centrifugation in plate spinner, or in centrifuge for 1 min at 1,000 × g.
-
Place plate in thermal cycler and run the following program (lid set to 105°C):
Step 1 (Reverse Transcription): 55°C for 10 min
Step 2 (Enzyme Inactivation): 80°C for 10 min
Step 3: 12°C hold
-
PCR pre-amplification
Prepare PCR PreAmp master mix (see Recipe 3).
Split PCR PreAmp master mix into 12 PCR tubes each containing 34.2 µL.
Using a multichannel pipette, pipette 7.6 µL of PCR PreAmp master mix out of the 12 PCR tubes prepared in the previous step, and add it to each reaction.
Seal plate with Microseal B adhesive film.
Mix by quick vortex and 1min centrifugation in plate spinner, or centrifuge for 1 min at 1,000 × g.
-
Place plate in thermal cycler and run the following program (lid set to 105°C):
Step 1 (Taq Activation): 98°C for 3 min
Step 2 (Denaturation): 98°C for 20 s
Step 3 (Annealing): 67°C for 15 s
Step 4 (Extension): 72°C for 6 min
(Repeat steps 2-4 for a total of 21 cycles*)
Step 5 (Final Extension): 72°C for 5 min
Step 6: 12°C hold
*Cycle number may need to be optimized, as samples with high starting amounts of RNA will require less cycles, and those with low starting amounts will require more.
It’s safe to stop and store PCR reactions at -20°C for at least 3 months.
Proceed to the next step once all 384 samples have been accumulated (~8 culture dishes).
-
Sample pooling and PCR purification
Let AMPure XP DNA Magnetic Beads stand at room temperature at least 30 min (see Note 3 for best practices when performing DNA purification using magnetic beads).
Pool 8 reactions derived from the same source (i.e., somata, dendrites, or empty cuts), but which were obtained from different RT index primers, into a single well in a DNA LoBind 96-well plate (pooling logic is described in detail in Note 4). This should result in a plate with 4 rows in which each well contains ~112 µL of pooled sample.
Add 112 µL of AMPure Magnetic Beads to each well. Seal plate. Mix well by vortexing.
Let plate stand at room temperature for 5 min.
Place plate in magnetic stand. Let stand for 5 min.
Prepare fresh 80% ethanol, considering that 48 pooled samples require ~22 mL of 80% ethanol.
Place 80% ethanol solution in 25 mL reservoir.
Discard supernatant from every well containing pooled samples, being careful not to disrupt the magnetic bead pellet.
Add 200 µL of freshly-made 80% ethanol to each well. Wait 30 s and remove.
Repeat previous step one more time, making sure to discard all ethanol at the end (may require additional pipetting out).
Let plate stand in magnetic stand for 5 min, or until the bead pellet looks dry. If cracks begin to appear in the pellet, proceed immediately to next step.
Remove plate from magnetic stand, and add 17.5 µL of Elution Buffer (EB) on top of pellet.
Resuspend pellet by vortexing and, if necessary, by repeatedly pipetting elution volume on top of pellet.
Wait 5 min.
Place tube back on magnetic stand and wait 2 min.
Transfer 15 µL of supernatant to wells in a new 96-well DNA LoBind plate.
Combine supernatant from 2 pooled samples derived from the same source (i.e., somata, dendrites, or empty cuts) that contained no overlap in the 8 indexes pooled in step 2 (see Figure 4). Thus, this mix will contain 16 different indexes, each representing a different sample.
-
Quality metrics of cDNA libraries
Check concentration of the 24 pooled samples using Qubit dsDNA BR Kit according to manufacturer’s instructions. Concentrations between 10-150 ng/µL per pooled sample are expected.
Check size distribution of the 24 pooled samples using the Agilent Bioanalyzer HS-DNA kit, according to manufacturer’s instructions. Libraries are expected to have few or no peaks below 500bp and a large peak between ~800 bp and ~5,000 bp, centered at ~2,000 bp (Figure 2A).
-
Tagmentation
In a new 96-well DNA LoBind plate, make 0.1 ng/µL dilutions for each pooled sample.
Add 10 µL of Nextera Tagment DNA Buffer to each well.
Add 5 µL of Nextera Amplicon Tagment Mix to each well.
Seal plate and centrifuge in plate spinner for 1 min.
Incubate samples for 5 min in a thermal cycler set to 55°C (heated lid 105°C).
