

Abstract
Since its selection as the method of the year in 2013, single-cell technologies have become mature enough to provide answers to complex research questions. With the growth of single-cell profiling technologies, there has also been a significant increase in data collected from single-cell profilings, resulting in computational challenges to process these massive and complicated datasets. To address these challenges, deep learning (DL) is positioned as a competitive alternative for single-cell analyses besides the traditional machine learning approaches. Here, we survey a total of 25 DL algorithms and their applicability for a specific step in the single cell RNA-seq processing pipeline. Specifically, we establish a unified mathematical representation of variational autoencoder, autoencoder, generative adversarial network and supervised DL models, compare the training strategies and loss functions for these models, and relate the loss functions of these models to specific objectives of the data processing step. Such a presentation will allow readers to choose suitable algorithms for their particular objective at each step in the pipeline. We envision that this survey will serve as an important information portal for learning the application of DL for scRNA-seq analysis and inspire innovative uses of DL to address a broader range of new challenges in emerging multi-omics and spatial single-cell sequencing.
Keywords: deep learning, single-cell RNA-seq, imputation, dimensionality reduction, clustering, batch correction, cell-type identificationfunctional prediction, visualization
Introduction
Single cell sequencing technology has been a rapidly developing area to study genomics, transcriptomics, proteomics, metabolomics and cellular interactions at the single cell level for cell-type identification, tissue composition and reprogramming [1, 2]. Specifically, sequencing of the transcriptome of single cells, or single-cell RNA-sequencing (scRNA-seq), has become the dominant technology in many frontier research areas such as disease progression and drug discovery [3, 4]. One particular area where scRNA-seq has made a tangible impact is cancer, where scRNA-seq is becoming a powerful tool for understanding invasion, intratumor heterogeneity, metastasis, epigenetic alterations, detecting rare cancer stem cells and therapeutic response [5, 6]. Currently, scRNA-seq is applied to develop personalized therapeutic strategies that are potentially useful in cancer diagnosis, therapy resistance during cancer progression and the survival of patients [5, 7]. The scRNA-seq has also been adopted to combat COVID-19 to elucidate how the innate and adaptive host immune system miscommunicates, worsening the immunopathology produced during the viral infection [8, 9].
These studies have led to a massive amount of scRNA-seq data deposited to public databases such as the 10× single-cell gene expression dataset, Human Cell Atlas and Mouse Cell Atlas. Expressions of millions of cells from 18 species have been collected and deposited, waiting for further analysis (Single Cell Expression Atlas, EMBL-EBI, October 2021). On the other hand, due to biological and technical factors, scRNA-seq data present several analytical challenges related to its complex characteristics such as missing expression values, high technical and biological variance, noise and sparse gene coverage, and elusive cell identities [1]. These characteristics make it difficult to directly apply commonly used bulk RNA-seq data analysis techniques and have called for novel statistical approaches for scRNA-seq data cleaning and computational algorithms for data analysis and interpretation. To this end, specialized scRNA-seq analysis pipelines such as Seurat [10] and Scanpy [11], along with a large collection of task-specific tools, have been developed to address the intricate technical and biological complexity of scRNA-seq data.
Recently, deep learning (DL) has demonstrated its significant advantages in natural language processing and speech and facial recognition with massive data [12–14]. Such advantages have initiated the application of DL in scRNA-seq data analysis as a competitive alternative to conventional machine learning (ML) approaches for uncovering cell clustering [15, 16], cell-type identification [15, 17], gene imputation [18–20] and batch correction [21] in scRNA-seq analysis. Compared with conventional ML approaches, DL is more powerful in capturing complex features of high-dimensional scRNA-seq data. It is also more versatile, where a single model can be trained to address multiple tasks or adapted and transferred to different tasks. Moreover, DL training scales more favorably with the number of cells in scRNA-seq data size, making it particularly attractive for handling the ever-increasing volume of single cell data. Indeed, the growing body of DL-based tools has demonstrated DL’s exciting potential as a learning paradigm to significantly advance the tools we use to interrogate scRNA-seq data.
In this paper, we present a comprehensive review of the recent advances of DL methods for solving the challenges in scRNA-seq data analysis (Table 1) from the quality control (QC), normalization/batch effect correction, dimensionality reduction, visualization, feature selection and data interpretation by surveying DL papers published up to April 2021. In order to maintain high quality for this review, we choose not to include any (bio)archival papers, although a proportion of these manuscripts contain important new findings that would be published after completing their peer-reviewed process. Previous efforts to review the recent advances in ML methods focused on efficient integration of single cell data [22, 23]. A recent review of DL applications on single cell data has summarized 21 DL algorithms that might be deployed in single cell studies [24]. It also evaluated the clustering and data correction effect of these DL algorithms using 11 datasets.
Table 1.
DL algorithms reviewed in the paper
| App | Algorithm | Models | Evaluation | Environment | Codes | Refs |
|---|---|---|---|---|---|---|
| Imputation | ||||||
| DCA | AE | DREMI | Keras, Tensorflow, scanpy | https://github.com/theislab/dca | [18] | |
| SAVER-X | AE + TL | t-SNE, ARI | R/sctransfer | https://github.com/jingshuw/SAVERX | [58] | |
| DeepImpute | DNN | MSE, Pearson’s correlation | Keras/Tensorflow | https://github.com/lanagarmire/DeepImpute | [20] | |
| LATE | AE | MSE | Tensorflow | https://github.com/audreyqyfu/LATE | [59] | |
| scGAMI | AE | NMI, ARI, HS and CS | Tensorflow | https://github.com/QUST-AIBBDRC/scGMAI/ | [60] | |
| scIGANs | GAN | ARI, ACC, AUC and F-score | PyTorch | https://github.com/xuyungang/scIGANs | [19] | |
| Batch correction | ||||||
| BERMUDA | AE + TL | KNN batch-effect test (kBET), the entropy of Mixing, SI | PyTorch | https://github.com/txWang/BERMUDA | [63] | |
| DESC | AE | ARI, KL | Tensorflow | https://github.com/eleozzr/desc | [67] | |
| iMAP | AE + GAN | kBET, Local Inverse Simpson’s Index (LISI) | PyTorch | https://github.com/Svvord/iMAP | [70] | |
| Clustering, latent representation, dimension reduction and data augmentation | ||||||
| Dhaka | VAE | ARI, Spearman Correlation | Keras/Tensorflow | https://github.com/MicrosoftGenomics/Dhaka | [72] | |
| scvis | VAE | KNN preservation, log-likelihood | Tensorflow | https://bitbucket.org/jerry00/scvis-dev/src/master/ | [75] | |
| scVAE | VAE | ARI | Tensorflow | https://github.com/scvae/scvae | [76] | |
| VASC | VAE | NMI, ARI, HS and CS | H5py, keras | https://github.com/wang-research/VASC | [77] | |
| scDeepCluster | AE | ARI, NMI, clustering accuracy | Keras, Scanpy | https://github.com/ttgump/scDeepCluster | [79] | |
| cscGAN | GAN | t-SNE, marker genes, MMD, AUC | Scipy, Tensorflow | https://github.com/imsb-uke/scGAN | [82] | |
| Multi-functional models (IM: imputation, BC: batch correction, CL: clustering) | ||||||
| scVI | VAE | IM: L1 distance; CL: ARI, NMI, SI; BC: Entropy of Mixing | PyTorch, Anndata | https://github.com/YosefLab/scvi-tools | [17] | |
| LDVAE | VAE | Reconstruction errors | Part of scVI | https://github.com/YosefLab/scvi-tools | [86] | |
| SAUCIE | AE | IM: R2 statistics; CL: SI; BC: modified kBET; Visualization: Precision/Recall | Tensorflow | https://github.com/KrishnaswamyLab/SAUCIE/ | [15] | |
| scScope | AE | IM:Reconstruction errors; BC: Entropy of mixing; CL: ARI | Tensorflow, Scikit-learn | https://github.com/AltschulerWu-Lab/scScope | [92] | |
| Cell-type Identification | ||||||
| DigitalDLSorter | DNN | Pearson correlation | R/Python/Keras | https://github.com/cartof/digitalDLSorter | [51] | |
| scCapsNet | CapsNet | Cell-type Prediction accuracy | Keras, Tensorflow | https://github.com/wanglf19/scCaps | [52] | |
| netAE | VAE | Cell-type Prediction accuracy, t-SNE for visualization | pyTorch | https://github.com/LeoZDong/netAE | [101] | |
| scDGN | DANN | Prediciton accuracy | pyTorch | https://github.com/SongweiGe/scDGN | [53] | |
| Function analysis | ||||||
| CNNC | CNN | AUROC, AUPRC and accuracy | Keras, Tensorflow | https://github.com/xiaoyeye/CNNC | [54] | |
| scGen | VAE | Correlation, visualization | Tensorflow | https://github.com/theislab/scgen | [114] | |
DL Model keywords: AE + TL: autoencoder with transfer learning, DANN: domain adversarial neural network, CapsNet: capsule neural network
In this review, we focus more on the DL algorithms with a much detailed explanation and comparison. Furthermore, to better understand the relationship of each surveyed DL model with the overall scRNA-seq analysis pipeline, we organize the surveys according to the challenge they address and discuss these DL models following the analysis pipeline. A unified mathematical description of the surveyed DL models is presented and the specific model features are discussed when reviewing each method. This will also shed light on the modeling connections among the surveyed DL methods and the recognization of the uniqueness of each model. Besides the models, we also summarize the evaluation matrics used by these DL algorithms and methods that each DL algorithm was compared with. The online location of the code, the development platform and the used datasets for each method are also cataloged to facilitate their utilization and additional effort to improve them. Finally, we also created a companion online version of the paper at https://huang-ai4medicine-lab.github.io/survey-of-DL-for-scRNA-seq-analysis/_book/, which includes expanded discussion as well as a survey of additional methods. We envision that this survey will serve as an important information portal for learning the application of DL for scRNA-seq analysis and inspire innovative use of DL to address a broader range of new challenges in emerging multi-omics and spatial single-cell sequencing.
Overview of the scRNA-seq processing pipeline
Various scRNA-seq techniques (such as SMART-seq, Drop-seq and 10× genomics sequencing) [25, 26] are available nowadays with their sets of advantages and disadvantages. Despite the differences in the scRNA-seq techniques, the data content and processing steps of scRNA-seq data are quite standard and conventional. A typical scRNA-seq dataset consists of three files: genes quantified (gene IDs), cells quantified (cellular barcode) and a count matrix (number of cells × number of genes), irrespective of the technology or pipeline used. A series of essential steps in the scRNA-seq data processing pipeline and optional tools for each step with both ML and DL approaches are illustrated in Figure 1.
Figure 1.

Single cell data analysis steps for both conventional ML methods (bottom) and DL methods (top). Depending on the input data and analysis objectives, major scRNA-seq analysis steps are illustrated in the center flow chart (color boxes) with conventional ML approaches along with optional analysis modules below each analysis step. DL approaches are categorized as DNN, GAN, VAE, and AE. For each DL approach, optional algorithms are listed on top of each step.
With the advantage of identifying each cell and unique molecular identifiers (UMIs) for expressions of each gene in a single cell, scRNA-seq data are embedded with increased technical noise and biases [27]. QC is the first and the key step to filter out dead cells, double-cells or cells with failed chemistry or other technical artifacts. The most commonly adopted three QC covariates include the number of counts (count depth) per barcode identifying each cell, the number of genes per barcode and the fraction of counts from mitochondrial genes per barcode [28].
Normalization is designed to eliminate imbalanced sampling, cell differentiation, viability and many other factors. Approaches tailored for scRNA-seq have been developed including the Bayesian-based method coupled with spike-in, or BASiCS [29], deconvolution approach, scran [30] and scTransfrom in Seurat where regularized Negative Binomial Regression was proposed [31]. Two important steps, batch correction and imputation, will be carried out if required by the analysis.
Batch Correction is a common source of technical variation in high-throughput sequencing experiments due to variant experimental conditions such as technicians and experimental time, imposing a major challenge in scRNA-seq data analysis. Batch effect correction algorithms include detection of mutual nearest neighbors (MNNs) [32], canonical correlation analysis (CCA) with Seurat [33] and Harmony algorithm through cell-type representation [34].
Imputation step is necessary to handle high sparsity data matrix, due to missing value or dropout in scRNA-seq data analysis. Several tools have been developed to ‘impute’ zero values in scRNA-seq data, such as SCRABBLE [35], SAVER [36] and scImpute [37].
Dimensionality reduction and visualization are essential steps to represent biologically meaningful variations and high dimensionality with significantly reduced computational cost. Dimensionality reduction methods, such as principal component analysis (PCA), are widely used in scRNA-seq data analysis to achieve that purpose. More advanced nonlinear approaches that preserve the topological structure and avoid overcrowding in lower dimension representation, such as LLE [38] (used in SLICER [39]), t-SNE [40] and UMAP [41], have also been developed and adopted as a standard in single-cell data visualization.
Clustering analysis is a key step to identify cell subpopulations or distinct cell types to unravel the extent of heterogeneity and their associated cell-type-specific markers. Unsupervised clustering is frequently used to categorize cells into clusters according to their similarity often measured in the aforementioned dimensionality-reduced representations. Some of popular algorithms include the community detection algorithm Louvain [42] and Leiden [43], and data-driven dimensionality reduction followed with k-Means cluster by SIMLR [44].
Feature selection is another important step in single-cell RNA-seq analysis to select a subset of genes, or features, for cell-type identification and functional enrichment of each cluster. This step is achieved by differential expression analysis designed for scRNA-seq, such as MAST that used linear model fitting and likelihood ratio testing [45]; SCDE that adopted a Bayesian approach with a Negative Binomial model for gene expression and Poisson process for dropouts [46], or DEsingle that utilized a Zero-Inflated Negative Binomial (ZINB) model to estimate the dropouts [47].
Besides these key steps, downstream analyses include cell-type identification, coexpression analysis, prediction of perturbation response, where DL has also been applied. Other advanced analyses including trajectory inference and velocity and pseudotime analysis are not discussed here because most of the approaches on these topics are non-DL based.
Overview of common DL models for scRNA-seq analysis
We start our review by introducing the general formulations of widely used DL models. As most of the tasks including batch correction, dimensionality reduction, imputation and clustering are unsupervised learning tasks, we will give special attention to unsupervised models including variational autoencoder (VAE), the autoencoder (AE) or generative adversarial networks (GAN). We will also discuss the general supervised and transfer learning formulations, which find their applications in cell-type predictions and functional studies. We will discuss these models in the context of scRNA-seq, detailing the different features and training strategies of each model and bringing attention to their uniqueness.
Variational autoencoder
Let  represent a
represent a 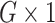 vector of expression levels (UMI counts or normalized, log-transformed expression) of
vector of expression levels (UMI counts or normalized, log-transformed expression) of 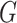 genes in cell
genes in cell 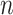 , where
, where 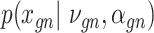 follows some distribution (e.g. ZINB or Gaussian), where
follows some distribution (e.g. ZINB or Gaussian), where 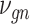 and
and 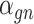 are distribution parameters (e.g. mean, variance or dispersion) (Figure 2A). We consider
are distribution parameters (e.g. mean, variance or dispersion) (Figure 2A). We consider 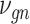 to be of particular interest (e.g. the mean counts) and is thus further modeled by a decoder neural network
to be of particular interest (e.g. the mean counts) and is thus further modeled by a decoder neural network 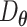 (Figure 2A) as
(Figure 2A) as
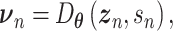 |
(1) |
where the  th element of
th element of  is
is 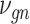 and
and  is a vector of decoder weights,
is a vector of decoder weights, 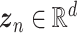 represents a latent representation of gene expression and is used for visualization and clustering and
represents a latent representation of gene expression and is used for visualization and clustering and 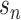 is an observed variable (e.g. the batch ID). For VAE,
is an observed variable (e.g. the batch ID). For VAE, 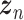 is commonly assumed to follow a multivariate standard Normal prior, i.e.
is commonly assumed to follow a multivariate standard Normal prior, i.e. 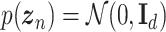 with
with 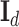 being a
being a  identity matrix. Furthermore,
identity matrix. Furthermore, 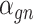 of
of 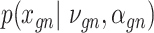 is a nuisance parameter, which has a prior distribution
is a nuisance parameter, which has a prior distribution 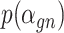 and can be either estimated or marginalized in variational inference. Now define
and can be either estimated or marginalized in variational inference. Now define  . Then,
. Then, 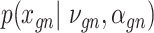 and (1) together define the likelihood
and (1) together define the likelihood 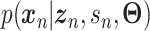 .
.
Figure 2.

Graphical models of the major surveyed DL models including (A) VAE, (B) AE and (C) GAN.
