

Abstract
The study of genetic minority variants is fundamental to the understanding of complex processes such as evolution, fitness, transmission, virulence, heteroresistance and drug tolerance in Mycobacterium tuberculosis (Mtb). We evaluated the performance of the variant calling tool LoFreq to detect de novo as well as drug resistance conferring minor variants in both in silico and clinical Mtb next generation sequencing (NGS) data. The in silico simulations demonstrated that LoFreq is a conservative variant caller with very high precision (≥96.7%) over the entire range of depth of coverage tested (30x to1000x), independent of the type and frequency of the minor variant. Sensitivity increased with increasing depth of coverage and increasing frequency of the variant, and was higher for calling insertion and deletion (indel) variants than for single nucleotide polymorphisms (SNP). The variant frequency limit of detection was 0.5% and 3% for indel and SNP minor variants, respectively. For serial isolates from a patient with DR-TB; LoFreq successfully identified all minor Mtb variants in the Rv0678 gene (allele frequency as low as 3.22% according to targeted deep sequencing) in whole genome sequencing data (median coverage of 62X). In conclusion, LoFreq can successfully detect minor variant populations in Mtb NGS data, thus limiting the need for filtering of possible false positive variants due to sequencing error. The observed performance statistics can be used to determine the limit of detection in existing whole genome sequencing Mtb data and guide the required depth of future studies that aim to investigate the presence of minor variants.
Keywords: M. tuberculosis, minor variant calling, low-frequency variant calling, LoFreq, whole genome sequencing, benchmarking
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is one of the top 10 causes of death worldwide and the leading cause of death from a single infectious agent [1]. Mtb continuously evolves through genomic acquisition of single nucleotide polymorphisms (SNPs), insertions and deletions (indels). The acquisition of genomic variants that confer drug resistance and the acquisition of compensatory mutations to overcome the fitness effect of these drug resistance causing mutations [2] have been widely studied. Understanding the patient’s isolate’s genomic drug resistance profile will help to define the optimal individualized treatment [3, 4]. Investigation of genomic variants in Mtb associated with other characteristics such as virulence, bacterial fitness, transmissibility and drug tolerance remains limited.
For decades, it was believed that Mtb infection was clonal with a single genome to be representative of an infection [5]. Advances in molecular biology have highlighted genomic diversity in Mtb and thereby frequent occurrence of polyclonal infections [6]. Within a single patient, multiple unrelated clones can be present from different infections (mixed infection) or multiple closely related clones can be present reflecting micro-evolution in a previously clonal Mtb population within a lesion. Variants that represent polyclonal infection can be present at different levels, showing frequencies anywhere between 0 and 100% and can become the only (fixed) variant of the Mtb population or become lost [7]. Confidently calling the presence of minor variants in Mtb whole genome sequence (WGS) data is essential to study population dynamics. For example, the dynamics of heteroresistance—where both wild type and mutant alleles co-occur in genes associated with resistance—is of clinical interest as this has been associated with poor treatment outcomes [8]. Understanding drug tolerance, by studying the change in Mtb populations in response to drug exposure, and gaining insight in how Mtb populations can overcome drug pressure in the absence of drug resistance mutations can help with the development of novel treatment strategies [9]. Improved sensitivity to characterize Mtb population structures can greatly benefit the accuracy of TB transmission studies, especially in high burden settings where mixed infections occur frequently [6].
Until recently, detection of minor variants in WGS data was difficult due to the inability of ad hoc trimming, filtering and threshold approaches to distinguish true low-frequency variants from sequencing error [10]. Minor variants are therefore usually excluded from bioinformatic analyses even though they may be biologically relevant and fundamental to the analysis of population evolution and dynamics [11]. Lately, a bioinformatic tool called BinoSNP has been developed to detect minor drug resistant variants in Mtb next generation sequencing (NGS) data [4]. BinoSNP evaluates a user-defined list of genomic positions using a binomial test procedure to determine the presence of low-frequency variants in Mtb NGS data. The tool can accurately distinguish between true variants and sequencing error at a 1% frequency with a coverage ≥400x [4], but is restricted to the detection of SNPs in (per default) resistance conferring genes. Consequently, tools such as BinoSNP are not suitable for detection of variants that are not pre-specified such as de novo detection of minor populations in genetic regions not associated to drug resistance.
LoFreq [12], a genome wide variant calling tool that models sequencing run-specific error rates and position-specific sequencing biases to call minor (<0.05%) variants overcomes these limitations as it allows detection of both low-frequency SNPs and indels in both pre-specified genetic regions such as resistance associated loci and previously unidentified genetic regions. The performance of LoFreq has been assessed for several pathogens (dengue virus [12], respiratory syncytial virus [10]). Unfortunately, as is the case for most variant-calling tools, performance evaluation has been restricted to the typical ‘average’ human or viral dataset, containing variants present at various frequencies and/or at a mixture of sequencing depths [10, 12–21]. In practice, however, researchers are interested in more precise information on the performance of bioinformatic tools that they can use to evaluate the performance of a tool for their own specific application. Due to differences in genome size, GC content, presence of highly repetitive regions and ploidy, the appropriateness of the assumptions and statistics used in variant callers may differ for microbial genomes [22]. Benchmarking of a WGS variant calling tool that is suitable to detect minor Mtb variants at different coverage depths and different variant frequencies remained to be done.
