

Abstract
Alzheimer’s disease (AD) has a strong genetic predisposition. However, its risk genes remain incompletely identified. We developed an Alzheimer’s brain gene network-based approach to predict AD-associated genes by leveraging the functional pattern of known AD-associated genes. Our constructed network outperformed existing networks in predicting AD genes. We then systematically validated the predictions using independent genetic, transcriptomic, proteomic data, neuropathological and clinical data. First, top-ranked genes were enriched in AD-associated pathways. Second, using external gene expression data from the Mount Sinai Brain Bank study, we found that the top-ranked genes were significantly associated with neuropathological and clinical traits, including the Consortium to Establish a Registry for Alzheimer’s Disease score, Braak stage score and clinical dementia rating. The analysis of Alzheimer’s brain single-cell RNA-seq data revealed cell-type-specific association of predicted genes with early pathology of AD. Third, by interrogating proteomic data in the Religious Orders Study and Memory and Aging Project and Baltimore Longitudinal Study of Aging studies, we observed a significant association of protein expression level with cognitive function and AD clinical severity. The network, method and predictions could become a valuable resource to advance the identification of risk genes for AD.
Keywords: brain gene network, multi-omics, disease gene prediction
1 Introduction
Alzheimer’s disease (AD) is a complex and progressive neurodegenerative disorder that accounts for the majority of all dementia cases [1–6]. AD is partly caused by genetic mutations [7–9]. Mutations in APP, PSEN1 and PSEN2 are associated with early-onset AD [7]. APOE-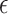 4 is a well-known risk allele for late-onset AD. Most known or putative AD-associated genes are discovered through genome-wide association studies (GWAS). Previously, GWAS identified CLU, CR1 and PICALM, along with approximately 20 more genes [7]. A meta-analysis of GWAS identified rare variants in PLCG2, ABI3 and TREM2 implicating microglia-related innate immunity in AD [10]. In addition, network approaches are used to identify AD-associated molecular networks or pathways [11]. For example, a module-trait network approach was proposed and applied to identify gene coexpression modules that were associated with cognitive function decline [12], whereas a large-scale proteomic analysis identified an energy metabolism-linked protein module, strongly associated with AD pathology [4]. AD stage-associated molecular networks were discovered by using a deep multilayer brain proteomics approach [13]. By leveraging a comprehensive collection of genome-wide epigenomic profiles, an approach called DIVAN is proposed, which can accurately and robustly identify disease-specific risk variants [14]. Despite the substantial advancements in understanding the genetic basis of AD, a large proportion of the phenotypic variance in AD cannot be explained by known risk genes [15–17], suggesting additional AD-associated genes that remain to be discovered. Since experimental approaches are often time-consuming and expensive, computational approaches provide a promising alternative to discovering AD-associated genes.
4 is a well-known risk allele for late-onset AD. Most known or putative AD-associated genes are discovered through genome-wide association studies (GWAS). Previously, GWAS identified CLU, CR1 and PICALM, along with approximately 20 more genes [7]. A meta-analysis of GWAS identified rare variants in PLCG2, ABI3 and TREM2 implicating microglia-related innate immunity in AD [10]. In addition, network approaches are used to identify AD-associated molecular networks or pathways [11]. For example, a module-trait network approach was proposed and applied to identify gene coexpression modules that were associated with cognitive function decline [12], whereas a large-scale proteomic analysis identified an energy metabolism-linked protein module, strongly associated with AD pathology [4]. AD stage-associated molecular networks were discovered by using a deep multilayer brain proteomics approach [13]. By leveraging a comprehensive collection of genome-wide epigenomic profiles, an approach called DIVAN is proposed, which can accurately and robustly identify disease-specific risk variants [14]. Despite the substantial advancements in understanding the genetic basis of AD, a large proportion of the phenotypic variance in AD cannot be explained by known risk genes [15–17], suggesting additional AD-associated genes that remain to be discovered. Since experimental approaches are often time-consuming and expensive, computational approaches provide a promising alternative to discovering AD-associated genes.
Previous studies have shown that functional gene networks (FGNs) are promising for predicting disease-associated genes [18, 19]. FGNs are constructed by integrating heterogeneous omics datasets mainly from public datasets [18, 20, 21]. In an FGN, a node represents a gene, and the edge connecting two genes represents the co-functional probability (CFP) that the two genes take participate in the same biological process or pathway [22]. For example, using a global (i.e. non-tissue specific) FGN for mice, novel candidate genes for bone-mineral density and thermal pain were predicted [23, 24]. Considering that gene interactions might be rewired in different tissues, tissue-specific networks were later proposed to capture gene interactions more accurately in tissues. Greene et al. proposed to leverage tissue-specific gene function annotation data to guide the construction of tissue-specific networks using a supervised learning approach; with this approach, they constructed 144 human tissue-specific networks and investigated these networks for the interpretation of gene functions and diseases [19, 20]. One limitation of these networks is that the gene expression data, which are taken as input features to construct the network for a given tissue (say brain), remain not tissue-specific; that is, expression data of not only the tissue under investigation but also other tissues were used. Therefore, the resulting network may have limited accuracy in modeling tissue-specific gene interactions due to the interference caused by other tissues. Later in our previous work, we built a brain gene network by integrating only microarray-based brain-specific gene expression data [25]. Existing networks were exclusively built with naïve Bayesian classifiers (NBC). NBC has the limitation that its feature independence assumption usually does not hold and it could not well capture the nonlinearity in real data.
Here we proposed to predict AD-associated genes by leveraging a brain FGN constructed with Alzheimer’s brain gene expression data. Considering the limitation of the NBC, the state-of-the-art XGBoost algorithm was adopted to construct the brain FGN because it does not assume feature independence and is suitable for nonlinear modeling. The resulting brain FGN (called ADBrainNexus for simplicity) is a weighted network, in which the weight of the edge represents the CFP that two genes participate in the same biological process in human brains. A machine learning model was trained to predict the association of each gene with AD by learning the functional pattern of known AD-associated genes collected from multiple data resources, including the GWAS Catalog and Online Mendelian Inheritance in Man (OMIM) databases. With this model, we scored all other human genes that were not used in model training. The higher the score is, the more likely the gene is associated with AD. Note that our predictions do not indicate any causality, that is, the predicted genes may be either directly or indirectly associated with AD. Using independent data from the Mount Sinai Brain Bank (MSBB) study, we found that the top-ranked genes were significantly associated with AD traits, including the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) score, Braak stage score and clinical dementia rating (CDR), suggesting the functional relevance of our predictions to AD. We observed cell-type-resolved association of predicted genes with early pathology of AD using brain single-cell RNA-sequencing (scRNA-seq) dataset. Further, we uncovered a significant association of the protein-level expression of top-ranked genes with both cognitive function and AD clinical severity using external proteomic data from the Religious Orders Study and Memory and Aging Project (ROSMAP) and Baltimore Longitudinal Study of Aging (BLSA) studies. The resulting predictions and pipeline could be valuable to advance the identification of risk genes for AD.