The goal of training is to compute the maximum likelihood estimate of 
 |
(2) |
where 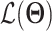 is the evidence lower bound (ELBO)
is the evidence lower bound (ELBO)
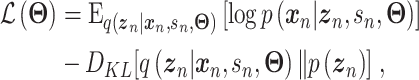 |
(3) |
and 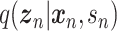 is an approximate to
is an approximate to 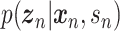 and assumed as
and assumed as
 |
(4) |
with 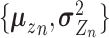 given by an encoder network
given by an encoder network 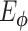 (Figure 2A) as
(Figure 2A) as
 |
(5) |
where  is the weights vector. Now,
is the weights vector. Now, 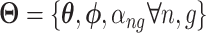 and Eq. (2) is solved by the stochastic gradient descent approach while a model is trained.
and Eq. (2) is solved by the stochastic gradient descent approach while a model is trained.
All the surveyed papers that deploy VAE follow this general modeling process. However, a more general formulation has a loss function defined as
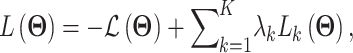 |
(6) |
where 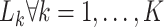 are losses for different functions (clustering, cell-type prediction, etc.) and
are losses for different functions (clustering, cell-type prediction, etc.) and 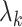 s are the Lagrange multipliers. With this general formulation, for each paper, we examined the specific choices of data distribution
s are the Lagrange multipliers. With this general formulation, for each paper, we examined the specific choices of data distribution  that define
that define 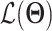 , different
, different 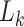 designed for specific functions, and how the decoder and encoder were applied to model different aspects of scRNA-seq data.
designed for specific functions, and how the decoder and encoder were applied to model different aspects of scRNA-seq data.
Autoencoders
AEs learn the low dimensional latent representation 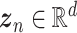 of expression
of expression 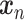 . The AE includes an encoder
. The AE includes an encoder 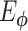 and a decoder
and a decoder 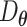 (Figure 2B) such that
(Figure 2B) such that
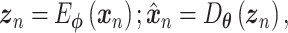 |
(7) |
where 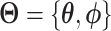 are encoder and decoder weight parameters, respectively, and
are encoder and decoder weight parameters, respectively, and  defines the parameters (e.g. mean) of the likelihood
defines the parameters (e.g. mean) of the likelihood 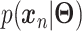 (Figure 2B) and is often considered as imputed and denoised expressions. Additional design can be included in an AE model for batch correction, clustering and other objectives.
(Figure 2B) and is often considered as imputed and denoised expressions. Additional design can be included in an AE model for batch correction, clustering and other objectives.
The training of an AE model is generally carried out by stochastic gradient descent algorithms to minimize the loss similar to Eq. (6) except 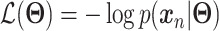 . When
. When 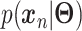 is the Gaussian,
is the Gaussian, 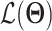 becomes the mean square error (MSE) loss
becomes the mean square error (MSE) loss
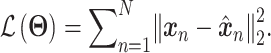 |
(8) |
Because different AE models differ in their AE architectures and loss functions, we will discuss the specific architecture and loss functions for each reviewed DL model in ****Section 4.
Generative adversarial networks
GANs have been used for imputation, data generation and augmentation of the scRNA-seq analysis. Without loss of generality, the GAN, when applied to scRNA-seq, is designed to learn how to generate gene expression profiles from 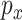 , the distribution of
, the distribution of 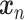 . The vanilla GAN consists of two deep neural networks (DNNs) [48]. The first network is the generator
. The vanilla GAN consists of two deep neural networks (DNNs) [48]. The first network is the generator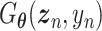 with parameter
with parameter 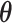 , a noise vector
, a noise vector  from the distribution
from the distribution 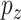 and a class label
and a class label 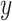 (e.g. cell type), and is trained to generate
(e.g. cell type), and is trained to generate 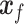 , a ‘fake’ gene expression (Figure 2C). The second network is the discriminator network
, a ‘fake’ gene expression (Figure 2C). The second network is the discriminator network 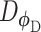 with parameters
with parameters 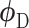 , trained to distinguish the ‘real’
, trained to distinguish the ‘real’ 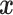 from fake
from fake 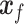 (Figure 2C). Both networks,
(Figure 2C). Both networks, 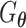 and
and 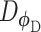 , are trained to outplay each other, resulting in a minimax game, in which
, are trained to outplay each other, resulting in a minimax game, in which 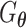 is forced by
is forced by 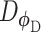 to produce better samples, which, when converge, can fool the discriminator
to produce better samples, which, when converge, can fool the discriminator  , thus becoming samples from
, thus becoming samples from 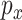 . The vanilla GAN suffers heavily from training instability and mode collapsing [49]. To that end, Wasserstein GAN (WGAN) [49] was developed with the WGAN loss [50]
. The vanilla GAN suffers heavily from training instability and mode collapsing [49]. To that end, Wasserstein GAN (WGAN) [49] was developed with the WGAN loss [50]
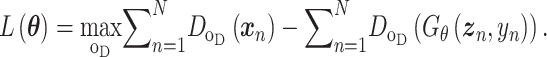 |
(9) |
Additional terms can also be added to Eq. (9) to constrain the functions of the generator. Training based on the WGAN loss in Eq. (9) amounts to a min-max optimization, which iterates between the discriminator and the generator, where each optimization is achieved by a stochastic gradient descent algorithm through back-propagation. The WGAN requires 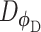 to be K-Lipschitz continuous [50], which can be satisfied by adding the gradient penalty to the WGAN loss [49]. Once the training is done, the generator
to be K-Lipschitz continuous [50], which can be satisfied by adding the gradient penalty to the WGAN loss [49]. Once the training is done, the generator 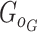 can be used to generate gene expression profiles of new cells.
can be used to generate gene expression profiles of new cells.
Supervised DL models
Supervised DL models, including DNNs, convolutional neural network (CNN) and capsule networks (CapsNet), have been used for cell-type identifications [51–53] and functional predictions [54]. The general supervised DL model 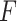 takes
takes 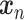 as an input and outputs
as an input and outputs 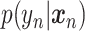 , the probability of phenotype label
, the probability of phenotype label 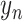 (e.g., a cell type) as
(e.g., a cell type) as
 |
(10) |
where  can be DNN, CNN or CapsNet. We omit the discussion of DNN and CNN as they are widely used in different applications and there are many excellent surveys on them [55]. We will focus our discussion on CasNet next.
can be DNN, CNN or CapsNet. We omit the discussion of DNN and CNN as they are widely used in different applications and there are many excellent surveys on them [55]. We will focus our discussion on CasNet next.
A CasNet takes an expression 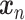 to first form a feature extraction network (consisting of
to first form a feature extraction network (consisting of 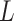 parallel single-layer neural networks) followed by a classification capsule network. Each of the
parallel single-layer neural networks) followed by a classification capsule network. Each of the 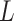 parallel feature extraction layers generates a primary capsule
parallel feature extraction layers generates a primary capsule 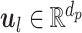 as
as
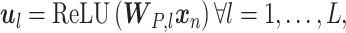 |
(11) |
where 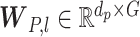 is the weight matrix. Then, the primary capsules are fed into the capsule network to compute
is the weight matrix. Then, the primary capsules are fed into the capsule network to compute  label capsules
label capsules 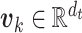 , one for each label, as
, one for each label, as
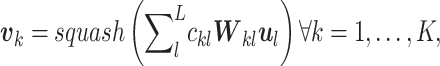 |
(12) |
where 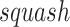 is the squashing function [56] to normalize the magnitude of its input vector to be less than one,
is the squashing function [56] to normalize the magnitude of its input vector to be less than one, 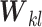 is another trainable weight matrix and
is another trainable weight matrix and 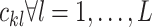 are the coupling coefficients that represent the probability distribution of each primary capsule’s impact on the predicted label
are the coupling coefficients that represent the probability distribution of each primary capsule’s impact on the predicted label 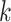 . Parameters
. Parameters 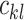 are not trained but computed through the dynamic routing process proposed in the original capsule networks [52]. The magnitude of each capsule
are not trained but computed through the dynamic routing process proposed in the original capsule networks [52]. The magnitude of each capsule  represents the probability of predicting label
represents the probability of predicting label  for input
for input 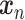 . Once trained, the important primary capsules for each label and then the most significant genes for each important primary capsule can be used to interpret biological functions associated with the prediction.
. Once trained, the important primary capsules for each label and then the most significant genes for each important primary capsule can be used to interpret biological functions associated with the prediction.
The training of the supervised models for classification overwhelmingly minimizes the cross-entropy loss by stochastic gradient descent computed by a back-propagation algorithm.
Survey of DL models for scRNA-seq analysis
In this section, we survey applications of DL models for scRNA-seq analysis. To better understand the relationship between the problems that each surveyed work addresses and the key challenges in the general scRNA-seq processing pipeline, we divide the survey into sections according to steps in the scRNA-seq processing pipeline illustrated in Figure 1. For each DL model, we present the model details under the general model framework introduced in Section 3 and discuss the specific loss functions. We also survey the evaluation metrics and summarize the evaluation results. To facilitate cross-references of the information, we summarized all algorithms reviewed in this section in Table 1 and tabulated the datasets and evaluation metrics used in each paper in Tables 3 and 8. We also listed in Figure 3 all other algorithms against which each surveyed method evaluated, highlighting the extensiveness that these algorithms were assessed for their performance.
Table 3.
Simulated single-cell data/algorithms
| Title | Algorithm | # Cells | Simulation methods | Reference |
|---|---|---|---|---|
| Splatter | DCA, DeepImpute, PERMUDA, scDeepCluster, scVI, scScope, solo | ~2000 | Splatter/R | [84] |
| CIDR | sclGAN | 50 | CIDR simulation | [61] |
| NB + dropout | Dhaka | 500 | Hierarchical model of NB/Gamma + random dropout | |
| Bulk RNA-seq | SAUCIE | 1076 | 1076 CCLE bulk RNAseq + dropout conditional on the expression level | |
| SIMLR | scScope | 1 million | SIMLR, high-dimensional data generated from latent vector | [44] |
| SUGAR | cscGAN | 3000 | Generating high dimensional data that follows a low dimensional manifold | [85] |
Table 8.
Evaluation metrics used in surveyed DL algorithms
| Evaluation Method | Equations | Explanation |
|---|---|---|
| Pseudobulk RNA-seq | Average of normalized (log2-transformed) scRNA-seq counts across cells is calculated and then correlation coefficient between the pseudobulk and the actual bulk RNA-seq profile of the same cell type is evaluated. | |
| Mean squared error (MSE) |
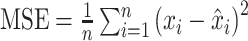
|
MSE assesses the quality of a predictor, or an estimator, from a collection of observed data x, with  being the predicted values. being the predicted values. |
| Pearson correlation |
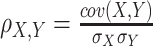
|
where cov() is the covariance, σX and σY are the standard deviation of X and Y, respectively. |
| Spearman correlation |
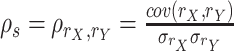
|
The Spearman correlation coefficient is defined as the Pearson correlation coefficient between the rank variables, where rX is the rank of X. |
| Entropy of accuracy, Hacc [21] |
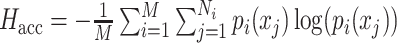
|
Measures the diversity of the ground-truth labels within each predicted cluster group. pi(xj) (or qi(xj)) are the proportions of cells in the jth ground-truth cluster (or predicted cluster) relative to the total number of cells in the ith predicted cluster (or ground-truth clusters), respectively. |
| Entropy of purity, Hpur [21] |
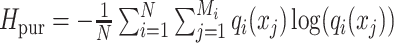
|
Measures the diversity of the predicted cluster labels within each ground-truth group. |
| Entropy of mixing [32] |

|
This metric evaluates the mixing of cells from different batches in the neighborhood of each cell. C is the number of batches, and 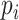 is the proportion of cells from batch is the proportion of cells from batch 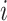 among N nearest cells. among N nearest cells. |
| MI [162] |
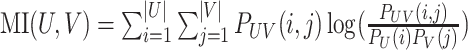
|
where 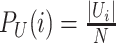 and and 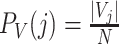 . Also, define the joint distribution probability is . Also, define the joint distribution probability is  . The MI is a measure of mutual dependency between two cluster assignments U and V. . The MI is a measure of mutual dependency between two cluster assignments U and V. |
| Normalized MI (NMI) [163] |

|
where  . The NMI is a normalization of the MI score between 0 and 1. . The NMI is a normalization of the MI score between 0 and 1. |
| KL divergence [164] |
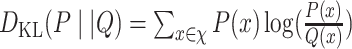
|
where discrete probability distributions P and Q are defined on the same probability space χ. This relative entropy is the measure for directed divergence between two distributions. |
| Jaccard Index |
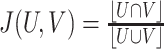
|
0 ≤ J(U,V) ≤ 1. J = 1 if clusters U and V are the same. If U are V are empty, J is defined as 1. |
| Fowlkes–Mallows Index for two clustering algorithms (FM) |
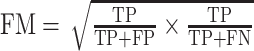
|
TP as the number of pairs of points that are present in the same cluster in both U and V; FP as the number of pairs of points that are present in the same cluster in U but not in V; FN as the number of pairs of points that are present in the same cluster in V but not in U and TN as the number of pairs of points that are in different clusters in both U and V. |
| Rand index (RI) |
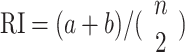
|
Measure of constancy between two clustering outcomes, where a (or b) is the count of pairs of cells in one cluster (or different clusters) from one clustering algorithm but also fall in the same cluster (or different clusters) from the other clustering algorithm. |
| Adjusted Rand index (ARI) [165] |
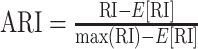
|
ARI is a corrected-for-chance version of RI, where E[RI] is the expected RI. |
| Silhouette index |
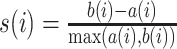
|
where a(i) is the average dissimilarity of ith cell to all other cells in the same cluster, and b(i) is the average dissimilarity of ith cell to all cells in the closest cluster. The range of s(i) is [−1,1], with 1 to be well-clustered and −1 to be completely misclassified. |
| MMD [64] |
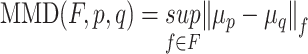
|
MMD is a non-parametric distance between distributions based on the reproducing kernel Hilbert space, or, a distance-based measure between two distribution p and q based on the mean embeddings μp and μq in a reproducing kernel Hilbert space F. |
| kBET [166] |
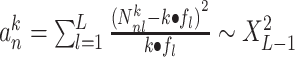
|
Given a dataset of 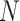 cells from cells from 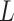 batches with batches with 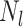 denoting the number of cells in batch denoting the number of cells in batch 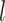 , , 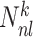 is the number of cells from batch is the number of cells from batch 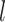 in the KNN s of cell in the KNN s of cell 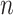 , fl is the global fraction of cells in batch , fl is the global fraction of cells in batch 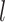 , or , or 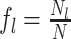 , and , and 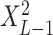 denotes the denotes the 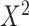 distribution with distribution with  degrees of freedom. It uses a degrees of freedom. It uses a 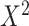 -based test for random neighborhoods of fixed size to determine the significance (‘well-mixed’). -based test for random neighborhoods of fixed size to determine the significance (‘well-mixed’). |
| LISI [34] |
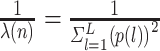
|
This is the inverse Simpson’s Index in the KNNs of cell 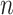 for all batches, where for all batches, where  denotes the proportion of batch denotes the proportion of batch 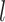 in the KNNs. The score reports the effective number of batches in the KNNs of cell in the KNNs. The score reports the effective number of batches in the KNNs of cell 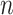 . . |
| Homogeneity |
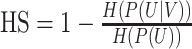
|
where H() is the entropy, and U is the ground-truth assignment and V is the predicted assignment. The HS range from 0 to 1, where 1 indicates perfectly homogeneous labeling. |
| Completeness |
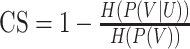
|
Its values range from 0 to 1, where 1 indicates all members from a ground-truth label are assigned to a single cluster. |
| V-Measure [167] |

|
where β indicates the weight of HS. V-Measure is symmetric, i.e. switching the true and predicted cluster labels does not change V-Measure. |
| Precision, recall |
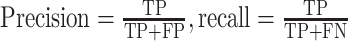
|
TP: true positive, FP: false positive, FN, false negative. |
| Accuracy |
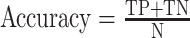
|
N: all samples tested, TN: true negative |
| F1-score |
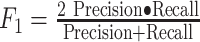
|
A harmonic mean of precision and recall. It can be extended to 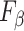 where where 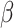 is a weight between precision and recall (similar to V-measure). is a weight between precision and recall (similar to V-measure). |
| AUC, RUROC |
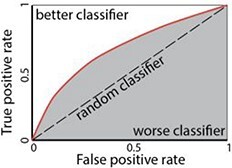
|
Area Under Curve (grey area). Receiver operating characteristic (ROC) curve (red line). A similar measure can be performed on the Precision-Recall curve (PRC) or AUPRC. PRCs summarize the trade-off between the true positive rate and the positive predictive value for a predictive model (mostly for an imbalanced dataset). |
Figure 3.

Algorithm comparison grid. DL methods surveyed in the paper are listed on the left-hand side, and some in the column. Algorithms selected to compare in each DL method are marked by ‘■’ at each cross-point.
Imputation
DCA: Deep count AE
DCA [18] is an AE for imputation (Figures 2B and 4B) and has been integrated into the Scanpy framework.
Figure 4.