We generated in silico (simulated) WGS datasets to assess the performance (sensitivity and precision) of LoFreq for the detection of SNPs and indels when present at a range of low-level frequencies and at a range of sequencing depths. We also applied the LoFreq tool to call minor variants in the Rv0678 gene in clinical Mtb WGS data and compared this to the findings obtained by targeted deep sequencing (TDS) [23].
Methods
Generation of in silico datasets
We used the ART next-generation sequencing simulator (Version 2.5.8) [24] to generate in silico sequencing reads in a way that mimics the technology-specific sequencing process. To simulate pair-end reads that would be obtained by sequencing Mtb DNA on the Illumina MiSeq v3 system, we used ART to generate reads with a length of 150 bp and a mean DNA fragment size of 350 bp with a standard deviation of 18.7 bp. ART’s default parameters were used except for masking that was turned off to include repetitive regions. Ten randomly mutated H37Rv reference genomes containing 1000 SNPs, 50 single base deletions and 50 single base insertions were generated to (1) guarantee sufficient statistical power, and (2) to reflect the observation that clinical samples typically differ 600–2600 SNPs from the H37Rv reference genome [25]. The performance of LoFreq to detect minor variants present at 0.1–20% frequency was investigated. Levels of coverage depth ranged from 30x to 1000x to explore the extremes of sequencing depth obtained when sequencing at minimalistic (30x) or deep sequencing (1000x) levels for Mtb WGS. In silico generated H37Rv (NC_000962.3) reference genome sequence reads were then merged with in silico generated random (but known) mutant sequence reads (SNPs and indels) to generate a WGS dataset of 640 Mtb genomes that represent the 64 combinations of minor variant frequency (0.1, 0.5, 1, 3, 5, 10, 15 and 20%) and depth of coverage (30, 50, 100, 200, 300, 400, 500 and 1000x). For example, to generate a dataset where a 3% variant is present at 400x depth, we merged 12x in silico generated mutant sequence reads with 388x in silico generated H37Rv (NC_000962.3) reference genome sequence reads.
Clinical WGS and TDS data
Clinical data used in this study have been published earlier by De Vos et al. [23] and are available online. WGS data from serial isolates from a previously reported case study were retrieved from the public European Nucleotide Archive (Project number: PRJEB32109) and TDS of the full Rv0678 region were retrieved from Bioproject at NCBI (project number PRJNA531707) [23].
Variant calling
Prior to LoFreq variant calling, in silico generated and clinical WGS fastq files were processed using the CompleX Bacterial Samples (XBS) pipeline to generate bam files [26]. Briefly, Fastq sequence data were mapped to the reference genome (H37Rv NC_000962.3) using BWA mem, after which the XBS pipeline performed identified read deduplication in the bam files using GATKMarkDuplicates (Picard) (Reference paper accepted, still to be added). Of note, unlike other Mtb pipelines, base quality score recalibration (BQSR) is not applied by XBS to avoid that variants in contaminant (non-Mtb) DNA are interpreted as systematic error by BQSR, which would result in reduced base qualities, including for genuine Mtb variants. The resulting bam files were indexed using the H37Rv reference genome and indel quality scores were assigned using LoFreq indelqual prior to variant calling. LoFreq (v2.1.2) was run using default parameters with the indel variant calling function turned on to evaluate the performance of both SNPs and indels [12]. Variant calling using Lofreq was performed on the whole Mtb genome, including highly repetitive regions such as PE/PPE regions.
For clinical isolates, variant frequencies called by LoFreq were compared to the variant frequencies earlier reported by De Vos et al. [23], where variant calling was performed (1) on TDS data (using the ASAP pipeline) and (2) on WGS data by means of a combination of GATK and a visual approach using Tablet (after preprocessing WGS data with Novoalign).
Statistical analysis of LoFreq’s performance
To assess the performance of LoFreq at each of the 64 combinations of variant frequency (0.1, 0.5, 1, 3, 5, 10, 15 and 20%) and depth of coverage (30, 50, 100, 200, 300, 400, 500 and 1000x), we compared the truth, i.e. the mutations introduced in the in silico generated mutated H37Rv reference genome, to the observed, i.e. the absence or presence of each minor variant as reported in the VCF files generated by LoFreq. This allowed us to calculate the number of true positive (TP), false positive (FP), and false negative (FN) variants reported by LoFreq in each of the 640 WGS datasets. Using these results, we assessed the performance of LoFreq by calculating the sensitivity (defined as the ratio of true positives over true positives plus false negatives) and precision (defined as the ratio of true positives over true positives plus false positives) at each of the 64 combinations. All analyses of the performance of LoFreq on in silico generated Mtb WGS data were done in Rstudio. Regression lines were constructed applying locally estimated scatterplot smoothing (LOESS) using the ggplot package in RStudio.
