

Abstract
IDO2 is one of two closely related tryptophan catabolizing enzymes induced under inflammatory conditions. In contrast to the immunoregulatory role defined for IDO1 in cancer models, IDO2 has a proinflammatory function in models of autoimmunity and contact hypersensitivity. In humans, two common single nucleotide polymorphisms have been identified that severely impair IDO2 enzymatic function, such that less than 25% of individuals express IDO2 with full catalytic potential. This, together with IDO2’s relatively weak enzymatic activity, suggests that IDO2 may have a role outside of its function in tryptophan catabolism. To determine if the enzymatic activity of IDO2 is required for its proinflammatory function, we used newly generated catalytically inactive IDO2 knock-in mice together with established models of contact hypersensitivity and autoimmune arthritis. Contact hypersensitivity was attenuated in catalytically inactive IDO2 knock-in mice. In contrast, induction of autoimmune arthritis was unaffected by the absence of IDO2 enzymatic activity. In pursuing this non-enzymatic IDO2 function, we identified GAPDH, Runx1, RANbp10, and Mgea5 as IDO2-binding proteins that do not interact with IDO1, implicating them as potential mediators of IDO2-specific function. Taken together, our findings identify a novel function for IDO2, independent of its tryptophan catabolizing activity, and suggest this non-enzymatic function could involve multiple signaling pathways. These data show that the enzymatic activity of IDO2 is required only for some inflammatory immune responses and provide the first evidence of a non-enzymatic role for IDO2 in mediating autoimmune disease.
INTRODUCTION
Two tryptophan (Trp) catabolizing enzymes, IDO1 and IDO2, have unexpected connections to immunity. While extensive research has focused on the role of IDO1 in T cell suppression and cancer development (1–3), IDO2 has been found to have an alternative, pro-inflammatory role in immune responses. Previous work established that IDO2 acts in B cells to promote inflammation in preclinical models of autoimmunity and contact hypersensitivity (4, 5). However, the mechanistic and cellular relationships between tryptophan catabolism, IDO2, and disease remain unresolved.
Both IDO1 and IDO2, along with the enzyme tryptophan-2,3-dioxygenase (TDO), can catabolize the first and rate-limiting step in the breakdown of the amino acid L-Trp to N-formylkynurenine (kyn). In comparison with IDO1 and TDO, however, IDO2 has relatively weak catalytic activity (6, 7). Indeed, IDO1 knockout (ko) mice, in which expression of the protein has been genetically disrupted, show a marked reduction in serum kyn, whereas IDO2 ko mice show no change in overall kyn levels, suggesting that IDO2 contributes little to the overall Trp catabolism, even in the context of inflammation (8). IDO2 may, in fact, catabolize some Trp derivatives such as 5-methoxytryptophan, with greater efficiency than Trp itself (9). The lack of association between IDO2 and overall kyn production suggests that IDO2 may be acting to catabolize Trp or Trp derivatives in only a small subset of cells or tissues, or that it may in fact have another, potentially non-enzymatic, role entirely. Non-enzymatic functions have been defined for IDO1, particularly via a TGF-β mediated process allowing a stable immune regulatory phenotype to be maintained in plasmacytoid dendritic cells (pDCs). This process is mediated by phosphorylation of two ITIM motifs in IDO1, triggering downstream processes affecting protein stability and affecting the balance between canonical and non-canonical NF-κB signaling (10–12).
There are two common Ido2 single nucleotide polymorphisms (SNPs) found in the human population that severely impair enzymatic function (6). The first (rs10109853), an Arginine to Tryptophan mutation at amino acid position 248 in human (235 in mouse), is located at a position analogous to R231 in human IDO1 that is critical for dioxygenase activity and may be directly involved in substrate recognition through hydrophobic interactions (13). The second (rs4503083) is a nonsense mutation, changing a Tyrosine to a stop codon at position 359 in human (346 in mouse), immediately prior to a conserved histidine residue known to be essential for catalytic activity in IDO1 (13, 14). Surprisingly, both inactivating polymorphisms are common in human populations, such that only ~25% of normal individuals appear to have a homozygous wild-type configuration of the gene conferring full catalytic potential (6). Both of these SNPs have been examined in the context of human disease and have been associated with multiple myeloma, pancreatic cancer, and aspergillosis in susceptible patients but not with multiple sclerosis or Crohn’s disease (15–21).
The prevalence of enzyme-inactivating IDO2 polymorphisms in the human population, together with the lack of effect of IDO2 deletion on serum kyn levels in mice, suggests that IDO2 may have functions outside of its catalytic activity. To directly examine the contribution of IDO2 enzyme activity to inflammatory immune responses, we generated IDO2 knock-in (ki) mice incorporating each of the two SNPs that catalytically impair IDO2 (R235W or Y346X). Given our previous findings defining the relationship between IDO2 and the inflammatory responses mediating autoimmune arthritis and contact hypersensitivity, we used these models to examine whether the enzymatic function of IDO2 was necessary for its proinflammatory effect.
MATERIALS AND METHODS
Mice.
KRN TCR transgenic C57BL/6 (22) and IDO2 deficient (IDO2 ko) (23) mice on a C57BL/6 background have been described. CRISPR/Cas9-based gene editing (performed by The Jackson Laboratory) was employed to introduce point mutations into the mouse Ido2 gene, which recapitulate the enzyme-inactivating C/T polymorphism at position 235 that introduces an R to W amino acid change in exon 8 and C/A polymorphism at position 346 that introduces a stop codon in exon 10. Sequences used for the CRISPR guides: Ido2ex8_sgRNA1: CTTACCCAGAGAGGAAGATC; Ido2ex8_sgRNA2: GTCATCCGGATCTTCCTCTC; Ido2ex8_sgRNA3: TCATCCGGATCTTCCTCTCT; Ido2ex10_sgRNA1: ATCTGGCCACGACATTGATGTGG; Ido2ex10_sgRNA2: CAGTTACCACATCAATGTCGTGG. Two founder lines on a C57BL/6 background were established for each polymorphism. Point mutations were confirmed by sequencing of genomic DNA using the primers 5’-CATACGGAACGGATTAACAGC-3’ and 5’-TTTGCCCCTTTTGGATTTAAG-3’ for sequencing of the R235W polymorphism and 5’-ATTTCCATGTGGAGCTATGACG-3’ and 5’-ACCACGTGGGTGAAGGATTG-3’ for the Y346X polymorphism. One founder line for each polymorphism was bred to homozygosity and expanded for further analysis.
Arthritic mice were generated by breeding KRN Tg C57BL/6 mice expressing the I-Ag7 MHC Class II molecule (KRN.g7). This process was repeated to generate arthritic mice homozygous for IDO2 ko, R235W, or Y346X. Homozygosity of the R235W and Y346X lines were confirmed by sequencing. KRN.g7 mice develop arthritis with similar kinetics as the original K/BxN mice (24). Both male and female mice were used in all experiments. Control and experimental groups were bred in-house and co-housed (when possible) under specific pathogen free conditions in the animal facility at the Lankenau Institute for Medical Research. Studies were performed in accordance with National Institutes of Health and Association for Assessment and Accreditation of Laboratory Animal Care guidelines with approval from the LIMR Institutional Animal Care and Use Committee.
T cell adoptive transfer arthritis.
CD4+ T cells from the spleens and lymph nodes (LN) of IDO2 ko KRN TCR tg B6 mice were purified by positive selection with anti-CD4 mouse MACS microbeads (Miltyeni Biotec). The elutant was purified over a second column to achieve higher purity (~90%). B cells from spleens of IDO2 wt, IDO2 ko, R235W, or Y346X I-Ag7/b mice were purified by anti-CD43 negative selection with MACS beads (Miltenyi). B cell purity was routinely ~97%. Following purification, 3.5×105 CD4+ T cells and 1×106 B cells were adoptively transferred i.v. into TCRα ko IDO2 ko B6g7/b hosts. Arthritis was measured starting at the day of cell transfer, as described below. Mice were sacrificed after 2 weeks.
Arthritis incidence.
The two rear ankles of KRN.g7, IDO2 ko KRN.g7, R235W KRN.g7, and Y346X KRN.g7 mice were measured starting at weaning (3 wk of age). Measurement of ankle thickness was made above the footpad axially across the ankle joint using a Fowler Metric Pocket Thickness Gauge. Ankle thickness was rounded off to the nearest 0.05mm. At 6wk of age, the rear paws were fixed in 10% buffered formalin for 48h, decalcified in 14% EDTA for 2wk, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Histology sections were imaged using a Zeiss Axioplan microscope with a Zeiss Plan-Apochromat 10x/0.32 objective and Zeiss AxioCam HRC camera using AxioVision 4.7.1 software. The images were then processed using Adobe Photoshop CC software.
ELISpot assay.
Anti-GPI antibody secreting cells were measured by ELISpot as described (25). Briefly, cells from the joint draining lymph nodes (axillary, brachial, and popliteal LNs) from 6 week-old KRN.g7, IDO2 ko KRN.g7, R235W KRN.g7, and Y346X KRN.g7 mice were plated at 4 × 105 cells per well and diluted serially 1:4 in Multiscreen HA mixed cellulose ester membrane plates (Millipore) coated with histidine (his)-tagged glucose-6-phosphate isomerase (GPI) (10μg/ml). The cells were incubated on the GPI-coated plates for 4hr at 37°C. The Ig secreted by the plated cells was detected by Alkaline Phosphatase-conjugated goat anti-mouse total Ig secondary Ab (Southern Biotechnology Associates) and visualized using NBT/BCIP substrate (nitroblue tetrazolium / 5-bromo-4-chloro-3-indolyl phosphate; Sigma).
Contact Hypersensitivity.
IDO2 ko, R235W, Y346X, and wt BALB/c mice were sensitized with 3% oxazolone (Sigma) in 100% ethanol on their shaved abdomen (100μl) and hind footpads (10μl each) 5 days prior to the start of the experiment. Five days later, mice were elicited with 20μl of 1% oxazolone in 100% ethanol, 10μl on each side of the ears. After 24h, ear thickness was measured using a dial gauge (Fowler) and harvested ears were fixed in 10% buffered formalin, embedded in paraffin, sectioned, and stained with H&E. Histology sections were imaged using a Zeiss Axioplan microscope wth a Zeiss Plan-Apochromat x10/0.32 objective and Zeiss AxioCam HRC camera using AxioVision 4.7.1 software. The images were then processed using Adobe Photoshop CC software. Trials performed in multiple mice were replicated at least 3 times.
Generation of R235W, Y346X, and wt IDO2 transfected cell lines.
