

Abstract
Protein-carbohydrate interactions are crucial for many cellular processes but can be challenging to biologically characterise. To improve our understanding and ability to model these molecular interactions, we used a carefully curated set of 370 protein-carbohydrate complexes with experimental structural and biophysical data in order to train and validate a new tool, cutoff scanning matrix (CSM)-carbohydrate, using machine learning algorithms to accurately predict their binding affinity and rank docking poses as a scoring function. Information on both protein and carbohydrate complementarity, in terms of shape and chemistry, was captured using graph-based structural signatures. Across both training and independent test sets, we achieved comparable Pearson’s correlations of 0.72 under cross-validation [root mean square error (RMSE) of 1.58 Kcal/mol] and 0.67 on the independent test (RMSE of 1.72 Kcal/mol), providing confidence in the generalisability and robustness of the final model. Similar performance was obtained across mono-, di- and oligosaccharides, further highlighting the applicability of this approach to the study of larger complexes. We show CSM-carbohydrate significantly outperformed previous approaches and have implemented our method and make all data freely available through both a user-friendly web interface and application programming interface, to facilitate programmatic access at http://biosig.unimelb.edu.au/csm_carbohydrate/. We believe CSM-carbohydrate will be an invaluable tool for helping assess docking poses and the effects of mutations on protein-carbohydrate affinity, unravelling important aspects that drive binding recognition.
Keywords: protein–carbohydrate complex, binding free energy, structure-based features, graph-based signatures
Introduction
Carbohydrates are found throughout biology, and beyond their metabolic roles are key drivers of many molecular interactions. This is particularly important on the cell surface, where protein-carbohydrate interactions drive viral infection [1], the immune response and cellular interactions [2, 3] and adhesion [4, 5]. Unlike many small-molecule interaction sites, which can show a high level of conservation, carbohydrate binding sites are structurally very diverse and found in a multitude of protein folds [6]. Although carbohydrate binding sites architecturally can vary significantly, their aliphatic nature typically means that their recognition is mediated by hydrogen bonding to the hydroxyl groups and aromatic interactions with the rings, particularly PI-stacking [7–13].
Experimentally characterising protein-carbohydrate interactions structurally and biophysically can be expensive and time-consuming, complicated by both the binding affinities and complexity of carbohydrate structures, which can range from mono-, di- and oligosaccharides (more than two monomers of saccharide) and their derivatives. To help guide these experimental efforts and insights, a range of computational approaches have been used, in particular studying the interactions through molecular dynamics and docking experiments [14–17]. In docking experiments, the predicted binding energy determined by a scoring function is key to guide the docking algorithm through the conformational landscape of poses. Over the year, a range of docking scoring functions have been proposed [18] including physics-based [19], empirical [20, 21] and knowledge-based [22, 23] approaches. The evolution of machine learning and accumulation of high-resolution structural information over the years has also enabled the development of a new class of scoring functions [24–27] that have been shown to outperform traditional approaches [28–30]. Machine learning-based scoring functions however have been, in general, optimised for protein and small molecules [31]. To overcome this, recently Gromiha and colleagues developed a novel scoring function to evaluate the binding affinities between proteins and carbohydrates [32]. This showed the potential for protein-carbohydrate specific scoring functions to overcome some of these limitations. Despite the significance of the effort, there is still room for improvement, particularly in terms of robust validation and improving generalisation capabilities. Significantly higher performance across the independent test in comparison with cross-validation (CV) results, might indicate a bias or potentially limit applicability.
We have previously shown that representing protein interactions as a graph-based signature allowed the accurate prediction of the effects of mutations on protein stability [33–35], dynamics [36, 37] and interactions [38–44], and prediction of protein-small molecule binding affinities [27]. Here, we have adapted our concept of graph-based signatures to capture the nature of carbohydrate interactions in order to accurately predict the binding affinity between proteins and carbohydrates. By comparing its performance with alternative methods, we show that CSM-carbohydrate, where CSM stands for cutoff scanning matrix, outperforms previous approaches and scoring functions and is efficient, facilitating its use as a part of large-scale analyses. The benchmark with machine-learning scoring functions has been expanded in the comparison with decoy structures. We also provide a freely accessible and user-friendly web interface and application programming interface (API) at http://biosig.unimelb.edu.au/csm_carbohydrate/.