For the patient samples, the detection of minor variants by LoFreq could not be compared to a known set of all true variants (as was done for in silico datasets). Instead, the detection of minor variants by LoFreq in the Rv0678 gene in the WGS data was compared to the variants identified by TDS of the Rv0678 gene of the same samples as previously described by De Vos et al. [23] In addition, we also compared the ability of Lofreq to detect these minor variants in the WGS data to what has been previously been reported by De Vos et al. [23], where a combination of GATK and a visual inspection was used. To compare the variant frequencies that were predicted by the different variant calling methods (TDS, GATK/Visual-method and LoFreq) two proportion Z-tests were performed.
Results
At very high Mtb depth of coverage of 1000x, the limit of detection of LoFreq to call minor variants was 3% for SNPs, with a sensitivity of 48.6% (95% CI 47.7%, 49.6%) (Figure 1, Supplementary Figure S1). At this depth, sensitivity increased to ≥98% for variants present at frequency ≥5%. For variants present at 10% frequency and higher, the sensitivity increased rapidly with increasing depth of coverage: at 50, 200 and ≥400 depth, sensitivities were 19.6% (95% CI 18.9–20.4%), 90.7% (95% CI 90.1–91.2%) and 98.5% (95% CI 98.2–98.7%), respectively.
Figure 1.
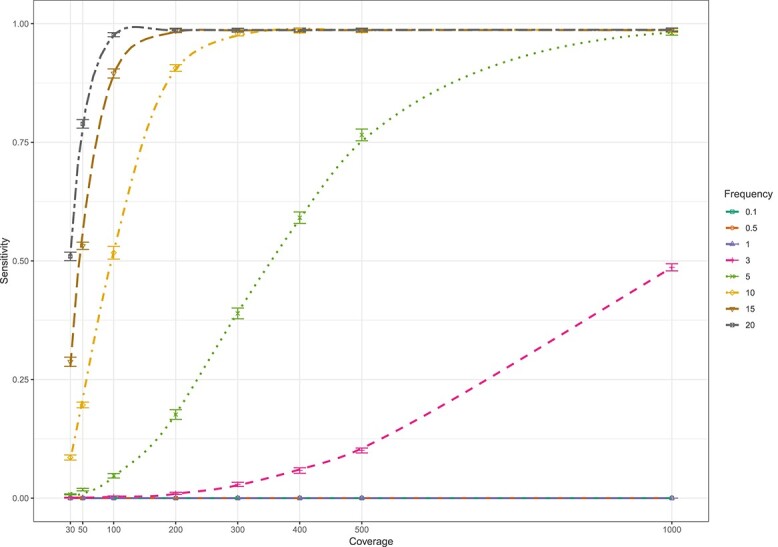
Sensitivity of the LoFreq tool for whole genome calling of minor SNP variants (present at 0.1–20%) at different levels of coverage (30–1000x) when evaluated on in silico whole genome sequence data sets.
Lowfreq’s limit of detection was lower for insertions than for SNPs. The sensitivity of LoFreq for the detection of insertions present at 0.5% frequency was 43.6% (95% CI 39.2–48.1%) at 1000x coverage. Insertions present at 1% were detected with a sensitivity of 56.6% (95% CI 52.1–61.0%) at 500x coverage and 92.2% (95% CI 89.5–94.4%) at 1000x coverage (Figure 2, Supplementary Figure S2). At higher minor variant frequencies (≥3%), sensitivity to detect insertions increased fast with increasing coverage before plateauing around 98.8%. At low coverage (30x), insertions present at 10, 15 or 20% could be detected with 59.4% (95% CI 55.0–63.7%), 78.8% (95% CI 75.0–82.3%) and 91.8% (95% CI 89.0–94.1%) sensitivity, respectively.
Figure 3.
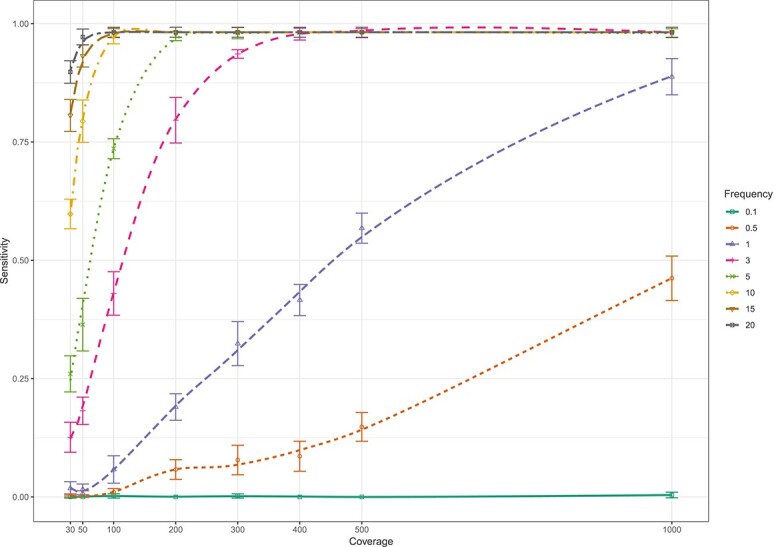
Sensitivity of the LoFreq tool for whole genome calling of minor deletion variants (present at 0.1–20%) at different levels of coverage (30–1000x) when evaluated on in silico whole genome sequence data sets.
Figure 2.