Stop Tagmentation by adding 5 µL of Nextera Neutralize Tagment Buffer to each well.
Seal plate and centrifuge in plate spinner for 1 min.
Incubate plate at room temperature for 5 min.
-
Amplification of sequencing-compatible fragments
Prepare Final PCR master mix (see Recipe 4).
Add 24 µL of Final PCR Amplification mix to each reaction.
To each reaction, add 1 of the 24 different Nextera i7 primers (N7XX).
Seal plate and centrifuge in plate spinner for 1 min.
-
Place plate in thermal cycler and run the following program (lid set to 105°C):
Step 1 (Pre-PCR Incubation): 98°C for 3 min
Step 2 (Denaturation): 95°C for 30 s
Step 3 (Denaturation): 95°C for 10 s
Step 4 (Annealing): 55°C for 30 s
Step 5 (Extension): 72°C for 30s
(Repeat steps 2-4 for a total of 12 cycles)
Step 6 (Final Extension): 72°C for 5 min
Step 6: 12°C hold
-
PCR cleanup and purification
Let AMPure XP DNA Magnetic Beads stand at room temperature at least 30 min.
Add 30 µL of AMPure XP DNA Magnetic Beads to each reaction. Mix well by vortexing.
Repeat steps 4-15 of section I to purify amplified DNA.
It’s safe to stop and store PCR reactions at -20°C. Sequence samples within 2 weeks.
-
Quality metrics
Check concentration of each sample using Qubit dsDNA HS Kit according to manufacturer’s instructions. Concentrations between 1-10 ng/µL per sample are expected.
Check size distribution of each sample using the Agilent Bioanalyzer HS-DNA kit, according to manufacturer’s instructions. Libraries are expected to show weak a large peak between ~300 bp and ~1,000 bp, centered at ~500 bp (Figure 2B).
-
Generation of 2 nM multiplexed library
Normalize the concentration of each library to 2 nM.
Combine 2 µL of each normalized library for a final volume of 48 µL. Mix well.
Confirm concentration of final multiplexed library using Qubit dsDNA HS Kit according to manufacturer’s instructions. Adjust concentration to 2 nM if necessary.
-
Paired-end sequencing
Perform paired-sequencing according to manufacturer’s protocol. Here, we describe the process for NextSeq 2000 using Nextseq 1000/2000 P2 reagents (100 cycles) v3.
Combine 12 µL of multiplexed sample with 12 µL of NextSeq RSB with Tween buffer. Vortex briefly and centrifuge for 1 min.
Combine 1.8 µLof Read1 CustomSeqB primer with 600 µL of HT1 buffer. Vortex and centrifuge.
Add 20 µL of diluted library to the bottom of the library well of the sequencing cartridge.
Load 550 µL of Read1 CustomSeqB primer dilution into well #1 of sequencing cartridge.
Follow manufactures instructions to start sequencing run.
Select custom 1 for Read 1.
Setup the following sequencing parameters and run: Read 1: 26 bp, Read 2: 82 bp, Read 1 Index: 8 bp.
-
Generation of files needed for Drop-seq core computational protocol (see Note 5).
-
Generate sequence dictionary using the following command:
java -jar /path/to/picard/picard.jar CreateSequenceDictionary \
REFERENCE=my.fasta \
OUTPUT= my.dict \
SPECIES=species_name
-
Generate refFlat annotation file using the following command:
/path/to/Drop-seq_tools/ConvertToRefFlat \
ANNOTATIONS_FILE=my.gtf \
SEQUENCE_DICTIONARY=my.dict \
OUTPUT=my.refFlat
-
Generate reduced GTF file using the following command:
/path/to/Drop-seq_tools/ReduceGtf \
GTF=my.gtf \
SEQUENCE_DICTIONARY=my.dict \
OUTPUT=my.reduced.gtf
-
Generate intervals files using the following command:
/path/to/Drop-seq_tools/CreateIntervalsFiles \
REDUCED_GTF=my.reduced.gtf \
SEQUENCE_DICTIONARY=my.dict \
PREFIX=my \
OUTPUT=/path/to/output/files \
MT_SEQUENCE=chrM
-
Generate genome directory for alignment process using the following command:
/path/to/STAR/STAR \
--runMode genomeGenerate \
--runThreadN 8 \
--genomeDir path/to/output/files\
--genomeFastaFiles path/to/FASTA/file \
--sjdbGTFfile path/to/GTF/file \
--sjdbOverhang 81
-
-
Data processing pipeline for the generation of digital gene expression tables.