2 Materials and methods
2.1 Construction of brain-specific FGNs
We constructed an Alzheimer’s disease brain FGN (called ADBrainNexus) by integrating heterogeneous genomic interaction data mainly including gene expression and DNA methylation. Gene expression data were obtained from the Gene Expression Omnibus (GEO) and Digital Expression Explorer 2 (DEE2) databases. DEE2 is a database of RNA-seq gene expression data generated using a unified pipeline [26]. From GEO, we searched datasets on human Alzheimer’s studies. Because for each dataset the correlation between every pair of genes needs to be calculated as features and the correlation could be spurious if the number of samples is small, we retained only the datasets containing at least 10 samples following the practice in our previous work [27]. For the dataset containing both AD and control samples, we partitioned the samples into the AD and control groups, thus forming two sub-datasets. In doing so, we obtained a total of 21 AD datasets and 13 control datasets. The control datasets will be used to build a healthy brain network for comparison. For these datasets, we calculated gene-level expression values by taking the mean of all its probes, which is the conventional practice. In addition, we also searched the DEE2 database and identified 17 healthy brain datasets, in which gene expression is measured with Fragments per Kilobase of exon model per million mapped reads (FPKM). In each dataset, lowly expressed genes (FPKM < 0.1 in more than 90 % samples, which is the same criteria used in our previous work [27]) were removed. We obtained 21 and 30 gene expression datasets for AD and control samples, respectively. For each gene expression dataset, Spearman correlation between gene pairs is calculated as features. In addition, we obtained human brain DNA methylation datasets from the ref [28, 29]. If a dataset contains both AD and control samples, the two groups of samples are separated. We obtained nine AD and five control DNA-methylation datasets. Gene methylation level is calculated as the average of the methylation level of the CpG sites that map to the gene. For each dataset, the Spearman correlation between methylation profiles of gene pairs is calculated as the feature. The accession ID of all above-described gene expression and DNA methylation datasets are provided in Supplementary Table 1. In addition, we also considered six pairwise genomic features, which were obtained from the GIANT website [20], including protein–protein interaction (PPI) from MINT, IntAct and BioGRID, the chemical and genetic perturbations, shared 3’ UTR microRNA binding motif from MSigDB and co-occurrence of transcription factor binding sites.
We generated functionally related (positives) and unrelated (negatives) gene pairs using the same method established in our previous work [18, 20, 23, 25]. Briefly, positive and negative gene pairs are obtained based on the functional annotation to Gene Ontology (GO) biological process or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. For GO terms, only the annotation with experimental evidence codes (EXP, IDA, IPI, IMP, IGI and IEP) were used for quality control. Because GO terms are hierarchical, we first propagate all genes of a GO term to its parent term recursively using the previously established methods [20]. Then, gene pairs that are co-annotated to the same GO term or KEGG pathway are considered as positives; gene pairs that are not co-annotated to any GO term or pathway are considered as negatives. The gene pairs generated from GO and KEGG were combined, and redundant ones were removed, resulting in 828 712 positive pairs. Negatives were generated using the same approach as established in the work [20, 23, 25]. Considering the limitation of the NBC as used in previous work, the state-of-the-art XGBoost algorithm was adopted to build the network prediction model because it does not assume feature independence, is suitable for nonlinear modeling, and scalable to large datasets. An XGBoost model was learned using the features and the labelled positive or negative gene pair as input. The learned model was then used to predict CFP for all possible gene pairs, resulting in the brain FGN (called ADBrainNexus for simplicity). As can be seen from the method to construct ADBrainNexus, the edge weight in the network represents the probability that two genes work in the same biological process or pathway, indicating that the network is a FGN based on previous studies [20, 23]. ADBrainNexus is a weighted network, in which the weight of an edge connecting two genes represents the CFP that two genes participate in the same biological process in human brains.
We tested the running time of building the network. We used a machine with 2 Intel(R) Xeon(R) Gold 6126 CPUs (2.60GHz, 48 cores) and 1T memory. The number of cores is set to 32 for running XGBoost. It takes 126 minutes to construct the ADBrainNexus network. This network is freely available at https://zenodo.org/record/5594149.
2.2 Model development for predicting AD-associated genes
First, we collected known AD-associated (positives) and non-AD (negatives) genes to build the machine learning model (see details in Supplementary Note 1). Briefly, to identify AD-associated genes, we performed intensive hand-curation of various disease gene resources, including AlzGene [30], AlzBase [31], OMIM [32], DisGenet [33], DistiLD [34] and UniProt [35], Open Targets [36], GWAS Catalog [37], differentially expressed genes (DEGs) in ROSMAP [38] and published literature. The genes curated from each resource along with the corresponding criteria to select genes are provided in Supplementary Note 1. As the AD-associated genes and their reliability vary across these resources, we used a voting strategy and selected only those genes that are included in at least two resources to ensure higher reliability. As a result, we obtained 147 genes that are associated with AD. To identify non-AD genes that had no known or minimal association with AD, functional enrichment and public databases were leveraged to remove any genes that exhibit potential associations with AD. The remaining were considered as non-AD genes.
For each gene, we extracted its CFP with each of the 147 collected AD-associated genes from ADBrainNexus as a feature based on a previously proposed method [39] (the weight between each gene and itself is set to 1). Consequently, each gene has 147 features and they are collected into a 147-dimensional feature vector. The feature data for the training set were represented by a matrix X. The labels (1 for positives and 0 for negatives) of these genes were stored in a vector y. The feature matrix of all other genes not in the training set was extracted. Then, a machine learning method is used to build models to predict AD-associated genes. The performance of the model is evaluated with 5-fold cross-validation.
2.3 Decile enrichment test for AD pathways and phenotypes
The decile enrichment test proposed in the previous study [19] is applied to statistically assess whether a larger proportion of a given AD-associated gene set falls into the first decile of predicted genes. The genes in the training set are first excluded. The remaining genes are ranked and split into 10 evenly binned deciles. Let P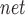 and P
and P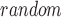 denote the proportion of a given gene set that falls into the first decile based on our prediction and random chance, respectively. The decile enrichment tests whether P
denote the proportion of a given gene set that falls into the first decile based on our prediction and random chance, respectively. The decile enrichment tests whether P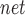 is significantly larger than P
is significantly larger than P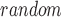 by using the binomial test (see details in [19]).
by using the binomial test (see details in [19]).
2.4 Statistical assessment of the association of top-ranked genes with AD using biological networks
We test whether top-ranked genes show significant functional associations with AD based on statistical analysis of functional genomic data. The method for the significance test is described below.
Let t denote the metric of interest calculated for a given list of genes based on given genomic data. For example, t can be the Pearson correlation coefficient (PCC) between a pair of genes calculated from gene expression data. Let t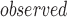 and t
and t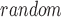 be the metrics calculated for a set of top-ranked k genes and randomly selected k genes, respectively. We test whether t
be the metrics calculated for a set of top-ranked k genes and randomly selected k genes, respectively. We test whether t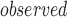 is significantly larger than t
is significantly larger than t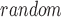 . By randomly generating 10
. By randomly generating 10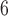 gene lists, 10
gene lists, 10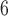 t
t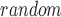 values can be obtained. Let N
values can be obtained. Let N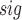 denote the number of t
denote the number of t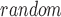 values that are larger than t
values that are larger than t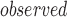 . Then, we calculate a P-value = N
. Then, we calculate a P-value = N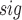 /10
/10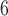 .
.
The details for calculating t are described separately for each type of genomic data in the following: (1) testing by sequence similarity networks. An identity score between each predicted gene and each known AD-associated gene is computed using BLAST. Then t is calculated as the maximum of the identity scores. (2) Testing by miRNA-target binding data. Only the miRNA that is associated with AD is considered. t denotes the number of AD-associated miRNAs that can bind to both known and predicted AD-associated genes. (3) Testing by gene coexpression networks. t represents the number of coexpressed gene pairs between predicted genes and the AD-associated genes. (4) Testing by PPI networks. t denotes the number of predicted genes that interact with at least one known AD-associated gene.
2.5 AD neuropathological and clinical traits in the MSBB study
We obtained an independent dataset with AD-associated neuropathological and clinical traits from the MSBB study [40]. The data from Brodmann area 36 (parahippocampal gyrus), one of the most vulnerable regions to AD [41], were used. This dataset contains gene expression data for 215 donors for which AD traits are available. These traits include the neuritic plaque density assessed by CERAD score, neurofibrillary tangle severity by Braak score and severity of dementia by CDR score. The dataset is publicly available at the AMP-AD portal on Synapse (Synapse ID: syn3159438). For each gene, its PCCs with the CERAD, Braak and CDR scores were calculated.