A plasmid (pReceiver-M08) containing the open reading frame of murine IDO2 (mIDO2) under the control of the CMV promoter was purchased from GeneCopoeia (Mm32442-M08). A 98bp fragment containing V5 and 6-his tags was inserted at the 3’ end of mIDO2, by insertion at the Not1 and Nhe1 sites in the pReceiver-M08 vector, to make wild-type (wt) mIDO2-V5his. Plasmids containing the R235W and Y346X polymorphisms were generated by site-directed mutagenesis of wt mIDO2-V5his using inverse PCR and In-Fusion® cloning technology (Takara Bio) following manufactures’ instructions. Primers used for inverse PCR were R235W: 5’-GGTCATCTGGATCTTCCTCTCTGGGTGGA-3’ and 5’-AAGATCCAGATGACCGAGTAAAATATGTCTGGG-3’, Y346X: 5’- GCGCAGTTAACACATCAATGTCGTGGCCAG-3’ and 5’- ATGTGTTAACTGCGCAGCTCTCCCAG-3’. To express the tagged Y346X truncated protein, the 0.2kb region between the new stop codon and the V5-his tag was removed. Correct mutations and tags were confirmed by sequencing. Wt, R235W, and Y346X IDO2 protein and V5 tag expression were confirmed by transient transfection into Human 293-T-REx™ cells, followed by Western blotting. Stable expression cell lines were generated by selection with 400μg/ml G418. Single high-expressing lines were subcloned by sorting single cells on a BD FACSAria III cell sorter.
Yeast Two-Hybrid Analysis.
Yeast two-hybrid screening was performed by Hybrigenics Services, S.A.S., Paris, France (http://www.hybrigenics-services.com). The coding sequence for the full length of Mus musculus - Ido2 (aa 1–405) (GenBank accession number gi: 170763487; https://www.ncbi.nlm.nih.gov/nuccore/170763487 [ncbi.nlm.nih.gov]) was PCR-amplified and cloned into pB27 as a C-terminal fusion to LexA (N-LexA-Ido2-C). The construct was checked by sequencing the entire insert and used as a bait to screen a random-primed mouse spleen cDNA library constructed into pP6. pB27 and pP6 derive from the original pBTM116 (26) and pGADGH (27) plasmids, respectively. 77.2 million clones (7-fold the complexity of the library) were screened using a mating approach with YHGX13 (Y187 ade2–101::loxP-kanMX-loxP, matα) and L40ΔGal4 (mata) yeast strains as previously described (28). 37 His+ colonies were selected on a medium lacking tryptophan, leucine and histidine. The prey fragments of the positive clones were amplified by PCR and sequenced at their 5’ and 3’ junctions. The resulting sequences were used to identify the corresponding interacting proteins in the GenBank database (NCBI) using a fully automated procedure. A confidence score (PBS, for Predicted Biological Score) was attributed to each interaction as previously described (29). The scores were divided into four categories, from A (highest confidence) to D (lowest confidence). The PBS scores have been shown to positively correlate with the biological significance of interactions (30, 31).
Co-immunoprecipitation (IP) and Western Blotting.
Livers were harvested from IDO2 ko, R235W, Y346X, or wt C57BL/6 mice. Human 293-T-REx™ cells stably transfected with V5-tagged IDO1 or IDO2 in a pcDNA4/TO vector (Invitrogen) were described previously (6). 293-T-REx™ cells expressing IDO1, wt IDO2, R235W IDO2, Y346X IDO2, or untransfected controls were transiently transfected with DDK/FLAG-tagged GAPDH, Runt-related transcription factor 1 (Runx1), RAN binding protein 10 (RANbp10), meningioma expressed antigen 5 (Mgea5) (Origene) or pyruvate kinase (PK) (32) (Addgene). IDO1 and wt IDO2 expression were induced with 2μg/ml doxycycline for 48 hours. Livers and transfected cells were lysed with RIPA buffer (Pierce) or IP lysis buffer (Thermo Scientific). For co-IP, IDO2 was immunoprecipitated with an anti-V5 antibody (T-REx cells) or rabbit anti-IDO2 (liver lysates) and Immunoprecipitation kit Dynabeads protein G (Thermo Scientific). For straight Western blots, whole cell lysates were run without prior IP. Protein was fractioned using standard SDS-PAGE and blotted to Immobilon-NC membranes (Millipore, USA). After blocking, blots were incubated at 4°C overnight with HRP or unconjugated primary antibodies. When needed, blots were then incubated 2h with HRP-conjugated secondary antibodies. Blots were developed with HYGLO Quickspray chemiluminescent HRP reagent (Denville Scientific) and analyzed using a ChemiDoc System with Image Lab Software (Biorad). IP antibodies to V5 (Invitrogen) or IDO2 (generated to peptide IDO2365-378 by GenScript) and primary antibodies to the following antigens were used: FLAG/DDK (BioLegend), V5 (Invitrogen); IDO2 (Santa Cruz); and α-tubulin (Biorad). HRP-conjugated anti-mouse Igκ (Jackson Immunoresearch) was used as a secondary antibody to detect α-tubulin.
Flow Cytometry.
Human 293-T-REx™ cells stably transfected with V5-tagged wild-type (wt) IDO2, R235W, or Y346X or untransfected controls (parental) were fixed in IC Fixation buffer (Invitrogen), permeabilized with 0.2% Tween-20, and stained with anti-V5-FITC (Invitrogen). Samples were gated for live cells using forward and side scatter. The samples were acquired on a BD FACSCanto II flow cytometer using FACSDiva Software and analyzed using FlowJo Software.
In vivo Kynurenine Assay.
Human 293-T-REx™ cells were stably transfected with V5-tagged wt IDO2, R235W, or Y346X or untransfected controls (parental). Cells were cultured in IMDM media with 10% FCS, 50μM 2-mercaptoethanol, 2mM glutamax (Gibco), and 50μg/ml gentamycin with the addition of 2mM L-tryptophan, 10μM hemin, and 4mM 5-aminolevulinic acid. Wt IDO2 expression in 293-T-REx™ cells was induced with 2μg/ml doxycycline for 48 hours. Harvested supernatants were analyzed for kynurenine levels using an Ehrlich’s Assay, as described (33, 34). Briefly, 200μl culture supernatant was incubated with 50μl 50% TCA for 30 minutes at 56°C to hydrolyze N-formylkynurenine to kynurenine. After centrifugation at 16,000 rpm for 10 min, 100μl of this supernatant was mixed with 100μl of freshly prepared Ehrlich reagent (2% 4-(dimethyamino) benzaldehyde w/v in glacial acetic acid), incubated 5 min at room temperature, and read at 490nm. Kynurenine levels were calculated by comparison to a standard curve.
In vitro Kynurenine Assay.
Human 293-T-REx™ cells, stably transfected with V5-tagged wt IDO2, R235W, or Y346X or untransfected controls (parental) were cultured in IMDM media with 10% FCS, 50μM 2-mercaptoethanol, 2mM glutamax (Gibco), and 50μg/ml gentamycin with the addition of 2mM L-tryptophan, 10μM hemin, and 4mM 5-aminolevulinic acid. Lysates containing IDO2 proteins were generated by cytolysis by resuspension of cells in purified, nuclease-free water at a ratio of 1×107 cells/ml. Lysates were subjected to 3 freeze-thaw cycles and 30s sonication to disrupt cell membranes, then spun at maximum speed 10 min at 4°C immediately prior to use to remove cellular debris. Cell lysates were immunoblotted with anti-V5 antibody to normalize IDO2 levels. Equal amounts of IDO2 were then assayed for tryptophan catabolic activity directly from this cell lysate by means of an in vitro enzymatic reaction to convert tryptophan to N-formylkynurenine in a mixture containing a final concentration of 50mM potassium phosphate, pH=6.5, 40mM sodium L-ascorbate, 400μg/ml catalase, 20μM methylene blue, and 2mM L-tryptophan for 1 h at 37°C. Products of this reaction were tested for kynurenine using the IDK® Kynurenine high sensitive ELISA (Immundiagnostik). Kynurenine levels were calculated by comparison to a standard curve.
Cycloheximide chase assay.
Human 293-T-REx™ cells stably transfected with V5-tagged wt R235W, or Y346X IDO2 were cultured with 300μg/ml cycloheximide (Sigma). Cells were harvested at 0, 3, 6, or 9 hours and lysed with RIPA buffer. Protein was fractionated using standard SDS-PAGE and blotted to Immobilon-NC membranes. After blocking, the blots were incubated with anti-V5-HRP (Invitrogen) and anti-GAPDH-Rhodamine (BioRad). Blots were developed with HYGLO Quickspray chemiluminescent HRP reagent (Denville Scientific) and analyzed using a ChemiDoc System with Image Lab Software (Biorad).
Statistical Analysis.
Statistical significance was determined using one or two way-ANOVA followed by comparison of means with Tukey’s post-hoc multiple comparison correction, an unpaired Student’s t test, or the Mann-Whitney nonparametric test using GraphPad Prism Software (GraphPad Software, Inc).
RESULTS
Generation of enzymatically inactive IDO2 knock-in mice
To determine if the tryptophan catabolizing activity of IDO2 was necessary for its function, we generated two new lines of enzymatically inactive IDO2 knock-in (ki) mice. CRISPR/Cas9 was used to introduce point mutations into the mouse IDO2 gene to recapitulate the enzyme-inactivating C/T polymorphism at position 235 that introduces an R to W amino acid change in exon 8 (R235W, Fig. 1A) and C/A polymorphism at position 346 that introduces a stop codon in exon 10 (Y346X, Fig. 1B). Successful knock-in of the Ido2 polymorphisms was confirmed by sequencing of genomic DNA (Fig. 1C,D).
Figure 1. Generation of catalytically inactive IDO2 polymorphism knock-in mice.

Point mutations were introduced into the mouse IDO2 gene using CRISPR/Cas9 to recapitulate the enzyme-inactivating (A) C/T polymorphism at position 235 that introduces an R to W amino acid change in exon 8 and (B) C/A polymorphism at position 346 that introduces a stop codon in exon 10. Sequences for the CRISPR guides are indicated. Successful knock-in of the Ido2 polymorphisms were confirmed for both founder and experimental mice by sequencing of genomic DNA. Representative traces for mice heterozygous and homozygous for (C) R235W and (D) Y346 polymorphisms are shown.
To verify lack of enzymatic activity, wild-type (wt), R235W, and Y346X mouse IDO2 V5-tagged constructs were stably transfected into 293-T-REx™ (T-REx) cells. IDO2 expression was measured by staining for the V5 tag by flow cytometry (Fig. 2A). R235W IDO2 expressed a similar level of IDO2 protein compared to wt IDO2; however, the level of Y346X IDO2 was significantly reduced compared to both wt and R235W IDO2 (Fig. 2A,B). To measure IDO2 enzymatic activity, the downstream catabolite kynurenine was measured in the supernatant using an Ehrlich’s Assay. Although reduced compared to IDO1, wt IDO2 does exhibit tryptophan catabolizing activity (5–7) that can be enhanced by the addition of elevated concentrations of tryptophan, hemin, and 5-aminolevulinic acid ((35) and Fig. 2C). In contrast, kynurenine was undetectable in cells expressing either the R235W or Y346X IDO2 polymorphism, even when normalized for IDO2 expression (Fig. 2B). Lack of tryptophan catabolizing activity of R235W and Y346X IDO2 enzymes was confirmed using an in vitro enzyme assay (Fig. 2D,E).