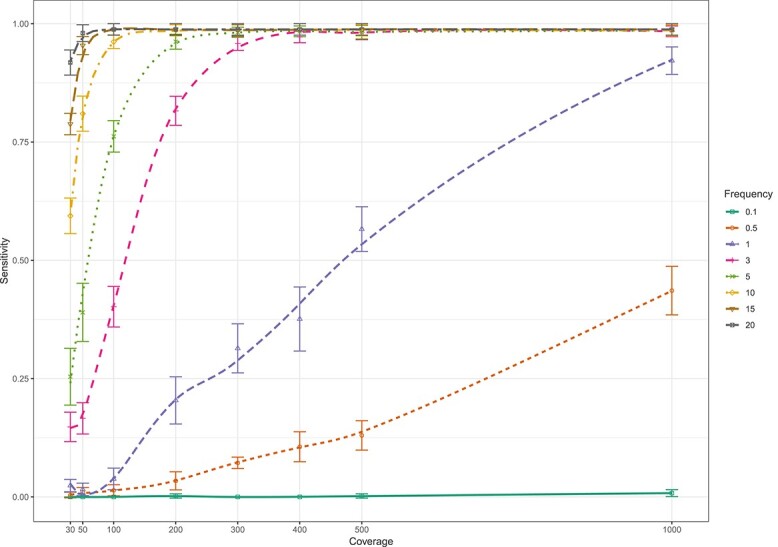
Sensitivity of the LoFreq tool for whole genome calling of minor insertion variants (present at 0.1–20%) at different levels of coverage (30–1000x) when evaluated on in silico whole genome sequence data sets.
Similar results were obtained for the detection of deletions. When present at 0.5% frequency, the sensitivity for detection of deletions was 46.2% (95% CI 41.8–50.7%) at 1000x coverage. The sensitivity to detect minor variants present at 1% was 56.8% (95% CI 52.3–61.2%) at 500x coverage and 88.8% (95% CI 85.7–91.4%) at 1000x coverage (Figure 3, Supplementary Figure S3). Sensitivity to detect deletions increased quickly with increasing coverage before plateauing around 98.2% for variants present at ≥3%. At low coverage (30x), the sensitivity to detect deletions present at 10, 15 and 20% was 59.8% (95% CI 55.4–64.1%), 80.6% (95% CI 76.9–84.0%) and 89.8% (95% CI 86.8–92.3%), respectively. In case of FP indel mutations, LoFreq reported 91.8% (45/49) of the FP deletions and 5.4% (2/37) of the FP insertions to contain a large (>10 base-pair) insertion or deletion region.
With regards to precision, our analysis of in silico data showed that LoFreq had a very low rate of calling false positives resulting in a precision of 1 for the detection of SNPs independent of frequency of the minor variant and depth of coverage (Supplementary Table S1). In the in silico datasets, a few false positive indels were reported, resulting in precision estimates between ≥99.5% for insertions and ≥96.7% for deletions (Supplementary Table S1).
In addition to simulation experiments, we assessed LoFreq’s ability to detect minor variants in clinical WGS data (at 62x average depth) using four serial Mtb samples collected from a patient with XDR-TB who failed a BDQ-containing treatment regimen [23] and compared results to when variant calling was performed by GATK followed by visual inspection using Tablet or on TDS data. LoFreq detected all variants in Rv0678 as detected by TDS, including the variants present at lower frequency (5.7% and 3.2%). In contrast, the GATK/visual detection method detected variants present at a frequency exceeding 25%, but missed the variants that occurred at a frequency of ≤5.7% (Table 1). Predicted variant frequencies also differed between TDS and LoFreq for all (four) high frequency (>65%) variants, with LoFreq systematically predicting a lower frequency of variants to be present. A similar observation was made for three out of four of these variants when comparing predicted variant frequency when variant calling was done using GATK and visual inspection using Tablet or by LoFreq (Table 1). For lower frequency variants no significant difference was observed when comparing predicted variant frequency between the different variant calling methods.
Table 1.
Minority populations identified in Rv0678 when variant calling was performed on TDS data or whole genome sequencing data with variant calling either performed by a combination of GATK and visual inspection (using Tablet) or by the LoFreq variant caller
| Sample accession number European nucleotide archive | Rv0678 variant | Variant frequency % (number of mutant/total reads) | P-value | ||||
|---|---|---|---|---|---|---|---|
| TDS | WGS GATK/Visual | WGS LoFreq | TDS versus GATK/Visual | TDS versus LoFreq | GATK/Visual versus LoFreq | ||
| SAMEA5562524 | 192 G ins | 96.66% (17 551/18 158) | 100% (56/56) | 87.06% (74/85) | 0.31 | <0.0001 | 0.013 |
| SAMEA5562526 | 138 GA ins | 97.52% (13 299/13 638) | 100% (75/75) | 80.91% (89/110) | 0.31 | <0.0001 | 0.0002 |
| SAMEA5562527 | 138 G ins | 65.48% (9317/14 230) | 63% (45/71) | 55.21% (53/96) | 0.81 | 0.046 | 0.37 |
| 138 GA ins | 28.35% (4034/14 230) | 25% (18/71) | 19.79% (19/96) | 0.67 | 0.08 | 0.50 | |
| 192 G ins | 3.22% (461/14 230) | No MV detected: data missing | 4.49% (4/89) | 0.71 | |||
| SAMEA5562528 | 138 G ins | 91.68% (13 029/14 212) | 96% (79/82) | 81.55% (84/103) | 0.18 | 0.0004 | 0.004 |
| 138 GA ins | 5.86% (832/14 212) | No MV detected:data missing | 6.80% (7/103) | 0.85 | |||
ins = insertion; WGS = whole genome sequencing; TDS = targeted deep sequencing; MV = minor variant.