Based on the CERAD score, we extracted control and AD samples using the criteria provided on https://www.synapse.org/Synapse:syn6101474; based on the Braak score, we followed the practice in [41] and divided samples into three groups in the ranges of [0, 2], [3, 4] and [5, 6], representing different levels of tau pathology; based on the CDR score, the samples were partitioned into three groups in the range of [0], [0.5, 2] and [3, 5] in the same way as used in [41], representing different degrees of severity of clinical dementia.
2.6 Proteomic and cognitive data in the ROSMAP study
ROSMAP are longitudinal clinical–pathologic cohort studies of aging and AD. The clinical and proteomic data for ROSMAP samples were downloaded from Synapse (accession ID: syn3219045). Based on the clinical variable ‘dcfdx_lv’, we identified individuals with no cognitive impairment (NCI, n = 174), mild cognitive impairment (MCI, n = 100) and ADs (n = 104). The protein expression was quantified with TMT labeling (Synapse: syn21266454). We considered the protein of which the expression increased or decreased monotonically across the three stages. Then, Kendall’s Tau-b test is applied to test whether the trend was significant.
2.7 Proteomic data for asymptomatic and symptomatic AD in BLSA study
Clinical and proteomic data for individuals in the BLSA study were downloaded from Synapse (accession ID: syn3606086). Based on the description, we identified controls (n = 13), asymptomatic AD (AsymAD) (n = 14) and AD (n = 20) individuals. The protein expression used in our analysis was publicly available (accession ID: syn4216216). We restricted our analysis to the protein of which the expression increased or decreased monotonically across the three stages. Kendall’s Tau-b test is applied to test whether protein expression is correlated with AD severity.
3 Results
3.1 Overview of ADBrainNexus-based prediction of AD-associated genes
Our approach predicts AD-associated genes by leveraging ADBrainNexus and 147 known AD-associated genes from multiple resources including OMIM and AD GWAS studies (Figure 1). ADBrainNexus was built by integrating 21 Alzheimer’s brain gene expression datasets, 9 Alzheimer’s brain DNA methylation datasets and 6 other functional genomic datasets (see Materials and Methods) using XGBoost; the network is accurate with area under the receiver operating characteristic curve (AUROC) = 0.9066. Non-AD genes were selected using a function enrichment-based method (Supplementary Note 1).
Figure 1.

Overview of the brain FGN (ADBrainNexus) based prediction of AD-associated genes and their functional characterization. (A) Construction of ADBrainNexus by integrating brain gene expression data, DNA methylation data and other interaction networks using XGBoost. (B) Selection of AD-associated genes. In total, 147 known AD-associated genes were collected from various resources, including OMIM, DisGeNet, Uniprot, DistiLD, AlzBase, AlzBase, AlzGene, literature report, Open Targets, ROSMAP-DEG and GWAS-catalog. The gene that was present in at least two resources was selected. The AD-associated genes, as well as potential positive genes inferred with a functional enrichment method, were then removed from the full set of all human genes. The remaining genes were treated as non-AD genes (negatives). (C) Predicting AD-associated genes using a machine learning model built by integrating ADBrainNexus and AD-associated genes. (D) Validation of predicted genes using heterogeneous functional genomic data based on their association with AD-associated traits.
First, we compared ADBrainNexus with the network built with healthy brain datasets based on their performance in predicting AD-associated genes (see Materials and Methods). For both networks, we tested lasso, ridge regression and ExtraTrees for building models. We found that ridge regression performed better than the other two methods on both networks (Table 1). We therefore chose ridge regression to build the model to predict AD-associated genes. The model based on ADBrainNexus has AUROC = 0.951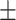 0.001 and area under the precision–recall curve (AUPRC) = 0.874
0.001 and area under the precision–recall curve (AUPRC) = 0.874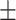 0.001, which is higher than that of the healthy brain-based network (AUROC = 0.922
0.001, which is higher than that of the healthy brain-based network (AUROC = 0.922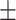 0.001, AURPC = 0.788
0.001, AURPC = 0.788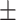 0.003). We also compared ADBrainNexus with the brain networks in the GIANT and BaiHui database; ADBrainNexus also achieved better performance (Table 1). This analysis suggests that ADBrainNexus is superior than the two existing networks in predicting AD-associated genes.
0.003). We also compared ADBrainNexus with the brain networks in the GIANT and BaiHui database; ADBrainNexus also achieved better performance (Table 1). This analysis suggests that ADBrainNexus is superior than the two existing networks in predicting AD-associated genes.
Table 1.
Comparison of the ADBrainNexus with the healthy brain network, the GIANT and BaiHui networks in predicting AD-associated genes
| Networks | Lasso | Ridge regression | ExtraTrees | |||
|---|---|---|---|---|---|---|
| AUROC | AUPRC | AUROC | AUPRC | AUROC | AUPRC | |
| GIANT | 0.690 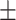 0.011 0.011 |
0.425 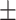 0.019 0.019 |
0.546 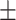 0.001 0.001 |
0.446 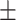 0.003 0.003 |
0.881 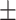 0.012 0.012 |
0.611 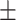 0.026 0.026 |
| BaiHui | 0.884 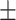 0.0005 0.0005 |
0.768 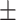 0.0005 0.0005 |
0.933 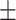 0.002 0.002 |
0.852 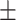 0.005 0.005 |
0.929 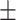 0.006 0.006 |
0.847 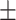 0.009 0.009 |
| Healthy Brain | 0.898 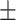 0.001 0.001 |
0.638 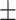 0.007 0.007 |
0.922 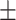 0.001 0.001 |
0.788 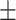 0.003 0.003 |
0.905 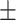 0.008 0.008 |
0.748 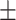 0.017 0.017 |
| ADBrainNexus | 0.926 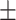 0.001 0.001 |
0.727 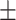 0.005 0.005 |
0.951 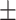 0.001 0.001 |
0.874 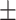 0.001 0.001 |
0.927 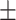 0.008 0.008 |
0.840 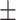 0.010 0.010 |
We validated the top-ranked genes using the top genes obtained by the gene-level GWAS P-value approach and the DIVAN approach [14] (the score for each gene is computed using cross-validation). First, we computed a gene-level P-value for each gene using the MAGMA software (v1.07). The GWAS summary statistics data from a large-scale AD study [42] are used as the input for MAGMA. Using the decile test method described in [19], we find that our top-ranked first-decile genes are significantly enriched in the top-ranked 100 genes obtained by the gene-level GWAS P-value method (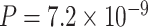 ). Second, for the disease-specific variant annotation approach, we obtained the variant scores for AD (https://sites.google.com/site/emorydivan/home), which are computed with the DIVAN method [14]. Then, we computed the sum of the score of all variants belonging to a gene as the gene score. Using the above-mentioned decile test method, we find that our top-ranked first-decile genes are significantly enriched in the top-ranked 100 genes obtained by DIVAN (P = 0.023).
). Second, for the disease-specific variant annotation approach, we obtained the variant scores for AD (https://sites.google.com/site/emorydivan/home), which are computed with the DIVAN method [14]. Then, we computed the sum of the score of all variants belonging to a gene as the gene score. Using the above-mentioned decile test method, we find that our top-ranked first-decile genes are significantly enriched in the top-ranked 100 genes obtained by DIVAN (P = 0.023).