Figure 2. R235W and Y346X IDO2 are enzymatically inactive.
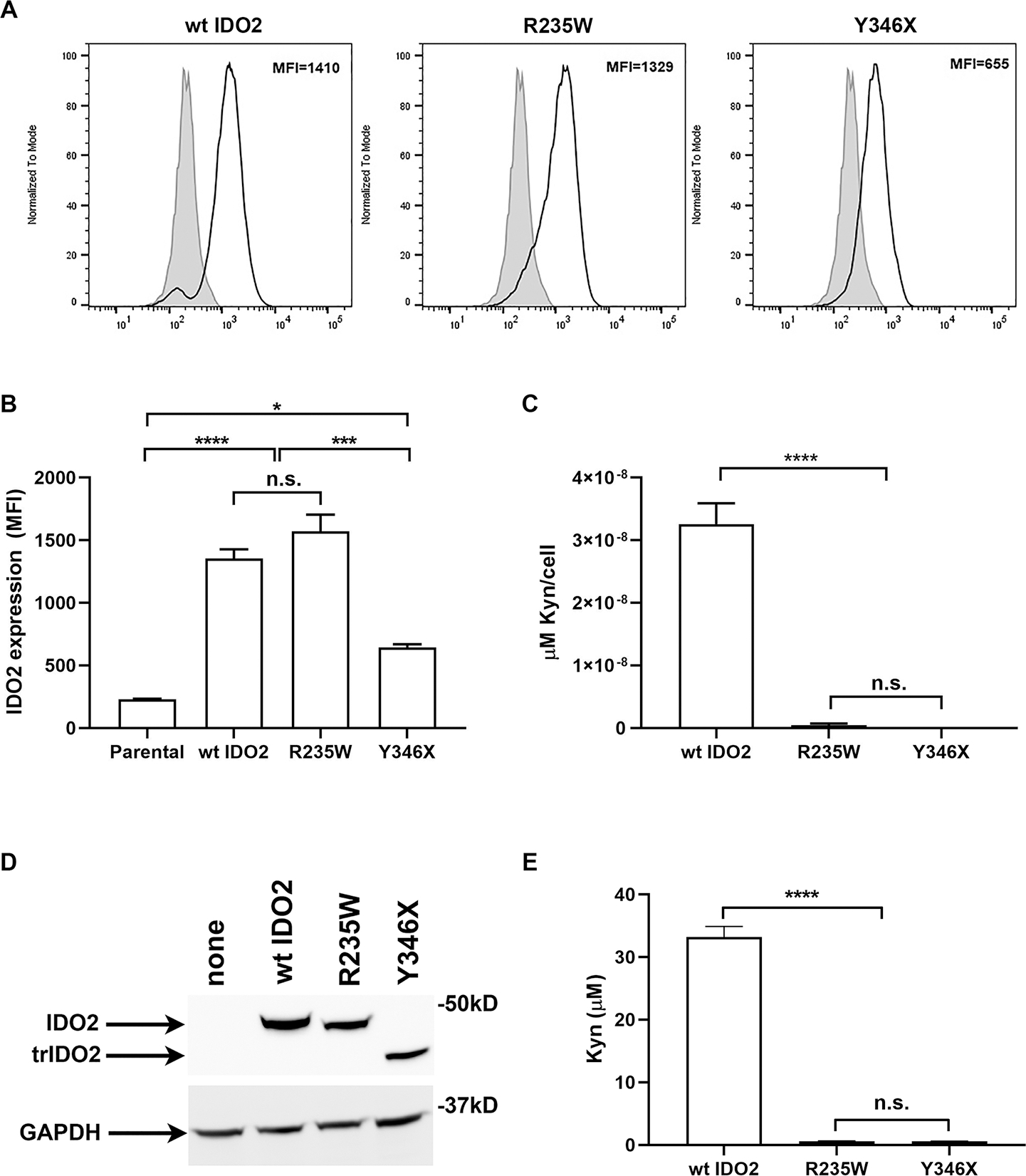
293-T-REx™ (Trex) cells were stably transfected with V5-tagged wt, R235W, or Y346X IDO2. (A) IDO2 expression in the transfected cells was measured by staining for the V5 tag using intracellular flow cytometry. Representative FACS plots show IDO2 levels in transfected (bold line) compared to untransfected Trex cells (filled histogram). (B) Graph shows the mean IDO2 expression ± SEM for n=3 each. (C) Kynurenine (Kyn) in the supernatants were measured by Ehrlich’s Assay. Graph indicates the mean amount of Kyn, normalized for IDO2 expression per cell, ± SEM for n=3 each. (D) Cell lysates were immunoblotted with V5 antibody to normalize IDO2 levels. GAPDH was used as a loading control. (E) Equal amounts of IDO2 were evaluated for tryptophan catabolic activity using an in vitro enzymatic assay. Kyn production was measured by ELISA. Graph shows mean amount of Kyn ± SD from n=3 each. *p<0.05, ***p<0.001, ****p<0.0001, n.s., not significant.
Mutations that impair IDO2 enzymatic activity do not attenuate arthritis
Previously, we showed that IDO2 played an important role in driving autoantibody production and joint inflammation in the KRN.g7 preclinical model of inflammatory arthritis (8). To determine if the enzymatic activity of IDO2 was necessary for its pro-arthritic effect, we bred the R235W and Y346X IDO2 ki alleles onto the KRN.g7 model and compared them to KRN.g7 mice expressing (wt) or lacking IDO2 (IDO2 ko). KRN.g7 mice expressing wt IDO2 developed robust arthritis between 3 and 4 weeks of age, whereas arthritis in IDO2 ko KRN.g7 mice was delayed in time of onset and reduced in overall severity ((8) and Fig. 3A,B). R235W KRN.g7 mice developed robust arthritis, with timing and severity indistinguishable from KRN.g7 mice expressing wt IDO2. In contrast, joint inflammation in Y346X KRN.g7 mice was reduced, similar to that seen in IDO2 ko KRN.g7 mice (Fig. 3A,B). Histological analysis of ankles demonstrated extensive immune cell infiltration, synovial hyperplasia, and cartilage erosion in R235W and IDO2 wt KRN.g7 mice and reduced in Y346X and IDO2 ko KRN.g7 mice, confirming the clinical analysis of joint inflammation (Fig. 3C).
Figure 3. IDO2 enzymatic activity is not required for joint inflammation or autoantibody production in KRN.g7 model of autoimmune arthritis.
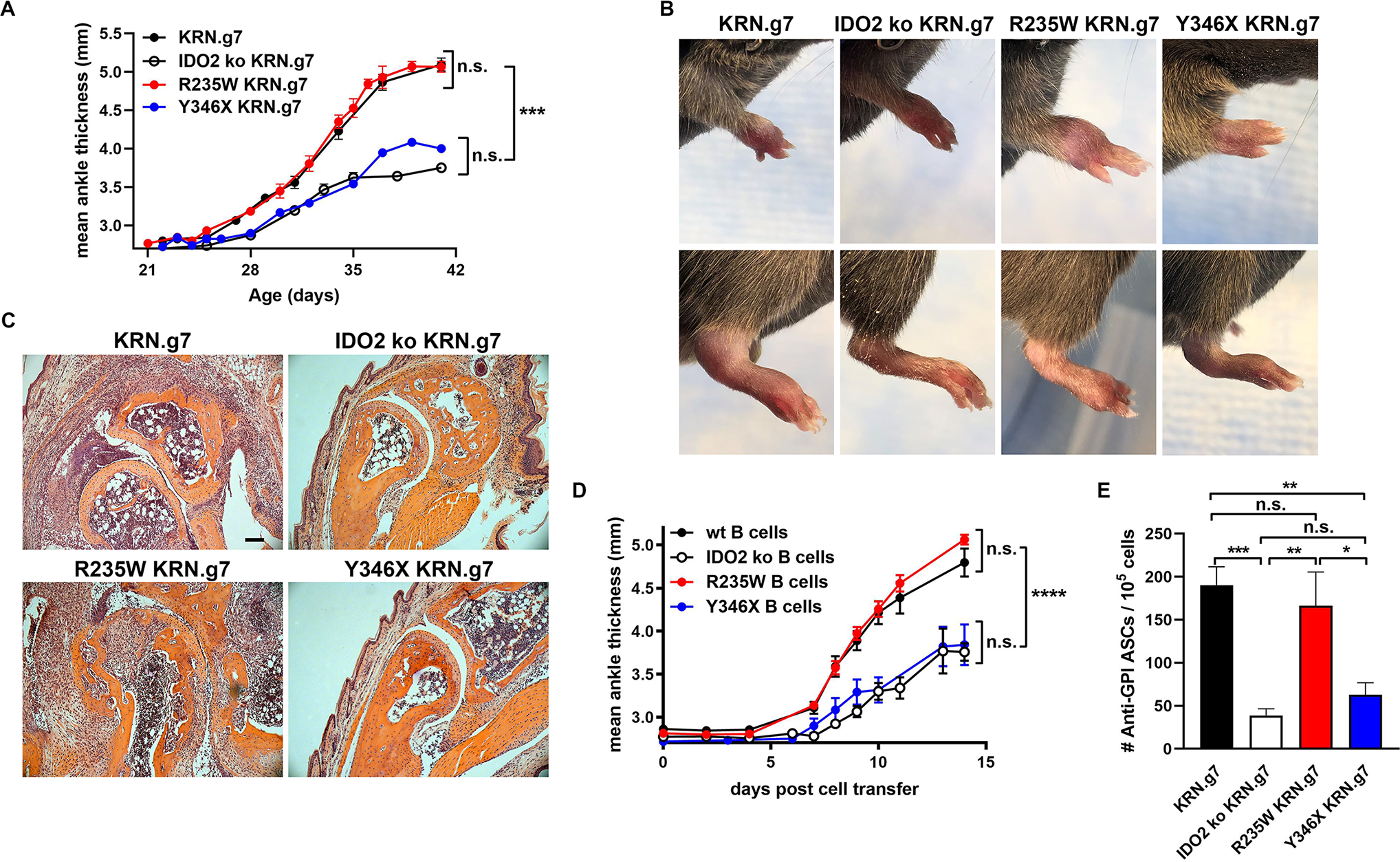
(A) Rear ankles were measured as an indication of arthritis and represented as the mean change in ankle thickness ± SEM from n=12 KRN.g7, n=14 IDO2 ko KRN.g7, n=9 R235W KRN.g7, and n=8 Y346X KRN.g7 mice, pooled from 4 independent litters for each genotype. At 6 wk of age, (B) pictures were taken of the front and rear paws and (C) rear paws were sectioned and stained with hematoxylin and eosin. Representative images from a total of n=8 mice per genotype. Scale bar = 100μm. (D) Arthritis was induced by adoptively transferring KRN IDO2 ko T cells into IDO2 ko TCRα ko B6g7/b mice together with B cells purified from B6g7/b (wt), IDO2 ko B6g7/b (IDO2 ko), R235W B6g7/b (R235W), or Y346X B6g7/b (Y346X) mice. Arthritis is represented as mean ankle thickness ± SEM. Data is from n=7 wt, n=7 IDO2 ko, n=14 R235W, and n=5 Y346X B cell transfers. (E) The number of anti-GPI ASCs from the joint dLNs was determined using an ELISpot assay. Data shows the mean number of ASCs ± SEM for n=29 KRN.g7, n=14 IDO2 ko KRN.g7, n=14 R235W KRN.g7, and n=14 Y346X KRN.g7 mice. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s., not significant.