Materials and methods
Figure 1 depicts the methodological steps of the present work, which involve data collection and curation, feature generation, model training and validation and web server development.
Figure 1.

CSM-carbohydrate methodological workflow. It involves data collection and curation, feature generation, model training and validation and web server development.
Data curation
Data were initially curated from previous resources, including ProCarbDB [45], ProCaff [46] and PCAPRED [32]. Entries were manually curated to check whether (i) the carbohydrates were correctly identified, (ii) there were any missing parts of the carbohydrates, (iii) the chains of the proteins were correctly identified and were biologically relevant (not due to crystallography symmetry) and (iv) to remove redundant data between training and independent test sets. Where available, we represented binding affinity as Gibbs Free Energy of binding (ΔG in Kcal/mol). Where ΔG was not available, we used the dissociation constant (KD) to calculate ΔG using the equation:
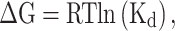 |
where R denotes the ideal gas constant (R = 0.0019872 Kcal.K−1.mol−1), and T is the room temperature in Kelvin.
Our final dataset had 370 protein-carbohydrate complexes with both experimental and biophysical information on the interaction. This was divided into a training dataset of 327 protein-carbohydrate interactions, and an independent, independent test of 43 protein-carbohydrate complexes, which was derived from PCAPRED, and was non-redundant at complex level. The training and independent test sets are available for download at http://biosig.unimelb.edu.au/csm_carbohydrate/datasets.
Feature generation
CSM-carbohydrate was trained and tested using three main classes of features as evidence, including (i) graph-based signatures, (ii) protein-carbohydrate interatomic interactions and (iii) molecular surface area of the interaction (Supplementary Table 1).
Graph-based signatures
The graph-based signatures model the molecular environment of proteins and their interaction partners by quantifying the physicochemical properties of atoms (modelled as nodes) and their interactions (modelled as edges) [33]. Nodes are labelled based on the physicochemical natures of the atoms. Seven atom types were used, namely: hydrophobic, aromatic, positive, negative, hydrogen bond acceptor, hydrogen bond donor and neutral [33]. The original graph-based signatures were used in CSM-lig [27] to predict the binding affinity of arbitrary ligands. In order to better capture the unique chemistry of carbohydrates, additional information on carbon and oxygen atoms of the carbohydrates was also incorporated as node labels. Features are extracted from this representation within the CSM algorithm [47]. In short, the algorithm starts by creating a matrix with the pairwise Euclidean distances between nodes, followed by a scanning step, where a cumulative distribution of interactions (frequency, per node label) is generated based on a maximum distance and a distance step. This cumulative distribution is represented as an array.
Protein-carbohydrate interactions
Intra- and inter-molecule interactions were calculated using Arpeggio [48] and were represented as a vector denoting the number of interactions established for that protein-carbohydrate complex, per interactions type (Arpeggio currently considers 13 types of interactions, including clash, covalent, vdw_clash, vdw, proximal, hbond, weak hbond, halogen bond, ionic, metal, aromatic, hydrophobic and carbonyl).
Molecular surface area
The molecular surface area of the carbohydrate was calculated from van der Waal surface area using Chem.MolSurf function of rdkit (Open-source cheminformatics; http://www.rdkit.org).
Machine learning scoring function
Two machine learning scoring functions, namely NNScore [25, 49] and RFScore [24, 50, 51] were estimated using ODDT (Open drug discovery tool; https://oddt.readthedocs.io/en/latest/index.html).
Machine learning methods
To develop the CSM-carbohydrate model, we evaluated nine different supervised machine learning algorithms across 5-, 10- and 20-fold cross validation (CV), including Gradient Boosting, Extreme Gradient Boosting, Random Forest, Extremely Randomized Trees, AdaBoost, K-Nearest Neighbour, Support Vector Regressor, Gaussian Processes and Neural Networks, using the implementations available on the Scikit-learn library. The best performing model, based on Pearson’s correlation coefficient (PCC) and root mean square error (RMSE) on the training set, was Gradient Boosting (n_estimators = 300, random_state = 1 and default values for all other parameters, using DecisionTreeRegressor as base learner. Further details on parameter tunning can be found in Supplementary Materials and Supplementary Figure 1). A traditional bottom-up greedy feature selection approach was used to optimise and simplify the model. The final predicted model included 12 representative features (detailed information of the 12 features can be found in Supplementary Table 2). Mean absolute error (MAE), Spearman’s, and Kendal’s correlations were also considered and reported. To examine the effect of potential outliers, model performance was evaluated on 100% and also on 90% of the data, which can be interpreted as the full data set minus the 10% worst predicted data points (i.e. the points further away from the regression line).