Furthermore, we evaluated the model based on the hypothesis that top-ranked genes were likely involved in AD if they show sequence similarity to or interact with known AD-associated genes. To do so, we analyzed external biological network data, including protein sequence similarity networks, brain gene coexpression from Mayo RNA-seq study, PPI from Human Reference Interactome and STRING, miRNA–target interaction from mirTarBase (Release 7.0), which were not used in building the ADBrainNexus. This analysis showed that the top-ranked genes showed significant sequence similarity to, were significantly co-expressed, had more interactions and shared more miRNA binding sites with known AD-associated genes (see Materials and Methods; Supplementary Figure 1).
Second, having validated the predictive value of ADBrainNexus on these external network data, we further updated the model to predict AD-associated genes by integrating the features extracted from both the external networks and ADBrainNexus (see details in Supplementary Figure 2). Compared with using the features only from the ADBrainNexus network, the updated model achieved higher prediction performance (AUROC = 0.969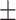 0.001, AUPRC = 0.921
0.001, AUPRC = 0.921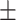 0.002) (Supplementary Figure 2). This model predicts a score for each gene. A higher score indicates that a gene is more likely to be associated with AD, and vice versa. We found that 18 of the top-ranked 20 genes were associated with AD based on the literature report (Supplementary Table 2), suggesting that our model captured the molecular signature of AD-associated genes and was able to make confident predictions. The source codes for building the model are freely available at https://github.com/genemine/ADBrainNexus. The scores for the top-ranked 200 genes (excluding know AD-associated genes) are provided in Supplementary Table 3 (see the top-ranked 2000 genes at https://github.com/genemine/ADBrainNexus).
0.002) (Supplementary Figure 2). This model predicts a score for each gene. A higher score indicates that a gene is more likely to be associated with AD, and vice versa. We found that 18 of the top-ranked 20 genes were associated with AD based on the literature report (Supplementary Table 2), suggesting that our model captured the molecular signature of AD-associated genes and was able to make confident predictions. The source codes for building the model are freely available at https://github.com/genemine/ADBrainNexus. The scores for the top-ranked 200 genes (excluding know AD-associated genes) are provided in Supplementary Table 3 (see the top-ranked 2000 genes at https://github.com/genemine/ADBrainNexus).
3.2 Top-ranked genes are enriched in AD-associated biological processes
We tested the enrichment of predicted genes in AD-associated biological processes (after excluding genes in the training set). We collected AD-associated biological processes from the GO database through enrichment analysis as follows. First, using the Panther web server, we performed GO enrichment analysis of our collected 147 AD-associated genes and obtained 858 GO biological processes. Second, from a recent review paper on AD [43], we identified AD-related risk factors, biological processes or phenotypes, which include amyloid-beta, memory, neuroinflammation, synapse-related functions, APP, reactive oxygen species in central nervous systems. Third, we manually went through the 858 enriched GO terms and selected those terms that are involved in the above-described AD-related risk factors, biological processes or phenotypes. Finally, we obtained 41 AD-associated biological processes. We performed the analysis using the decile enrichment test established in [19], which tests whether the genes in a biological process of interest are enriched in the top-ranked first-decile (i.e.10% ) of predicted genes (see Materials and Methods). We observed that the top-ranked genes were significantly enriched in all gene sets. Examples of enriched biological processes include cognition (GO:0050890, False Discovery Rate (FDR) = 2.8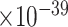 ), learning or memory (GO:0007611, FDR = 1.2
), learning or memory (GO:0007611, FDR = 1.2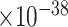 ), neuroinflammatory response (GO:0150076, FDR = 2.5
), neuroinflammatory response (GO:0150076, FDR = 2.5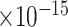 ), regulation of synaptic plasticity (GO:0048167, FDR = 1.5
), regulation of synaptic plasticity (GO:0048167, FDR = 1.5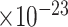 ), response to amyloid-beta (GO:1904645, FDR = 8.5
), response to amyloid-beta (GO:1904645, FDR = 8.5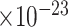 ), amyloid-beta clearance (GO:0097242, FDR = 5.3
), amyloid-beta clearance (GO:0097242, FDR = 5.3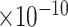 ) (Figure 2). The enrichment results for all 41 biological processes are provided in Supplementary Table 4.
) (Figure 2). The enrichment results for all 41 biological processes are provided in Supplementary Table 4.
Figure 2.

Enrichment of the top-ranked first-decile predictions in AD-associated gene sets or pathways using the decile enrichment test (described in Materials and Methods).
We tested whether the top-ranked first-decile genes overlapped with gene modules that were associated with AD in published studies. We obtained two gene sets from a recently published network association study on AD [13]. The first was a set of 28 kinases that were possibly implicated in AD; 20 of them were enriched in the first decile of our predictions (P-value < 0.0001). The second was the A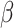 -correlated cascade gene set (14 genes) (after removing CLU because it was in the training set); seven ranked in the first decile (P-value < 0.0001). These results support the association of our predicted genes with AD.
-correlated cascade gene set (14 genes) (after removing CLU because it was in the training set); seven ranked in the first decile (P-value < 0.0001). These results support the association of our predicted genes with AD.
3.3 Expression of top-ranked genes are correlated with neuropathological and clinical traits on independent datasets
We tested whether the expression of top-ranked genes was associated with AD using the independent MSBB RNA-seq dataset (see Materials and Methods). For each gene, we calculated its PCC with the CERAD, Braak and CDR score (see Materials and Methods). We then evaluated the correlation of the predicted ranks of the genes with the three traits. This analysis showed that higher ranks (higher predicted scores) were associated with higher mean PCC values for all three phenotypes. The predicted ranks were well correlated with the CERAD (r = -0.915), Braak (r = -0.902) and CDR (r = -0.931) score (Figure 3A). We assessed the association of top-ranked genes as a module with the three traits. The eigengenes (i.e. the first principal component) for the top-ranked 100, 200 and 300 genes were all significantly correlated with the CERAD, Braak and CDR scores (Figure 3B; Supplementary Figure 3).
Figure 3.

Correlation of top-ranked genes with AD neuropathological and clinical traits on the MSBB dataset. (A) The correlation between predicted scores and the three AD traits, (i.e. the CERAD, Braak and CDR scores. All genes (after excluding genes in the training set) were sorted by their predicted scores and divided into 50 bins. For each gene, its absolute PCC with each trait was calculated. For the genes in each bin, their average score predicted by our method was plotted against their average correlation. The trend is fitted with a linear regression model. (B) The association of the eigengene (the first principal component) of the top-ranked 200 genes with the three traits. CERAD score is a measurement of neuritic plaque density. Braak score is a measurement of neurofibrillary tangle severity. CDR measures the severity of dementia. (C) The proportion of variance of each trait explained by the top-ranked genes. The proportion of variance explained by the number of APOE-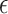 4 alleles (the baseline genetic risk factor) is also shown for comparison.
4 alleles (the baseline genetic risk factor) is also shown for comparison.
We then examined the correlations of top-ranked individual genes (those not included in the training set) with AD traits [12]. Among the top-ranked 200 genes, we identified 123, 120 and 135 genes that were significantly correlated with the CERAD, Braak and CDR score, respectively (FDR < 0.05). Of them, 107 were correlated with all three phenotypes (Supplementary Table 3). Taking PRKCB as an example, its correlations with CERAD, Braak and CDR scores were -0.38 (FDR = 1.43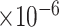 ), -0.35 (FDR = 2.22
), -0.35 (FDR = 2.22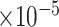 ) and -0.38 (FDR = 7.70
) and -0.38 (FDR = 7.70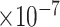 ). Another example is PLCB1, of which the PCC with the three traits were -0.40 (FDR = 3.39
). Another example is PLCB1, of which the PCC with the three traits were -0.40 (FDR = 3.39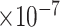 ), -0.40 (FDR = 1.64
), -0.40 (FDR = 1.64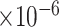 ) and -0.46 (FDR = 9.12
) and -0.46 (FDR = 9.12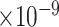 ), respectively. These results on independent datasets indicate the association of top-ranked genes with AD.