Using the T cell transfer model of arthritis and a cell “add-back” approach, we previously identified B cells as the critical IDO2-expressing cell mediating arthritis in the KRN.g7 model (4). To determine if IDO2 in B cells mediated arthritis through this non-enzymatic mechanism, we again used this T cell transfer model of arthritis. Purified populations of B cells isolated from IDO2 wt, ko, R235W, or Y346X C57BL/6g7/b mice were adoptively transferred into TCRα ko IDO2 ko C57BL/6g7/b mice and arthritis induced with IDO2 ko KRN T cells. Consistent with our findings in the spontaneous arthritis model, B cells from R235W mice induced robust arthritis; whereas B cells from Y346X mice induced only attenuated arthritis, similar to that generated by transfer of IDO2 wt and ko B cells, respectively (Fig. 3D). Taken together, these data demonstrate that IDO2’s enzymatic function is not required to drive B cell-mediated arthritis.
Mutations that impair IDO2 enzymatic activity do not attenuate autoreactive B cell responses
Arthritis in the KRN.g7 model is dependent on autoreactive B and T cell responses directed against the autoantigen glucose-6-phosphate isomerase (GPI) (36). Previous work from our lab demonstrated that GPI-specific B and T cell responses were reduced in IDO2 ko KRN.g7 mice (8). Subsequent studies using IDO1/2 double ko mice showed that the diminished autoreactive B cell responses were directly dependent on IDO2, whereas the effect on autoreactive T cell responses was an indirect effect of altered IDO1 expression in the IDO2 ko mice (5). To determine if IDO2’s enzymatic function affects the autoreactive B cell response, the number of anti-GPI autoantibody secreting cells (ASCs) was evaluated (Fig. 3E). Large numbers of anti-GPI ASCs were found in R235W KRN.g7 mice. In contrast, Y346X mice produced reduced numbers of anti-GPI ASCs, similar the level found in IDO2 ko mice. These data are consistent with the development of robust vs. attenuated arthritis in R235W and Y346X KRN.g7 mice, respectively, and demonstrate that the autoreactive B cell response in KRN.g7 mice is not dependent on IDO2’s enzymatic activity.
Reduced stability of truncated Y346X IDO2 protein
The large autoantibody response and robust arthritis that developed in the R235W KRN.g7 mice demonstrated that the enzymatic activity of IDO2 was not required for its pro-arthritic effect. However, mice expressing the Y346X polymorphism developed reduced autoantibodies and attenuated arthritis more consistent with IDO2 genetic deficiency. One potential reason for this discrepancy is that the truncated protein may be unstable and rapidly degraded, as suggested by the reduced expression in our transfected cells (Fig. 2). Consistent with this, IDO2 protein was undetectable in liver tissue from Y346X mice. IDO2 protein was detectable in livers from R235W mice, although reduced compared to wt controls (Fig. 4A,B). To measure the relative stability of the full length R235W and truncated Y346X IDO2 proteins compared to wt IDO2, we performed a cycloheximide chase assay using T-REx cells stably transfected with wt, R235W, or Y346X IDO2. Cells were incubated with cycloheximide to inhibit protein synthesis and the amount of IDO2 remaining at 3, 6, and 9 hours was determined by Western blotting (Fig. 5A). During the first 3 hours, wt and R235W IDO2 protein levels decreased approximately 30%, but remained stable over the next 6 hours (9 hours total). In contrast, Y346X protein levels decreased by 80% at 3 hours and >95% by 9 hours (Fig. 5B). These data demonstrate that the truncated Y346X protein is rapidly degraded and, together with the lack of IDO2 protein in Y346X mice, suggest that the difference in arthritis severity between the R235W and Y346X mice can be explained by reduced stability of the Y346X protein.
Figure 4. Truncated IDO2 protein absent in Y346 IDO2 ki mice.
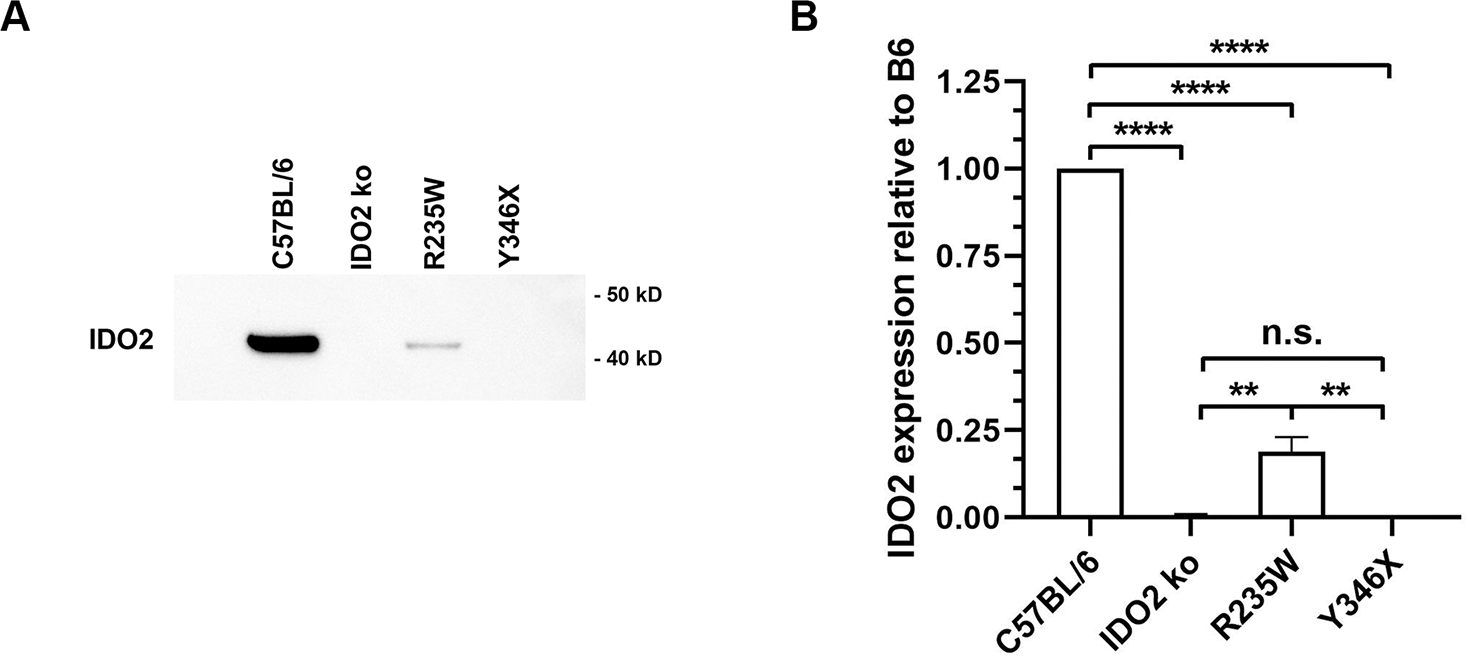
Whole liver lysates from IDO2 ko, R235W, Y346X, or wt C57BL/6 mice were immunoprecipitated with rabbit anti-IDO2 and then immunoblotted with HRP-conjugated mouse anti-IDO2. (A) A representative blot from total of 3 independent experiments is shown. (B) Graph shows the mean IDO2 protein expression relative to the C57BL/6 control SEM for n=3 samples per genotype. **p<0.01, ****p<0.0001, n.s., not significant
Figure 5. Reduced stability of truncated Y346X IDO2 protein.
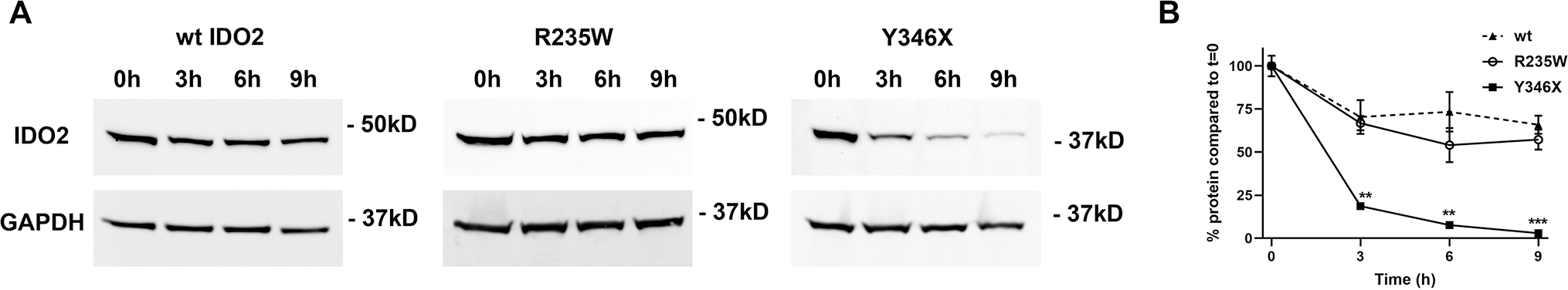
293-T-REx™ cells were stably transfected with V5-tagged wt, R235W, or Y346X IDO2. IDO2-transfected cells were cultured in the presence of cycloheximide to inhibit protein synthesis. (A) The cells were harvested after 0, 3, 6, or 9 hours and levels of IDO2 expression in the protein lysates were measured by Western blotting. GAPDH was used as a loading control. (B) Graph shows the percentage of IDO2 expression relative to time 0 for n=3 samples for each cell line/time point. **p<0.01, ***p<0.001
Polymorphisms that impair IDO2 enzymatic activity inhibit contact hypersensitivity
The finding that IDO2’s enzymatic function was not involved in its proinflammatory role driving arthritis led us to ask whether IDO2’s enzymatic function was required to drive inflammation in any context. A second model where inflammation has been shown to be dependent on IDO2 is the contact hypersensitivity (CHS) model (23). To determine if IDO2 enzymatic function was required for CHS, we sensitized wt, IDO2 ko, R235W, and Y346X BALB/c mice with oxazolone on the footpads and abdomen, challenged them 5 days later with oxazolone on the ears, and then measured swelling after 24 hours. Consistent with our previously published data, wt mice showed marked swelling, whereas the CHS response in IDO2 ko mice was significantly reduced ((23) and Fig. 6). In contrast to their robust inflammatory response in the KRN.g7 arthritis model, R235W mice had a greatly diminished CHS inflammatory response, similar to the reduced response in IDO2 ko and Y346X mice (Fig. 6A). Histological analysis confirmed the minimal inflammatory cell infiltration and swelling in the ears from R235W, Y346X, and IDO2 ko mice compared to the robust infiltration of inflammatory cells and swelling seen in wt mice (Fig. 6B). Therefore, despite a clear non-enzymatic function for IDO2 in the KRN.g7 arthritis model, IDO2’s enzymatic activity does play an important role in mediating inflammation in the context of CHS.