-
Demultiplex i7 indexes using the following command:
bcl2fastq -runfolder-dir /path/to/rawdata/folder/ \
- output-dir /path/to/output/folder/ \
--no-lane-splitting \
--loading-threads 8 \
-- writing-threads 8 \
--minimum-trimmed-read-length 0 \
--mask-short-adapter-reads 0 \
--sample-sheet /path/to/sample/sheet/
-
Evaluate quality of sequencing data for all files using the following command:
/path/to/fastqc *.fastq.gz
(go to https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/3%20Analysis%20Modules/ , for information on how to interpret results of FastQC quality metrics)
-
For each i7-index sample, convert Fastq file to Sam file while merging R1 and R2 files using the following command:
java -jar /path/to/picard/picard.jar CreateSequenceDictionary \
F1= SampleX_R1.fastq.gz \
F2= SampleX_R2.fastq.gz \
O= SampleX.bam
SM=SampleX
-
Extract the RT index sequence of each read using the following command:
/path/to/Drop-seq_tools/TagBamWithReadSequenceExtended \
INPUT=SampleX.bam \
OUTPUT=Indexed_SampleX.bam \
SUMMARY= Indexed_SampleX.summary \
BASE_RANGE= 9-16 \
BASE_QUALITY=10 \
DISCARD_READ=False \
TAG_NAME=XC \
NUM_BASES_BELOW_QUALITY=1
-
Extract the molecular barcode sequence of each read using the following command:
/path/to/Drop-seq_tools/TagBamWithReadSequenceExtended \
INPUT=Indexed_SampleX.bam \
OUTPUT=UMIed_SampleX.bam \
SUMMARY= UMIed_SampleX.summary \
BASE_RANGE= 1-8 \
BASE_QUALITY=10 \
DISCARD_READ=True \
TAG_NAME=XM \
NUM_BASES_BELOW_QUALITY=1
-
Remove reads with low quality RT index or molecular barcode sequences using the following command:
/path/to/Drop-seq_tools/FilterBam \
TAG_REJECT=XQ \
INPUT=UMIed_SampleX.bam \
OUTPUT= Filtered_SampleX.bam
-
Trim reads containing part of the template switch oligo using the following command:
/path/to/Drop-seq_tools/TrimStartingSequence \
INPUT=Filtered_SampleX.bam \
OUTPUT= Trimmed_SampleX.bam \
OUTPUT_SUMMARY= Trimmed_SampleX.summary \
SEQUENCE= AAGCAGTGGTATCAACGCAGAGTGAATGGG \
MISMATCHES=0 \
NUM_BASES=5
-
Trim polyA tails within reads using the following command:
/path/to/Drop-seq_tools/PolyATrimmer \
INPUT=Trimmed_SampleX.bam \
OUTPUT= PolyATrimmed_SampleX.bam \
OUTPUT_SUMMARY= PolyATrimmed_SampleX.summary \
SEQUENCE= AAGCAGTGGTATCAACGCAGAGTGAATGGG \
MISMATCHES=0 \
NUM_BASES=6 \
USE_NEW_TRIMMER =true
-
Convert bam files back to Fastq format using the following command:
java jar /path/to/picard/picard.jar SamToFastq \
INPUT=PolyATrimmed_SampleX.bam \
FASTQ= PolyATrimmed_SampleX.fastq
-
Align reads to genome using the following command:
/path/to/STAR/STAR \
--runMode alignReads \
--runTreadN 8 \
--genomeDir path/to/genome/folder/ \
--readFilesIn PolyATrimmed_SampleX.fastq \
--outSAMtype BAM \
--SortedByCoordinate \
--alignSoftClipAtReferenceEnds No \
--outFilterScoreMinOverLread 0.66 \
--outFilterMatchNminOverLread 0.66
-
Calculate quality metrics for RNA-sequencing using the following command:
java jar /path/to/picard/picard.jar CollectRNASeqMetrics \
I=Aligned_SampleX.bam \
O= SampleX.RNA_Metrics \
REF_FLAT = my.refFlat \
STRAND=FIRST_READ_TRANSCRIPTION_STRAND \
CHART_OUTPUT=SampleX_Metagene.plot \
RRNA_FRAGMENT_PERCENTAGE=0.8 \
MINIMUM_LENGTH=500 \
RIBOSOMAL_INTERVALS=/path/to/my.intervals/rRNA.intervals
(we expect the overwhelming majority of bases to map to mRNAs, and the metagene plot should show a strong 3’ bias as seen in Figure 3.)