DL model network illustration. (A) DNN, (B) AE, (C) VAE, (D) AE with recursive imputer, (E) GAN, (F) CNN and (G) domain adversarial neural network.
Model
DCA models UMI counts with missing values using the ZINB distribution
 |
(13) |
where 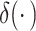 is a Dirac delta function,
is a Dirac delta function,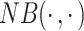 denotes the negative binomial distribution and
denotes the negative binomial distribution and 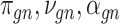 , representing dropout rate, mean and dispersion, respectively, are functions of the output (
, representing dropout rate, mean and dispersion, respectively, are functions of the output (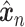 ) of the decoder in the DCA as follows:
) of the decoder in the DCA as follows:
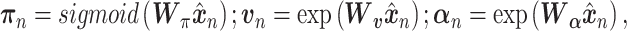 |
(14) |
where 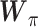 ,
,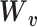 and
and 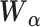 are additional weights to be estimated. The DCA encoder and decoder follow the general AE formulation as in Eq. (7) but the encoder takes the normalized, log-transformed expression as input. To train the model, DCA uses a constrained log-likelihood as the loss function
are additional weights to be estimated. The DCA encoder and decoder follow the general AE formulation as in Eq. (7) but the encoder takes the normalized, log-transformed expression as input. To train the model, DCA uses a constrained log-likelihood as the loss function
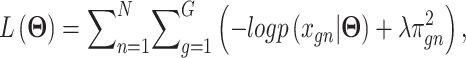 |
(15) |
with 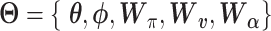 . Once the DCA is trained, the mean counts
. Once the DCA is trained, the mean counts 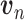 are used as the denoised and imputed counts for cell
are used as the denoised and imputed counts for cell 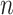 .
.
Results
For evaluation, DCA was compared with other methods using simulated data (using Splatter R package), and real bulk transcriptomics data from a developmental Caenorhabditis elegans time-course experiment were used with added simulating single-cell specific noise. Gene expression was measured from 206 developmentally synchronized young adults over a 12-h period (C. elegans). Single-cell specific noise was added in silico by genewise subtracting values drawn from the exponential distribution such that 80% of values were zeros. The paper analyzed the Bulk contains less noise than single-cell transcriptomics data and can thus aid in evaluating single-cell denoising methods by providing a good ground truth model. The authors also did a comparison of other methods including SAVER [36], scImpute [37] and MAGIC [57]. DCA denoising recovered original time-course gene expression pattern while removing single-cell specific noise. Overall, DCA demonstrated the strongest recovery of the top 500 genes most strongly associated with development in the original data without noise; DCA was shown to outperform other existing methods in capturing cell population structure in real data using PBMC, CITE-seq, runtime scales linearly with the number of cells.
SAVER-X: Single-cell analysis via expression recovery harnessing external data
SAVER-X [58] is an AE model (Figures 2B and 4B) developed to denoise and impute scRNA-seq data with transfer learning from other data resources.
Model
SAVER-X decomposes the variation in the observed counts 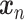 with missing values into three components: (i) predictable structured component representing the shared variation across genes, (ii) unpredictable cell-level biological variation and gene-specific dispersions and (iii) technical noise. Specifically,
with missing values into three components: (i) predictable structured component representing the shared variation across genes, (ii) unpredictable cell-level biological variation and gene-specific dispersions and (iii) technical noise. Specifically, 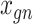 is modeled as a Poisson–Gamma hierarchical model
is modeled as a Poisson–Gamma hierarchical model
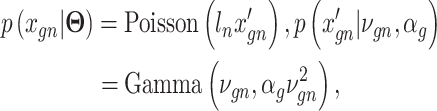 |
(16) |
where 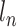 is the sequencing depth of cell n,
is the sequencing depth of cell n, 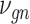 is the mean and
is the mean and  is the dispersion. This Poisson–Gamma mixture is an equivalent expression to the NB distribution, and thus, the ZINB distribution as Eq. (13) is adopted to model missing values.
is the dispersion. This Poisson–Gamma mixture is an equivalent expression to the NB distribution, and thus, the ZINB distribution as Eq. (13) is adopted to model missing values.
The loss is similar to Eq. (15). However,  is initially learned by an AE pre-trained using external datasets from an identical or similar tissue and then transferred to
is initially learned by an AE pre-trained using external datasets from an identical or similar tissue and then transferred to 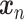 to be denoised. Such transfer learning can be applied to data between species (e.g. human and mouse in the study), cell types, batches and single-cell profiling technologies. After
to be denoised. Such transfer learning can be applied to data between species (e.g. human and mouse in the study), cell types, batches and single-cell profiling technologies. After 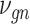 is inferred, SAVER-X generates the final denoised data
is inferred, SAVER-X generates the final denoised data 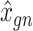 by an empirical Bayesian shrinkage.
by an empirical Bayesian shrinkage.
Results
SAVER-X was applied to multiple human single-cell datasets of different scenarios: (i) T-cell subtypes, (ii) a cell type (CD4+ regulatory T cells) that was absent from the pretraining dataset, (iii) gene-protein correlations of CITE-seq data and (iv) immune cells of primary breast cancer samples with a pretraining on normal immune cells. SAVER-X with pretraining on HCA and/or PBMCs outperformed the same model without pretraining and other denoising methods, including DCA [28], scVI [17], scImpute [37] and MAGIC [57]. The model achieved promising results even for genes with very low UMI counts. SAVER-X was also applied for a cross-species study in which the model was pre-trained on a human or mouse dataset and transferred to denoise another. The results demonstrated the merit of transferring public data resources to denoise in-house scRNA-seq data even when the study species, cell types or single-cell profiling technologies are different.
DeepImpute: DNN imputation
DeepImpute [20] imputes genes in a divide-and-conquer approach, using a bank of DNN models (Figure 4A) with 512 output, each to predict gene expression levels of a cell.
Model
For each dataset, DeepImpute selects to impute a list of genes or highly variable genes (variance over mean ratio, default = 0.5). Each sub-neural network aims to understand the relationship between the input genes and a subset of target genes. Genes are first divided into 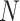 random subsets of 512 target genes. For each subset, a two-layer DNN is trained where the input includes genes that are among the top 5 best-correlated genes to target genes but not part of the target genes in the subset. The loss is defined as the weighted MSE
random subsets of 512 target genes. For each subset, a two-layer DNN is trained where the input includes genes that are among the top 5 best-correlated genes to target genes but not part of the target genes in the subset. The loss is defined as the weighted MSE
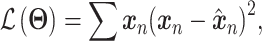 |
(17) |
which gives higher weights to genes with higher expression values.
Result
DeepImpute had the highest overall accuracy and offered shorter computation time with less demand on computer memory than other methods such as MAGIC, DrImpute, ScImpute, SAVER, VIPER and DCA. Using simulated and experimental datasets (Table 3), DeepImpute showed benefits in improving clustering results and identifying significantly differentially expressed genes. DeepImpute and DCA show overall advantages over other methods and between which DeepImpute performs even better. The properties of DeepImpute contribute to its superior performance include (i) a divide-and-conquer approach which contrary to an AE as implemented in DCA, resulting in a lower complexity in each sub-model and stabilizing neural networks and (ii) the subnetworks are trained without using the target genes as the input which reduces overfitting while enforcing the network to understand true relationships between genes.
LATE: Learning with AE
LATE [59] is an AE (Figures 2B and 4B) whose encoder takes the log-transformed expression as input.
Model
LATE sets zeros for all missing values and generates the imputed expressions. LATE minimizes the MSE loss as Eq. (8). One issue is that some zeros could be real and reflect the actual lack of expressions.
Result
Using synthetic data generated from pre-imputed data followed by random dropout selection at different degrees, LATE outperforms other existing methods such as MAGIC, SAVER, DCA and scVI, particularly when the ground truth contains only a few or no zeros. However, when the data contain many zero expression values, DCA achieved a lower MSE than LATE, although LATE still has a smaller MSE than scVI. This result suggests that DCA likely does a better job identifying true zero expressions, partly because LATE does not make assumptions on the statistical distributions of the single-cell data that potentially have inflated zero counts.
scGMAI
Technically, scGMAI [60] is a model for clustering but it includes an AE (Figures 2B and 4B) in the first step to combat dropout.
Model
To impute the missing values, scGMAI applies an AE like LATE to reconstruct log-transformed expressions with dropout but chooses a smoother Softplus activation function instead. The MSE loss as in Eq. (8) is adopted.
After imputation, scGMAI uses fast independent component analysis (ICA) on the AE reconstructed expressions to reduce the dimension and then applies a Gaussian mixture model on the ICA reduced data to perform the clustering.
Results
To assess the performance, the AE in scGMAI was replaced by five other imputation methods including SAVER [36], MAGIC [57], DCA [28], scImpute [37] and CIDR [61]. A scGMAI implementation without AE was also compared. Seventeen scRNA-seq data (part of them are listed in Tables 4 and 5 as marked) were used to evaluate cell clustering performances. The results indicated that the AEs significantly improved the clustering performance in 8 of 17 scRNA-seq datasets.
Table 4.
Human single-cell data sources used by different DL algorithms
| Title | Algorithm | Cell origin | # Cells | Data sources | Reference |
|---|---|---|---|---|---|
| 68 k PBMCs | DCA SAVER-X LATE, scVAE, scDeepCluster, scCapsNet, scDGN | Blood | 68 579 | 10× Single Cell Gene Expression Datasets | |
| Human pluripotent | DCA | hESCs | 1876 | GSE102176 | [128] |
| CITE-seq | SAVER-X | Cord blood mononuclear cells | 8005 | GSE100866 | [129] |
| Midbrain and Dopaminergic Neuron Development | SAVER-X | Brain/ embryo ventral midbrain cells | 1977 | GSE76381 | [130] |
| HCA | SAVER-X | Immune cell, Human Cell Atlas | 500 000 | HCA data portal | |
| Breast tumor | SAVER-X | Immune cell in tumor micro-environment | 45 000 | GSE114725 | [131] |
| 293 T cells | DeepImpute, iMAP | Embryonic kidney | 13 480 | 10X Single Cell Gene Expression Datasets | |
| Jurkat | DeepImpute, iMAP | Blood/ lymphocyte | 3200 | 10X Single Cell Gene Expression Datasets | |
| ESC, Time-course | scGAN | ESC | 350 758 | GSE75748 | [132] |
| Baron-Hum-1 | scGMAI, VASC | Pancreatic islets | 1937 | GSM2230757 | [133] |
| Baron-Hum-2 | scGMAI, VASC | Pancreatic islets | 1724 | GSM2230758 | [133] |
| Camp | scGMAI, VASC | Liver cells | 303 | GSE96981 | [134] |
| CEL-seq2 | PERMUDA, DESC | Pancreas/Islets of Langerhans | GSE85241 | [135] | |
| Darmanis | scGMAI, sclGAN, VASC | Brain/cortex | 466 | GSE67835 | [136] |
| Tirosh-brain | Dhaka, scvis | Oligodendroglioma | >4800 | GSE70630 | [137] |
| Patel | Dhaka | Primary glioblastoma cells | 875 | GSE57872 | [138] |
| Li | scGMAI, VASC | Blood | 561 | GSE146974 | [67] |
| Tirosh-skin | scvis | melanoma | 4645 | GSE72056 | [73] |
| xenograft 3, and 4 | Dhaka | Breast tumor | ~250 | EGAS00001002170 | [74] |
| Petropoulos | VASC/netAE | Human embryos | 1529 | E-MTAB-3929 | |
| Pollen | scGMAI, VASC | 348 | SRP041736 | [139] | |
| Xin | scGMAI, VASC | Pancreatic cells (α-, β-, δ-) | 1600 | GSE81608 | [140] |
| Yan | scGMAI, VASC | embryonic stem cells | 124 | GSE36552 | [141] |
| PBMC3k | VASC, scVI | Blood | 2700 | SRP073767 | [99] |
| CyTOF, Dengue | SAUCIE | Dengue infection | 11 M, ~42 antibodies | Cytobank, 82 023 | [15] |
| CyTOF, ccRCC | SAUCIE | Immunue profile of 73 ccRCC patients | 3.5 M, ~40 antibodies | Cytobank: 875 | [142] |
| CyTOF, breast | SAUCIE | 3 patients | Flow Repository: FR-FCM-ZYJP | [131] | |
| Chung, BC | DigitalDLSorter | Breast tumor | 515 | GSE75688 | [93] |
| Li, CRC | DigitalDLSorter | Colorectal cancer | 2591 | GSE81861 | [94] |
| Pancreatic datasets | scDGN | Pancreas | 14 693 | SeuratData | |
| Kang, PBMC | scGen | PBMC stimulated by INF-β | ~15 000 | GSE96583 | [115] |
Table 5.
Mouse single-cell data sources used by different DL algorithms
| Title | Algorithm | Cell origin | # Cells | Data Sources | Reference |
|---|---|---|---|---|---|
| Brain cells from E18 mice | DCA, SAUCIE | Brain Cortex | 1 306 127 | 10×: Single Cell Gene Expression Datasets | |
| Midbrain and Dopaminergic Neuron Development | SAVER-X | Ventral Midbrain | 1907 | GSE76381 | [130] |
| Mouse cell atlas | SAVER-X | 405 796 | GSE108097 | [143] | |
| neuron9k | DeepImpute | Cortex | 9128 | 10×: Single Cell Gene Expression Datasets | |
| Mouse Visual Cortex | DeepImpute | Brain cortex | 114 601 | GSE102827 | [144] |
| murine epidermis | DeepImpute | Epidermis | 1422 | GSE67602 | [145] |
| myeloid progenitors | LATE DESC, SAUCIE | Bone marrow | 2730 | GSE72857 | [146] |
| Cell-cycle | sclGAN | mESC | 288 | E-MTAB-2805 | [147] |
| A single-cell survey | Intestine | 7721 | GSE92332 | [119] | |
| Tabula Muris | iMAP | Mouse cells | >100 K | ||
| Baron-Mou-1 | VASC | Pancreas | 822 | GSM2230761 | [133] |
| Biase | scGMAI, VASC | Embryos/SMARTer | 56 | GSE57249 | [148] |
| Biase | scGMAI, VASC | Embryos/Fluidigm | 90 | GSE59892 | [148] |
| Deng | scGMAI, VASC | Liver | 317 | GSE45719 | [149] |
| Klein | VASC scDeepCluster sclGAN | Stem Cells | 2717 | GSE65525 | [150] |
| Goolam | VASC | Mouse Embryo | 124 | E-METAB-3321 | [151] |
| Kolodziejczyk | VASC | mESC | 704 | E-MTAB-2600 | [152] |
| Usoskin | VASC | Lumbar | 864 | GSE59739 | [153] |
| Zeisel | VASC, scVI, SAUCIE, netAE | Cortex, hippocampus | 3005 | GSE60361 | [154] |
| Bladder cells | scDeepCluster | Bladder | 12 884 | GSE129845 | [155] |
| HEMATO | scVI | Blood cell | >10 000 | GSE89754 | [156] |
| retinal bipolar cells | scVI, scCapsNet SAUCIE | retinal | ~25 000 | GSE81905 | [100] |
| Embryo at 9 time points | LDAVE | embryos from E6.5 to E8.5 | 116 312 | GSE87038 | [157] |
| Embryo at 9 time points | LDAVE | embryos from E9.5 to E13.5 | ~2 millions | GSE119945 | [158] |
| CyTOF, | SAUCIE | Mouse thymus | 200 K, ~38 antibodies | Cytobank: 52942 | [159] |
| Nestorowa | netAE | hematopoietic stem and progenitor cells | 1920 | GSE81682 | [160] |
| small intestinal epithelium | scGen | Infected with Salmonella and worm H. polygyrus | 1957 | GSE92332 | [119] |
scIGANs
Imputation approaches based on information from cells with similar expressions suffer from oversmoothing, especially for rare cell types. scIGANs [19] is a GAN-based imputation algorithm (Figures 2C and 4E), which overcomes this problem by training a GAN model to generate samples with imputed expressions.
Model
scIGAN takes the image-like reshaped gene expression data 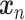 as input. The model follows a BEGAN [62] framework, which replaces the GAN discriminator
as input. The model follows a BEGAN [62] framework, which replaces the GAN discriminator 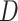 with a function
with a function 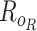 to compute the reconstruction MSE. Then, the Wasserstein distance loss between the reconstruction errors between the real and generated samples is computed
to compute the reconstruction MSE. Then, the Wasserstein distance loss between the reconstruction errors between the real and generated samples is computed
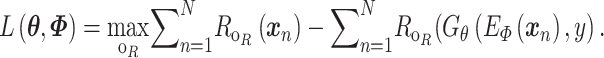 |
(18) |
This framework forces the model to meet two computing objectives, i.e. reconstructing the real samples and discriminating between real and generated samples. Proportional Control Theory was applied to balance these two goals during the training [62].
After training, the decoder 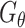 is used to generate new samples of a specific cell type. Then, the k-nearest neighbors (KNNs) approach is applied to the real and generated samples to impute the real samples’ missing expressions.
is used to generate new samples of a specific cell type. Then, the k-nearest neighbors (KNNs) approach is applied to the real and generated samples to impute the real samples’ missing expressions.