Figure 6. Polymorphisms that impair IDO2 enzymatic activity inhibit contact hypersensitivity.
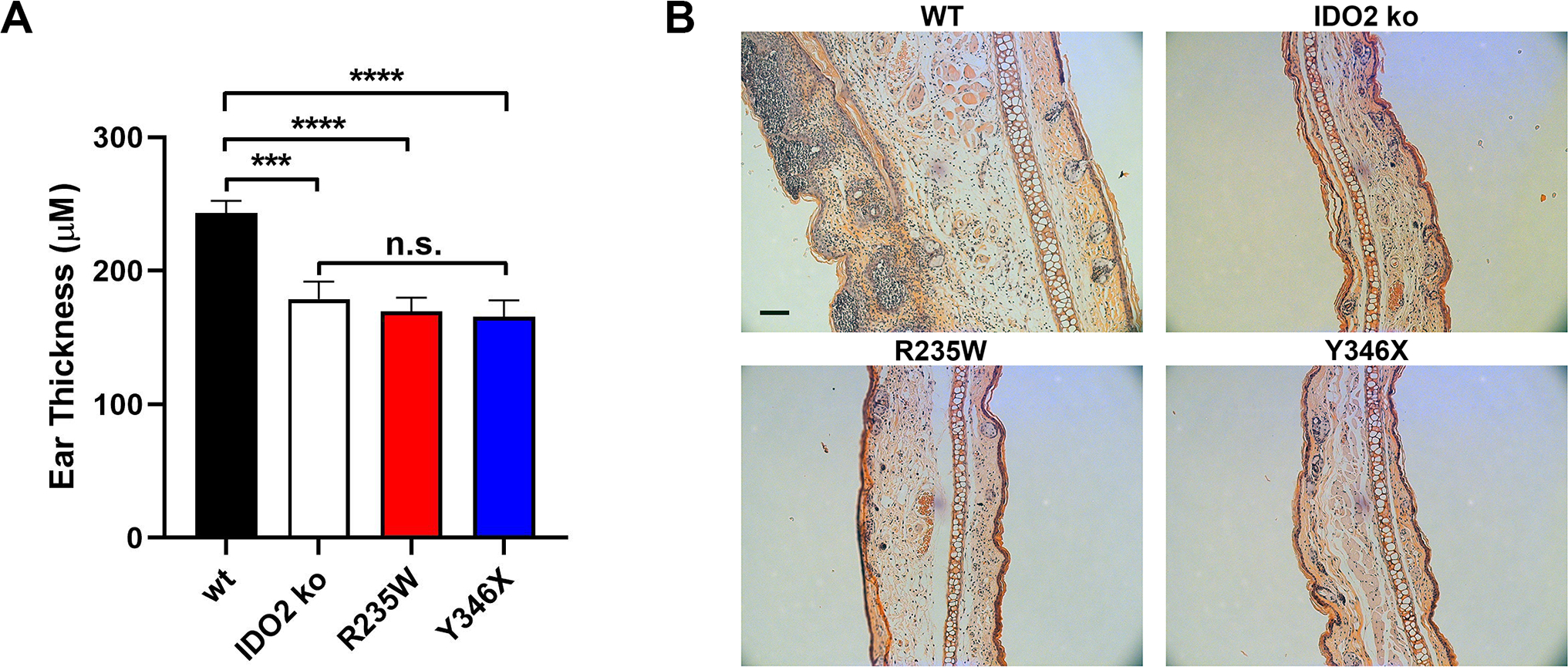
Wt, IDO2ko, R235W, and Y346X BALB/c mice were sensitized with oxazolone. Five days later, a CHS response was elicited by painting of oxazolone on the ears. (A) Graphs show the change in mean ear thickness ± SEM 24 h after elicitation, relative to ear thickness prior to elicitation. n=20 wt, n=13 IDO2 ko, n=13 R235W and n=9 Y346X mice. ***p<0.001, ****p<0.0001, n.s., not significant. (B) 24 hr post-elicitation, ears were sectioned and stained with hematoxylin and eosin. Representative images are shown from a total of n=5 mice per genotype. Scale bar = 100μm.
IDO2 interacts with multiple unique binding partners
Our data suggest that in addition to tryptophan catabolism, IDO2 also has a non-enzymatic function that mediates its proinflammatory activity in arthritis. As a first step to determine the molecular signaling pathway mediated by IDO2, we performed a yeast two-hybrid screen using full-length mouse IDO2 as bait to screen a random-primed mouse spleen cDNA library (Hybrigenics Services; Paris, France) to identify proteins that directly interact with IDO2 (28). From a total of 77.2 million interactions, 37 positive clones were isolated and sequenced to identify 4 unique proteins (Table I). To confirm the interaction between IDO2 and these candidate binding proteins, T-REx cells stably expressing V5-tagged IDO2 were transfected with DDK/FLAG-tagged GAPDH (Fig. 7A), Runx1 (Fig. 7B), Mgea5 (Fig. 7C), RANbp10 (Fig. 7D) or PK as a negative control (Fig. 7E) and the interaction between IDO2 and each candidate protein was evaluated by IP/Western using antibodies to the V5 and FLAG tags. IDO2 co-immunoprecipitated with GAPDH, Runx1, RANbp10 and Mgea5, but not PK, providing an initial validation of the two-hybrid results (Fig. 7). Given the distinct roles of IDO2 and IDO1 in mediating autoreactive B cell responses and arthritis in the KRN model (5), we next determined if any of these IDO2-interacting proteins also bound to IDO1. We hypothesized that proteins that bind to IDO2, but not IDO1, would be potential components of the non-enzymatic pathway IDO2 uses to drive arthritis. To distinguish proteins that bind to IDO2 but not IDO1, we transfected the DDK/FLAG-tagged candidate proteins into T-REx cells stably expressing IDO1 and repeated the Co-IP experiment. Importantly, none of the IDO2-binding proteins bound to IDO1 (Fig. 7), identifying them as potential intermediators of IDO2 function.
Table I. IDO2 interacting proteins.
Full-length cDNA for mouse IDO2 was cloned as a C-terminal fusion to LexA. The construct was screened against a mouse spleen cDNA library using a mating approach with YHGX13 and L40ΔGal4 yeast strains. His+ colonies were selected on a medium lacking tryptophan, leucine, and histidine. Predicted biological score (PBS): A: Very high confidence interaction; B: High confidence interaction; C: Good confidence interaction; D: Moderate confidence interaction.
| PBS | Prey Protein |
|---|---|
|
| |
| B | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) |
| D | Runt related transcription factor 1 (Runx1) |
| D | Meningioma expressed antigen 5 (Mgea5) |
| B | RAN binding protein 10 (Ranbp10) |
Figure 7. IDO2-binding protein interactions suggest potential non-enzymatic pathway mediated by IDO2.
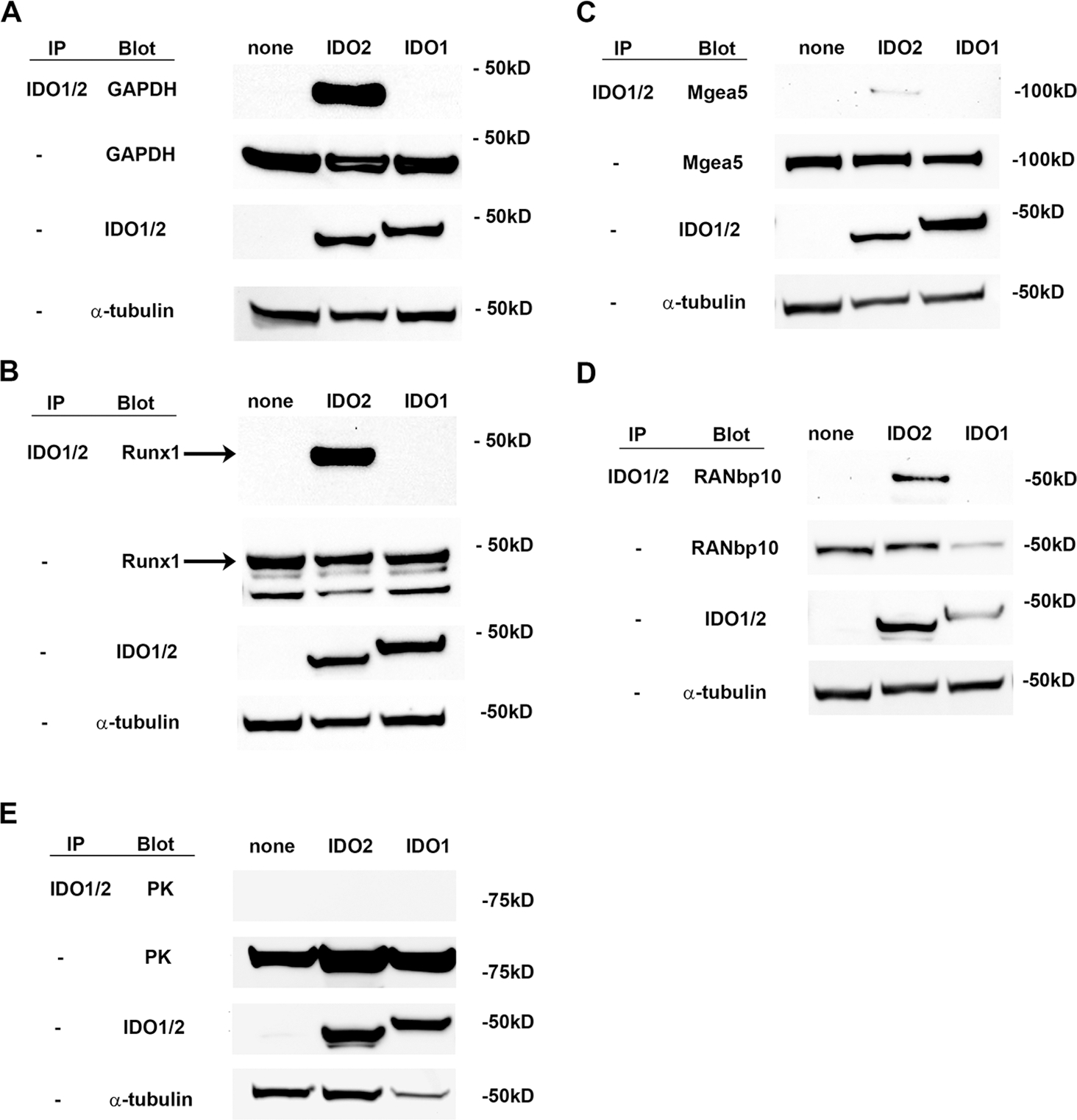
293-T-REx™ cells, stably transfected with V5-tagged IDO2, IDO1 or no IDO (none), were transiently transfected with FLAG-tagged (A) GAPDH, (B) Runx1, (C) Mgea5, (D) RANbp10, or (E) PK. Lysates were immunoprecipitated with anti-V5 resin and then immunoblotted with anti-FLAG-HRP. Whole lysates were also directly blotted with anti-V5 or anti-FLAG to verify expression of the transfected proteins. Although the same plasmid backbone was used to express all four test proteins, there was variation in the expression level between the individual proteins (GAPDH > Runx1 and Mgea5 >RANbp10). However, each individual protein was expressed at approximately the same level between Trex cells expressing IDO2, IDO1, or no IDO. Representative blots are shown from a total of 2 independent experiments. Arrow indicates Runx1 band.