Scoring function methods
We benchmarked our CSM-carbohydrate with three available scoring function methods, namely NNscore [25], RFscore [52] and PLECscore [26], using the ODDT programme [53]. These three methods are widely used for ranking docking poses. We also benchmarked CSM-carbohydrate with CSM-lig [27], a method that assesses protein-small molecule affinities.
Protein-carbohydrate docking
A bind redocking was performed for each protein-carbohydrate complex, aiming to assess the ability of CSM-carbohydrate to correctly identify the binding site. For this purpose, the grid box was defined as 5 Å from the surface of the proteins, and docking was performed using Autodock Vina and EasyVS [54, 55].
Web server development
CSM-carbohydrate was implemented via a user-friendly web server and API freely available at http://biosig.unimelb.edu.au/csm_carbohydrate. The front end of the CSM-carbohydrate server was built using Bootstrap framework version 3.3.7, whereas the backend was built in Python via the Flask framework (Version 0.10.1), running on a Linux server with Apache.
Results
Distribution of carbohydrate types and binding affinities
The distribution of carbohydrate types (Supplementary Figure 2) and binding affinities (Supplementary Figure 3) shows that both the training and independent test sets have comparable distributions of carbohydrate types and affinities. Most carbohydrates in both the training and independent test sets were monosaccharides (53% and 54%, respectively), whereas few carbohydrates were oligosaccharides (16 and 21%, respectively). No correlation between the carbohydrate size (denoted as the number of heavy atoms) and experimental binding energy was observed (Supplementary Figure 4). This reflects some of the experimental difficulties in characterising larger carbohydrate complexes.
The distribution of binding affinities ranged from −14 Kcal/mol to −1 Kcal/mol, with a peak at ~ −5 Kcal/mol (Supplementary Figure 3). Protein-carbohydrate interactions are usually weaker than protein–protein interactions where the distribution of the latter has a peak at ~ −10 Kcal/mol [32].
Performance under CV
We evaluated the performance of CSM-carbohydrate via multiple forms of CV (Supplementary Table 2). Using 5-, 10- and 20-fold CV, CSM-carbohydrate achieved Pearson’s, Spearman’s, and Kendal’s correlations of up to 0.72, 0.69, and 0.51 respectively, with small deviations across repetitions (<0.01; Figure 2A), MAE of 1.23 Kcal/mol and RMSE of 1.58 Kcal/mol (Supplementary Table 2). Performance increased following removal of 10% of the worst predicted data points, achieving Pearson’s, Spearman’s, and Kendal’s correlations of up to 0.81, 0.79 and 0.59 (MAE = 1.05 Kcal/mol, and RMSE = 1.32 Kcal/mol). These outliers were distributed evenly across the dataset (ΔG ranging from −12.28 to −4.09 Kcal/mol) and were not associated with a particular structural fold or carbohydrate structure.
Figure 2.

Regression plot between experimental and predicted binding affinity (in Kcal/mol) for all saccharides (A) during CV. CSM-carbohydrate obtained a Pearson’s correlation of 0.72. The performance of the model against complexes containing Mono- (B), Di- (C), and Oligosaccharide (D) are shown, highlighting the accuracy and applicability of CSM-carbohydrate to handle different types of carbohydrate molecules. The overall PCCs, including outliers, are shown in red; with the correlation after removing outliers (10%) shown in black. The Pearson’s, Spearman’s, and Kendall’s correlations are written in abbreviations as p, s and k, respectively.
The performance of the CSM-carbohydrate model was evaluated across the different carbohydrate classes, mono-, di-, and oligosaccharides. Pearson’s correlation values of CSM-carbohydrate on the protein-carbohydrate complexes were 0.75, 0.68, and 0.64, for mono-, di- and oligosaccharides, respectively (Figure 2B–D). This indicates that, even though the data set is biased towards monosaccharides, it still translates robustly to more complex carbohydrates. After 10% outlier removal, the corresponding Pearson’s correlations increased to 0.85, 0.73 and 0.74 for mono-, di- and oligosaccharides, respectively.