-
Merge aligned bam file with the Indexed and UMIed bam file using the following command:
java jar /path/to/picard/picard.jar MergeBamAlignment \
REFERENCE_SEQUENCE= /path/to/Genome/fasta \
UNMAPPED_BAM=UMIed_SampleX.bam \
ALIGNED_BAM= Aligned_SampleX.bam \
OUTPUT= Merged_SampleX.bam \
INCLUDE_SECONDARY_ALIGNMENTS=false \
PAIRED_RUN =false
-
Tag reads with gene name using the following command:
/path/to/Drop-seq_tools/TagReadWithGeneFunction \
I=Merged_SampleX.bam \
O= GeneTagged_SampleX.bam \
ANNOTATIONS_FILE= my.refFlat
-
Generate digital gene expression table using the following command:
/path/to/Drop-seq_tools/DigitalExpression \
I=GeneTagged_SampleX.bam \
O= SampleX.DGE.gz \
STRAND_STRATEGY=SENSE \
SUMMARY=SampleX.DGE.summary.txt \
CELL_BC_FILE=RT_Indexes*
*This is a text file listing line-by-line the 8nt sequences of all indexes used.
-
Video 1. Delineation and laser capture microdissection of soma and dendritic processes of five single neurons.
Figure 4. Sample pooling workflow.

A. Pooling performed in section I, step 2. Two 96-well plates containing 48 samples each (only rows A-D are shown, as rows E-H are unoccupied). Somatic samples are placed in columns 1, 3, 5, and 7, and their respective dendrites are placed in columns 2, 4, 6 and 8. Control somata from less accessible areas are collected in columns 9, 10, and 11. Column 12 contains empty cuts (negative controls). Each row of the plate contains 1 of 16 different RT-UMI-Index primers. In section I step 2, all samples within the same columns of two complementary plates (containing a non-overlapping set of indexes) are pooled together. B. Pooling performed in section I, step 17. Two complementary samples within the same column are pooled together to reduce sample number to 24. C. Pooling performed in section N, step 2. After adding Nextera indexes, all samples are pooled together to reduce sample number to 1.
Figure 2. Expected size distribution of cDNA libraries.

A. Bioanalyzer electropherogram from a pooled of dendritic samples after section J, step 2. B. Bioanalyzer electropherogram from same sample after section N, step 2.
Figure 3. scRNA-seq data 3’ bias.

Metagene plot showing the expected distribution of reads (normalized coverage) across the length of an mRNA, where 0 represents the 5’ most region and 100 the 3’ most region.
Data analysis
A dataset generated using this method on rat primary hippocampal neurons can be found in the NCBI Gene Expression Omnibus under the accession code GSE157204. A digital expression table generated from the data and a metadata file describing the experimental design can be found in Supplementary File 1 and Supplementary File 2 of our publication (Perez et al., 2021). The R source code used in analyses of the data can be found in our GitHub repository DOI: 10.5281/zenodo.4384479 .
The ERCCs Spike-Ins included in the protocol can be used to quantify the accuracy of the experiment, by comparing the input number of individual ERCCs with their average number in the sequenced results. A Pearson correlation >0.8 is expected. Also, the sensitivity of the experiment (how many of the mRNAs present in the lysis reaction were detected) can be calculated by determining the detection probability of ERCCs with different input values. On average, we detected 1 out of every 4 molecules present.
Before analyzing the digital gene expression table, additional quality evaluations and data cleaning are necessary. First, ERCCs should be used to evaluate the quality of library preparation and sequencing: outliers with little to no ERCCs sequenced should be discarded. Second, somatic or dendritic samples with a number of RNA molecules comparable to that of empty cuts, should also be discarded. Finally, as described below, unsupervised dimensionality methods can reveal cell types present in the dataset. In our experience, this may reveal samples enriched in markers for apoptotic or glial cells, both of which should be discarded.
To classify samples into types, use unsupervised dimensionality reduction methods such as UMAP or tSNE, and nearest neighbor approaches such as k-clustering, all of which are available in the Seurat package ( Stuart et al., 2019 ). As dendritic transcriptomes are usually shallower than somatic ones, we perform cell type classifications based only on the somatic samples, and later extrapolate this information to their corresponding dendrites.