Lastly, we determined whether the IDO2-interacting proteins bound to IDO2 expressing R235W or Y346X polymorphisms (Fig. 8). To do this, we transfected the DDK/FLAG-tagged candidate proteins into T-REx cells stably expressing wt, R235W, or Y346X IDO2 and again repeated the Co-IP experiment. The R235W polymorphism did not affect IDO2’s ability to bind to GAPDH and only moderately reduced its binding to Mgea5. In contrast, IDO2’s interactions with Runx1 and RANbp10 were more affected by the R235W polymorphism (Fig. 8). IDO2 expressing the Y346X polymorphism bound to Runx1, and RANbp10 with similar efficacy as wt IDO2, whereas binding to GAPDH was moderately reduced and binding to Mgea5 was greatly reduced (Fig. 8). Taken together, these data show that all four identified IDO2 interacting proteins retain at least some binding to both R235W and Y346X IDO2, suggesting the non-enzymatic function of IDO2 could involve multiple signaling pathways.
Figure 8. Varying effect of R235W and Y346X polymorphisms on binding to IDO2-interacting proteins.
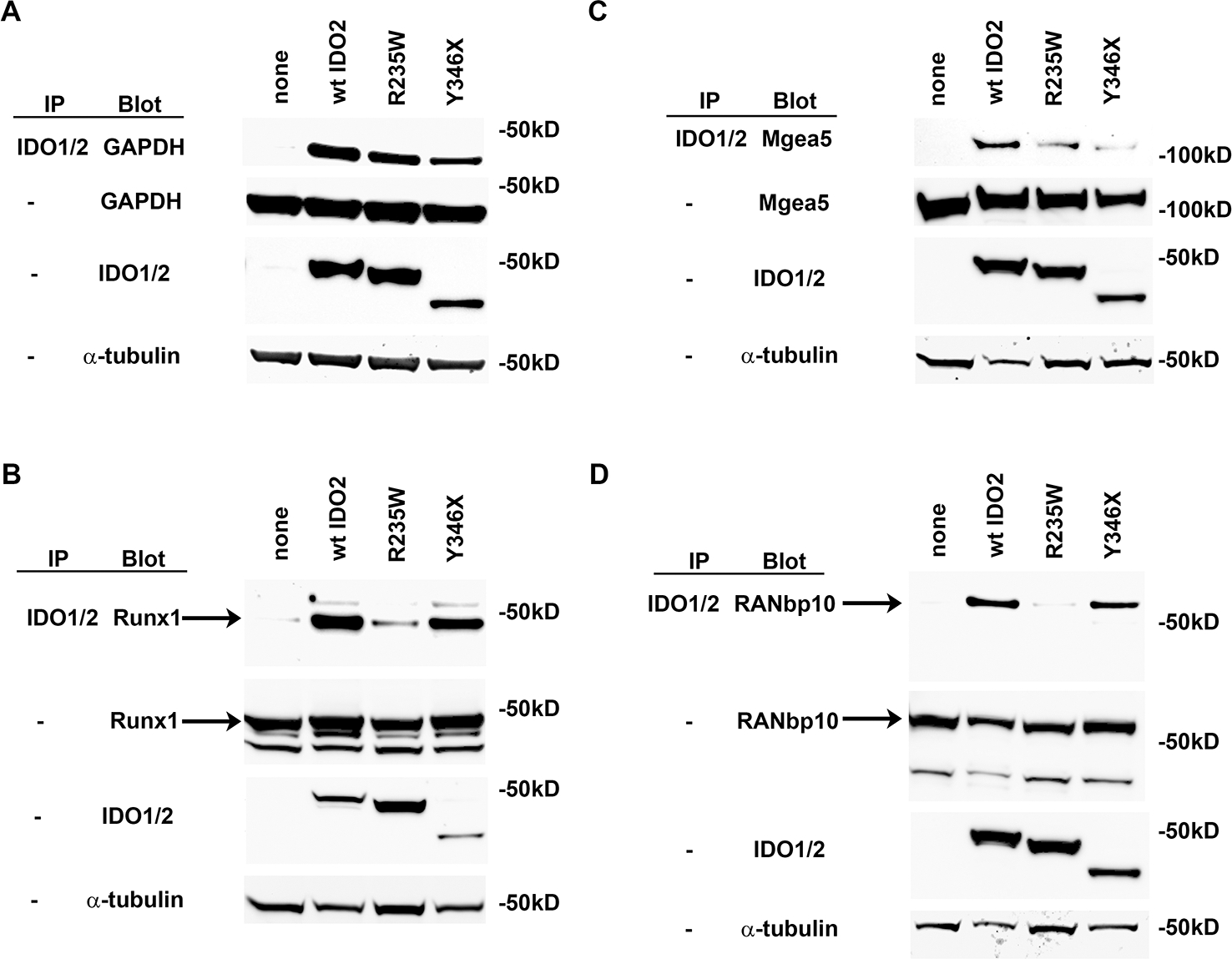
293-T-REx™ cells, stably transfected with V5-tagged wt IDO2, R235W IDO2, Y346X IDO2 or no IDO2 (none), were transiently transfected with FLAG-tagged (A) GAPDH, (B) Runx1, (C) Mgea5, (D) RANbp10. Lysates were immunoprecipitated with anti-V5 resin and then immunoblotted with anti-FLAG-HRP. Whole lysates were also directly blotted with anti-V5 or anti-FLAG to verify expression of the transfected proteins. Representative blots are shown from a total of 3 independent experiments. Arrows indicate Runx1 and RANbp10 bands.
DISCUSSION
The IDO pathway has long been connected to inflammatory immunity; however, there has been debate in the literature about the relative importance of IDO2 to the autoimmune response (23, 24, 37–41). IDO2 has greatly reduced tryptophan catabolic activity compared to IDO1. In addition, single nucleotide polymorphisms affecting tryptophan catalysis by IDO2 are extremely common, with only a minority of people carrying two copies of wt IDO2. This suggests one of two possibilities, either that IDO2 has limited relevance to disease or that the main role of IDO2 lies outside of its enzymatic function. Here, using catalytically inactive IDO2 ki mice, we demonstrate that the enzymatic function of IDO2 is required to mediate some inflammatory immune responses. Importantly, our findings also provide the first evidence of a novel non-enzymatic role for IDO2 in mediating inflammatory autoimmune disease.
Mice expressing enzyme-inactivating polymorphisms in IDO2 develop a reduced contact hypersensitivity response, similar to that seen in IDO2 ko mice. This suggests an important role for tryptophan catabolism in mediating inflammation in this model, although an additional effect of the R235W mutation on IDO2 conformation cannot be excluded. In contrast, the robust arthritis in KRN mice expressing the R235W polymorphism is indistinguishable from that in mice expressing wt IDO2, demonstrating that inflammatory autoimmunity can be generated in the absence of IDO2 catalytic activity. The differential requirement for IDO2 enzymatic function in CHS vs. arthritis is reminiscent of the contrasting dependence on IDO1 expression in the two models. CHS responses are reduced in both IDO1 and IDO2 ko mice, suggesting a common, but non-redundant, requirement for tryptophan catabolism to mediate inflammatory immunity in this model (23). In contrast, joint inflammation in the KRN arthritis model is attenuated in IDO2 ko, but not IDO1 ko, mice, providing further support for a novel non-enzymatic role for IDO2 in mediating inflammatory autoimmunity (8).
IDO2 inactivating polymorphisms have been associated with some cancers and autoimmune/inflammatory conditions, suggesting the non-enzymatic role identified in mouse models may also be relevant to human disease. Specifically, familial pancreatic cancer is positively associated with the Y359X (rs4503083) but not R248W (rs10109853) polymorphism (21). In Crohn’s disease, Y359X is associated with a reduced risk of disease, while R248W has no effect (17). In these cases, as we see in our KRN.g7 arthritis model, loss of catalytic activity alone (R/W) does not affect disease susceptibility compared to wt IDO2, whereas truncation associated with reduced protein stability (Y/X) does affect disease state, suggesting that full-length but catalytically inactive IDO2 has a relevant signaling role in these conditions. In contrast, both polymorphisms are associated with increased risk of aspergillosis in hematopoietic stem cell transfer recipients (18), radiotherapy responsiveness in pancreatic cancer patients (19), and R/W is associated with susceptibility to multiple myeloma (20), signifying a potential role for IDO2 catalytic activity in these circumstances.
The two IDO enzymes have been primarily studied in the context of their direct relationship to tryptophan catalysis and several established connections between tryptophan, the IDO pathway, and immunity have been described. These include 1) sensing of Trp depletion as mediated by GCN2 activation to induce amino acid insufficiency signaling and alter levels of immune-modulating cytokines such as IL-6 (42, 43); 2) sensing of Trp depletion by mTOR suppression to block amino acid sufficiency signaling, resulting in subsequent downstream effects on autophagy (3, 44, 45); and 3) activation of the aryl hydrocarbon receptor (AhR) by kynurenine and other Trp derivatives, which promotes the generation of T regulatory cells that suppress adaptive immunity (46). In addition, a non-tryptophan associated function of IDO1 has been described involving the activation of the non-canonical NFκB pathway and subsequent induction of TGF-β and type I interferons leading to suppression of T cell activation and development of regulatory T cells (11). Our data suggest that IDO2 is also likely involved in multiple molecular pathways, only some of which require enzymatic function. As such, the non-enzymatic functions of IDO2 must be directly examined when investigating the immunomodulatory properties of this protein.