In terms of speed, we found LoFreq’s runtime to scale linearly with sequencing depth. Using a single core QEMU Virtual CPU version 0.12 operating at 2.5GHz the runtime for LoFreq was roughly 10 min for assigning indel quality scores using the LoFreq Indelqual command and approximately 2 h to perform the variant calling using the LoFreq call command for a single sample where the full Mtb genome (size = 4.4 Mbp) was sequenced at 1000× coverage.
Discussion
The finding that LoFreq’s sensitivity to detect minor variants increases with sequencing depth and variant frequency is consistent with the general assumption that sequencing populations at higher coverages reduces the uncertainties associated with random sequencing errors [27]. This finding is in sheer contrast with two previous studies where similar coverage ranges where investigated and LoFreq’s sensitivity was not found to be significantly affected by depth of coverage [10, 13]. Moreover, our results indicate that LoFreq’s performance depends on the type of variant to be detected, with greater sensitivity to detect indel mutations than SNPs. This likely reflects that random sequencing errors generated by short-read sequencers are mostly SNPs rather than indels [28], allowing LoFreq to more rapidly and confidently call an observed indel variant as true as opposed to a sequencing error.
Congruent with what is reported in literature, our in silico assessment confirms LoFreq to be a conservative variant caller with high precision, minimizing the need for subsequent filtering of false positive variants and potentially losing a significant proportion of true positive variants [10]. In contrast to the perfect precision of 1 when variant-calling is performed on in silico introduced SNPs, in some cases a precision smaller than 1 (but >0.96) for calling indel variants was observed, which is in agreement with what has been previously reported and has been attributed to mis-alignment of the indel supporting reads [14]. We further observed that for a considerable proportion of FP indels (91.8% for deletions and 5.4% for insertions) LoFreq reported considerably large (>10 bp) indels to be present, suggesting that additional filtering of indel variants on length might further decrease FP-rate.
From a clinical perspective, previous publications have suggested that only variants occurring at a frequency of ≥19% become fixed in Mtb populations [7, 11]. This clinically relevant variant frequency threshold is further supported by the observation that the presence of low-frequency resistance mutations (<5%) does not affect treatment outcomes of patients infected with drug-susceptible TB [29]. Our in silico results indicate high specificity (>0.97) to detect minor Mtb variants at such clinically relevant frequency levels (20%) when sequenced at ongoing sequencing depth (100X), supporting LoFreq to be a clinically relevant variant calling tool for Mtb WGS data.
On the other hand, from a biological perspective, it can be expected that variants with biological advantages (such as drug resistance, drug tolerance or higher fitness) may be selected even when initially occurring at very low frequencies [30]. For variants occurring below or at the detection limit (3% for SNP calling and 0.5% for indel calling) for which—even at high coverage (1000X)—sensitivity of LoFreq is low (<50%), very high coverages currently seem indispensable, favoring a targeted sequencing approach.
The study by McCrone et al. [15] found LoFreq’s sensitivity to call variants in data resembling clinical samples to be lower than what was expected from previous benchmarking, pointing out potential variation in LoFreq’s performance on clinical data. In contrast to this study (performed on viral populations with very low to low (0.16–5%) frequency variant populations), we found LoFreq able to detect all TDS-determined minor variants in WGS data from clinical isolates. In our setup, LoFreq detected two low-frequency variants (3.22% and 5.86%) that could not be identified by a combination of GATK and visual inspection. The ability of LoFreq to detect these low-frequency variants at a median coverage of only 62X indicates that the results of in silico simulations described above may underestimate sensitivity compared to when clinical data are used. In our hands, LoFreq did not have lower sensitivity to detect variants in clinical data compared to in silico datasets. Other studies have shown that sequential sequencing of serial patient samples generates a large number of transient variants. It is however unclear whether such variants are genuine or the result of sequencing error [7]. The fact that for our clinical dataset LoFreq finds exactly and exclusively the same variants that are found by TDS (which is known to be precise and where called variants are unlikely to be due to sequencing error when present >1%), suggests that variants called by LoFreq are genuine rather than sequencing error. For the transient variants described in the clinical study referred to in this paper, we would thus argue that variants are thus genuine rather than sequencing error.
For resistance-associated genes, variants are often categorized into micro- (<5%) and macro- (5–95%) heteroresistant variants [30]. However, our observation that absolute variant frequencies diverge between the chosen variant caller for multiple samples indicates that such categorization should be done with care. Multiple factors could result in discrepant variant frequencies: (1) stochasticity of the sequenced reads (which decreases with sequencing depth and thus favors variant frequency as predicted by TDS (>13.000X) as compared to WGS (62X)); (2) Some forms of bias tend to push variants to above 50% allele frequency, particularly at higher coverages and when more selective events occur. Forms of PCR biases present in both TDS and WGS library preparations could thus underly the observed variation in variant frequency and explain why lower frequency variants increase and higher frequency variants decrease when variant calling is done using TDS compared to WGS.