), respectively. These results on independent datasets indicate the association of top-ranked genes with AD.
We further compared the predicted genes with the number of APOE-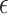 4 alleles based on the proportion of the variance of the three traits that they can explain using the method established in [44]. The number of APOE-
4 alleles based on the proportion of the variance of the three traits that they can explain using the method established in [44]. The number of APOE-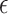 4 alleles was considered as the baseline genetic risk factor [44]. We observed that the expression of the top-ranked genes explained comparable or more variance for the CERAD, Braak, and CDR scores compared with APOE-
4 alleles was considered as the baseline genetic risk factor [44]. We observed that the expression of the top-ranked genes explained comparable or more variance for the CERAD, Braak, and CDR scores compared with APOE-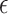 4 (Figure 3C). Taking the result for CERAD as an example, PLCB1 explained the highest proportion (16.2 % ) of its variance, whereas APOE-
4 (Figure 3C). Taking the result for CERAD as an example, PLCB1 explained the highest proportion (16.2 % ) of its variance, whereas APOE-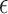 4 explains only 4.1 % . Of interest, PLCB1 also explained the highest proportion of variance for the Braak (15.9 % ) and CDR (20.7 % ) scores, implying that it might be a potential candidate gene.
4 explains only 4.1 % . Of interest, PLCB1 also explained the highest proportion of variance for the Braak (15.9 % ) and CDR (20.7 % ) scores, implying that it might be a potential candidate gene.
3.4 Brain single-cell analysis identifies cell-type-specific transcriptional changes associated with early pathology of AD
We examined whether the cell-type-specific expression of predicted genes was associated with the early pathology of AD. We overlapped the top-ranked 200 genes with the AD-associated DEGs identified in six major cell types in a large-scale single-cell transcriptomic study of Alzheimer’s brains [45]. The study analyzed three subgroups of individuals: no-pathology, early-pathology and late-pathology [45]. The cell types are astrocytes (Ast), oligodendrocytes (Oli), oligodendrocyte precursor cells (Opc), microglia (Mic), excitatory (Ex) and inhibitory (In) neurons.
Of the top-ranked genes, we found that 3, 11, 2, 48 and 30 were DEGs between no-pathology and early-pathology subgroups in Ast, Oli, Opc, Ex and In, respectively (Figure 4A; Supplementary Table 3). We observed that these DEGs were highly cell-type specific; most of them were differential in only neurons (Ex or In) (Figure 4A). For example, PRKX was upregulated in oligodendrocytes (log2fold change = 1.15, FDR = 2.6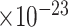 ). Another example was FOS, which was downregulated in excitatory neurons in early-pathology individuals (log2fold change = -1.67, FDR=3.85
). Another example was FOS, which was downregulated in excitatory neurons in early-pathology individuals (log2fold change = -1.67, FDR=3.85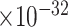 ). This analysis suggests that our predicted genes are likely involved in the early stages of AD. To examine the functions of these DEGs, we performed GO enrichment analysis using the Panther web server and found that they were enriched in AD-associated biological processes such as neuron death (GO:1901214, FDR = 4.32
). This analysis suggests that our predicted genes are likely involved in the early stages of AD. To examine the functions of these DEGs, we performed GO enrichment analysis using the Panther web server and found that they were enriched in AD-associated biological processes such as neuron death (GO:1901214, FDR = 4.32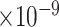 ) and aging (GO:0007568, FDR = 2.02
) and aging (GO:0007568, FDR = 2.02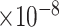 ) (Supplementary Table 5).
) (Supplementary Table 5).
Figure 4.
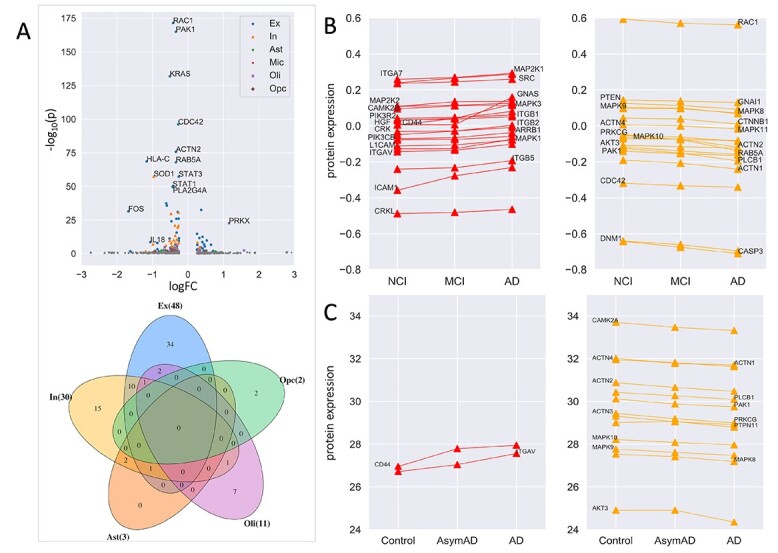
Association of the top-ranked 200 genes with early-pathology AD, cognitive function, and AD clinical severity. (A) Differential expression of the top-ranked genes between no-pathology and early-pathology subgroups of individuals for each cell type using the brain scRNA-seq data. The cell types are astrocytes (Ast), oligodendrocytes (Oli), oligodendrocyte precursor cells (Opc), microglia (Mic), excitatory (Ex) and inhibitory (In) neurons. Colored and gray dots indicate significant and insignificant differential expression, respectively. The Venn diagram shows the overlap of the DEGs among different cell types. (B) Protein expression of 40 genes showed monotonously upregulation (n = 21) or downregulation (n = 19) from NCI to MCI to AD based on the ROSMAP proteomic data. (C) Protein expression encoded by 15 genes showed monotonously upregulation (n=2) or downregulation (n=13) across controls, AsymAD and AD based on the BLSA proteomic data.
Further, we found that 46% of the DEGs were still differential between early-pathology and late-pathology subgroups (Supplementary Table 3). This observation implied that expression dysregulation observed in the late stages of AD could have happened early before symptoms could be observed. These DEGs might be valuable for AD risk prediction.
3.5 Protein expression of top-ranked genes are associated with cognitive function and AD clinical severity
First, we investigated whether expression changes of proteins encoded by top-ranked genes were associated with cognitive function. We obtained clinical and protein expression data from the ROSMAP study (see Materials and Methods). We identified three groups of individuals with different levels of cognitive function: NCI (n = 174), MCI (n = 100) and AD (n = 104). We focused on the top-ranked 200 genes, of which 131 have protein expression available. This analysis identified 40 genes (31%) of which the protein expression levels were significantly correlated with cognitive function (FDR < 0.05; see Materials and Methods; Supplementary Figure 4). Of them, 21 showed positive correlations and the remaining 19 showed negative correlations (Figure 4B). For example, the expression of MAPK1 and PLCB1 was positively and negatively correlated with cognitive function, respectively.
Second, we examined whether protein expression of top-ranked genes was associated with AD clinical severity. Asymptomatic AD (AsymAD) was defined as a preclinical state by the international working group [46]. Clinical and protein expression data were obtained from the BLSA study. We identified control (n = 13), AsymAD (n = 14) and AD (n = 20) samples. 57 of the top-ranked 200 genes had protein expression available and were considered for this analysis. This analysis identified 15 (26%) proteins exhibiting a significant correlation with AD clinical severity (Supplementary Figure 5). Of these, two genes showed positive correlation and the remaining showed negative correlation (Figure 4C).