While our experiments here provide in vivo evidence for the non-enzymatic role of IDO2 in mediating autoimmune disease, the mechanism by which this occurs has yet to be elucidated. Given that IDO2 lacks the functional ITIM sites that are essential for IDO1’s non-enzymatic signaling, IDO2 is unlikely to share the mechanistic pathways identified for IDO1 (11, 47, 48). A yeast-2-hybrid screen identified four diverse proteins with the potential to bind IDO2, and which may be involved in immune signaling pathways. Though there is no previously established link to the IDO pathway for Runx1, GAPDH, RANbp10, or Mgea5, we found that each of the four successfully co-immunoprecipitates specifically with IDO2 but not IDO1. The RUNT-related transcription factor Runx1 has been implicated in lymphoid, myeloid, and megakaryocyte lineage maturation (49) and is genetically associated with autoimmunity in RA, systemic lupus erythematosus (SLE), and psoriasis (50–52). GAPDH is a glycolytic enzyme expressed in all cells. In addition to its critical role in the glucose metabolism, GAPDH has been implicated in a diverse array of alternative functions, including oligomerization, posttranslational modification, and subcellular localization (53). GAPDH has also been reported to bind heme and may have heme sensing or chaperone functions (54), a role that could be investigated in the context of the heme binding properties of IDO2. Additionally, antibodies against GAPDH have been associated with SLE (55, 56). RANbp10 is a small GTPase important in microtubule dynamics, particularly in platelets, and may have a role in coupling membrane receptors to intracellular signaling pathways (57), though it has been little studied compared to other RAN binding proteins (58). Mgea5 encodes the protein O-GlcNAcase, which is involved in the post-translational cycling of the O-linked β-N-acetyl-D-glucosamine (O-GlcNAc) monosaccharide moiety on proteins (59). O‑GlcNAcylation has been implicated in both B and T cell activation and is associated with autoimmunity, particularly as a pro-inflammatory mediator of diabetes and insulin resistance (60–63). There are hundreds of proteins subject to O-GlcNAc modification, with far reaching implications for regulation of immunity, though no known direct links to IDO pathways. The wide range of protein functions associated with each of these IDO2 interacting proteins suggests that the non-enzymatic function of IDO2 may occur through multiple independent signaling pathways.
Comparison of the binding of GAPDH, Runx1, Mgea5, and RANbp10 between wt IDO2 and IDO2 expressing the R235W or Y346X polymorphisms provided further insight into the molecular basis of the interactions. Although both R235W and Y346X retained some binding to all four proteins, R235W interacted more strongly with GAPDH and Mgea5, whereas Y346X bound better to RANbp10 and Runx1. As the Y346X polymorphism truncates the IDO2 protein prior to the C terminus, this suggests that Mgea5 and GAPDH bind IDO2 near the C terminus and Runx1 and RANbp10 bind upstream of the truncation at position 346. Furthermore, the reduced binding of Runx1 and RANbp10 with R235W suggests that their interaction with IDO2 either occurs close to the catalytic site or is dependent on protein conformation that is disrupted by the R235W mutation. As the binding protein interactions were evaluated using transient transfections and an overexpression system, future experiments will be needed to explore how these potential IDO2 binding partners impact IDO2 function in vivo.
Approaches to therapeutically target the IDO pathway have historically been designed to inhibit catalytic activity. Preclinical and early clinical studies suggested that inhibitors of IDO1 enzymatic activity would provide a therapeutic benefit in cancer (64–68), but similar approaches yielded conflicting results in models of autoimmunity (24, 38, 39, 69, 70). Our findings demonstrating that IDO2 enzyme function is not necessary for arthritis development in the KRN model suggest that effective therapeutic strategies for targeting IDO2 need to address its non-enzymatic signaling capability. Two such approaches, monoclonal antibody and siRNA-based modalities have been shown to be effective at ameliorating disease in the KRN model (71, 72).
In summary, our findings demonstrating that IDO2 enzyme function is not necessary for arthritis development but is required for a contact hypersensitivity response suggest that IDO2 has multifaceted, context-dependent roles in immunity. The discovery of a differential requirement for tryptophan catabolizing activity may help to clarify seemingly conflicting findings in the literature showing pro vs. anti-inflammatory roles for the IDO pathway in models of autoimmune disease. Given the reported nonenzymatic roles of IDO1 and the potential involvement of newly established IDO2 binding partners described in this study, future studies are warranted to determine the specific non-enzymatic pathway mediating IDO2’s function driving autoimmune disease.
KEY POINTS.
IDO2 has both enzymatic and non-enzymatic functions.
IDO2 mediates autoimmune arthritis via a non-enzymatic mechanism.
Acknowledgments
This project was supported in part by the Giorgi Family Foundation (LM-N), the Zuckermann Autoimmune Disorder Research Fund (LM-N) and the National Cancer Institute of the National Institutes of Health under award number R01CA191191 (GCP).
3. Nonstandard abbreviations:
- ASCs
antibody secreting cells
- Cα ko
TCR alpha chain deficient
- CHS
contact hypersensitivity
- EIF2α
eukaryotic translation initiation factor 2A
- GCN2
general control nonderepressible 2
- GPI
glucose-6-phosphate isomerase
- his
histidine
- IP
immunoprecipitation
- ki
knock-in
- ko
knock-out
- kyn
N-formylkynurenine
- LN
lymph node
- Mgea5
meningioma expressed antigen 5
- mTOR
mammalian target of rapamycin
- O-GlcNAc
O-linked β-N-acetyl-D-glucosamine
- PK
pyruvate kinase
- PKC
protein kinase C
- RA
rheumatoid arthritis
- RANbp10
RAN binding protein 10
- Runx1
Runt-related transcription factor 1 (Runx1)
- SLE
systemic lupus erythematosus
- SNPs
single nucleotide polymorphisms
- Trp
tryptophan
- TDO
tryptophan-2,3-dioxygenase
- T-REx
293-T-REx™ cell line expressing the tetracycline repressor protein
- wt
wild-type
REFERENCES
- 1.Banerjee T, Duhadaway JB, Gaspari P, Sutanto-Ward E, Munn DH, Mellor AL, Malachowski WP, Prendergast GC, and Muller AJ 2008. A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene 27: 2851–2857. [DOI] [PubMed] [Google Scholar]
- 2.Prendergast GC, Chang MY, Mandik-Nayak L, Metz R, and Muller AJ 2011. Indoleamine 2,3-dioxygenase as a modifier of pathogenic inflammation in cancer and other inflammation-associated diseases. Curr. Med. Chem 18: 2257–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, and Muller AJ 2014. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother 63: 721–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merlo LM, DuHadaway J, Grabler S, Prendergast GC, Muller AJ, and Mandik-Nayak L 2016. IDO2 modulates T cell-dependent autoimmune responses through a B cell-intrinsic mechanism. J. Immunol 196: 4487–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merlo LMF, DuHadaway JB, Montgomery JD, Peng WD, Murray PJ, Prendergast GC, Caton AJ, Muller AJ, and Mandik-Nayak L 2020. Differential Roles of IDO1 and IDO2 in T and B Cell Inflammatory Immune Responses. Front Immunol 11: 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, and Prendergast GC 2007. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res 67: 7082–7087. [DOI] [PubMed] [Google Scholar]
- 7.Ball HJ, Sanchez-Perez A, Weiser S, Austin CJ, Astelbauer F, Miu J, McQuillan JA, Stocker R, Jermiin LS, and Hunt NH 2007. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 396: 203–213. [DOI] [PubMed] [Google Scholar]
- 8.Merlo LM, Pigott E, Duhadaway JB, Grabler S, Metz R, Prendergast GC, and Mandik-Nayak L 2014. IDO2 Is a Critical Mediator of Autoantibody Production and Inflammatory Pathogenesis in a Mouse Model of Autoimmune Arthritis. J. Immunol 192: 2082–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pantouris G, Serys M, Yuasa HJ, Ball HJ, and Mowat CG 2014. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino Acids 46: 2155–2163. [DOI] [PubMed] [Google Scholar]
- 10.Pallotta MT, Orabona C, Bianchi R, Vacca C, Fallarino F, Belladonna ML, Volpi C, Mondanelli G, Gargaro M, Allegrucci M, Talesa VN, Puccetti P, and Grohmann U 2014. Forced IDO1 expression in dendritic cells restores immunoregulatory signalling in autoimmune diabetes. J Cell Mol Med 18: 2082–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, Mazza EM, Boon L, Grassi F, Fioretti MC, Fallarino F, Puccetti P, and Grohmann U 2011. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nature immunology 12: 870–878. [DOI] [PubMed] [Google Scholar]
- 12.Pallotta MT, Rossini S, Suvieri C, Coletti A, Orabona C, Macchiarulo A, Volpi C, and Grohmann U 2021. Indoleamine 2,3-dioxygenase 1 (IDO1): an up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugimoto H, Oda S, Otsuki T, Hino T, Yoshida T, and Shiro Y 2006. Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. U. S. A 103: 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Littlejohn TK, Takikawa O, Truscott RJ, and Walker MJ 2003. Asp274 and his346 are essential for heme binding and catalytic function of human indoleamine 2,3-dioxygenase. J. Biol. Chem 278: 29525–29531. [DOI] [PubMed] [Google Scholar]
- 15.Agliardi C, Guerini FR, Zanzottera M, Rovaris M, Caputo D, and Clerici M 2017. Indoleamine-2,3-dioxygenase(IDO)2 polymorphisms are not associated with multiple sclerosis in Italians. J. Neurol. Sci 377: 31–34. [DOI] [PubMed] [Google Scholar]