Results
scIGANs were first tested on simulated samples with different dropout rates. Performance of rescuing the correct clusters was compared with 11 existing imputation approaches including DCA, DeepImpute, SAVER, scImpute, MAGIC, etc. scIGANs reported the best performance for all metrics. scIGAN was next evaluated for its ability to correctly cluster cell types on the Human brain scRNA-seq data, which showed superior performance than existing methods again. scIGANs were then evaluated for identifying cell-cycle states using scRNA-seq datasets from mouse embryonic stem cells. The results showed that scIGANs outperformed competing existing approaches for recovering subcellular states of cell cycle dynamics. scIGANs were further shown to improve the identification of differentially expressed genes and enhance the inference of cellular trajectory using time-course scRNA-seq data from the differentiation from H1 ESC to definitive endoderm cells (DECs). Finally, scIGAN was also shown to scale to scRNA-seq methods and data sizes.
Batch effect correction
BERMUDA: Batch Effect ReMoval Using Deep AEs
BERMUDA [63] deploys a transfer-learning method (Figures 2B and 4B) to remove the batch effect. It performs correction to the shared cell clusters among batches and therefore preserves batch-specific cell populations.
Model
BERMUDA is an AE that takes normalized, log-transformed expression as input. Its consists of two parts as
 |
(19) |
where  is the MSE loss and
is the MSE loss and 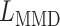 is the maximum mean discrepancy (MMD) [64] loss that measures the differences in distributions between pairs of similar cell clusters shared among batches as
is the maximum mean discrepancy (MMD) [64] loss that measures the differences in distributions between pairs of similar cell clusters shared among batches as
 |
(20) |
where  is the latent variable of
is the latent variable of 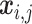 , the input expression of a cell from cluster
, the input expression of a cell from cluster 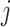 of batch
of batch 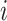 ,
, 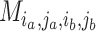 is
is 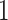 if cluster
if cluster 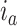 of batch
of batch 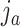 and cluster
and cluster  of batch
of batch 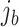 are determined to be similar by MetaNeighbor [65] and
are determined to be similar by MetaNeighbor [65] and 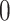 , otherwise. The
, otherwise. The 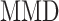 equals zero when the underlying distributions of the observed samples are the same.
equals zero when the underlying distributions of the observed samples are the same.
Results
BERMUDA was shown to outperform other methods such as MNNCorrect [32], BBKNN [66], Seurat [10] and scVI [17] in removing batch effects on simulated and human pancreas data while preserving batch-specific biological signals. BERMUDA provides several improvements compared with existing methods: (i) capable of removing batch effects even when the cell population compositions across different batches are vastly different and (ii) preserving batch-specific biological signals through transfer-learning which enables discovering new information that might be hard to extract by analyzing each batch individually.
DESC: Batch correction based on clustering
DESC [67] is an AE model (Figures 2B and 4B) that removes batch effect through clustering with the hypothesis that batch differences in expressions are smaller than true biological variations between cell types, and, therefore, properly performing clustering on cells across multiple batches can remove batch effects without the need to define batches explicitly.
Model
DESC has a conventional AE architecture. Its encoder takes normalized, log-transformed expression and uses decoder output,  as the reconstructed gene expression, which is equivalent to a Gaussian data distribution with
as the reconstructed gene expression, which is equivalent to a Gaussian data distribution with  being the mean. The loss function is similar to Eq. (19) and except that the second loss
being the mean. The loss function is similar to Eq. (19) and except that the second loss 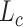 is the clustering loss that regularizes the learned feature representations to form clusters as in the deep embedded clustering [68]. The model is first trained to minimize
is the clustering loss that regularizes the learned feature representations to form clusters as in the deep embedded clustering [68]. The model is first trained to minimize 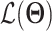 only to obtain the initial weights before minimizing the combined loss. After the training, each cell is assigned with a cluster ID.
only to obtain the initial weights before minimizing the combined loss. After the training, each cell is assigned with a cluster ID.
Table 6.
Single-cell data derived from other species
| Title | Algorithm | Species | Tissue | # Cells | SRA/GEO | Reference |
|---|---|---|---|---|---|---|
| Worm neuron cells a | scDeepCluster | C. elegans | Neuron | 4186 | GSE98561 | [161] |
| Cross species, stimulation with LPS and dsRNA | scGen | Mouse, rat, rabbit and pig | bone marrow-derived phagocyte | 5000–10 000 /species | 13 accessions in ArrayExpress | [120] |
aProcessed data are available at https://github.com/ttgump/scDeepCluster/tree/master/scRNA-seq%20data
Results
DESC was applied to the macaque retina dataset, which includes animal level, region level and sample-level batch effects. The results showed that DESC is effective in removing the batch effect, whereas CCA [33], MNN [32], Seurat 3.0 [10], scVI [17], BERMUDA [63] and scanorama [69] were all sensitive to batch definitions. DESC was then applied to human pancreas datasets to test its ability to remove batch effects from multiple scRNA-seq platforms and yielded the highest ARI among the comparing approaches mentioned above. When applied to human PBMC data with interferon-beta stimulation, where biological variations are compounded by batch effect, DESC was shown to be the best in removing batch effect while preserving biological variations. DESC was also shown to remove batch effect for the monocytes and mouse bone marrow data, and DESC was shown to preserve the pseudotemporal structure. Finally, DESC scales linearly with the number of cells, and its running time is not affected by the increasing number of batches.
iMAP: Integration of Multiple single-cell datasets by Adversarial Paired-style transfer networks
iMAP [70] combines AE (Figures 2B and 4B) and GAN (Figures 2C and 4E) for batch effect removal. It is designed to remove batch biases while preserving dataset-specific biological variations.
Model
iMAP consists of two processing stages, each including a separate DL model. In the first stage, a special AE, whose decoder combines the output of two separate decoders 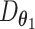 and
and  , is trained such that
, is trained such that
 |
(21) |
where 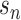 is the one-hot encoded batch number of cell
is the one-hot encoded batch number of cell 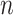 .
. 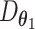 can be understood as decoding the batch noise, whereas
can be understood as decoding the batch noise, whereas 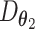 reconstructs batch-removed expression from the latent variable
reconstructs batch-removed expression from the latent variable 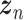 . The training minimizes the loss in Eq. (19) except the 2nd loss is the content loss
. The training minimizes the loss in Eq. (19) except the 2nd loss is the content loss
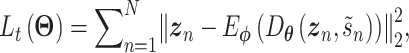 |
(22) |
where 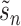 is a random batch number. Minimizing
is a random batch number. Minimizing 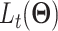 further ensures that the reconstructed expression
further ensures that the reconstructed expression 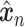 would be batch agnostic and has the same content as
would be batch agnostic and has the same content as 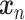 .
.
Table 7.
Large single-cell data source used by various algorithms
| Title | Sources | Notes |
|---|---|---|
| 10× Single-cell gene expression dataset | https://support.10xgenomics.com/single-cell-gene-expression/datasets | Contains large collection of scRNA-seq dataset generated using 10× system |
| Tabula Muris | https://tabula-muris.ds.czbiohub.org/ | Compendium of scRNA-seq data from mouse |
| HCA | https://data.humancellatlas.org/ | Human single-cell atlas |
| MCA | https://figshare.com/s/865e694ad06d5857db4b, or GSE108097 | Mouse single-cell atlas |
| scQuery | https://scquery.cs.cmu.edu/ | A web server cell-type matching and key gene visualization. It is also a source for scRNA-seq collection (processed with common pipeline) |
| SeuratData | https://github.com/satijalab/seurat-data | List of datasets, including PBMC and human pancreatic islet cells |
| cytoBank | https://cytobank.org/ | Community of big data cytometry |
However, due to the limitation of AE, this step is still insufficient for batch removal. Therefore, a second stage is included to apply a GAN model to make expression distributions of the shared cell type across different baches indistinguishable. To identified the shared cell types, an MNNs strategy adapted from [32] was developed to identify MNN pairs across batches using batch effect independent  as opposed to
as opposed to  . Then, a mapping generator
. Then, a mapping generator 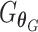 is trained using MNN pairs based on GAN such that
is trained using MNN pairs based on GAN such that 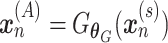 , where
, where  and
and 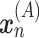 are the MNN pairs from batch
are the MNN pairs from batch  and an anchor batch
and an anchor batch 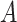 . The WGAN-GP loss as in Eq. (9) was adopted for the GAN training. After training,
. The WGAN-GP loss as in Eq. (9) was adopted for the GAN training. After training, 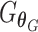 is applied to all cells of a batch to generate batch-corrected expression.
is applied to all cells of a batch to generate batch-corrected expression.
Results
iMAP was first tested on benchmark datasets from human dendritic cells and Jurkat and 293 T cell lines and then human pancreas datasets from five different platforms. All the datasets contain both batch-specific cells and batch-shared cell types. iMAP was shown to separate the batch-specific cell types but mix batch shared cell types and outperformed nine other existing batch correction methods including Harmony, scVI, fastMNN, Seurat, etc. iMAP was then applied to the large-scale Tabula Muris datasets containing over 100 K cells sequenced from two platforms. iMAP could not only reliably integrate cells from the same tissues but identify cells from platform-specific tissues. Finally, iMAP was applied to datasets of tumor-infiltrating immune cells and shown to reduce the dropout ratio and the percentage of ribosomal genes and non-coding RNAs, thus improving the detection of rare cell types and ligand-receptor interactions. iMAP scales with the number of cells, showing minimal time cost increase after the number of cells exceeds thousands. Its performance is also robust against model hyperparameters.
Dimensionality reduction, latent representation, clustering and data augmentation
Dimensionality reduction is indispensable for many type of scRNA-seq data analysis, considering the limited number of cell types in each biospecimen. Furthermore, biological processes of interests often involve the complex coordination of many genes, therefore, latent representations which capture biological variation in reduced dimensions are useful in interpreting experiment conditions and cell heterogeneity. Both AE- and VAE-based are capable of learning latent representations. VAE-based models have the benefit of regularity of the latent space and generative factors. The GAN-based models can produce augmented data that may in return to enhance the clustering, e.g. due to low representation of certain cell types.
Dimensionality reduction by AEs with gene-interaction constrained architecture
This study [71] considers AEs (Figures 2B and 4B) for learning the low-dimensional representation and specifically explores the benefit of incorporating prior biological knowledge of gene–gene interactions to regularize the AE network architecture.
Model
Several AE models with single or two hidden layers that incorporate gene interactions reflecting transcription factor (TF) regulations and protein–protein interactions (PPIs) are implemented. The models take normalized, log-transformed expressions and follow the general AE structure, including dimension-reducing and reconstructing layers, but the network architectures are not symmetrical. Specifically, gene interactions are incorporated such that each node of the first hidden layer represented a TF or a protein in the PPI; only genes that are targeted by TFs or involved in the PPI were connected to the node. Thus, the corresponding weights of 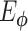 and
and 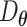 are set to be trainable and otherwise fixed at zero throughout the training process. Both unsupervised (AE-like) and supervised (cell-type label) learning were studied.
are set to be trainable and otherwise fixed at zero throughout the training process. Both unsupervised (AE-like) and supervised (cell-type label) learning were studied.
Results
Regularizing encoder connections with TF and PPI information considerably reduced the model complexity by almost 90% (7.5–7.6 M to 1.0–1.1 M). The clusters formed on the data representations learned from the models with or without TF and PPI information were compared with those from PCA, NMF, ICA, t-SNE and SIMLR [44]. The model with TF/PPI information and two hidden layers achieved the best performance by five of the six measures and the best average performance. In terms of the cell-type retrieval of single cells, the encoder models with and without TF/PPI information achieved the best performance in four and three cell types, respectively. PCA yielded the best performance in only two cell types. The DNN model with TF/PPI information and two hidden layers again achieved the best average performance across all cell types. In summary, this study demonstrated a biologically meaningful way to regularize AEs by the prior biological knowledge for learning the representation of scRNA-seq data for cell clustering and retrieval.
Dhaka: A VAE-based dimension reduction model
Dhaka [72] was proposed to reduce the dimension of scRNA-seq data for efficient stratification of tumor subpopulations.
Model
Dhaka adopts a general VAE formulation (Figures 2A and 4C). It takes the normalized, log-transformed expressions of a cell as input and outputs the low-dimensional representation.
Result
Dhaka was first tested on the simulated dataset. The simulated dataset contains 500 cells, each including 3 K genes, clustered into 5 different clusters with 100 cells each. The clustering performance was compared with other methods including t-SNE, PCA, SIMLR, NMF, an AE, MAGIC and scVI. Dhaka was shown to have an ARI higher than most other comparing methods. Dhaka was then applied to the Oligodendroglioma data and could separate malignant cells from non-malignant microglia/macrophage cells. It also uncovered the shared glial lineage and differentially expressed genes for the lineages. Dhaka was also applied to the Glioblastoma data and revealed an evolutionary trajectory of the malignant cells where cells gradually evolve from a stemlike state to a more differentiated state. In contrast, other methods failed to capture this underlying structure. Dhaka was next applied to the Melanoma cancer dataset [73] and uncovered two distinct clusters that showed the intra-tumor heterogeneity of the Melanoma samples. Dhaka was finally applied to copy number variation data [74] and shown to identify one major and one minor cell clusters, of which other methods could not find.
scvis: A VAE for capturing low-dimensional structures
scvis [75] is a VAE network (Figures 2A and 4C) that learns that the low-dimensional representations capture both local and global neighboring structures in scRNA-seq data.
Model
scvis adopts the generic VAE formulation described in section 3.1. However, it has a unique loss function defined as
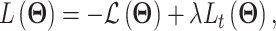 |
(23) |
where 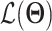 is ELBO as in Eq. (3) and
is ELBO as in Eq. (3) and 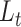 is a regularizer using non-symmetrized t-SNE objective function [75], which is defined as
is a regularizer using non-symmetrized t-SNE objective function [75], which is defined as
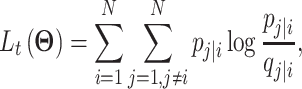 |
(24) |
where 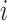 and
and  are two different cells,
are two different cells, 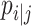 measures the local cell relationship in the data space and
measures the local cell relationship in the data space and  measures such relationship in the latent space. Because t-SNE algorithm preserves the local structure of high dimensional space,
measures such relationship in the latent space. Because t-SNE algorithm preserves the local structure of high dimensional space, 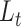 learns local structures of cells.
learns local structures of cells.
Results
scvis was tested on the simulated data and outperformed t-SNE in a nine-dimensional space task. scvis preserved both local structure and global structure. The relative positions of all clusters were well kept but outliers were scattered around clusters. Using simulated data and comparing to t-SNE, scvis generally produced consistent and better patterns among different runs, while t-SNE could not. scvis also presented good results on adding new data to an existing embedding, with median accuracy on new data at 98.1% for K = 5 and 94.8% for K = 65, when train K cluster on original data then test the classifier on new-generated sample points. The scvis was subsequently tested on four real datasets including metastatic melanoma, oligodendroglioma, mouse bipolar and mouse retina datasets. In each dataset, scvis was showed to preserve both the global and local structure of the data.
scVAE: VAE for single-cell gene expression data
scVAE [76] includes multiple VAE models (Figures 2A and4C) for denoising gene expression levels and learning the low-dimensional latent representation of cells. It investigates different choices of the likelihood functions in the VAE model to model different data sets.
Model
scVAE is a conventional fully connected network. However, different distributions have been discussed for 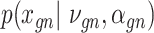 to model different data behaviors. Specifically, scVAE considers Poisson, constrained Poisson and negative binomial distributions for count data, piece-wise categorical Poisson for data including both high and low counts, and zero-inflated version of these distributions to model missing values. To model multiple modes in cell expressions, a Gaussian mixture is also considered for
to model different data behaviors. Specifically, scVAE considers Poisson, constrained Poisson and negative binomial distributions for count data, piece-wise categorical Poisson for data including both high and low counts, and zero-inflated version of these distributions to model missing values. To model multiple modes in cell expressions, a Gaussian mixture is also considered for 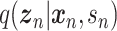 , resulting in a GMVAE. The inference process still follows that of a VAE as discussed in section 3.1.
, resulting in a GMVAE. The inference process still follows that of a VAE as discussed in section 3.1.
Results
scVAEs were evaluated on the PBMC data and compared with factor analysis (FA) models. The results showed that GMVAE with negative binomial distribution achieved the highest lower bound and ARI. Zero-inflated Poisson distribution performed the second best. All scVAE models outperformed the baseline linear FA model, which suggested that a non-linear model is needed to capture single-cell genomic features. GMVAE was also compared with Seurat and shown to perform better using the withheld data. However, scVAE performed no better than scVI [17] or scvis [75], both are VAE models.
VASC: VAE for scRNA-seq
VASC [77] is another VAE (Figures 2A and 4C) for dimension reduction and latent representation but it models dropout.