Strengths and Limitations
To our knowledge, this is the first study benchmarking a WGS variant calling tool suitable to detect minor Mtb variants. This study yielded precise in silico as well as clinical information on the performance of bioinformatic tools, allowing researchers to evaluate the performance of tools for their own specific application.
One limitation of the in silico validation performed in this study is that the introduced indel mutations were limited to single nucleotide insertion or deletions, while the power to sensitively detect indels has been reported to decrease with indel-length [14]. This is particularly relevant as longer indel regions (>50 bp) have shown to be present in the Mtb genome [31]. Moreover, indels have been reported to occur with increased frequency in low-complexity regions in the Mtb genome where indel-calling is known to be more error-prone, while in silico generated indels were randomly distributed throughout the genome [32]. Therefore, the sensitivity of LoFreq to detect longer indels and indels present in low-complexity regions requires further investigation.
Another limitation of the study is that the small clinical sample size did not allow to statistically validate LoFreq’s in silico predicted performance metrics when applied on clinical sequencing data. For the same reason, our serial data (corresponding to a single patient) do not allow us to properly address the debated question whether transient variants observed upon sequential sampling are in general genuine (as suggested by our data) or due to sequencing error. Similar studies containing larger sample size would be required to generalize our findings. In addition, all variants present in the clinical data were insertions. Further validation of LoFreq’s performance on larger clinical sample sizes containing both SNP and indel mutations thus remains to be done and would be highly complementary to the in silico findings reported in this paper.
Key Points
A benchmarked genome wide minor variant calling tool is currently missing for Mtb.
Sensitivity of LoFreq to detect minor variants ranges from up to 98.8%, improving with increasing frequency of minor variants in the Mtb genome and increasing levels of coverage depth.
Sensitivity of LoFreq is found to be higher for calling indel mutations than single nucleotide polymorphisms.
LoFreq shows to be a highly precise, conservative variant caller, limiting the need for subsequent filtering of false positive variants.
LoFreq is a clinically valuable whole genome sequence (WGS) variant calling tool.
Performance statistics as reported in this study are required to guide future studies aiming to investigate the presence of minor variants in Mtb WGS datasets, such findings are necessary to improve our understanding on various—currently understudied—tuberculosis topics including bacterial transmissibility, bacterial fitness, virulence and drug tolerance.
Supplementary Material
Sander Goossens: completed his Master of Science in Genetics, Cell and Development Biology at the Vrije Universiteit Brussel in 2014. The three subsequent years he worked as a Biology and Physics teacher at the International Montessori School of Brussels. In October 2018, he started his PhD at the faculty of medicine and health sciences at the University of Antwerp. Sander uses whole genome sequencing and RNA sequencing data in order to respectively investigate the within-host MTB population dynamics and MTB transcriptional responses under drug pressure. Next he is also studying the effect of epigenetic modifications in MTB under drug pressure. Sander is currently working on his PhD dissertation, titled: ‘Drug tolerance mechanisms and micro-evolution in Mycobacterium tuberculosis.’
Tim Heupink: is an evolutionary geneticist with an interest in NGS and real-time evolutionary dynamics across space and time. Tim is part of the Center for Whole Genome Sequencing of Mycobacterium tuberculosis. He works on longitudinal data sets to study the evolutionary dynamics of MTB within individuals and populations. The aim of his research is to get a better understanding of MTB’s evolution in hyper-endemic settings and genetic response to different interventions.
Elise De Vos: applies her background in infectious disease epidemiology to gain knowledge with regards to emergence, transmission dynamics and control of drug resistant TB. Elise is currently working on her PhD dissertation, titled: ‘Application of advanced epidemiological methods to improve the study of genomic-guided management of drug resistant tuberculosis.’
Anzaan Dippenaar: is a post-doctoral research fellow at the University of Antwerp and interested in using whole genome sequencing approaches to investigate the microevolution of M. tuberculosis during transmission, the mycobacterial genomics of treatment response during tuberculosis disease, and to explore the genomic characteristics of various mycobacterial strains causing tuberculosis in a variety of animal host species.
Margaretha de Vos: is a molecular biologist and currently a scientific officer in the TB program at FIND. She has over 10 years of research experience in the field of Mycobacteriology, and currently leads multiple diagnostic accuracy studies for new TB and drug-resistant TB diagnostic tests. Margaretha graduated with a PhD in Molecular Biology at Stellenbosch University in 2013 and continued her research there as a post-doctoral fellow. Her research focus at Stellenbosch included the development and validation of new diagnostics for the identification of tuberculosis and antibiotic resistance. She also used next generation technologies to study the microevolution of M. tuberculosis during treatment and acquisition of drug resistance with the aim to identify molecular markers and other risk factors for the prediction of treatment failure in regimens that include the new anti-tuberculosis drugs bedaquiline and delamanid.