We further assessed the robustness of the CSM-carbohydrate model using a stringent low-redundancy approach, via leave-similar-targets-out CV (i.e. those complexes having the same protein or its homolog were grouped and used iteratively as a test set). CSM-carbohydrate was able to achieve Pearson’s, Spearman’s, and Kendal’s correlations of up to 0.60, 0.60 and 0.42, respectively (MAE = 1.55 Kcal/mol, and RMSE = 2.00 Kcal/mol, Supplementary Table 3), consistent with its original performance, and providing confidence in the robustness of the model.
We next assessed the robustness of CSM-carbohydrate using a stringent low-redundancy approach, via leave-saccharide-type-out CV (i.e. those ligands having the same saccharide class were grouped and used iteratively as a test set). CSM-carbohydrate was able to achieve Pearson’s, Spearman’s, and Kendal’s correlations of up to 0.61, 0.59 and 0.43, respectively (MAE = 1.55 Kcal/mol, and RMSE = 1.94 Kcal/mol, Supplementary Table 4), consistent with its original performance, and providing confidence in the robustness of the model. The best performance was on Di-Phospho-Mono-sugar-Mono-base, followed by Sub-Tri and Disaccharide. We also assessed the performance of CSM-carbohydrate when all the Di-Phospho-Mono-sugar-Mono-base and Di-Phospho-Di-sugar-Mono-base were left out of training. CSM-carbohydrate performance (Pearson’s, Spearman’s and Kendal’s correlations of 0.72, 0.71 and 0.52, respectively, and MAE = 1.26 Kcal/mol, and RMSE = 1.62 Kcal/mol, Supplementary Figure 5) was consistent with its original performance.
Performance on an independent test set
The generalisation capability of the predicted model was evaluated using an independent data set of 43 protein-carbohydrate complexes. Across the independent test set, our final model achieved Pearson’s, Spearman’s, and Kendall’s correlations of 0.67, 0.64 and 0.48, respectively (MAE = 1.29 Kcal/mol and RMSE = 1.72 Kcal/mol) (Supplementary Figure 6A), consistent with its performance in CV, further demonstrating that the model is generalising rather than memorising.
Assessing feature importance
We next assessed the importance of the 12 features selected and how they contribute to our final model (Supplementary Figure 7). Although 10 out of 12 features have very low Pearson’s correlations with the experimental binding affinity, ranging from −0.35 to −0.05 (Supplementary Figure 8, Supplementary Table 5), the final model achieved a high Pearson’s correlation (0.72 on training and 0.67 on independent test). The largest contribution comes from interactions between negative charged atoms at up to 10 Å (Neg:Neg-10.00). This reflects the role of ionic interactions in protein-carbohydrate recognition, even for long-range interactions. The next two interaction contributions are from the interactions between donor atoms and either negatively or positively charged atoms (Don:Neg-9.00, and Don:Pos-5.00). The scoring functions NNscore and RFscore are the next contributors. The following contributions are from H-donor and aromatic interactions (Aro:Don-8.00), followed by neutral, aromatic, H-acceptor, positively charged interactions either within the protein or between protein and carbohydrate. A feature modelling van der Waals surfaces (SlogP_VSA5) is also included in our model.
Comparison with other available methods
We benchmarked CSM-carbohydrate with the available protein-carbohydrate binding affinity prediction method, namely PCAPRED, and scoring function for the purpose of ranking docking poses, namely NNscore, RFscore and PLECscore. We used the 43 complexes in our independent test set and performed the prediction in the PCAPRED server. Our Pearson’s, Spearman’s, and Kendall’s correlations on the independent test set were 0.67, 0.64 and 0.48 (MAE = 1.29 Kcal/mol and RMSE = 1.72 Kcal/mol), which significantly higher than the corresponding values of all other methods (P value <0.001—Table 1). PCAPRED achieved a Pearson’s coefficient of 0.46, whereas the remaining methods achieved 0.28 (Autodock Vina), 0.20 (CSM-lig), 0.40 (NNscore), 0.36 (RFscore) and 0.49 (PLECscore) (Supplementary Figure 6). CSM-carbohydrate has outperformed all five benchmarked methods, including four generic scoring functions which are not limited to assessing carbohydrate poses, namely NNscore, RFscore, PLECscore and Autodock Vina.