Model
VASC’s input is the log-transformed expression but rescaled in the range [0,1]. A dropout layer (dropout rate of 0.5) is added after the input layer to force subsequent layers to learn to avoid dropout noise. The encoder network has three layers fully connected and the first layer uses linear activation, which acts like an embedded PCA transformation. The next two layers use the ReLU activation, which ensures a sparse and stable output. This model’s novelty is the zero-inflation layer (ZI layer), which is added after the decoder to model scRNA-seq dropout events. The probability of dropout event is defined as 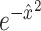 where
where 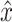 is the recovered expression value obtained by the decoder network. Since back-propagation cannot deal with a stochastic network with categorical variables, a Gumbel-softmax distribution [78] is introduced to address the difficulty of the ZI layer. The loss function of the model takes the form
is the recovered expression value obtained by the decoder network. Since back-propagation cannot deal with a stochastic network with categorical variables, a Gumbel-softmax distribution [78] is introduced to address the difficulty of the ZI layer. The loss function of the model takes the form 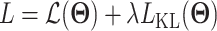 , where
, where  is the binary entropy because the input is scaled to [0 1], and
is the binary entropy because the input is scaled to [0 1], and 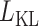 a loss performed using Kullback–Leibler (KL) divergence on the latent variables. After the model is trained, the latent code can be used as the dimension-reduced feature for downstream tasks and visualization.
a loss performed using Kullback–Leibler (KL) divergence on the latent variables. After the model is trained, the latent code can be used as the dimension-reduced feature for downstream tasks and visualization.
Results
VASC was compared with PCA, t-SNE, ZIFA and SIMLR on 20 datasets. In the study of embryonic development from zygote to blast cells, all methods roughly re-established the development stages of different cell types in the dimension-reduced space. However, VASC showed the better performance to model embryo developmental progression. In the Goolam, Biase and Yan datasets, scRNA-seq data were generated through embryonic development stages from zygote to blast, VASC re-established development stage from 1, 2, 4, 8, 16 to blast, while other methods failed. In the Pollen, Kolodziejczyk and Baron datasets, VASC formed an appropriate cluster, either with homogeneous cell type, preserved proper relative positions, or minimal batch influence. Interestingly, when tested on the PBMC dataset, VASC was shown to identify the major global structure (B cells, CD4+, CD8+ T cells, NK cells, Dendritic cells) and detect subtle differences within monocytes (FCGR3A+ versus CD14+ monocytes), indicating the capability of VASC handling a large number of cells or cell types. Quantitative clustering performance in NMI, ARI, homogeneity and completeness was also performed. VASC always ranked top two in all the datasets. In terms of NMI and ARI, VASC best performed on 15 and 17 out of 20 datasets, respectively.
scDeepCluster
scDeepCluster [79] is an AE network (Figures 2B and 4B) that simultaneously learns feature representation and performs clustering via explicit modeling of cell clusters as in DESC.
Model
Similar to DCA, scDeepCluster adopts a ZINB distribution for 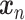 as in Eqs. (13) and (15). The loss is similar to Eq. (19) except that the first term is the negative log-likelihood of the ZINB data distribution as defined in Eq. (15) and the second
as in Eqs. (13) and (15). The loss is similar to Eq. (19) except that the first term is the negative log-likelihood of the ZINB data distribution as defined in Eq. (15) and the second 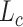 is a clustering loss performed using KL divergence as in DESC algorithm. Compared with ***csvis, scDeepcluster focuses more on clustering assignment due to the KL divergence.
is a clustering loss performed using KL divergence as in DESC algorithm. Compared with ***csvis, scDeepcluster focuses more on clustering assignment due to the KL divergence.
Results
scDeepCluster was first tested on the simulation data and compared with other seven methods including DCA [18], two multi-kernel spectral clustering methods MPSSC [80] and SIMLR [44], CIDR [61], PCA + k-mean, scvis [75] and DEC [81]. In different dropout rate simulations, scDeepCluster significantly outperformed the other methods consistently. In signal strength, imbalanced sample size and scalability simulations, scDeepcluster outperformed all other algorithms and scDeepCluster and most notably advantages for weak signals, robust against different data imbalance levels and scaled linearly with the number of cells. scDeepCluster was then tested on four real datasets (10X PBMC, Mouse ES cells, Mouse bladder cells, Worm neuron cells) and shown to outperform all other comparing algorithms. Particularly, MPSSC and SIMLR failed to process the full datasets due to quadratic complexity.
cscGAN: Conditional single-cell generative adversarial neural networks
cscGAN [82] is a GAN model (Figures 2C and 4E) designed to augment the existing scRNA-seq samples by generating expression profiles of specific cell types or subpopulations.
Model
Two models, csGAN and cscGAN, were developed following the general formulation of WGAN described in section 3.3. The difference between the two models is that cscGAN is a conditional GAN such that the input to the generator also includes a class label 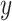 or cell type, i.e.
or cell type, i.e. 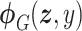 . The projection-based conditioning (PCGAN) method [83] was adopted to obtain the conditional GAN. For both models, the generator (three layers of 1024, 512 and 256 neurons) and discriminator (three layers of 256, 512 and 1024 neurons) are fully connected DNNs.
. The projection-based conditioning (PCGAN) method [83] was adopted to obtain the conditional GAN. For both models, the generator (three layers of 1024, 512 and 256 neurons) and discriminator (three layers of 256, 512 and 1024 neurons) are fully connected DNNs.
Results
The performance of scGAN was first evaluated using PBMC data. The generated samples were shown to capture the desired clusters and the real data’s regulons. Additionally, the AUC performance for classifying real from generated samples by a Random Forest classifier only reached 0.65, performance close to 0.5. Finally, scGAN’s generated samples had a smaller MMD than those of Splatter, a state-of-the-art scRNA-seq data simulator [84]. Even though a large MMD was observed for scGAN when compared with that of SUGAR, another scRNA-seq simulator, SUGAR [85] was noted for prohibitively high runtime and memory. scGAN was further trained and assessed on the bigger mouse brain data and shown to model the expression dynamics across tissues. Then, the performance of cscGAN for generating cell-type-specific samples was evaluated using the PBMC data. cscGAN was shown to generate high-quality scRAN-seq data for specific cell types. Finally, the real PBMC samples were augmented with the generated samples. This augmentation improved the identification of rare cell types and the ability to capture transitional cell states from trajectory analysis.
Multi-functional models
Given the versatility of AE and VAE in addressing different scRAN-seq analysis challenges, DL models possessing multiple analysis functions have been developed. We survey these models in this section.
scVI: Single-cell variational inference
scVI [17] is designed to address a range of fundamental analysis tasks, including batch correction, visualization, clustering and differential expression.
Model
scVI is a VAE (Figures 2A and 4C) that models the counts of each cell from different batches. scVI adopts a ZINB distribution for 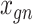
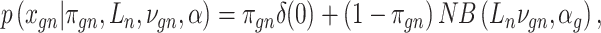 |
(25) |
which is defined similarly as Eq. (14) in DCA, except that 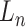 denotes the scaling factor for cell
denotes the scaling factor for cell 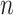 , which follows a log-Normal (
, which follows a log-Normal (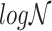 ) prior as
) prior as 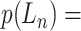
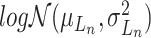 ; therefore,
; therefore, 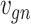 represents the mean counts normalized by
represents the mean counts normalized by  . Now, let
. Now, let  be the batch ID of cell
be the batch ID of cell  with
with 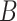 being the total number of batches. Then,
being the total number of batches. Then,  and
and 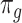 are further modeled as functions of the
are further modeled as functions of the  -dimension latent variable
-dimension latent variable 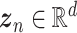 and the batch ID
and the batch ID 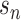 by the decoder networks
by the decoder networks 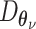 and
and 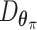 as
as
 |
(26) |
where the 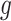 th element of
th element of  and
and 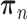 are
are 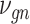 and
and 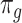 , respectively, and
, respectively, and 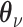 and
and 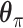 are the decoder weights. Note that the lower layers of the two decoders are shared. For inference, both
are the decoder weights. Note that the lower layers of the two decoders are shared. For inference, both 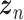 and
and 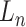 are considered as latent variables, and therefore,
are considered as latent variables, and therefore, 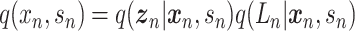 is a mean-field approximate to the intractable posterior distribution
is a mean-field approximate to the intractable posterior distribution 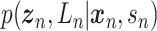 and
and
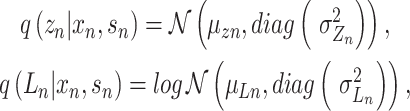 |
(27) |
whose means and variances  and
and  are defined by the encoder networks
are defined by the encoder networks 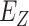 and
and 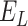 applied to
applied to 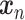 and
and  as
as
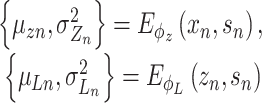 |
(28) |
where 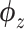 and
and 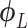 are the encoder weights. Note that, like the decoders, the lower layers of the two encoders are also shared. Overall, the model parameters to be estimated by the variational optimization is
are the encoder weights. Note that, like the decoders, the lower layers of the two encoders are also shared. Overall, the model parameters to be estimated by the variational optimization is 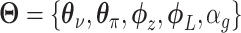 . After inference,
. After inference,  are used for visualization and clustering;
are used for visualization and clustering;  provides a batch-corrected, size-factor normalized estimate of gene expression for each gene
provides a batch-corrected, size-factor normalized estimate of gene expression for each gene 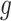 in each cell
in each cell 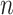 . An added advantage of the probabilistic representation by scVI is that it provides a natural probabilistic treatment of the subsequent differential analysis, resulting in lower variance in the adopted hypothesis tests.
. An added advantage of the probabilistic representation by scVI is that it provides a natural probabilistic treatment of the subsequent differential analysis, resulting in lower variance in the adopted hypothesis tests.
Results
scVI was evaluated for its scalability, the performance of imputation. For scalability, ScVI was shown to be faster than most non-DL algorithms and scalable to handle twice as many cells as non-DL algorithms with a fixed memory. For imputation, ScVI, together with other ZINB-based models, performed better than methods using alternative distributions. However, it underperformed for the dataset (HEMATO) with fewer cells. For the latent space, scVI was shown to provide a comparable stratification of cells into previously annotated cell types. Although scVI failed to ravel SIMLR, it is among the best in capturing biological structures (hierarchical structure, dynamics, etc.) and recognizing noise in data. For batch correction, it outperforms ComBat. For normalizing sequencing depth, the size factor inferred by scVI was shown to be strongly correlated with the sequencing depth. Interestingly, the negative binomial distribution in the ZINB was found to explain the proportions of zero expressions in the cells, whereas the zero probability 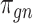 is found to be more correlated with alignment errors. For differential expression analysis, scVI was shown to be among the best.
is found to be more correlated with alignment errors. For differential expression analysis, scVI was shown to be among the best.
LDVAE: Linearly decoded VAE
LDVAE [86] is an adaption of scVI to improve the model interpretability but it still benefits from the scalability and efficiency of scVI. Also, this formulation applies to general VAE models and thus is not restricted to scRNA-seq analysis.
Model
LDVAE follows scVI’s formulation but replaces the decoder 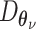 in Eq. (26) by a linear model
in Eq. (26) by a linear model
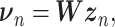 |
(29) |
where 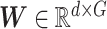 is the weight matrix. Being the linear decoder provides interpretability in the sense that the relationship between latent representation
is the weight matrix. Being the linear decoder provides interpretability in the sense that the relationship between latent representation 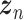 and gene expression
and gene expression 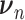 can be readily identified. LDVAE still follows the same loss and non-linear inference scheme as scVI.
can be readily identified. LDVAE still follows the same loss and non-linear inference scheme as scVI.
Results
LDVAE’s latent variable 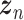 could be used for clustering of cells with similar accuracy as a VAE. Although LDVAE had a higher reconstruction error than VAE, due to the linear decoder, the variations along the different axes of
could be used for clustering of cells with similar accuracy as a VAE. Although LDVAE had a higher reconstruction error than VAE, due to the linear decoder, the variations along the different axes of 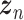 establish direct linear relationships with input genes. As an example from analyzing mouse embryo scRNA-seq,
establish direct linear relationships with input genes. As an example from analyzing mouse embryo scRNA-seq, 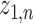 , the second element of
, the second element of 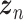 , is shown to relate to simultaneous variations in the expression of gene
, is shown to relate to simultaneous variations in the expression of gene  and
and 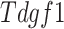 . In contrast, such interpretability would be intractable without approximation for a VAE. LDVAE was also shown to induce fewer correlations between latent variables and to improve the grouping of the regulatory programs. LDVAE is capable to scale to a large dataset with ~2 M cells.
. In contrast, such interpretability would be intractable without approximation for a VAE. LDVAE was also shown to induce fewer correlations between latent variables and to improve the grouping of the regulatory programs. LDVAE is capable to scale to a large dataset with ~2 M cells.
SAUCIE
SAUCIE [15] is an AE (Figures 2B and 4B) designed to perform multiple functions, including clustering, batch correlation, imputation and visualization. SAUCIE is applied to the normalized data instead of count data.
Model
SAUCIE includes multiple model components designed for different functions.
(i) Clustering: SAUCIE first introduced a ‘digital’ binary encoding layer
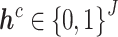 in the decoder
in the decoder  that functions to encode the cluster ID. To learn this encoding, an entropy loss is introduced
that functions to encode the cluster ID. To learn this encoding, an entropy loss is introduced
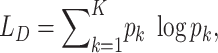 |
(30) |
where 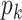 is the probability (proportion) of activation on neuron
is the probability (proportion) of activation on neuron 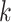 by the previous layer. Minimizing this entropy loss promotes sparse neurons, thus forcing a binary encoding. To encourage clustering behavior, SAUCIE also introduced an intracluster loss as
by the previous layer. Minimizing this entropy loss promotes sparse neurons, thus forcing a binary encoding. To encourage clustering behavior, SAUCIE also introduced an intracluster loss as
 |
(31) |
which computes the distance  between the expressions of a pair of cells (
between the expressions of a pair of cells ( ) that have the same cluster ID (
) that have the same cluster ID ( ).
).
(ii) Batch correction: To correct the batch effect, an MMD loss is introduced to measure the differences in terms of the distribution between batches in the latent space
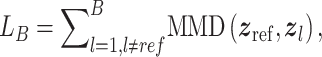 |
(32) |
where 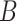 is the total number of batches and
is the total number of batches and 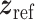 is the latent variable of an arbitrarily chosen reference batch.
is the latent variable of an arbitrarily chosen reference batch.
(iii) Imputation and visualization: The output of the decoder is taken by SAUCIE as an imputed version of the input gene expression. To visualize the data without performing an additional dimension reduction directly, the dimension of the latent variable
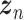 is forced to 2.
is forced to 2.
Training the model includes two sequential runs. In the first run, an AE is trained to minimize the loss 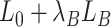 with
with 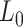 being the MSE reconstruction loss defined in Eq. (9) so that a batch-corrected, imputed input
being the MSE reconstruction loss defined in Eq. (9) so that a batch-corrected, imputed input 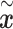 can be obtained at the output of the decoder. In the second run, the bottleneck layer of the encoder from the first run is replaced by a 2D latent code for visualization and a digital encoding layer is also introduced. This model takes the cleaned
can be obtained at the output of the decoder. In the second run, the bottleneck layer of the encoder from the first run is replaced by a 2D latent code for visualization and a digital encoding layer is also introduced. This model takes the cleaned  as the input and is trained for clustering by minimizing the loss
as the input and is trained for clustering by minimizing the loss 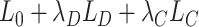 . After the model is trained,
. After the model is trained, 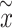 is the imputed, batch-corrected gene expression. The 2D latent code is used for visualization and the binary encoder encodes the cluster ID.
is the imputed, batch-corrected gene expression. The 2D latent code is used for visualization and the binary encoder encodes the cluster ID.
Results
SAUCIE was evaluated for clustering, batch correction, imputation and visualization on both simulated and real scRNA-seq and scCyToF datasets. The performance was compared with minibatch kmeans, Phenograph [87] and 22 for clustering; MNN [32] and CCA [33] for batch correction; PCA, Monocle2 [88], diffusion maps, UMAP [89], t-SNE [90] and PHATE [91] for visualization; and MAGIC [57], scImpute [37] and nearest neighbors completion (NN completion) for imputation. Results showed that SAUCIE had a better or comparable performance with other approaches. Also, SAUCIE has better scalability and faster runtimes than any of the other models. SAUCIE’s results on the scCyToF dengue dataset were further analyzed in greater detail. SAUCIE was able to identify subtypes of the T cell populations and demonstrated distinct cell manifold between acute and healthy subjects.
scScope
scScope [92] is an AE (Figures 2B and 4D) with recurrent steps designed for imputation and batch correction.
Model
scScope has the following model design for batch correction and imputation.
(i) Batch correction: A batch correction layer is applied to the input expression as
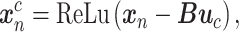 |
(33) |
where  is the ReLu activation function,
is the ReLu activation function,  is the batch correction matrix,
is the batch correction matrix,  is an indicator vector with entry 1 indicates the batch of the input and
is an indicator vector with entry 1 indicates the batch of the input and 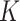 is the total number of batches.
is the total number of batches.