In summary, this proteomic analysis showed that our top predictions were associated with cognitive function and AD clinical severity, suggesting that they might be promising risk genes for AD.
3.6 Case studies of top-ranked genes supported by multiple lines of evidence
In the above sections, we showed that the top-ranked genes were associated with AD supported by different types of functional genomic evidence. To identify genes with multiple evidence, we considered the following six lines of individual gene-level evidence: the correlation with the CERAD, Braak and CDR score based on the MSBB dataset, the differential expression between no-pathology and early-pathology individuals based on the Alzheimer’s brain scRNA-seq data, the association with cognitive function based on the ROSMAP data and the association with AD clinical severity based on the BLSA data. The evidence of the top-ranked 200 genes was visualized in the circular plot (Figure 5). We considered the genes with at least four lines of evidence. In total, 59 such genes were identified (Supplementary Table 3). Specifically, three genes (PLCB1, PAK1, ACTN2) were supported by all six lines of evidence. This analysis provided a set of multiple evidence-based candidate genes to the community for further experimental verification.
Figure 5.
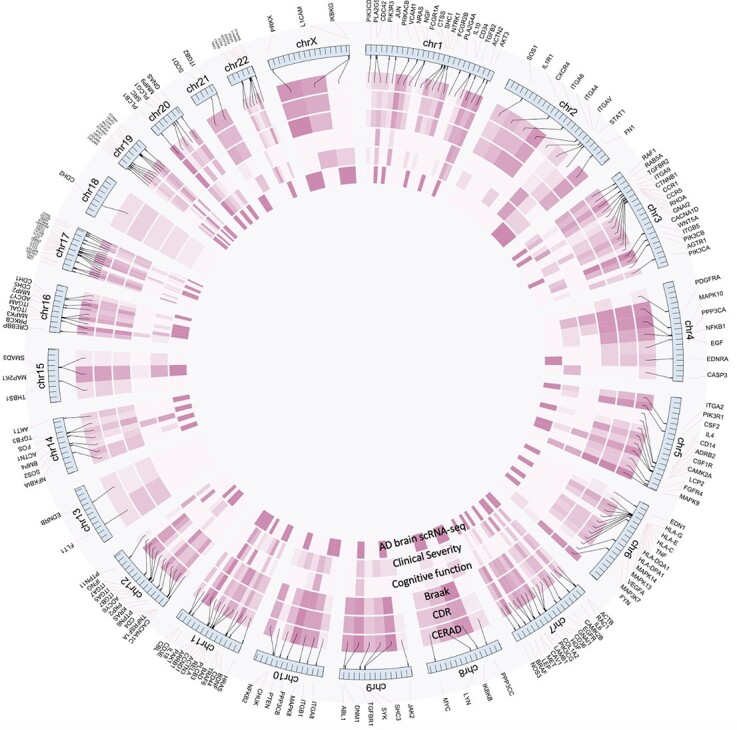
Visualization of the six lines of evidence supporting the functional association of the top-ranked 200 genes with AD. The six circles show the strength of the six lines of evidence, including the Pearson correlation with the CERAD, Braak and CDR scores based on the MSBB dataset, the differential expression between controls and early-pathology AD based on the human brain scRNA-seq data, the association with cognitive function based on the ROSMAP proteomic data and the association with AD clinical severity based on the BLSA proteomic data. The darker the color is, the stronger the association is.
We selected PLCB1 (phospholipase C beta 1) for illustration because it was supported by six lines of evidence (Figure 6). The gene expression was correlated with the CERAD (PCC = -0.37), Braak (PCC = -0.35) and CDR (PCC = -0.37) score (FDR < 0.001) (Figure 6A–C). Its expression was upregulated in inhibitory neurons and downregulated in oligodendrocytes in individuals with early-pathology AD compared with that in healthy controls (Figure 6D). Increased protein expression of PLCB1 was associated with a higher level of cognitive function declining (Figure 6E) and AD clinical severity (Figure 6F). Previous work showed that PLCB1 was genetically associated with AD with suggestive evidence based on GWAS Catalog database (P-value = 5.0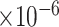 for rs3859675 [47]; P-value = 2.0
for rs3859675 [47]; P-value = 2.0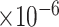 for rs117019330 [48]). Our analysis at the gene and protein expression level provides further evidence supporting the association of PLCB1 with AD, making it a promising candidate risk gene.
for rs117019330 [48]). Our analysis at the gene and protein expression level provides further evidence supporting the association of PLCB1 with AD, making it a promising candidate risk gene.
Figure 6.
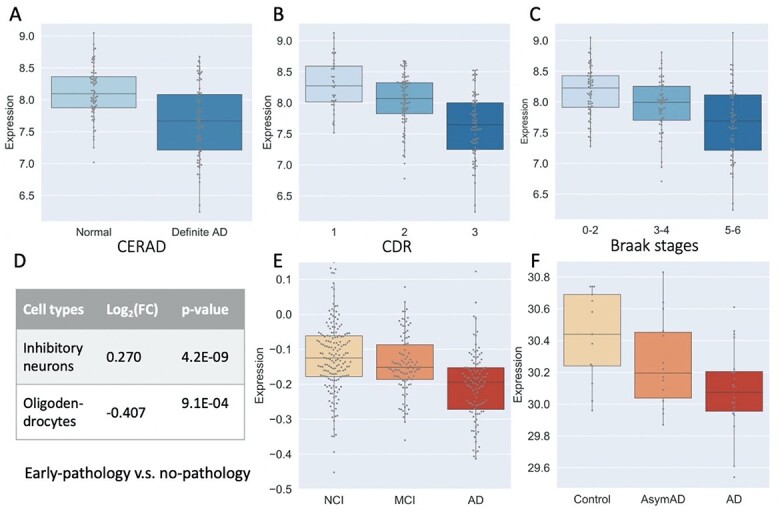
Multiple lines of evidence supporting the association of PLCB1 expression with AD traits. (A, B and C) The gene expression was associated with the CERAD, CDR and Braak stage scores using the MSBB dataset. (D) The expression in inhibitory neurons and oligodendrocytes was differentially expressed between no-pathology and early-pathology individuals based on the brain scRNA-seq study. A positive sign of log(FC) indicates that the gene expression is upregulated in early-pathology individuals compared with no-pathology individuals. In contrast, a negative sign indicates that the gene expression is downregulated in early-pathology individuals compared with no-pathology individuals. For this comparison, PLCB1 is upregulated in inhibitory neurons (thus giving a positive sign of log(FC)) but downregulated in oligodendrocytes (thus a negative sign of log(FC)). (E) Correlation of protein expression level with cognitive function based on the ROSMAP data. (F) Correlation of protein expression level with AD clinical severity using the BLSA data.
4 Discussion
AD is a neurodegenerative disease with heterogeneous pathologies [16, 49, 50]. However, predicting AD-associated genes remains a challenge because AD is caused mainly by common variants of multiple genes and by the disruption of complex pathways. FGNs are an important model for characterizing complex functional relationships between genes and have been successfully applied to predict candidate genes for complex diseases, including autism [19] and Parkinson’s disease [51]. Since AD is caused by gene dysregulation in the brain, we considered and constructed an Alzheimer’s brain gene network, called ADBrainNexus, for predicting AD-associated genes. The key idea of our approach was to learn the pattern of AD-associated genes from ADBrainNexus using machine learning methods. Our model assigns each gene a probabilistic score, which reflects the likelihood that the gene is associated with AD.