Differential expression analyses can be performed both between the somata or dendrites of different cell types, or between the soma and dendrites of single neurons. For these, we recommend test based on logistic regression ( Ntranos et al., 2019 ), or a Poisson generalized linear model ( Stuart et al., 2019 ). When comparing soma versus dendrites, we recommend a paired-differential expression analysis using cell of origin as a latent variable.
Notes
During the experimental design phase, several factors should be considered when choosing an appropriate N. Power analyses should be perform considering the types of tests and analyses that will be eventually performed with the data, as the required N to achieve statistical power varies from test to test. For differential expression analyses between cell types or between subcellular compartments, we suggest tools optimized for single cell datasets such as powsimR ( Vieth et al., 2017 ) or scPower ( Schmid et al., 2020 ). The number of samples that can be profiled in a single sequencing run depends on the multiplexing capacity of the experimental setup. In this protocol, the RT primer carries 1 of 16 different indexes while the i7 Nextera PCR step adds 1 of 24 indexes to each of the previous ones, allowing a maximum of (16×24) 384 samples to be simultaneously profiled. For us, this number provided sufficient sequencing depth per sample. However, it is certainly possible to increase the number of indexes and therefore the multiplexing capacity per run. In principle, all samples could be derived from a single plate but this is not recommended. As LCM occurs at room temperature, and only one sample is collected at a time, RNA integrity will significantly decrease overtime. We observed a trend to less RNA molecules detected per sample (suggestive of lower RNA integrity) after 3 h of collection. Thus, we suggest to limit the number of samples collected per dish to an amount that can be safely collected in 3 h (in our case, 48). We recommend using dishes from multiple primary neurons preparations, since variability between preparations occurs, and thus dishes from the same preparation do not yield fully independent samples.
In addition to paired somatic and dendritic samples derived from the same single neurons, we recommend including two types of controls. To have a sense of the potential bias introduced by the selection of cells amenable for laser capture, we suggest collecting somata-only samples from less accessible neurons in the same dish. Secondly, as a negative control, we include samples in which Laser Pressure Catapulting is applied to regions in the dish that are devoid of soma and/or dendrites. The size of these cuts should be comparable to the area occupied by somata and/or dendrites. These samples serve to control for potentially contaminating extracellular RNAs, and can help set an expression cutoff to include a sample.
When using magnetic beads to purify DNA, it is important to thoroughly resuspend beads by vortexing, before mixing them with the samples. It is also important to accurately pipette the desired volume in section I-step 3, as inaccuracies in volume can increase or decrease the presence of smaller DNA fragments. If possible, use low-binding tips at this step. It is also important to monitor that the pellet does not become too dry and begins to show cracks in section I-step 11, as this will result in reduced DNA concentrations after resuspension.
To go from 384 collected samples to sequencing a single multiplexed sample, we incrementally pool at key steps of the protocol, namely steps 2 and 17 of section I, and step 2 of section N. To avoid pooling samples with the same index combination, and to perform the process rapidly and efficiently, we suggest the organization of samples based on the 96-well plate format illustrated in Figure 4.
Commands in sections P and Q are to be used in a command-line-interface in the directory where the analysis is performed.
Recipes
Cell Lysis Master Mix (Table 1)
RT Master Mix (Table 2)
PCR PreAmp Master Mix (Table 3)
Final PCR Master Mix (Table 4)
Table 1. Cell Lysis Master Mix.
| Cell Lysis Master Mix | [Units] | [Stock] | [Final] | 1 Rx (µl) | 12Rx Master Mix (µl)1 |
|---|---|---|---|---|---|
| Nuclease-free H2O | - | - | - | 1.602 | 21.1 |
| SingleShot Lysis Buffer | X | 6.25 | 1 | 0.48 | 6.3 |
| Qiagen Protease | mAU/ml | 900 | 45 | 0.15 | 2 |
| dNTPs | mM | 10 | 1 | 0.64 | 8.4 |
| RT-UMI-Index(1-16) Primer | µM | 100 | 1 | 0.064 | 0.8 |
| Diluted ERCCs2 | X | 100 | 1 | 0.064 | 0.8 |
| - | -- | - | Total | 3 | 39.4 |
Table 2. RT Master Mix.