Table 1.
Comparative performance between available methods and CSM-carbohydrate. Pearson’s, Spearman’s, and Kendall’s correlation, MAE and RMSE of six methods predicting protein-carbohydrate binding affinity. CSM-carbohydrate significantly outperforms all alternative methods *(P value <0.001, by Fisher r-to-z transformation test)
| Method | Pearson’s | Spearman’s | Kendall’s | MAE | RMSE |
|---|---|---|---|---|---|
| CSM-carbohydrate | 0.67* | 0.64 | 0.48 | 1.29 | 1.72 |
| PCAPRED | 0.46 | 0.58 | 0.42 | 1.79 | 2.73 |
| CSM-lig | 0.20 | 0.14 | 0.10 | 4.04 | 5.07 |
| NNscore (pdbbind2016) | 0.40 | 0.36 | 0.25 | 2.01 | 2.49 |
| RFscore (v3 pdbbind2016) | 0.36 | 0.33 | 0.25 | 1.78 | 2.19 |
| PLEC score (pdbbind2016) | 0.49 | 0.56 | 0.39 | 1.77 | 2.49 |
| Autodock Vina | 0.28 | 0.29 | 0.22 | 1.86 | 2.54 |
CSM-carbohydrate as a docking scoring function
We next assessed the adequacy of CSM-carbohydrate as a docking scoring function tailored for protein-carbohydrate complexes. To evaluate this, ligands were blindly docked into the apo structures, and the ability of the docking scoring function to identify the experimental binding site and pose was assessed. NNscore achieved the highest number of correctly identify the binding site (20 cases out of 43), followed by CSM-carbohydrate (12 cases), RFscore (8 cases), PLECscore (7 cases) and Autodock Vina (5 cases) (Supplementary Table 6). The results are consistent with the conclusion that the machine-learning scoring functions outperform classical scoring functions 28–30]. CSM-carbohydrate was ranked the second most accurate in identifying the correct binding site, and the most accurate in predicting the binding values (Supplementary Figure 6). The docking was performed as a redock, where the ligand was docked blindly into its crystal structure, to evaluate whether the correct binding site and mode could be identified. Cross-docking was not performed as CSM-Carbohydrate does not consider non-carbohydrate ligands or decoys. To perform redocking, we only considered the native ligands as decoys. It is important to clarify that this performance highlights the applicability of CSM-carbohydrate to understand and assess carbohydrate interactions; however, the method might not be adequate for virtual screening, as it was not designed for non-carbohydrate ligands.
Web server
CSM-carbohydrate is implemented through an easy-to-use web server. To predict protein-carbohydrate binding affinity, users need to provide (i) either a PDB file or a PDB code of the protein-carbohydrate complex, (ii) chain of the carbohydrate (consistent with the provided PDB) and (iii) residue numbers of the carbohydrate as in the PDB file. The prediction is also available via a RESTful API. CSM-carbohydrate predicts the numerical values of the Gibbs Binding Free Energy (ΔG in Kcal/mol) in tabular format, which is made available to download as a comma-separated file.
Users can visualize their uploaded PDB file with its carbohydrate environment from the server via the GLmol molecular viewer, and a Pymol session file showing all the intra- and inter-molecular interactions made by the surrounding environment of carbohydrate, calculated by Arpeggio, is available for download and viewing in Pymol for preparation of publication quality figures and to allow further analysis (Supplementary Figure 9).
Conclusions
In this study, we show that CSM-carbohydrate is an accurate and high-throughput approach to predict the binding affinity of protein-carbohydrate complexes, outperforming alternative approaches. We believe CSM-carbohydrate will be a valuable tool for the study and design of protein-carbohydrate complexes.
Data availability
The training and independent test sets used on CSM-carbohydrate are freely available for download at http://biosig.unimelb.edu.au/csm_carbohydrate/datasets. The CSM-carbohydrate predictive model is available as a user-friendly web interface at http://biosig.unimelb.edu.au/csm_carbohydrate. An API has also been made available to facilitate its programmatic access and integration with Bioinformatics pipelines.