We demonstrated that the top-ranked genes predicted by our approach were functionally relevant to AD by interrogating multiple lines of genomic evidence. First, we showed that AD-associated pathways or phenotypes were enriched for top-ranked genes using the decile enrichment test. Second, based on the analyses of the independent MSBB data, we observed that the top-ranked genes were correlated with AD-associated neuropathological (CERAD and Braak scores) and clinical (CDR) traits, suggesting that they were likely associated with AD. Third, the analysis of an AD brain scRNA-seq dataset found that a large proportion of the top-ranked genes were differentially expressed between no-pathology and early-pathology individuals, implying their association with early-stage AD. Intersecting our predicted genes with the DEGs identified in different cell types revealed cell-type-resolved association of top-ranked genes with AD. Fourth, using external data from the ROSMAP and BLSA studies, we showed that the protein expression of top-ranked genes was associated with the degrees of cognitive function (NCI, MCI and AD) and AD clinical severity (controls, AsymAD, AD). Taken together, the above multi-omics analysis of molecular, neuropathological and clinical data provided evidence that our predictions were reliable, and the top-ranked genes were promising candidates for further experimental verification.
Our contributions are 3-fold. First, we constructed ADBrainNexus, a brain-specific FGN by integrating multiple AD brain RNA-seq datasets and several other gene interaction networks. We showed that this network showed better performance in predicting AD-associated genes than existing networks. Second, we collected a set of genes that were likely associated to AD by performing an intensive, stringent hand curation of multiple resources, providing a potential resource for the community. Third, we predicted novel candidate genes and showed that the top-ranked genes exhibit significant associations with AD through functional enrichment analysis and multi-omics analysis of multiple external datasets. These genes were found to be correlated with AD-associated traits such as early-pathology, neuropathological traits, cognitive function and clinical severity. We narrowed down the top-ranked 200 genes by taking advantage of the multiple lines of evidence. This resulted in a list of 59 AD-associated genes supported by at least four lines of evidence, providing a set of promising candidates to the community for further experimental testing.
Although our predictions are promising, our model to predict AD-associated genes could be improved in several ways. First, our predictions were made at the gene level without differentiating splice isoforms generated from the same gene through alternative splicing [6, 52, 53]. This is essential because isoforms of the same gene might have different or even opposite functions. Isoforms have been implicated in diseases such as ovarian cancers [54]. The prediction of AD-associated genes at the isoform level could have the potential to promote our understanding of AD. Second, the human brain consists of multiple heterogeneous structures, each of which may contain many cell types. ADBrainNexus is not cell-type-resolved yet. Building cell-type-specific FGN by leveraging single-cell genomic data [55–58] could be helpful to address this question. Lastly, our predictions do not implicate causality. Our predictions represent only statistical association.
In summary, we predicted novel AD-associated genes and provided multiple lines of evidence at different molecular levels supporting their association with AD. Further studies are needed to experimentally test the validity of our predictions. The developed method for prediction and validation is generic and can be readily extended to other complex diseases, such as Parkinson’s disease, cancers and heart diseases. We expect that the predicted genes might become a useful resource for therapeutic target discovery for AD.
Key Points
We constructed a brain-specific functional gene network (called ADBrainNexus for short), which achieved better performance in predicting Alzheimer’s disease (AD)-associated genes than existing networks.
A set of known AD-associated genes are collected by intensive manual curation of various disease gene resources, including OMIM, GWAS Catalog, DisGenet and AD-associated publications.
We built a model by mining ADBrainNexus and predicted novel candidate genes for AD. The association of top-ranked genes with AD were validated using genetic, transcriptomic and proteomic data from multiple external datasets.
Supplementary Material
Funding
National Natural Science Foundation of China [No. U1909208, 61972423, 61732009 to J.X.W.]; 111 Project [No. B18059 to J.X.W.]; Hunan Provincial Science and Technology Program (2018WK4001 to J.X.W.). The results published here are in part based on data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org/). The Mayo RNA-seq data were provided by the following sources: The Mayo Clinic Alzheimer’s Disease Genetic Studies, led by Dr Nilufer Ertekin-Taner and Dr Steven G. Younkin, Mayo Clinic, Jacksonville, FL using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimer’s Disease Research Center and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, NINDS grant R01 NS080820, CurePSP Foundation, and support from Mayo Foundation. Study data include samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders); the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center); the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center); the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. The MSBB data were generated from postmortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by Dr. Eric Schadt from Mount Sinai School of Medicine.
Cui-Xiang Lin is a PhD student in the School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China. Her research interests include bioinformatics and computational medicine.
Hong-Dong Li is an associate professor in the School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China. His research interests include bioinformatics, computational medicine and systems biology.
Chao Deng is a master student in Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China. His research interests include bioinformatics and machine learning.
Weisheng Liu is a master student in School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China. His research interests include bioinformatics and machine learning.
Shannon Erhardt is a master student in Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, Texas, USA. Her research interests include biology and bioinformatics.
Fang-Xiang Wu is a professor in the Division of Biomedical Engineering, University of Saskatchewan, Saskatoon, SKS7N5A9, Canada. His research interests include computer algorithms and bioinformatics.
Xing-Ming Zhao is a professor in the Institute of Science and Technology for Brain-Inspired Intelligence, Fudan University, Shanghai, China. His research interests include disease omics and bioinformatics.
Yuanfang Guan is an Associate Professor in the Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan, United States. Her research interests include machine learning and computational medicine.
Jun Wang is a professor in the Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, Texas, USA. Her research interests include disease biology.
Daifeng Wang is a professor in the Department of Biostatistics and Medical Informatics and Waisman Center, University of Wisconsin-Madison, Madison, Wisconsin, USA. His research interests include systems biology of complex diseases.
Bin Hu is a professor in the Institute of Engineering Medicine, Beijing Institute of Technology, Beijing, China. His research interests include computational biology for neurologic diseases.
Jianxin Wang is a Professor in the School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China. His research interests include computational genomics and proteomics.
Contributor Information
Cui-Xiang Lin, School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China.
Hong-Dong Li, School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China.
Chao Deng, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China.
Weisheng Liu, School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China.
Shannon Erhardt, Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, Texas, USA.
Fang-Xiang Wu, Division of Biomedical Engineering, University of Saskatchewan, Saskatoon, SKS7N5A9, Canada.
Xing-Ming Zhao, Institute of Science and Technology for Brain-Inspired Intelligence, Fudan University, Shanghai, China.
Yuanfang Guan, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan, United States.
Jun Wang, Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, Texas, USA.
Daifeng Wang, Department of Biostatistics and Medical Informatics and Waisman Center, University of Wisconsin-Madison, Madison, Wisconsin, USA.
Bin Hu, Institute of Engineering Medicine, Beijing Institute of Technology, Beijing, China.
Jianxin Wang, School of Computer Science and Engineering, Central South University, Changsha, Hunan 410083, P. R. China; Hunan Provincial Key Lab of Bioinformatics, Central South University, Changsha, Hunan 410083, P. R. China.