(ii) Recursive imputation: Instead of using the reconstructed expression
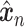 as the imputed expression like in SAUCIE, scScope adds an imputer to
as the imputed expression like in SAUCIE, scScope adds an imputer to 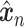 to recursively improve the imputation result. The imputer is a single-layer AE, whose decoder performs the imputation as
to recursively improve the imputation result. The imputer is a single-layer AE, whose decoder performs the imputation as
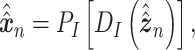 |
(34) |
where 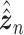 is the output of the imputer encoder,
is the output of the imputer encoder, 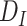 is the imputer decoder and
is the imputer decoder and 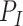 is a masking function that sets the elements in
is a masking function that sets the elements in 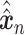 that corresponds to the non-missing values to zero. Then,
that corresponds to the non-missing values to zero. Then, 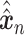 will be fed back to fill the missing value in the batch corrected input as
will be fed back to fill the missing value in the batch corrected input as 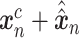 , which will be passed on to the main AE. This recursive imputation can iterate multiple cycles as selected.
, which will be passed on to the main AE. This recursive imputation can iterate multiple cycles as selected.
The loss function is defined as
 |
(35) |
where  is the total number of recursion,
is the total number of recursion,  is the reconstructed expression at
is the reconstructed expression at  th recursion,
th recursion,  is another masking function that forces the loss to compute only the non-missing values in
is another masking function that forces the loss to compute only the non-missing values in  .
.
Results
scScope was evaluated for its scalability, clustering, imputation and batch correction. It was compared with PCA, MAGIC, ZINB-WaVE, SIMLR, AE, scVI and DCA. For scalability and training speed, scScope was shown to offer scalability (for >100 K cells) with high efficiency (faster than most of the approaches). For clustering results, scScope outperformed most of the algorithms on small simulated datasets but offered similar performance on large simulated datasets. For batch correction, scScope performed comparably with other approaches but with faster runtime. For imputation, scScope produced smaller errors consistently across a different range of expression. scScope was further shown to be able to identify rear cell populations from a large mix of cells.
Automated cell-type identification
scRNA-seq can catalog cell types in complex tissues under different conditions. However, the commonly adopted manual cell typing approach based on known markers is time-consuming and less reproducible. We survey DL models of automated cell-type identification.
DigitalDLSorter
DigitalDLSorter [51] was proposed to identify and quantify the immune cells infiltrated in tumors captured in bulk RNA-seq, utilizing single-cell RNA-seq data.
Model
DigitalDLSorter is a 4-layer DNN (Figure 4A) (2 hidden layers of 200 neurons each and an output of 10 cell types). The DigitalDLSorter is trained with two single-cell datasets: breast cancers [93] and colorectal cancers [94]. For each cell, it is determined to be tumor cell or non-tumor cell using RNA-seq based CNV method [93], followed by using xCell algorithm [95] to determine immune cell types for non-tumor cells. Different pseudo bulk (from 100 cells) RNA-seq datasets were prepared with known mixture proportions to train the DNN. The output of DigitalDLSorter is the predicted proportions of cell types in the input bulk sample.
Result
DigitalDLSorter was first tested on simulated bulk RNA-seq samples. DigitalDLSorter achieved excellent agreement (linear correlation of 0.99 for colorectal cancer, and good agreement in quadratic relationship for breast cancer) at predicting cell types proportion. The proportion of immune and non-immune cell subtypes of test bulk TCGA samples was predicted by DigitalDLSorter and the results showed a very good correlation to other deconvolution tools including TIMER [93], ESTIMATE [96], EPIC [97] and MCPCounter [98]. Using DigitalDLSorter predicted CD8+ (good prognosis for overall and disease-free survival) and Monocytes-Macrophages (MM, indicator for protumoral activity) proportions, it is found that patients with higher CD8+/MM ratio had better survival for both cancer types than those with lower CD8+/MM ratio. Both EPIC and MCPCounter yielded non-significant survival associations using their cell proportion estimate.
scCapsNet
scCapsNet [52] is an interpretable capsule network designed for cell-type prediction. The paper showed that the trained network could be interpreted to inform marker genes and regulatory modules of cell types.
Model
scCapsNet takes log-transformed, normalized expressions as input and follows the general CapsNet model described in Section 3.4. Capsule 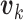 represents the probability of a single cell
represents the probability of a single cell  belonging to cell type
belonging to cell type 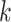 , which will be used for cell-type classification. Once trained, the interpretation of marker genes and regulatory modules can be achieved by determining first the important primary capsules for each cell type and then the most significant genes for each important primary capsule (identified based on
, which will be used for cell-type classification. Once trained, the interpretation of marker genes and regulatory modules can be achieved by determining first the important primary capsules for each cell type and then the most significant genes for each important primary capsule (identified based on 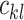 directly). To determine the genes that are important for an important primary capsule
directly). To determine the genes that are important for an important primary capsule 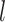 , genes are ranked base on the scores of the first principal component computed from the columns of
, genes are ranked base on the scores of the first principal component computed from the columns of 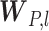 in Eq. (15) and then the markers are obtained by a greedy search along with the ranked list for the best classification performance.
in Eq. (15) and then the markers are obtained by a greedy search along with the ranked list for the best classification performance.
Results
scCapsNet’s performance was evaluated on human PBMCs [99] and mouse retinal bipolar cells [100] datasets and shown to have comparable accuracies (99% and 97%, respectively) with DNN and other popular ML algorithms (SVM, random forest, LDA and nearest neighbor). However, the interpretability of scCapsNet was demonstrated extensively. First, examining the coupling coefficients for each cell type showed that only a few primary capsules have high values and thus are effective. Subsequently, a set of core genes were identified for each effective capsule using the greedy search on the PC-score ranked gene list. GO enrichment analysis showed that these core genes were enriched in cell-type-related biological functions. Mapping the expression data into space spanned by PCs of the columns of 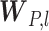 corresponding to all core genes uncovered regulatory modules that would be missed by the t-SNE of gene expressions, which demonstrated the effectiveness of the embeddings learned by scCapsNet in capturing the functionally important features.
corresponding to all core genes uncovered regulatory modules that would be missed by the t-SNE of gene expressions, which demonstrated the effectiveness of the embeddings learned by scCapsNet in capturing the functionally important features.
netAE: Network-enhanced AE
netAE [101] is a VAE-based semi-supervised cell-type prediction model (Figures 2A and 4C) that deals with scenarios of having a small number of labeled cells.
Model
netAE works with UMI counts and assumes a ZINB distribution for 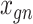 as in Eq. (25) in scVI. However, netAE adopts the general VAE loss as in Eq. (6) with two function-specific loss as
as in Eq. (25) in scVI. However, netAE adopts the general VAE loss as in Eq. (6) with two function-specific loss as
 |
(36) |
where  is a set of indices for all cells and
is a set of indices for all cells and  is a subset of
is a subset of 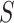 for only cells with cell-type labels,
for only cells with cell-type labels, 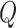 is modified Newman and Girvan modularity [102] that quantifies cluster strength using
is modified Newman and Girvan modularity [102] that quantifies cluster strength using 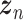 ,
, 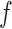 is the softmax function and
is the softmax function and 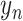 is the cell-type label. The second loss in Eq. (36) functions as a clustering constraint and the last term is the cross-entropy loss that constrains the cell-type classification.
is the cell-type label. The second loss in Eq. (36) functions as a clustering constraint and the last term is the cross-entropy loss that constrains the cell-type classification.
Results
netAE was compared with popular dimension reduction methods including scVI, ZIFA, PCA and AE as well as a semi-supervised method scANVI [103]. For different dimensionality reduction methods, cell-type classification from latent features of cells was carried out using KNN and logistic regression. The effect of different labeled samples sizes on classification performance was also investigated, where the sample size varied from as few as 10 cells to 70% of all cells. Among 3 test datasets (mouse brain cortex, human embryo development and mouse hematopoietic stem and progenitor cells), netAE outperformed most of the baseline methods. Latent features were visualized using t-SNE and cell clusters by netAE were tighter than those by other embedding spaces. netAE also showed consistency of better cell-type classification with improved cell-type clustering. This suggested that the latent spaces learned with added modularity constraint in the loss helped identify clusters of similar cells. Ablation study by removing each of the three loss terms in Eq. (36) showed a drop of cell-type classification accuracy, suggesting that all three were necessary for the optimal performance.
scDGN: supervised adversarial alignment of single-cell RNA-seq data
scDGN [53], or Single Cell Domain Generalization Network (Figure 4G), is a domain adversarial network that aims to accurately assign cell types of single cells while performing batch removal (domain adaptation) at the same time. It benefits from the superior ability of domain adversarial learning to learn representations that are invariant to technical confounders.
Model
scDGN takes the log-transformed, normalized expression as the input and has three main modules: (i) an encoder (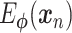 ) for dimension reduction of scRNA-seq data, (ii) cell-type classifier,
) for dimension reduction of scRNA-seq data, (ii) cell-type classifier,  with parameters
with parameters 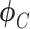 and (iii) domain (batch) discriminator,
and (iii) domain (batch) discriminator, 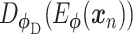 . The model has a Siamese design and the training takes a pair of cells
. The model has a Siamese design and the training takes a pair of cells 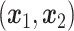 , each from the same or different batches. The encoder network contains two hidden layers with 1146 and 100 neurons.
, each from the same or different batches. The encoder network contains two hidden layers with 1146 and 100 neurons. 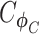 classifies the cell type and
classifies the cell type and 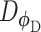 predicts whether
predicts whether 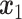 and
and 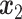 are from the same batch or not. The overall loss is denoted by
are from the same batch or not. The overall loss is denoted by
 |
(37) |
where 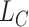 is the cross-entropy loss, and
is the cross-entropy loss, and 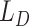 is a contrastive loss as described in [104]. Notice that Eq. (37) has an adversarial formulation and minimizing this loss maximizes the misclassification of cells from different batches, thus making them indistinguishable. Similar to GAN training, scDGN is trained to iteratively solve:
is a contrastive loss as described in [104]. Notice that Eq. (37) has an adversarial formulation and minimizing this loss maximizes the misclassification of cells from different batches, thus making them indistinguishable. Similar to GAN training, scDGN is trained to iteratively solve: 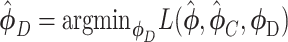 and
and 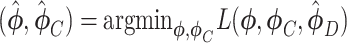 .
.
Results
scDGN was tested for classifying cell types and aligning batches ranging in size from 10 to 39 cell types and from 4 to 155 batches. The performance was compared with a series of DL and traditional ML methods, including Lin et al. DNN [71], CaSTLe [105], MNN [32], scVI [17] and Seurat [10]. scDGN outperformed all other methods in the classification accuracy on a subset of scQuery datasets (0.29), PBMC (0.87) and four of the six Seurat pancreatic datasets (0.86–0.95). PCA visualization of the learned data representations demonstrated that scDGN overcame the batch differences and clearly separated cell clusters based on cell types, while other methods were vulnerable to batch effects. In summary, scDGN is a supervised adversarial alignment method to eliminate the batch effect of scRNA-seq data and create cleaner representations of cell types.
Biological function prediction
Predicting biological functions and responses to treatment at single cell level or cell types is critical to understand cellular system functioning and potent responses to stimulations. DL models are capable of capture gene–gene relationship and their property in the latent space. Several surveyed models demonstrate exciting results to learn complex biological functions and outcomes.
CNN for coexpression
CNNC [54] is proposed to infer causal interactions between genes from scRNA-seq data.
Model
CNNC is a CNN (Figure 4F), one of the most popular DL models. CNNC takes expression levels of two genes from many cells and transforms them into a 32 × 32 image-like normalized empirical probability function (NEPDF), which measures the probabilities of observing different coexpression levels between the two genes. CNNC includes six convolutional layers, three max-pooling layers, one flatten layer and one output layer. All convolution layers have 32 kernels of size 3 × 3. Depending on the application, the output layer can be designed to predict the state of interaction (Yes/No) between the genes or the causal interaction between the input genes (no interaction, gene A regulates gene B or gene B regulates gene A).
Result
CNNC was trained to predict TF-Gene interactions using the mESC data from scQuery [106], where the ground truth interactions were obtained from the ChIP-seq dataset from the GTRD database [107]. The performance was compared with DNN, count statistics [108] and mutual information (MI)-based approach [109]. CNNC was shown to have more than 20% higher AUPRC than other methods and reported almost no false-negative for the top 5% predictions. CNNC was also trained to predict the pathway regulator-target gene pairs. The positive regulator-gene pairs were obtained from KEGG [110] and Reactome [111], and negative samples were genes pairs that appeared in pathways but do not interacted. CNNC was shown to have better performance of predicting regulator-gene pairs on both KEGG and Reactome pathways than other methods including Pearson correlation, count statistics, GENIE3 [112], MI, Bayesian-directed network (BDN) and DREMI [109]. CNNC was also applied for causality prediction between two genes, that is if two genes regulate each other and if they do, which gene is the regulator. The ground truth causal relationships were also obtained from the KEGG and Reactome datasets. Again, CNNC reported better performance than BDN, the common method developed to learn casual relationships from gene expression data. CNNC was finally trained to assign three essential cell functions (cell cycle, circadian rhythm and immune system) to genes. This is achieved by training CNNC to predict pairs of genes from the same function (e.g. Cell Cycle defined by mSigDB from gene set enrichment analysis [113]) as 1 and all other pairs as 0. The performance was compared with ‘guilt by association’ and DNN, and CNNC was shown to have more than 4% higher AUROC and reported all positives for the top 10% predictions.
scGen, a generative model to predict perturbation response of single cells across cell types
scGen [114] is designed to learn cellular responses to certain perturbations such as drug treatment and gene knockout from single-cell expression data, and then predict cellular responses to the same perturbation for a new sample or a new cell type. The novelty of scGen is that it learns the cellular response in the latent space instead of the expression data space.
Model
ScGen follows the general VAE (Figures 2A and 4C) for scRNA-seq data but uses the ‘latent space arithmetics’ to learn perturbations’ response. Given scRNA-seq samples of perturbed (denoted as p) and unperturbed cells (denoted as unp), a VAE model is trained. Then, the latent space representation 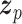 and
and  are obtained for the perturbed and unperturbed cells. Following the notion that VAE could map nonlinear operations (e.g. perturbation) in the data space to linear operations in the latent space, ScGen estimates the response in the latent space as
are obtained for the perturbed and unperturbed cells. Following the notion that VAE could map nonlinear operations (e.g. perturbation) in the data space to linear operations in the latent space, ScGen estimates the response in the latent space as  , where
, where  is the average representation of samples from the same cell type or different cell types. Then, given the latent representation
is the average representation of samples from the same cell type or different cell types. Then, given the latent representation  of an unperturbed cell for a new sample from the same or different cell type, the latent representation of the corresponding perturbed cell can be predicted as
of an unperturbed cell for a new sample from the same or different cell type, the latent representation of the corresponding perturbed cell can be predicted as  =
=  . The expression of the perturbed cell can also be estimated by feeding
. The expression of the perturbed cell can also be estimated by feeding 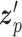 into the VAE decoder. The scGen can also be expanded to samples and treatment across two species (using orthologues between species). When scGen is trained for species 1 (s1) with both perturbed and unperturbed cells and species 2 (s2) with only unperturbed cells, the latent code for the perturbed cells from s2 can be predicted as
into the VAE decoder. The scGen can also be expanded to samples and treatment across two species (using orthologues between species). When scGen is trained for species 1 (s1) with both perturbed and unperturbed cells and species 2 (s2) with only unperturbed cells, the latent code for the perturbed cells from s2 can be predicted as 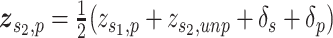 where
where 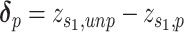 captures the response of perturbation and
captures the response of perturbation and 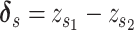 represents the difference between species.
represents the difference between species.
Result
scGen was applied to predict the perturbation of out-of-samples response in human PBMCs data, and scGen showed a higher average correlation (R2 = 0.948) between predicted and real data for six cell types [115]. Compared with other methods including CVAE [116], style transfer GAN [117], linear approaches based on vector arithmetics (VA) similar in [118] and PCA + VA, scGen predicted full distribution of ISG15 gene (strongest regulated gene by IFN-β) response to IFN-β [115], while others might predict mean (CAVE and style transfer GAN) but failed to produce the full distribution. scGen was also tested on predicting the intestinal epithelial cells’ response to infections [119]. For early transit-amplifying cells, scGen showed good prediction (R2 = 0.98) for both Heligmosomoides polygyrus and Salmonella infections. Finally, scGen was evaluated for perturbation across species using scRNA-seq data set by Hagai et al. [120], which comprises bone marrow-derived mononuclear phagocytes from mice, rats, rabbits and pigs perturbed with lipopolysaccharide (LPS). scGen’s predictions of LPS perturbation responses were shown to be highly correlated (R2 = 0.91) with the real responses.