Key Points
CSM-carbohydrate is a new method to accurately predict the binding affinity and rank docking poses for protein-carbohydrate complexes.
CSM-carbohydrate uses the concept of graph-based signatures to train and test predictive methods using supervised learning.
We show CSM-carbohydrate performs robustly in internal and external validation, outperforming five alternative methods.
Supplementary Material
Acknowledgements
The authors wish to thank Prof M. Michael Gromiha, and N.R. Siva Shanmugam for the help of running PCAPRED locally.
T.B. Nguyen is currently a postdoctoral research fellow at the Baker Institute of Heart and Diabetes and the University of Melbourne. Her interest is on computational biology, particularly understanding protein structures, and their interaction.
D.E.V. Pires is a Senior Lecturer in Digital Health with the School of Computing and Information Systems at the University of Melbourne. He is a computer scientist and bioinformatician specialising in machine learning and AI.
D.B. Ascher is Deputy Director of Biotechnology at The University of Queensland, and head of Computational Biology and Clinical Informatics at the Baker Institute and Systems and Computational Biology at Bio21 Institute. His groups research interest is in developing and applying the next generation of computational tools to assist leveraging clinical and omics data for drug discovery and personalised medicine.
Contributor Information
Thanh Binh Nguyen, Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute, Melbourne, Victoria, Australia; Systems and Computational Biology, Bio21 Institute, University of Melbourne, Melbourne, Victoria, Australia; School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Australia.
Douglas E V Pires, Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute, Melbourne, Victoria, Australia; Systems and Computational Biology, Bio21 Institute, University of Melbourne, Melbourne, Victoria, Australia; School of Computing and Information Systems, University of Melbourne, Melbourne, Victoria, Australia.
David B Ascher, Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute, Melbourne, Victoria, Australia; Systems and Computational Biology, Bio21 Institute, University of Melbourne, Melbourne, Victoria, Australia; School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Australia; Department of Biochemistry, University of Cambridge, Cambridge, UK.
Funding
Medical Research Council (MR/M026302/1 to D.B.A. and D.E.V.P.); the National Health and Medical Research Council of Australia (GNT1174405 to D.B.A.); the Wellcome Trust (093167/Z/10/Z); and the Victorian Government’s Operational Infrastructure Support Program.
References
- 1. Karlsson KA. Pathogen-host protein-carbohydrate interactions as the basis of important infections. Adv Exp Med Biol 2001;491:431–43. [DOI] [PubMed] [Google Scholar]
- 2. De Schutter K, Van Damme EJ. Protein-carbohydrate interactions as part of plant defense and animal immunity. Molecules 2015;20:9029–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brewer CF, Miceli MC, Baum LG. Clusters, bundles, arrays and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr Opin Struct Biol 2002;12:616–23. [DOI] [PubMed] [Google Scholar]
- 4. Miura Y, Hoshino Y, Seto H. Glycopolymer Nanobiotechnology. Chem Rev 2016;116:1673–92. [DOI] [PubMed] [Google Scholar]
- 5. Zeng X, Andrade CA, Oliveira MD, et al. Carbohydrate-protein interactions and their biosensing applications. Anal Bioanal Chem 2012;402:3161–76. [DOI] [PubMed] [Google Scholar]
- 6. Loris R. Principles of structures of animal and plant lectins. Biochim Biophys Acta 2002;1572:198–208. [DOI] [PubMed] [Google Scholar]
- 7. Abayakoon P, Jin Y, Lingford JP, et al. Structural and biochemical insights into the function and evolution of sulfoquinovosidases. ACS Cent Sci 2018;4:1266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sernee MF, Ralton JE, Nero TL, et al. A family of dual-activity glycosyltransferase-phosphorylases mediates Mannogen turnover and virulence in leishmania parasites. Cell Host Microbe 2019;26:385–399 e389. [DOI] [PubMed] [Google Scholar]
- 9. Hudson KL, Bartlett GJ, Diehl RC, et al. Carbohydrate-aromatic interactions in proteins. J Am Chem Soc 2015;137:15152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamelryck TW, Loris R, Bouckaert J, et al. Carbohydrate binding, quaternary structure and a novel hydrophobic binding site in two legume lectin oligomers from Dolichos biflorus. J Mol Biol 1999;286:1161–77. [DOI] [PubMed] [Google Scholar]