References
- 1. Nativio R, Lan Y, Donahue G, et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat Genet 2020;52(10):1024–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Association A. Alzheimer’s disease facts and figures. Alzheimers Dement 2021;17:327–406. [DOI] [PubMed] [Google Scholar]
- 3. Neff RA, Wang M, Vatansever S, et al. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci Adv 2021;7(2):eabb5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnson EC, Dammer EB, Duong DM, et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med 2020;26(5):769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Higginbotham L, Ping L, Dammer E, et al. Integrated Proteomics Reveals Brain-Based Cerebrospinal Fluid Biomarkers in Asymptomatic and Symptomatic Alzheimer’s Disease. Sci Adv 2020;6(43):eaaz9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li HD, Funk CC, McFarland K, et al. Integrative functional genomic analysis of intron retention in human and mouse brain with Alzheimer’s disease. Alzheimers Dement 2021;17(6):984–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sims R, Hill M, Williams J. The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci 2020;23(3):311–22. [DOI] [PubMed] [Google Scholar]
- 8. Kim YW, Al-Ramahi I, Koire A, et al. Harnessing the paradoxical phenotypes of apoe 2 and apoe 4 to identify genetic modifiers in Alzheimer’s disease. Alzheimers Dement 2021;17(5):831–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Llibre-Guerra JJ, Li Y, Allegri RF, et al. Dominantly inherited Alzheimer’s disease in Latin America: genetic heterogeneity and clinical phenotypes. Alzheimers Dement 2021;17(4):653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sims R, Van Der Lee SJ, Naj AC, et al. Rare coding variants in plcg2, abi3, and trem2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 2017;49(9):1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allen M, Wang X, Burgess J, et al. Conserved brain myelination networks are altered in Alzheimer’s and other neurodegenerative diseases. Alzheimers Dement 2018;14:352–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mostafavi S, Gaiteri C, Sullivan SE, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 2018;21(6):811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bai B, Wang X, Li Y, et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 2020;105(6):975–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen L, Jin P, Qin ZS. Divan: accurate identification of non-coding disease-specific risk variants using multi-omics profiles. Genome Biol 2016;17(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ridge PG, Mukherjee S, Crane PK, et al. Alzheimer’s disease: analyzing the missing heritability. PLoS One 2013;8(11):e79771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cuyvers E, Sleegers K. Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. Lancet Neurol 2016;15(8):857–68. [DOI] [PubMed] [Google Scholar]
- 17. Ridge PG, Hoyt KB, Boehme K, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging 2016;41:200–e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guan Y, Myers CL, Lu R, et al. A genomewide functional network for the laboratory mouse. PLoS Comput Biol 2008;4(9):e1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krishnan A, Zhang R, Yao V, et al. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat Neurosci 2016;19(11):1454–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greene CS, Krishnan A, Wong AK, et al. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet 2015;47(6):569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun X, Pittard WS, Xu T, et al. Omicseq: a web-based search engine for exploring omics datasets. Nucleic Acids Res 2017;45(W1):W445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Troyanskaya OG, Dolinski K, Owen AB, et al. A bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc Natl Acad Sci 2003;100(14):8348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guan Y, Ackert-Bicknell CL, Kell B, et al. Functional genomics complements quantitative genetics in identifying disease-gene associations. PLoS Comput Biol 2010;6(11):e1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Recla JM, Robledo RF, Gatti DM, et al. Precise genetic mapping and integrative bioinformatics in diversity outbred mice reveals hydin as a novel pain gene. Mamm Genome 2014;25(5):211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li HD, Bai T, Sandford E, et al. Baihui: cross-species brain-specific network built with hundreds of hand-curated datasets. Bioinformatics 2019;35(14):2486–8. [DOI] [PubMed] [Google Scholar]
- 26. Ziemann M, Kaspi A, El-Osta A. Digital expression explorer 2: a repository of uniformly processed RNA sequencing data. Gigascience 2019;8(4):giz022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li HD, Menon R, Govindarajoo B, et al. Functional networks of highest-connected splice isoforms: from the chromosome 17 human proteome project. J Proteome Res 2015;14(9):3484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang Y, Sun X, Jiang H, et al. A machine learning approach to brain epigenetic analysis reveals kinases associated with Alzheimer’s disease. Nat Commun 2021;12(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith RG, Pishva E, Shireby G, et al. A meta-analysis of epigenome-wide association studies in Alzheimer’s disease highlights novel differentially methylated loci across cortex. Nat Commun 2021;12(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bertram L, McQueen MB, Mullin K, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the alzgene database. Nat Genet 2007;39(1):17–23. [DOI] [PubMed] [Google Scholar]
- 31. Bai Z, Han G, Xie B, et al. Alzbase: an integrative database for gene dysregulation in Alzheimer’s disease. Mol Neurobiol 2016;53(1):310–9. [DOI] [PubMed] [Google Scholar]
- 32. Hamosh A, Scott AF, Amberger JS, et al. Online Mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005;33(suppl_1):D514–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Piñero J, Queralt-Rosinach N, Bravo A, et al. Disgenet: a discovery platform for the dynamical exploration of human diseases and their genes. Database 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palleja A, Horn H, Eliasson S, et al. Distild database: diseases and traits in linkage disequilibrium blocks. Nucleic Acids Res 2012;40(D1):D1036–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu CH, Apweiler R, Bairoch A, et al. The universal protein resource (uniprot): an expanding universe of protein information. Nucleic Acids Res 2006;34(suppl_1):D187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carvalho-Silva D, Pierleoni A, Pignatelli M, et al. Open targets platform: new developments and updates two years on. Nucleic Acids Res 2019;47(D1):D1056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019;47(D1):D1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Canchi S, Raao B, Masliah D, et al. Integrating gene and protein expression reveals perturbed functional networks in Alzheimer’s disease. Cell Rep 2019;28(4):1103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Duda M, Zhang H, Li HD, et al. Brain-specific functional relationship networks inform autism spectrum disorder gene prediction. Transl Psychiatry 2018;8(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang K, Wang R, Liu G, et al. Hergepred: heterogeneous network embedding representation for disease gene prediction. IEEE J Biomed Health Inform 2018;23(4):1805–15. [DOI] [PubMed] [Google Scholar]
- 41. Wang M, Roussos P, McKenzie A, et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med 2016;8(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates a, tau, immunity and lipid processing. Nat Genet 2019;51(3):414–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 2021;17(3):157–72. [DOI] [PubMed] [Google Scholar]
- 44. Dube U, Del-Aguila JL, Li Z, et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat Neurosci 2019;22(11):1903–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mathys H, Davila-Velderrain J, Peng Z, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019;570(7761):332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010;9(11):1118–27. [DOI] [PubMed] [Google Scholar]
- 47. Dumitrescu L, Barnes LL, Thambisetty M, et al. Sex differences in the genetic predictors of Alzheimer’s pathology. Brain 2019;142(9):2581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu C, Yu J. Genome-wide association studies for cerebrospinal fluid soluble trem2 in Alzheimer’s disease. Front Aging Neurosci 2019;11:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cummings J, Feldman HH, Scheltens P. The “rights” of precision drug development for Alzheimer’s disease. Alzheimer’s Res Therapy 2019;11(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at clu and cr1 associated with Alzheimer’s disease. Nat Genet 2009;41(10):1094–9. [DOI] [PubMed] [Google Scholar]
- 51. Yao V, Kaletsky R, Keyes W, et al. An integrative tissue-network approach to identify and test human disease genes. Nat Biotechnol 2018;36(11):1091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li HD, Menon R, Omenn GS, et al. The emerging era of genomic data integration for analyzing splice isoform function. Trends Genet 2014;30(8):340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol 2017;18(7):437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barrett CL, DeBoever C, Jepsen K, et al. Systematic transcriptome analysis reveals tumor-specific isoforms for ovarian cancer diagnosis and therapy. Proc Natl Acad Sci 2015;112(23):E3050–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fetahu IS, Ma D, Rabidou K, et al. Epigenetic signatures of methylated DNA cytosine in Alzheimer’s disease. Sci Adv 2019;5(8):eaaw2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zheng R, Li M, Liang Z, et al. Sinnlrr: a robust subspace clustering method for cell type detection by non-negative and low-rank representation. Bioinformatics 2019;35(19):3642–50. [DOI] [PubMed] [Google Scholar]
- 57. Cao J, Spielmann M, Qiu X, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019;566(7745):496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin CX, Li HD, Deng C, et al. TissueNexus: a database of human tissue functional gene networks built with a large compendium of curated RNA-seq data. Nucleic Acids Res. 2021;gkab1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.