Rob Warren: is a distinguished Professor at the University Of Stellenbosch. Under his guidance the study of the molecular epidemiology of M. tuberculosis in a high incidence setting (Cape Town, South Africa) was brought to the forefront of international tuberculosis research. This study now represents the largest molecular epidemiological data set in the developing world and has been referred to as a national heritage. Much of this work has provided new understanding, which has allowed long standing dogmas to be challenged. He has published more than 230 papers in international peer reviewed journals in the fields of molecular epidemiology, drug resistance and bacterial evolution since 1996. These studies have given me excellent experience in managing grant-related outputs and established infrastructure for conduct of ongoing research. In January 2017, he was appointed at the Unit director for the South African Medical Research flagship Centre for Tuberculosis, which is housed within the Division.
Annelies Van Rie: a physician-scientist, epidemiologist and public health specialist whose expertise lies in the study of tuberculosis and HIV in resource poor settings and translate the knowledge gained into practice and action. Her research encompasses clinical, epidemiological, implementation and translational research. Over the past 20 years, she has been the PI or co-investigator on several NIH, CDC and USAID funded research.
Contributor Information
Sander N Goossens, Family Medicine and Population Health (FAMPOP), Faculty of Medicine and Health Sciences, University of Antwerp, Wilrijk, Belgium.
Tim H Heupink, Family Medicine and Population Health (FAMPOP), Faculty of Medicine and Health Sciences, University of Antwerp, Wilrijk, Belgium.
Elise De Vos, Family Medicine and Population Health (FAMPOP), Faculty of Medicine and Health Sciences, University of Antwerp, Wilrijk, Belgium.
Anzaan Dippenaar, Family Medicine and Population Health (FAMPOP), Faculty of Medicine and Health Sciences, University of Antwerp, Wilrijk, Belgium.
Margaretha De Vos, FIND, Geneva, Switzerland.
Rob Warren, Department of Science and Innovation-National Research Foundation Centre for Excellence for Biomedical Tuberculosis Research, South African Medical Research Council Centre for Tuberculosis Research, Division of Molecular Biology and Human Genetics, Faculty of Medicine and Health Sciences, Stellenbosch University, Tygerberg, South Africa.
Annelies Van Rie, Family Medicine and Population Health (FAMPOP), Faculty of Medicine and Health Sciences, University of Antwerp, Wilrijk, Belgium.
Conclusion
We found that sensitivity of LoFreq to detect minor variants ranged from up to 98.8%, improving with increasing frequency of minor variants in the Mtb genome and increasing levels of coverage depth. Sensitivity of LoFreq was found to be higher for calling indel mutations than SNPs. LoFreq shows to be a highly precise, conservative variant caller, limiting the need for subsequent filtering of false positive variants. Despite its low sensitivity (<50%) for detection of low-frequency variants (≤3% for SNP calling and ≤0.5% for indel calling), even at high coverage (1000X), we report LoFreq to be a clinically valuable WGS variant calling tool, as in silico results indicate high sensitivity (>0.97) to detect minor variants at clinically relevant frequency levels (20%) when sequenced at easily achievable sequencing depth (100X). LoFreq’s ability to detect all minor variants that were detected by TDS in the clinical data analyzed further supports LoFreq’s clinical applicability. Performance statistics reported in this study can guide future studies aiming to investigate the presence of minor variants in Mtb WGS datasets by providing the required sequencing depth to detect minor variant populations with the desired specificity at a given variant frequency. We believe these findings will contribute to accelerating and improving our understanding on various—currently understudied—TB topics including bacterial transmissibility, bacterial fitness, virulence and drug tolerance.
Funding Statement
This work was supported by the Research Foundation Flanders (FWO) (G0F8316N, FWO Odysseus).
References
- 1. WHO . Global Tuberculosis Report, 2020.
- 2. Gygli SM, Borrell S, Trauner A, et al. Antimicrobial resistance in mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol Rev 2017;41:354–73. 10.1093/femsre/fux011. [DOI] [PubMed] [Google Scholar]
- 3. Phelan JE, O’Sullivan DM, Machado D, et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med 2019;11(1):41. 10.1186/s13073-019-0650-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dreyer V, Utpatel C, Kohl TA, et al. Detection of low-frequency resistance-mediating SNPs in next-generation sequencing data of mycobacterium tuberculosis complex strains with binoSNP. Sci Rep 2020;10(1):7874. 10.1038/s41598-020-64708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garcia de Viedma D, Marin M, Ruiz MJ, et al. Analysis of clonal composition of mycobacterium tuberculosis isolates in primary infections in children. J Clin Microbiol 2004;42:3415–8. 10.1128/JCM.42.8.3415-3418.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moreno-Molina M, Shubladze N, Khurtsilava I, et al. Genomic analyses of mycobacterium tuberculosis from human lung resections reveal a high frequency of polyclonal infections. Nat Commun 2021;12(1):2716. 10.1038/s41467-021-22705-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nimmo C, Brien K, Millard J, et al. Dynamics of within-host mycobacterium tuberculosis diversity and heteroresistance during treatment. EBioMedicine 2020;55:102747. 10.1016/j.ebiom.2020.102747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hofmann-Thiel S, van Ingen J, Feldmann K, et al. Mechanisms of heteroresistance to isoniazid and rifampin of mycobacterium tuberculosis in Tashkent. Uzbekistan Eur Respir J 2009;33(2):368–74. 10.1183/09031936.00089808. [DOI] [PubMed] [Google Scholar]