| RT Master Mix | [Units] | [Stock] | [Final]2 | 1 Rx (µl) | 48Rx Master Mix (µl)1 |
|---|---|---|---|---|---|
| Nuclease-free H2O | - | - | - | 0.15 | 8.5 |
| SSIV buffer | X | 5 | 1 | 1.28 | 72 |
| MgCl2 | mM | 1000 | 6 | 0.04 | 2.2 |
| DTT | mM | 300 | 5 | 0.11 | 6 |
| Betaine | M | 5 | 1 | 1.28 | 72 |
| Template Switch Oligo | µM | 100 | 1 | 0.06 | 3.6 |
| RNase Inhibitor | U/µl | 40 | 1 | 0.16 | 9 |
| SuperScript IV | U/µl | 200 | 10 | 0.32 | 18 |
| - | - | - | Total | 3.4 | 191.3 |
Table 3. PCR PreAmp Master Mix.
| PCR PreAmp Master Mix | [Units] | [Stock] | [Final]4 | 1 Rx (µl) | 48Rx Master Mix (µl)1 |
|---|---|---|---|---|---|
| Nuclease-free H2O | - | - | - | 0.59 | 33 |
| KAPA HiFi HotStart Mix | X | 2 | 1 | 7 | 393.8 |
| ISPCR Primer | µM | 100 | 0.1 | 0.014 | 0.8 |
| - | - | - | Totals | 7.6 | 427.5 |
Table 4. Final PCR Master Mix.
| Final PCR Master Mix | [Units] | [Stock] | [Final]5 | 1 Rx (µl) | 24Rx Master Mix (µl)1 |
|---|---|---|---|---|---|
| Nuclease-free H2O | - | - | - | 8 | 250 |
| Nextera PCR Master Mix | X | 3.3333 | 1 | 15 | 468.75 |
| P5-ISPCR Primer | µM | 10 | 0.2 | 1 | 31.25 |
| - | - | - | Totals | 24 | 750 |
1. Master mix volume is calculated to account for pipetting errors.
2. To make 100× ERCC solution, dilute ERCC RNA Spike-In Mix 1, 1:200,000.
3. Final concentrations for RT master mix reagents are calculated based on a final volume of 6.4 µL (3 µL of cell lysis mix + 3.4 µL RT mix).
4. Final concentrations for PCR PreAmp master mix reagents are calculated based on a final volume of 14 µL (6.4 µL of RT reaction + 7.6 µL of PCR PreAmp mix).
5. Final concentrations for Final PCR master mix reagents are calculated based on a final volume of 50 µL (26 µL of Tagmentation reaction + 24 µL of Final PCR mix).
Acknowledgments
This work was supported by the Max Planck Society, and the Advanced Investigator award from the European Research Council (grant 743216), DFG CRC 1080: Molecular and Cellular Mechanisms of Neural Homeostasis, and DFG CRC 902: Molecular Principles of RNA-based Regulation. We thank Dr. Susanne tom Dieck, Ivy CW. Chan and current and past members of the Schuman lab for helpful discussions and advice. This protocol is derived from our previous work ( Perez et al., 2021 ; DOI: 10.7554/eLife.63092).
Competing interests
The authors declare no conflict of interest.
Ethics
The procedures involving animal care are conducted in conformity with the institutional guidelines that are in compliance with the national and international laws and policies (DIRECTIVE2010/63/EU; German animal welfare law, FELASA guidelines) and approved by and reported to the local governmental supervising authorities (Regierungsprasidium Darmstadt). The animals were euthanized according to annex 2 of 2 Abs. 2 Tierschutz-Versuchstier-Verordnung.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
Supplementary Data.