CONCLUSIONS
We systematically survey 25 DL models according to the challenges they address. We categorize major DL model statements into VAE, AE and GAN with a unified mathematic formulation in Section 3 (graphic model representation in Figure 2), which will guide readers to focus on the DL model selection, training strategies and loss functions in each algorithm. Specifically, the differences in loss functions are highlighted in each DL model’s applications to meet specific objectives. DL/ML models that 25 surveyed models are evaluated against are presented in Figure 3, providing a straightforward way for readers to pick up the most suitable DL model at a specific step for their own scRNA-seq data analysis. All evaluation methods are listed in Table 8, as we foresee Table 8 to be an easy recipe book for researchers to establish their scRNA-seq pipeline. In addition, a summary of all the 25 DL models concerning their DL models, evaluation metrics, implementation environment, downloadable source codes, features and application notes is presented in Tables 1 and 2. Taken together, this survey provides a rich resource to select a DL model for proper research applications, and we expect to inspire new DL model developments for scRNA-seq analysis.
Table 2.
Comparison of DL algorithms reviewed in the paper
| App | Algorithm | Feature | Application notes |
|---|---|---|---|
| Imputation | |||
| DCA |
|
|
|
| SAVER-X |
|
|
|
| DeepImpute |
|
|
|
| LATE |
|
|
|
| scGAMI |
|
|
|
| scIGANs |
|
|
|
| Batch correction | |||
| BERMUDA |
|
|
|
| DESC |
|
|
|
| iMAP |
|
|
|
| Clustering, latent representation, dimension reduction and data augmentation | |||
| Dhaka |
|
|
|
| scvis |
|
|
|
| scVAE |
|
|
|
| VASC |
|
|
|
| scDeepCluster |
|
|
|
| cscGAN |
|
|
|
| Multi-functional models | |||
| scVI |
|
|
|
| LDVAE |
|
|
|
| SAUCIE |
|
|
|
| scScope |
|
|
|
| Cell type Identification | |||
| DigitalDLSorter |
|
|
|
| scCapsNet |
|
|
|
| netAE |
|
|
|
| scDGN |
|
|
|
| Function analysis | |||
| CNNC |
|
|
|
| scGen |
|
|
|
Abbreviation: NB: negative binomial distribution.
One advantage of DL for scRNA-seq repeatedly demonstrated in many of these surveyed papers is DL’s ability to scale to a large number of cells, thanks to the stochastic gradient descent algorithm. For imputation, DCA shows linear scalability with the number of cells, and scIGAN and DeepInpute are demonstrated to scale to 100-K cells, while non-DL algorithms including SAVER and SCRABBLE fail due to excessive memory usage and runtime [19]. A similar favorable scalability result has been echoed for batch normalization by DESC and iMAP, clustering by scDeepCluster, and multi-functional analysis by scVI, LDVAE, SAUCIE and scScope. Overall, the advantage of DL in scalability becomes more apparent over non-DL approaches after the number of cells exceeds thousands. However, many of these comparisons exclude the time for determining DL models’ hyperparameters. Although iMAP shows that the model is robust against model hyperparameters, determinination of optimal hyperparameters in DL models has not been comprehensively studied for these scRNA-seq tasks.
This review focuses on surveying common DL models, such as AE, VAE and GAN, and their model variations or combinations to address single-cell data analysis challenges. With the advancement of multi-omics single-cell technologies, new single-cell data types and DL models will be introduced to the single-cell analysis pipeline, such as cyTOF using SAUCIE [15], spatial transcriptome using DNN [121] and CITE-seq that simultaneously generates read counts for surface protein expression along with gene expression [122, 123]. Other than three most common unsupervised DL models using AE, VAE and GAN, this review also discusses supervised network frameworks including CapsNet (e.g. scCapsNet [52]), CNN (e.g. CNNC [54]) and domain adaption learning (e.g. scDGN [53]). It is expected that more DL models and learning paradigms will be developed and implemented for the most challenging steps for scRNA-seq data, including but not limited to, multi-omics data integration and data interpretation. For example, integrating PPI graphs into DL models has been shown for its advantages of biological knowledge and nonlinear interactions embedded in the graphs [124–126]. Indeed, a recently published scRNA-seq analysis pipeline, scGNN [127], incorporates three iterative AEs (including one graph AE) and successfully demonstrated Alzheimer’s disease-related neural development and differentiation mechanisms. We expect that our careful organization of this review will provide a basic understanding of DL models for scRNA-seq and inspire innovative applications of DL models for single cell analysis.
Authors’ contributions
Y.H., Y.C., M.F. and Y.F.J. conceived the study. M.F., Z.L., T.Z., M.M.H., Y.C.C., Z.Y., K.P., S.J., J.Z., S.J.G., Y.F.J., Y.C. and Y.H. summarized resources, wrote and approved the final version of the paper.
Key Points
Single cell RNA sequencing technology generates a large collection of transcriptomic profiles of up to millions of cells, enabling biological investigation of hidden expression functional structures or cell types, predicting their effects or responses to treatment more precisely or utilizing subpopulations to address unanswered hypotheses.
Twenty-five deep learning-based approaches for single cell RNA seq data analysis are systematically reviewed in this paper according to the challenge they address and their roles in the analysis pipeline.
A unified mathematical description of the surveyed DL models is presented and the specific model features were discussed when reviewing each approach.
A comprehensive summary of the evaluation metrics, comparison algorithms and datasets by each approach is presented.
Mario Flores, PhD, is an Assistant Professor in the Department of Electrical and Computer Engineering at the University of Texas at San Antonio. His research focuses on DNA and RNA sequence methods, transcriptomics analysis (including scRNA-seq), epigenetics, comparative genomics and deep learning to study the mechanisms of gene regulation.
Zhentao Liu is a PhD student in the Department of Electrical and Computer Engineering, The University of Texas at San Antonio. His research focuses on deep learning for cancer genomics and drug response prediction.
Tinghe Zhang is a PhD student in the Department of Electrical and Computer Engineering, The University of Texas at San Antonio. His research focuses on deep learning for cancer genomics and drug response prediction.
Md Musaddaqui Hasib is a PhD student in the Department of Electrical and Computer Engineering, The University of Texas at San Antonio. His research focuses on interpretable deep learning for cancer genomics.
Yu-Chiao Chiu, PhD, is a Postdoctoral Fellow at the Greehey Children’s Cancer Research Institute at The University of Texas Health San Antonio. His postdoctoral research is focused on developing deep learning models for pharmacogenomic studies.
Zhenqing Ye, PhD, is an Assistant Professor in the Department of Population Health Sciences and the bioinformatics facility manager at Greehey Children’s Cancer Research Institute at The University of Texas Health San Antonio. His research focuses on computational methods on next-generation sequencing and single-cell RNA-seq data analysis.
Karla Paniagua is a PhD student in the Department of Electrical and Computer Engineering, The University of Texas at San Antonio. Her research focuses on applications of deep learning algorithms.
Sumin Jo is a PhD student in the Department of Electrical and Computer Engineering, The University of Texas at San Antonio. Her research focuses on m6A mRNA methylation and deep learning for biomedical applications.
Jianqiu Zhang, PhD, is an Associate Professor in the Department of Electrical and Computer Engineering at the University of Texas at San Antonio. Her current research focuses on deep learning for biomedical applications such as m6A mRNA methylation.
Shou-Jiang Gao, PhD, is a Professor in the UPMC Hillman Cancer Center and the Department of Microbiology and Molecular Genetics, University of Pittsburgh. His current research interests include Kaposi’s sarcoma-associate herpesvirus, AIDS-related malignancies, translational and cancer therapeutics, and systems biology.
Yu-Fang Jin, PhD, is a Professor in the Department of Electrical and Computer Engineering at the University of Texas at San Antonio. Her research focuses on mathematical modeling of cellular responses in immune systems, data-driven modeling and analysis of macrophage activations, and deep learning applications.
Yidong Chen, PhD, is a Professor in the Department of Population Health Sciences and the director of Computational Biology and Bioinformatics at Greehey Children’s Cancer Research Institute at the University of Texas Health San Antonio. His research interests include bioinformatics methods in next-generation sequencing technologies, integrative genomic data analysis, genetic data visualization and management, and machine learning in translational cancer research.
Yufei Huang, PhD, is a Professor in the Department of Medicine, University of Pittsburgh School of Medicine and Leader in AI Research at UPMC Hillman Cancer Center. His current research interests include uncovering the functions of m6A mRNA methylation, cancer virology and medical AI & deep learning.
Contributor Information
Mario Flores, Department of Electrical and Computer Engineering, the University of Texas at San Antonio, San Antonio, TX 78249, USA.
Zhentao Liu, Department of Electrical and Computer Engineering, the University of Texas at San Antonio, San Antonio, TX 78249, USA.
Tinghe Zhang, Department of Electrical and Computer Engineering, the University of Texas at San Antonio, San Antonio, TX 78249, USA.
Md Musaddaqui Hasib, Department of Electrical and Computer Engineering, the University of Texas at San Antonio, San Antonio, TX 78249, USA.
Yu-Chiao Chiu, Greehey Children’s Cancer Research Institute, University of Texas Health San Antonio, San Antonio, TX 78229, USA.
Zhenqing Ye, Greehey Children’s Cancer Research Institute, University of Texas Health San Antonio, San Antonio, TX 78229, USA; Department of Population Health Sciences, University of Texas Health San Antonio, San Antonio, TX 78229, USA.
Karla Paniagua, Department of Electrical and Computer Engineering, the University of Texas at San Antonio, San Antonio, TX 78249, USA.
Sumin Jo, Department of Electrical and Computer Engineering, the University of Texas at San Antonio, San Antonio, TX 78249, USA.
Jianqiu Zhang, Department of Electrical and Computer Engineering, the University of Texas at San Antonio, San Antonio, TX 78249, USA.
Shou-Jiang Gao, Department of Microbiology and Molecular Genetics, University of Pittsburgh, Pittsburgh, Pennsylvania, PA 15232, USA; UPMC Hillman Cancer Center, University of Pittsburgh, PA 15232, USA.
Yu-Fang Jin, Department of Electrical and Computer Engineering, the University of Texas at San Antonio, San Antonio, TX 78249, USA.
Yidong Chen, Greehey Children’s Cancer Research Institute, University of Texas Health San Antonio, San Antonio, TX 78229, USA; Department of Population Health Sciences, University of Texas Health San Antonio, San Antonio, TX 78229, USA.
Yufei Huang, Department of Medicine, School of Medicine, University of Pittsburgh, PA 15232, USA; UPMC Hillman Cancer Center, University of Pittsburgh, PA 15232, USA.
Funding
National Institutes of Health (grant no. CTSA 1UL1RR025767-01 to Y.C., R01GM113245 to Y.H., NCI Cancer Center Shared Resources P30CA54174 to Y.C., K99CA248944 to Y.C.C.); National Science Foundation (grant no. 2051113 to Y.F.J.); Cancer Prevention and Research Institute of Texas (grant no. RP160732 to Y.C., RP190346 to Y.C. and Y.H.); Fund for Innovation in Cancer Informatics (ICI Fund to Y.C.C. and Y.C.). The funding sources had no role in the design of the study; collection, analysis and interpretation of data; or in writing the manuscript.
References
- 1. Lahnemann D, Koster J, Szczurek E, et al. . Eleven grand challenges in single-cell data science. Genome Biol 2020;21(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vitak SA, Torkenczy KA, Rosenkrantz JL, et al. . Sequencing thousands of single-cell genomes with combinatorial indexing. Nat Methods 2017;14(3):302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu H, Wang C, Wu S. Single-cell sequencing for drug discovery and drug development. Curr Top Med Chem 2017;17(15):1769–77. [DOI] [PubMed] [Google Scholar]
- 4. Kinker GS, Greenwald AC, Tal R, et al. . Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52(11):1208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Navin NE. The first five years of single-cell cancer genomics and beyond. Genome Res 2015;25(10):1499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suva ML, Tirosh I. Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol Cell 2019;75(1):7–12. [DOI] [PubMed] [Google Scholar]
- 7. Mannarapu M, Dariya B, Bandapalli OR. Application of single-cell sequencing technologies in pancreatic cancer. Mol Cell Biochem 2021;476(6):2429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wauters E, Van Mol P, Garg AD, et al. . Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res 2021;31(3):272–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bost P, Giladi A, Liu Y, et al. . Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 2020;181(7):1475–1488.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stuart T, Butler A, Hoffman P, et al. . Comprehensive integration of single-cell data. Cell 2019;177(7):1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 2018;19(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vaswani A, Shazeer N, Parmar N, et al. . Attention is all you need. In: Advances in neural information processing systems, 2017, 5998–6008.
- 13. Karpathy A, Toderici G, Shetty S, Leung T, Sukthankar R, Fei-Fei L: Large-scale video classification with convolutional neural networks. In: Proceedings of the IEEE conference on Computer Vision and Pattern Recognition, 2014. 1725–32.
- 14. Deng L, Liu Y. Deep learning in natural language processing. Springer, 2018. [Google Scholar]
- 15. Amodio M, Dijk D, Srinivasan K, et al. . Exploring single-cell data with deep multitasking neural networks. Nat Methods 2019;16(11):1139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Srinivasan S, Leshchyk A, Johnson NT, et al. . A hybrid deep clustering approach for robust cell type profiling using single-cell RNA-seq data. RNA 2020;26(10):1303–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lopez R, Regier J, Cole MB, et al. . Deep generative modeling for single-cell transcriptomics. Nat Methods 2018;15(12):1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eraslan G, Simon LM, Mircea M, et al. . Single-cell RNA-seq denoising using a deep count autoencoder. Nat Commun 2019;10(1):390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu Y, Zhang Z, You L, et al. . scIGANs: single-cell RNA-seq imputation using generative adversarial networks. Nucleic Acids Res 2020;48(15):e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arisdakessian C, Poirion O, Yunits B, et al. . DeepImpute: an accurate, fast, and scalable deep neural network method to impute single-cell RNA-seq data. Genome Biol 2019;20(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tran HTN, Ang KS, Chevrier M, et al. . A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol 2020;21(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Petegrosso R, Li Z, Kuang R. Machine learning and statistical methods for clustering single-cell RNA-sequencing data. Brief Bioinform 2020;21(4):1209–23. [DOI] [PubMed] [Google Scholar]
- 23. Abdelaal T, Michielsen L, Cats D, et al. . A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol 2019;20(1):194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang J, Zou Q, Lin C. A comparison of deep learning-based pre-processing and clustering approaches for single-cell RNA sequencing data. Brief Bioinform 2021. 10.1093/bib/bbab345. [DOI] [PubMed] [Google Scholar]
- 25. Picelli S, Bjorklund AK, Faridani OR, et al. . Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 2013;10(11):1096–8. [DOI] [PubMed] [Google Scholar]
- 26. Macosko EZ, Basu A, Satija R, et al. . Highly parallel genome-wide expression profiling of individual cells using Nanoliter droplets. Cell 2015;161(5):1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eisenstein M. Single-cell RNA-seq analysis software providers scramble to offer solutions. Nat Biotechnol 2020;38(3):254–7. [DOI] [PubMed] [Google Scholar]
- 28. Chen G, Ning B, Shi T. Single-cell RNA-Seq technologies and related computational data analysis. Front Genet 2019;10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vallejos CA, Marioni JC, Richardson S. BASiCS: Bayesian analysis of single-cell sequencing data. PLoS Comput Biol 2015;11(6):e1004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lun AT, Bach K, Marioni JC. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol 2016;17:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 2019;20(1):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haghverdi L, Lun ATL, Morgan MD, et al. . Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 2018;36(5):421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Butler A, Hoffman P, Smibert P, et al. . Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36(5):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Korsunsky I, Millard N, Fan J, et al. . Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 2019;16(12):1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peng T, Zhu Q, Yin P, et al. . SCRABBLE: single-cell RNA-seq imputation constrained by bulk RNA-seq data. Genome Biol 2019;20(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang M, Wang J, Torre E, et al. . SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods 2018;15(7):539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li WV, Li JJ. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat Commun 2018;9(1):997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roweis ST, Saul LK. Nonlinear dimensionality reduction by locally linear embedding. Science 2000;290(5500):2323–6. [DOI] [PubMed] [Google Scholar]
- 39. Welch JD, Hartemink AJ, Prins JF. SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol 2016;17(1):106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Linderman GC, Rachh M, Hoskins JG, et al. . Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat Methods 2019;16(3):243–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Becht E, McInnes L, Healy J, et al. . Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2019;37(1):38–44. [DOI] [PubMed] [Google Scholar]
- 42. Subelj L, Bajec M. Unfolding communities in large complex networks: combining defensive and offensive label propagation for core extraction. Phys Rev E Stat Nonlin Soft Matter Phys 2011;83(3):036103. [DOI] [PubMed] [Google Scholar]
- 43. Traag VA, Waltman L, Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 2019;9(1):5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang B, Zhu J, Pierson E, et al. . Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat Methods 2017;14(4):414–6. [DOI] [PubMed] [Google Scholar]
- 45. Finak G, McDavid A, Yajima M, et al. . MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 2015;16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods 2014;11(7):740–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miao Z, Deng K, Wang X, et al. . DEsingle for detecting three types of differential expression in single-cell RNA-seq data. Bioinformatics 2018;34(18):3223–4. [DOI] [PubMed] [Google Scholar]
- 48. Goodfellow I, Pouget-Abadie J, Mirza M, et al. . Generative adversarial networks. Commun ACM 2020;63(11):139–44. [Google Scholar]
- 49. Gulrajani I, Ahmed F, Arjovsky M, et al. . Improved training of wasserstein gans. arXiv preprint arXiv:170400028. 2017.