- 9. Goossens SN, Sampson SL, Van Rie A. Mechanisms of drug-induced tolerance in mycobacterium tuberculosis. Clin Microbiol Rev 2021;34(1):e00141–20. 10.1128/CMR.00141-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Said Mohammed K, Kibinge N, Prins P, et al. Evaluating the performance of tools used to call minority variants from whole genome short-read data. Wellcome Open Res 2018;3:21. 10.12688/wellcomeopenres.13538.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vargas R, Freschi L, Marin M, et al. In-host population dynamics of mycobacterium tuberculosis complex during active disease. Elife 2021;10:e61805. 10.7554/eLife.61805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilm A, Aw PPK, Bertrand D, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 2012;40(22):11189–201. 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sandmann S, de Graaf AO, Karimi M, et al. Evaluating variant calling tools for non-matched next-generation sequencing data. Sci Rep 2017;7(1):43169. 10.1038/srep43169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Narzisi G, Corvelo A, Arora K, et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun Biol 2018;1(1):20. 10.1038/s42003-018-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCrone JT, Lauring AS. Measurements of Intrahost viral diversity are extremely sensitive to systematic errors in variant calling. J Virol 2016;90:6884–95. 10.1128/JVI.00667-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bush SJ, Foster D, Eyre DW, et al. Genomic diversity affects the accuracy of bacterial single-nucleotide polymorphism-calling pipelines. Gigascience 2020;9(2):1–21. 10.1093/gigascience/giaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ura H, Togi S, Niida Y. Dual deep sequencing improves the accuracy of low-frequency somatic mutation detection in cancer gene panel testing. Int J Mol Sci 2020;21:3530. 10.3390/ijms21103530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andrews TD, Jeelall Y, Talaulikar D, et al. DeepSNVMiner: a sequence analysis tool to detect emergent, rare mutations in subsets of cell populations. PeerJ 2016;4:e2074. 10.7717/peerj.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Borght K, Thys K, Wetzels Y, et al. QQ-SNV: single nucleotide variant detection at low frequency by comparing the quality quantiles. BMC Bioinformatics 2015;16(1):379. 10.1186/s12859-015-0812-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang HW, Program NCS, Mullikin JC, et al. Evaluation of variant detection software for pooled next-generation sequence data. BMC Bioinformatics 2015;16:235. 10.1186/s12859-015-0624-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leung RK, Dong ZQ, Sa F, et al. Quick, sensitive and specific detection and evaluation of quantification of minor variants by high-throughput sequencing. Mol Biosyst 2014;10(2):206–14. 10.1039/c3mb70334g. [DOI] [PubMed] [Google Scholar]
- 22. Olson ND, Lund SP, Colman RE, et al. Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front Genet 2015;6:235. 10.3389/fgene.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vos M, Ley SD, Wiggins KB, et al. Bedaquiline microheteroresistance after cessation of tuberculosis treatment. N Engl J Med 2019;380(22):2178–80. 10.1056/NEJMc1815121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang W, Li L, Myers JR, et al. ART: a next-generation sequencing read simulator. Bioinformatics 2012;28:593–4. 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gomez-Gonzalez PJ, Andreu N, Phelan JE, et al. An integrated whole genome analysis of Mycobacterium tuberculosis reveals insights into relationship between its genome, transcriptome and methylome. Sci Rep 2019;9(1):5204. 10.1038/s41598-019-41692-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heupink T, H VL, Warren RM, et al. Comprehensive and accurate genetic variant identification from contaminated and low coverage Mycobacterium tuberculosis whole genome sequencing data. Microb Genom 2021;7:000689. 10.1099/mgen.0.000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zukurov JP, do Nascimento-Brito S, Volpini AC, et al. Estimation of genetic diversity in viral populations from next generation sequencing data with extremely deep coverage. Algorithms Mol Biol 2016;11(1):2. 10.1186/s13015-016-0064-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Albers CA, Lunter G, MacArthur DG, et al. Dindel: accurate indel calls from short-read data. Genome Res 2011;21(6, 6):961–73. 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shin SS, Modongo C, Baik Y, et al. Mixed Mycobacterium tuberculosis-strain infections are associated with poor treatment outcomes among patients with newly diagnosed tuberculosis, independent of pretreatment heteroresistance. J Infect Dis 2018;218:1974–82. 10.1093/infdis/jiy480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Metcalfe JZ, Streicher E, Theron G, et al. Cryptic Microheteroresistance explains Mycobacterium tuberculosis phenotypic resistance. Am J Respir Crit Care Med 2017;196(9):1191–201. 10.1164/rccm.201703-0556OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Godfroid M, Dagan T, Merker M, et al. Insertion and deletion evolution reflects antibiotics selection pressure in a Mycobacterium tuberculosis outbreak. PLoS Pathog 2020;16(9):e1008357. 10.1371/journal.ppat.1008357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gupta A, Alland D. Reversible gene silencing through frameshift indels and frameshift scars provide adaptive plasticity for Mycobacterium tuberculosis. Nat Commun 2021;12:4702. 10.1038/s41467-021-25055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.