- 11. Fisher SZ, Schantz L, Hakansson M, et al. Neutron crystallographic studies reveal hydrogen bond and water-mediated interactions between a carbohydrate-binding module and its bound carbohydrate ligand. Biochemistry 2015;54:6435–8. [DOI] [PubMed] [Google Scholar]
- 12. Kapoor M, Thomas CJ, Bachhawat-Sikder K, et al. Exploring kinetics and mechanism of protein-sugar recognition by surface plasmon resonance. Methods Enzymol 2003;362:312–29. [DOI] [PubMed] [Google Scholar]
- 13. Tateno H, Nakamura-Tsuruta S, Hirabayashi J. Frontal affinity chromatography: sugar-protein interactions. Nat Protoc 2007;2:2529–37. [DOI] [PubMed] [Google Scholar]
- 14. Fadda E, Woods RJ. Molecular simulations of carbohydrates and protein-carbohydrate interactions: motivation, issues and prospects. Drug Discov Today 2010;15:596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bryce RA, Hillier IH, Naismith JH. Carbohydrate-protein recognition: molecular dynamics simulations and free energy analysis of oligosaccharide binding to concanavalin a. Biophys J 2001;81:1373–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kerzmann A, Fuhrmann J, Kohlbacher O, et al. BALLDock/SLICK: a new method for protein-carbohydrate docking. J Chem Inf Model 2008;48:1616–25. [DOI] [PubMed] [Google Scholar]
- 17. Nance ML, Labonte JW, Adolf-Bryfogle J, et al. Development and evaluation of GlycanDock: a protein-glycoligand docking refinement algorithm in Rosetta. J Phys Chem B 2021;125:6807–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li J, Fu A, Zhang L. An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdisciplinary Sciences: Comput Life Sci 2019;11:320–8. [DOI] [PubMed] [Google Scholar]
- 19. Uehara S, Tanaka S. AutoDock-GIST: incorporating thermodynamics of active-site water into scoring function for accurate protein-ligand docking. Molecules 2016;21:1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eldridge MD, Murray CW, Auton TR, et al. Empirical scoring functions: I. the development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des 1997;11:425–45. [DOI] [PubMed] [Google Scholar]
- 21. Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem 2006;49:6177–96. [DOI] [PubMed] [Google Scholar]
- 22. Neudert G, Klebe G. DSX: a knowledge-based scoring function for the assessment of protein–ligand complexes. J Chem Inf Model 2011;51:2731–45. [DOI] [PubMed] [Google Scholar]
- 23. Huang S-Y, Zou X. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res 2014;42:e55–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ballester PJ, Mitchell JB. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 2010;26:1169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durrant JD, McCammon JA. NNScore 2.0: a neural-network receptor-ligand scoring function. J Chem Inf Model 2011;51:2897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wojcikowski M, Kukielka M, Stepniewska-Dziubinska MM, et al. Development of a protein-ligand extended connectivity (PLEC) fingerprint and its application for binding affinity predictions. Bioinformatics 2019;35:1334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pires DE, Ascher DB. CSM-lig: a web server for assessing and comparing protein-small molecule affinities. Nucleic Acids Res 2016;44:W557–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ain QU, Aleksandrova A, Roessler FD, et al. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip Rev Comput Mol Sci 2015;5:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ballester PJ. Selecting machine-learning scoring functions for structure-based virtual screening. Drug Discov Today Technol 2019;32-33:81–7. [DOI] [PubMed] [Google Scholar]
- 30. Li H, Sze K-H, Lu G, et al. Machine Learning Scoring Functions for Structure-Based Virtual Screening. Wiley Interdisciplinary Reviews: Computational Molecular Science, 2021;11:e1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frank M. Computational docking as a tool for the rational design of carbohydrate-based drugs. In: Seeberger PH, Rademacher C (eds). Carbohydrates as Drugs. Cham: Springer International Publishing, 2014, 53–72. [Google Scholar]
- 32. Siva Shanmugam NR, Jino Blessy J, Veluraja K, et al. Prediction of protein-carbohydrate complex binding affinity using structural features. Brief Bioinform 2020;22:bbaa319. [DOI] [PubMed] [Google Scholar]