- 50. Arjovsky M, Chintala S, Bottou L. Wasserstein Gan. arXiv 2017arXiv preprint arXiv:170107875;2017:30. [Google Scholar]
- 51. Torroja C, Sanchez-Cabo F. Digitaldlsorter: deep-learning on scRNA-Seq to deconvolute gene expression data. Front Genet 2019;10:978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang L, Nie R, Yu Z, et al. . An interpretable deep-learning architecture of capsule networks for identifying cell-type gene expression programs from single-cell RNA-sequencing data. Nat Mach Intell 2020;2:693–703. [Google Scholar]
- 53. Ge S, Wang H, Alavi A, et al. . Supervised adversarial alignment of single-cell RNA-seq data. J Comput Biol 2021;28(5):501–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yuan Y, Bar-Joseph Z. Deep learning for inferring gene relationships from single-cell expression data. Proc Natl Acad Sci U S A 2019;116(52):27151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eraslan G, Avsec Z, Gagneur J, et al. . Deep learning: new computational modelling techniques for genomics. Nat Rev Genet 2019;20(7):389–403. [DOI] [PubMed] [Google Scholar]
- 56. Patel ND, Nguang SK, Coghill GG. Neural network implementation using bit streams. IEEE Trans Neural Netw 2007;18(5):1488–504. [DOI] [PubMed] [Google Scholar]
- 57. Dijk D, Sharma R, Nainys J, et al. . Recovering gene interactions from single-cell data using data diffusion. Cell 2018;174(3):716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang J, Agarwal D, Huang M, et al. . Data denoising with transfer learning in single-cell transcriptomics. Nat Methods 2019;16(9):875–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Badsha MB, Li R, Liu B, et al. . Imputation of single-cell gene expression with an autoencoder neural network. Quant Biol 2020;8(1):78–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu B, Chen C, Qi R, et al. . scGMAI: a Gaussian mixture model for clustering single-cell RNA-Seq data based on deep autoencoder. Brief Bioinform 2021;22(4). [DOI] [PubMed] [Google Scholar]
- 61. Lin P, Troup M, Ho JW. CIDR: ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol 2017;18(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Berthelot D, Schumm T, Metz L. BEGAN: boundary equilibrium generative adversarial networks. arXiv 2017. [Google Scholar]
- 63. Wang T, Johnson TS, Shao W, et al. . BERMUDA: a novel deep transfer learning method for single-cell RNA sequencing batch correction reveals hidden high-resolution cellular subtypes. Genome Biol 2019;20(1):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Borgwardt KM, Gretton A, Rasch MJ, et al. . Integrating structured biological data by kernel maximum mean discrepancy. Bioinformatics 2006;22(14):e49–57. [DOI] [PubMed] [Google Scholar]
- 65. Crow M, Paul A, Ballouz S, et al. . Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nat Commun 2018;9(1):884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Polanski K, Young MD, Miao Z, et al. . BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 2020;36(3):964–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li X, Wang K, Lyu Y, et al. . Deep learning enables accurate clustering with batch effect removal in single-cell RNA-seq analysis. Nat Commun 2020;11(1):2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guo X, Gao L, Liu X, Yin J. Improved deep embedded clustering with local structure preservation. In: Proc 26th International Joint Conference on Artificial Integlligence, 2017:1753–9.
- 69. Hie B, Bryson B, Berger B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat Biotechnol 2019;37(6):685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang D, Hou S, Zhang L, et al. . iMAP: integration of multiple single-cell datasets by adversarial paired transfer networks. Genome Biol 2021;22(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lin C, Jain S, Kim H, et al. . Using neural networks for reducing the dimensions of single-cell RNA-Seq data. Nucleic Acids Res 2017;45(17):e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rashid S, Shah S, Bar-Joseph Z, et al. . Dhaka: Variational autoencoder for unmasking tumor heterogeneity from single cell genomic data. Bioinformatics 2021;37(11):1535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tirosh I, Izar B, Prakadan SM, et al. . Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352(6282):189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zahn H, Steif A, Laks E, et al. . Scalable whole-genome single-cell library preparation without preamplification. Nat Methods 2017;14(2):167–73. [DOI] [PubMed] [Google Scholar]
- 75. Ding J, Condon A, Shah SP. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat Commun 2018;9(1):2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gronbech CH, Vording MF, Timshel PN, et al. . scVAE: variational auto-encoders for single-cell gene expression data. Bioinformatics 2020;36(16):4415–22. [DOI] [PubMed] [Google Scholar]
- 77. Wang D, Gu J. VASC: dimension reduction and visualization of single-cell RNA-seq data by deep variational autoencoder. Genomics Proteomics Bioinformatics 2018;16(5):320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jang E. GSaPB: categorical reparameterization with gumbel-softmax. arXiv 2016. [Google Scholar]
- 79. Tian T, Wan J, Song Q, et al. . Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat Mach Intell 2019;1(4):191–8. [Google Scholar]
- 80. Regev A, Teichmann SA, Lander ES, et al. . The human cell atlas. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xie J, Girshick R, Farhadi A: Unsupervised deep embedding for clustering analysis. In: International conference on machine learning, 2016. 478–87. PMLR. [Google Scholar]
- 82. Marouf M, Machart P, Bansal V, et al. . Realistic in silico generation and augmentation of single-cell RNA-seq data using generative adversarial networks. Nat Commun 2020;11(1):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Miyato TaK M. cGANs with projection discriminator. In press. 2018.
- 84. Zappia L, Phipson B, Oshlack A. Splatter: simulation of single-cell RNA sequencing data. Genome Biol 2017;18(1):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lindenbaum O, Stanley JS, Wolf G, et al. . Geometry-based data generation. In: Advances in Neural Information Processing Systems, 2018.
- 86. Svensson V, Gayoso A, Yosef N, et al. . Interpretable factor models of single-cell RNA-seq via variational autoencoders. Bioinformatics 2020;36(11):3418–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Levine JH, Simonds EF, Bendall SC, et al. . Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 2015;162(1):184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Qiu X, Mao Q, Tang Y, et al. . Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 2017;14(10):979–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. McInnes L, Healy J, Melville J. Umap: uniform manifold approximation and projection for dimension reduction. ArXiv 2018. [Google Scholar]
- 90. Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn 2008;9:2579–605. [Google Scholar]
- 91. Moon KR, Dijk DV, Wang Z, et al. . PHATE: a dimensionality reduction method for visualizing trajectory structures in high-dimensional biological data. bioRxiv 2017. [Google Scholar]
- 92. Deng Y, Bao F, Dai Q, et al. . Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Nat Methods 2019;16(4):311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chung W, Eum HH, Lee HO, et al. . Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 2017;8:15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Li H, Courtois ET, Sengupta D, et al. . Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet 2017;49(5):708–18. [DOI] [PubMed] [Google Scholar]
- 95. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18(1):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yoshihara K, Shahmoradgoli M, Martinez E, et al. . Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Racle J, Jonge K, Baumgaertner P, et al. . Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Becht E, Giraldo NA, Lacroix L, et al. . Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17(1):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zheng GX, Terry JM, Belgrader P, et al. . Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shekhar K, Lapan SW, Whitney IE, et al. . Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell 2016;166(5):1308–1323.e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dong Z, Alterovitz G. netAE: semi-supervised dimensionality reduction of single-cell RNA sequencing to facilitate cell labeling. Bioinformatics 2021;37(1):43–9. [DOI] [PubMed] [Google Scholar]
- 102. Newman ME. Modularity and community structure in networks. Proc Natl Acad Sci U S A 2006;103(23):8577–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Xu C, Lopez R, Mehlman E, et al. . Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Mol Syst Biol 2021;17(1):e9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hadsell R, Chopra S, Le Cun Y: Dimensionality reduction by learning an invariant mapping. In: 2006 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR'06), 2006. 1735–42. IEEE. [Google Scholar]
- 105. Lieberman Y, Rokach L, Shay T. CaSTLe–classification of single cells by transfer learning: harnessing the power of publicly available single cell RNA sequencing experiments to annotate new experiments. PLoS One 2018;13(10):e0205499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Alavi A, Ruffalo M, Parvangada A, et al. . A web server for comparative analysis of single-cell RNA-seq data. Nat Commun 2018;9(1):4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yevshin I, Sharipov R, Valeev T, et al. . GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res 2016;45(D1):D61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wang YXR, Waterman MS, Huang HY. Gene coexpression measures in large heterogeneous samples using count statistics. P Natl Acad Sci USA 2014;111(46):16371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Krishnaswamy S, Spitzer MH, Mingueneau M, et al. . Systems biology. Conditional density-based analysis of T cell signaling in single-cell data. Science 2014;346(6213):1250689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kanehisa M, Furumichi M, Tanabe M, et al. . KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017;45(D1):D353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Fabregat A, Jupe S, Matthews L, et al. . The Reactome pathway knowledgebase. Nucleic Acids Res 2018;46(D1):D649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Huynh-Thu VA, Irrthum A, Wehenkel L, et al. . Inferring regulatory networks from expression data using tree-based methods. PLoS One 2010;5(9):e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Subramanian A, Tamayo P, Mootha VK, et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lotfollahi M, Wolf FA, Theis FJ. scGen predicts single-cell perturbation responses. Nat Methods 2019;16(8):715–21. [DOI] [PubMed] [Google Scholar]
- 115. Kang HM, Subramaniam M, Targ S, et al. . Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 2018;36(1):89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Duvenaud D, Maclaurin D, Iparraguirre J, et al. In: Cortes C, Lawrence ND, Lee DDet al. (eds). Advances in Neural Information Processing Systems 28, 2015, 2224–32.
- 117. Amodio M, Krishnaswamy S: MAGAN: Aligning biological manifolds. In: International Conference on Machine Learning, 2018. 215–23. PMLR. [Google Scholar]
- 118. Ghahramani A, Watt FM, Luscombe NM. Generative adversarial networks simulate gene expression and predict perturbations in single cells. bioRxiv 2018;262501. [Google Scholar]
- 119. Haber AL, Biton M, Rogel N, et al. . A single-cell survey of the small intestinal epithelium. Nature 2017;551(7680):333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hagai T, Chen X, Miragaia RJ, et al. . Gene expression variability across cells and species shapes innate immunity. Nature 2018;563(7730):197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Maseda F, Cang Z, Nie Q. DEEPsc: a deep learning-based map connecting single-cell transcriptomics and spatial imaging data. Front Genet 2021;12:636743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Musu Y, Liang C, Deng M. Clustering single cell CITE-seq data with a canonical correlation based deep learning method. bioRxiv 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhou Z, Ye C, Wang J, et al. . Surface protein imputation from single cell transcriptomes by deep neural networks. Nat Commun 2020;11(1):651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ramirez R, Chiu Y-C, Hererra A, et al. . Classification of cancer types using graph convolutional neural networks. Frontiers in Physics 2020;8(203). 10.3389/fphy.2020.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ramirez R, Chiu YC, Zhang S, et al. . Prediction and interpretation of cancer survival using graph convolution neural networks. Methods 2021;192:120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Battaglia PW, Hamrick JB, Bapst V, et al. . Relational inductive biases, deep learning, and graph networks arXiv preprint arXiv:180601261 . 2018.
- 127. Wang J, Ma A, Chang Y, et al. . scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses. Nat Commun 2021;12(1):1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Peng Y, Baulier E, Ke Y, et al. . Human embryonic stem cells extracellular vesicles and their effects on immortalized human retinal Muller cells. PLoS One 2018;13(3):e0194004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Stoeckius M, Hafemeister C, Stephenson W, et al. . Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 2017;14(9):865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. La Manno G, Gyllborg D, Codeluppi S, et al. . Molecular diversity of midbrain development in mouse, human, and stem cells. Cell 2016;167(2):566–580 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Azizi E, Carr AJ, Plitas G, et al. . Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018;174(5):1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Chu LF, Leng N, Zhang J, et al. . Single-cell RNA-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biol 2016;17(1):173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Baron M, Veres A, Wolock SL, et al. . A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst 2016;3(4):346–360.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Camp JG, Sekine K, Gerber T, et al. . Multilineage communication regulates human liver bud development from pluripotency. Nature 2017;546(7659):533–8. [DOI] [PubMed] [Google Scholar]
- 135. Muraro MJ, Dharmadhikari G, Grun D, et al. . A single-cell transcriptome atlas of the human pancreas. Cell Syst 2016;3(4):385–394.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Darmanis S, Sloan SA, Zhang Y, et al. . A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A 2015;112(23):7285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tirosh I, Venteicher AS, Hebert C, et al. . Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016;539(7628):309–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Patel AP, Tirosh I, Trombetta JJ, et al. . Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pollen AA, Nowakowski TJ, Shuga J, et al. . Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol 2014;32(10):1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Xin Y, Kim J, Okamoto H, et al. . RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab 2016;24(4):608–15. [DOI] [PubMed] [Google Scholar]
- 141. Yan L, Yang M, Guo H, et al. . Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol 2013;20(9):1131–9. [DOI] [PubMed] [Google Scholar]
- 142. Chevrier S, Levine JH, Zanotelli VRT, et al. . An immune atlas of clear cell renal cell carcinoma. Cell 2017;169(4):736–749.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Han X, Wang R, Zhou Y, et al. . Mapping the mouse cell atlas by microwell-Seq. Cell 2018;172(5):1091–1107.e17. [DOI] [PubMed] [Google Scholar]
- 144. Hrvatin S, Hochbaum DR, Nagy MA, et al. . Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neurosci 2018;21(1):120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Joost S, Zeisel A, Jacob T, et al. . Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst 2016;3(3):221–237.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Paul F, Arkin Y, Giladi A, et al. . Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 2015;163(7):1663–77. [DOI] [PubMed] [Google Scholar]
- 147. Buettner F, Natarajan KN, Casale FP, et al. . Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol 2015;33(2):155–60. [DOI] [PubMed] [Google Scholar]
- 148. Biase FH, Cao X, Zhong S. Cell fate inclination within 2-cell and 4-cell mouse embryos revealed by single-cell RNA sequencing. Genome Res 2014;24(11):1787–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Deng Q, Ramskold D, Reinius B, et al. . Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014;343(6167):193–6. [DOI] [PubMed] [Google Scholar]
- 150. Klein AM, Mazutis L, Akartuna I, et al. . Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015;161(5):1187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Goolam M, Scialdone A, Graham SJL, et al. . Heterogeneity in Oct4 and Sox2 targets biases cell fate in 4-cell mouse embryos. Cell 2016;165(1):61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kim JK, Kolodziejczyk AA, Ilicic T, et al. . Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression. Nat Commun 2015;6:8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Usoskin D, Furlan A, Islam S, et al. . Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015;18(1):145–53. [DOI] [PubMed] [Google Scholar]
- 154. Zeisel A, Munoz-Manchado AB, Codeluppi S, et al. . Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015;347(6226):1138–42. [DOI] [PubMed] [Google Scholar]
- 155. Yu Z, Liao J, Chen Y, et al. . Single-cell transcriptomic map of the human and mouse bladders. J Am Soc Nephrol 2019;30(11):2159–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Tusi BK, Wolock SL, Weinreb C, et al. . Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 2018;555(7694):54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Pijuan-Sala B, Griffiths JA, Guibentif C, et al. . A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 2019;566(7745):490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Cao J, Spielmann M, Qiu X, et al. . The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019;566(7745):496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Setty M, Tadmor MD, Reich-Zeliger S, et al. . Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat Biotechnol 2016;34(6):637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Nestorowa S, Hamey FK, Pijuan Sala B, et al. . A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016;128(8):e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Cao J, Packer JS, Ramani V, et al. . Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017;357(6352):661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Strehland A, Ghosh J. Cluster ensembles---a knowledge reuse framework for combining multiple partitions. J Mach Learn Res 2002;3:583–617. [Google Scholar]
- 163. McDaid AF, Greene D, Hurley N. Normalized mutual information to evaluate overlapping community finding algorithms arXiv preprint arXiv:11102515. 2011.
- 164. MacKay DJ, Mac Kay DJ. Information Theory, Inference and Learning Algorithms. Cambridge University Press, 2003. [Google Scholar]
- 165. Hubert L, Arabie P. Comparing partitions. J Classif 1985;2(1):193–218. [Google Scholar]
- 166. Buttner M, Miao Z, Wolf FA, et al. . A test metric for assessing single-cell RNA-seq batch correction. Nat Methods 2019;16(1):43–9. [DOI] [PubMed] [Google Scholar]
- 167. Rosenberg A, Hirschberg J. V-measure: a conditional entropy-based external cluster evaluation measure. In: Proceedings of the 2007 Joint Conference on Empirical Methods in Natural Language Processing and Computational Natural Language Learning (EMNLP-CoNLL), 2007. 410–20.