References
- 1. Aakalu G., Smith W. B., Nguyen N., Jiang C. and Schuman E. M.(2001). Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 30(2): 489-502. [DOI] [PubMed] [Google Scholar]
- 2. Alon S., Goodwin D. R., Sinha A., Wassie A. T., Chen F., Daugharthy E. R., Bando Y., Kajita A., Xue A. G., Marrett K., et al.(2021). Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems . Science 371(6528). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cajigas I. J., Tushev G., Will T. J., S. tom Dieck, Fuerst N. and Schuman E. M.(2012). The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74(3): 453-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glock C., Biever A., Tushev G. and Bartnik I.(2020). The mRNA translation landscape in the synaptic neuropil. bioRxiv. doi: 10.1101/2020.06.09.141960. [DOI] [Google Scholar]
- 5. Gumy L. F., Yeo G. S., Tung Y. C., Zivraj K. H., Willis D., Coppola G., Lam B. Y., Twiss J. L., Holt C. E. and Fawcett J. W.(2011). Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17(1): 85-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hafner A. S., Donlin-Asp P. G., Leitch B., Herzog E. and Schuman E. M.(2019). Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science 364(6441): eaau3644. [DOI] [PubMed] [Google Scholar]
- 7. Hagemann-Jensen M., Abdullayev I., Sandberg R. and Faridani O. R.(2018). Small-seq for single-cell small-RNA sequencing. Nat Protoc 13(10): 2407-2424. [DOI] [PubMed] [Google Scholar]
- 8. Holt C. E., Martin K. C. and Schuman E. M.(2019). Local translation in neurons: visualization and function. Nat Struct Mol Biol 26(7): 557-566. [DOI] [PubMed] [Google Scholar]
- 9. Macosko E. Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A. R., Kamitaki N., Martersteck E. M., et al.(2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161(5): 1202-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mazaré N., Oudart M. and Cohen-Salmon M.(2021). Local translation in perisynaptic and perivascular astrocytic processes- a means to ensure astrocyte molecular and functional polarity? J Cell Sci 134(2): jcs251629-11. doi: 10.1242/jcs.251629. [DOI] [PubMed] [Google Scholar]
- 11. Middleton S. A., Eberwine J. and Kim J.(2019). Comprehensive catalog of dendritically localized mRNA isoforms from sub-cellular sequencing of single mouse neurons. BMC Biol 17(1): 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moor A. E., Golan M., Massasa E. E., Lemze D., Weizman T., Shenhav R., Baydatch S., Mizrahi O., Winkler R., Golani O., et al.(2017). Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science 357(6357): 1299-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ntranos V., Yi L., Melsted P. and Pachter L.(2019). A discriminative learning approach to differential expression analysis for single-cell RNA-seq. Nat Methods 16(2): 163-166. [DOI] [PubMed] [Google Scholar]
- 14. Perez J. D., Dieck S. T., Alvarez-Castelao B., Tushev G., Chan I. C. and Schuman E. M.(2021). Subcellular sequencing of single neurons reveals the dendritic transcriptome of GABAergic interneurons. Elife 10: e63092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Picelli S., Faridani O. R., Bjorklund A. K., Winberg G., Sagasser S. and Sandberg R.(2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9(1): 171-181. [DOI] [PubMed] [Google Scholar]
- 16. Poon M. M., Choi S. H., Jamieson C. A., Geschwind D. H. and Martin K. C.(2006). Identification of process-localized mRNAs from cultured rodent hippocampal neurons. J Neurosci 26(51): 13390-13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakers K., Lake A. M., Khazanchi R., Ouwenga R., Vasek M. J., Dani A. and Dougherty J. D.(2017). Astrocytes locally translate transcripts in their peripheral processes. Proc Natl Acad Sci U S A 114(19): E3830-E3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schmid K. T., Cruceanu C., Böttcher A., Lickert H., Binder E. B., Theis F. J. and Heinig M.(2020). Design and power analysis for multi-sample single cell genomics experiments. bioRxiv 2020.04.01.019851. doi: 10.1101/2020.04.01.019851. [DOI] [Google Scholar]
- 19. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. 3rd Hao Y., Stoeckius M., Smibert P. and Satija R.(2019). Comprehensive Integration of Single-Cell Data. Cell 177(7): 1888-1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tóth E. N., Lohith A., Mondal M., Guo J., Fukamizu A. and Pourmand N.(2018). Single-cell nanobiopsy reveals compartmentalization of mRNAs within neuronal cells. J Biol Chem 293(13): 4940-4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vieth B., Ziegenhain C., Parekh S., Enard W. and Hellmann I.(2017). powsimR: power analysis for bulk and single cell RNA-seq experiments. Bioinformatics 33(21): 3486-3488. [DOI] [PubMed] [Google Scholar]
- 22. Wang G., Ang C. E., Fan J., Wang A., Moffitt J. R. and Zhuang X.(2020). Spatial organization of the transcriptome in individual neurons. bioRxiv 101:123–45.. doi: 10.1101/2020.12.07.414060. [DOI] [Google Scholar]
- 23. Zhong J., Zhang T. and Bloch L. M.(2006). Dendritic mRNAs encode diversified functionalities in hippocampal pyramidal neurons. BMC Neurosci 7: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.