- 33. Pires DE, Ascher DB, Blundell TL. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014;30:335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pires DE, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 2014;42:W314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pires DEV, Rodrigues CHM, Ascher DB. mCSM-membrane: predicting the effects of mutations on transmembrane proteins. Nucleic Acids Res 2020;48:W147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rodrigues CH, Pires DE, Ascher DB. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 2018;46:W350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rodrigues CHM, Pires DEV, Ascher DB. DynaMut2: assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci 2021;30:60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Myung Y, Pires DEV, Ascher DB. mmCSM-AB: guiding rational antibody engineering through multiple point mutations. Nucleic Acids Res 2020;48:W125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Myung Y, Rodrigues CHM, Ascher DB, et al. mCSM-AB2: guiding rational antibody design using graph-based signatures. Bioinformatics 2020;36:1453–9. [DOI] [PubMed] [Google Scholar]
- 40. Pires DE, Ascher DB. mCSM-AB: a web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res 2016;44:W469–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pires DE, Blundell TL, Ascher DB. mCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci Rep 2016;6:29575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pires DEV, Ascher DB. mCSM-NA: predicting the effects of mutations on protein-nucleic acids interactions. Nucleic Acids Res 2017;45:W241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rodrigues CHM, Myung Y, Pires DEV, et al. mCSM-PPI2: predicting the effects of mutations on protein-protein interactions. Nucleic Acids Res 2019;47:W338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodrigues CHM, Pires DEV, Ascher DB. mmCSM-PPI: predicting the effects of multiple point mutations on protein-protein interactions. Nucleic Acids Res 2021;49:W417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Copoiu L, Torres PHM, Ascher DB, et al. ProCarbDB: a database of carbohydrate-binding proteins. Nucleic Acids Res 2020;48:D368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Siva Shanmugam NR, Jino Blessy J, Veluraja K, et al. ProCaff: protein-carbohydrate complex binding affinity database. Bioinformatics 2020;36:3615–7. [DOI] [PubMed] [Google Scholar]
- 47. Pires DE, Melo-Minardi RC, Santos MA, et al. Cutoff scanning matrix (CSM): structural classification and function prediction by protein inter-residue distance patterns. BMC Genomics 2011;12(Suppl 4):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jubb HC, Higueruelo AP, Ochoa-Montano B, et al. Arpeggio: a web server for calculating and visualising interatomic interactions in protein structures. J Mol Biol 2017;429:365–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Durrant JD, McCammon JA. BINANA: a novel algorithm for ligand-binding characterization. J Mol Graph Model 2011;29:888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H, Leung KS, Wong MH, et al. Improving AutoDock Vina using random Forest: the growing accuracy of binding affinity prediction by the effective exploitation of larger data sets. Mol Inform 2015;34:115–26. [DOI] [PubMed] [Google Scholar]
- 51. Ballester PJ, Schreyer A, Blundell TL. Does a more precise chemical description of protein-ligand complexes lead to more accurate prediction of binding affinity? J Chem Inf Model 2014;54:944–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li H, Peng J, Leung Y, et al. The impact of protein structure and sequence similarity on the accuracy of machine-learning scoring functions for binding affinity prediction. Biomolecules 2018;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wojcikowski M, Zielenkiewicz P, Siedlecki P. Open drug discovery toolkit (ODDT): a new open-source player in the drug discovery field. J Chem 2015;7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pires DEV, Veloso WNP, Myung Y, et al. EasyVS: a user-friendly web-based tool for molecule library selection and structure-based virtual screening. Bioinformatics 2020;36:4200–2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The training and independent test sets used on CSM-carbohydrate are freely available for download at http://biosig.unimelb.edu.au/csm_carbohydrate/datasets. The CSM-carbohydrate predictive model is available as a user-friendly web interface at http://biosig.unimelb.edu.au/csm_carbohydrate. An API has also been made available to facilitate its programmatic access and integration with Bioinformatics pipelines.
Key Points
CSM-carbohydrate is a new method to accurately predict the binding affinity and rank docking poses for protein-carbohydrate complexes.
CSM-carbohydrate uses the concept of graph-based signatures to train and test predictive methods using supervised learning.
We show CSM-carbohydrate performs robustly in internal and external validation, outperforming five alternative methods.