- 16.Witkiewicz AK, Costantino CL, Metz R, Muller AJ, Prendergast GC, Yeo CJ, and Brody JR 2009. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. J. Am. Coll. Surg 208: 781–787; discussion 787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee A, Kanuri N, Zhang Y, Sayuk GS, Li E, and Ciorba MA 2014. IDO1 and IDO2 non-synonymous gene variants: correlation with crohn’s disease risk and clinical phenotype. PloS one 9: e115848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Napolioni V, Pariano M, Borghi M, Oikonomou V, Galosi C, De Luca A, Stincardini C, Vacca C, Renga G, Lucidi V, Colombo C, Fiscarelli E, Lass-Florl C, Carotti A, D’Amico L, Majo F, Russo MC, Ellemunter H, Spolzino A, Mosci P, Brancorsini S, Aversa F, Velardi A, Romani L, and Costantini C 2019. Genetic Polymorphisms Affecting IDO1 or IDO2 Activity Differently Associate With Aspergillosis in Humans. Front Immunol 10: 890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nevler A, Muller AJ, Sutanto-Ward E, DuHadaway JB, Nagatomo K, Londin E, O’Hayer K, Cozzitorto JA, Lavu H, Yeo TP, Curtis M, Villatoro T, Leiby BE, Mandik-Nayak L, Winter JM, Yeo CJ, Prendergast GC, and Brody JR 2019. Host IDO2 Gene Status Influences Tumor Progression and Radiotherapy Response in KRAS-Driven Sporadic Pancreatic Cancers. Clin. Cancer Res 25: 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasamatsu T, Hashimoto N, Sakaya N, Awata-Shiraiwa M, Ishihara R, Murakami Y, Masuda Y, Gotoh N, Nagai K, Oda T, Yokohama A, Saitoh T, Handa H, Tsukamoto N, Hayashi K, and Murakami H 2021. IDO2 rs10109853 polymorphism affects the susceptibility to multiple myeloma. Clin Exp Med 21: 323–329. [DOI] [PubMed] [Google Scholar]
- 21.Nevler A, Muller AJ, Cozzitorto JA, Goetz A, Winter JM, Yeo TP, Lavu H, Yeo CJ, Prendergast GC, and Brody JR 2018. A Sub-Type of Familial Pancreatic Cancer: Evidence and Implications of Loss-of-Function Polymorphisms in Indoleamine-2,3-Dioxygenase-2. J. Am. Coll. Surg 226: 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, and Mathis D 1996. Organ-specific disease provoked by systemic autoimmunity. Cell 87: 811–822. [DOI] [PubMed] [Google Scholar]
- 23.Metz R, Smith C, DuHadaway JB, Chandler P, Baban B, Merlo LMF, Pigott E, Keough MP, Rust S, Mellor AL, Mandik-Nayak L, Muller AJ, and Prendergast GC 2014. IDO2 is critical for IDO1-mediated T cell regulation and exerts a non-redundant function in inflammation. Int. Immunol 26: 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott GN, DuHadaway J, Pigott E, Ridge N, Prendergast GC, Muller AJ, and Mandik-Nayak L 2009. The immunoregulatory enzyme IDO paradoxically drives B cell-mediated autoimmunity. J. Immunol 182: 7509–7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandik-Nayak L, Wipke BT, Shih FF, Unanue ER, and Allen PM 2002. Despite ubiquitous autoantigen expression, arthritogenic autoantibody response initiates in the local lymph node. Proc. Natl. Acad. Sci. U. S. A 99: 14368–14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vojtek AB, and Hollenberg SM 1995. Ras-Raf interaction: two-hybrid analysis. Methods Enzymol 255: 331–342. [DOI] [PubMed] [Google Scholar]
- 27.Bartel PL, Cheien C-T, Sternglanz R, and Fields S 1993. Using the two-hybrid system to detect protein-protein interactions. In Cellular Interactions in Development: A Practical Approach. Hartley DA, ed. Oxford University Press, Oxford. 153–179. [Google Scholar]
- 28.Fromont-Racine M, Rain JC, and Legrain P 1997. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet 16: 277–282. [DOI] [PubMed] [Google Scholar]
- 29.Formstecher E, Aresta S, Collura V, Hamburger A, Meil A, Trehin A, Reverdy C, Betin V, Maire S, Brun C, Jacq B, Arpin M, Bellaiche Y, Bellusci S, Benaroch P, Bornens M, Chanet R, Chavrier P, Delattre O, Doye V, Fehon R, Faye G, Galli T, Girault JA, Goud B, de Gunzburg J, Johannes L, Junier MP, Mirouse V, Mukherjee A, Papadopoulo D, Perez F, Plessis A, Rosse C, Saule S, Stoppa-Lyonnet D, Vincent A, White M, Legrain P, Wojcik J, Camonis J, and Daviet L 2005. Protein interaction mapping: a Drosophila case study. Genome Res 15: 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schachter V, Chemama Y, Labigne A, and Legrain P 2001. The protein-protein interaction map of Helicobacter pylori. Nature 409: 211–215. [DOI] [PubMed] [Google Scholar]
- 31.Wojcik J, Boneca IG, and Legrain P 2002. Prediction, assessment and validation of protein interaction maps in bacteria. J. Mol. Biol 323: 763–770. [DOI] [PubMed] [Google Scholar]
- 32.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, and Cantley LC 2008. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: 230–233. [DOI] [PubMed] [Google Scholar]
- 33.Littlejohn TK, Takikawa O, Skylas D, Jamie JF, Walker MJ, and Truscott RJ 2000. Expression and purification of recombinant human indoleamine 2, 3-dioxygenase. Protein Expr. Purif 19: 22–29. [DOI] [PubMed] [Google Scholar]
- 34.Flick HE, Lalonde JM, Malachowski WP, and Muller AJ 2013. The Tumor-Selective Cytotoxic Agent beta-Lapachone is a Potent Inhibitor of IDO1. International journal of tryptophan research : IJTR 6: 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winters M, DuHadaway JB, Pham KN, Lewis-Ballester A, Badir S, Wai J, Sheikh E, Yeh SR, Prendergast GC, Muller AJ, and Malachowski WP 2019. Diaryl hydroxylamines as pan or dual inhibitors of indoleamine 2,3-dioxygenase-1, indoleamine 2,3-dioxygenase-2 and tryptophan dioxygenase. Eur J Med Chem 162: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumoto I, Staub A, Benoist C, and Mathis D 1999. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 286: 1732–1735. [DOI] [PubMed] [Google Scholar]
- 37.Gurtner GJ, Newberry RD, Schloemann SR, McDonald KG, and Stenson WF 2003. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology 125: 1762–1773. [DOI] [PubMed] [Google Scholar]
- 38.Sakurai K, Zou JP, Tschetter JR, Ward JM, and Shearer GM 2002. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J. Neuroimmunol 129: 186–196. [DOI] [PubMed] [Google Scholar]
- 39.Szanto S, Koreny T, Mikecz K, Glant TT, Szekanecz Z, and Varga J 2007. Inhibition of indoleamine 2,3-dioxygenase-mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis research & therapy 9: R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu H, Oriss TB, Fei M, Henry AC, Melgert BN, Chen L, Mellor AL, Munn DH, Irvin CG, Ray P, and Ray A 2008. Indoleamine 2,3-dioxygenase in lung dendritic cells promotes Th2 responses and allergic inflammation. Proc. Natl. Acad. Sci. U. S. A 105: 6690–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Bubnoff D, and Bieber T 2012. The indoleamine 2,3-dioxygenase (IDO) pathway controls allergy. Allergy 67: 718–725. [DOI] [PubMed] [Google Scholar]
- 42.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, and Mellor AL 2005. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22: 633–642. [DOI] [PubMed] [Google Scholar]
- 43.Dey S, Mondal A, DuHadaway JB, Sutanto-Ward E, Laury-Kleintop LD, Thomas S, Prendergast GC, Mandik-Nayak L, and Muller AJ 2021. IDO1 Signaling through GCN2 in a Subpopulation of Gr-1(+) Cells Shifts the IFNgamma/IL6 Balance to Promote Neovascularization. Cancer Immunol Res 9: 514–528. [DOI] [PubMed] [Google Scholar]
- 44.Wu H, Gong J, and Liu Y 2018. Indoleamine 2, 3-dioxygenase regulation of immune response (Review). Mol Med Rep 17: 4867–4873. [DOI] [PubMed] [Google Scholar]
- 45.Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, Link CJ, and Prendergast GC 2012. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 1: 1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Platten M, von Knebel Doeberitz N, Oezen I, Wick W, and Ochs K 2014. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front Immunol 5: 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Albini E, Rosini V, Gargaro M, Mondanelli G, Belladonna ML, Pallotta MT, Volpi C, Fallarino F, Macchiarulo A, Antognelli C, Bianchi R, Vacca C, Puccetti P, Grohmann U, and Orabona C 2017. Distinct roles of immunoreceptor tyrosine-based motifs in immunosuppressive indoleamine 2,3-dioxygenase 1. J Cell Mol Med 21: 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fallarino F, Grohmann U, and Puccetti P 2012. Indoleamine 2,3-dioxygenase: from catalyst to signaling function. Eur. J. Immunol 42: 1932–1937. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Newton RC, Friedman SM, and Scherle PA 2009. Indoleamine 2,3-dioxygenase, an emerging target for anti-cancer therapy. Current cancer drug targets 9: 938–952. [DOI] [PubMed] [Google Scholar]
- 50.Tokuhiro S, Yamada R, Chang X, Suzuki A, Kochi Y, Sawada T, Suzuki M, Nagasaki M, Ohtsuki M, Ono M, Furukawa H, Nagashima M, Yoshino S, Mabuchi A, Sekine A, Saito S, Takahashi A, Tsunoda T, Nakamura Y, and Yamamoto K 2003. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet 35: 341–348. [DOI] [PubMed] [Google Scholar]
- 51.Helms C, Cao L, Krueger JG, Wijsman EM, Chamian F, Gordon D, Heffernan M, Daw JA, Robarge J, Ott J, Kwok PY, Menter A, and Bowcock AM 2003. A putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is associated with susceptibility to psoriasis. Nat. Genet 35: 349–356. [DOI] [PubMed] [Google Scholar]
- 52.Prokunina L, Castillejo-Lopez C, Oberg F, Gunnarsson I, Berg L, Magnusson V, Brookes AJ, Tentler D, Kristjansdottir H, Grondal G, Bolstad AI, Svenungsson E, Lundberg I, Sturfelt G, Jonssen A, Truedsson L, Lima G, Alcocer-Varela J, Jonsson R, Gyllensten UB, Harley JB, Alarcon-Segovia D, Steinsson K, and Alarcon-Riquelme ME 2002. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat. Genet 32: 666–669. [DOI] [PubMed] [Google Scholar]
- 53.Tristan C, Shahani N, Sedlak TW, and Sawa A 2011. The diverse functions of GAPDH: views from different subcellular compartments. Cell. Signal 23: 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hannibal L, Collins D, Brassard J, Chakravarti R, Vempati R, Dorlet P, Santolini J, Dawson JH, and Stuehr DJ 2012. Heme binding properties of glyceraldehyde-3-phosphate dehydrogenase. Biochemistry (Mosc) 51: 8514–8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun J, Li X, Zhou H, Liu X, Jia J, Xie Q, Peng S, Sun X, Wang Q, and Yi L 2019. Anti-GAPDH Autoantibody Is Associated with Increased Disease Activity and Intracranial Pressure in Systemic Lupus Erythematosus. J Immunol Res 2019: 7430780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takasaki Y, Kaneda K, Matsushita M, Yamada H, Nawata M, Matsudaira R, Asano M, Mineki R, Shindo N, and Hashimoto H 2004. Glyceraldehyde 3-phosphate dehydrogenase is a novel autoantigen leading autoimmune responses to proliferating cell nuclear antigen multiprotein complexes in lupus patients. Int. Immunol 16: 1295–1304. [DOI] [PubMed] [Google Scholar]
- 57.Yudin D, and Fainzilber M 2009. Ran on tracks--cytoplasmic roles for a nuclear regulator. J. Cell Sci 122: 587–593. [DOI] [PubMed] [Google Scholar]
- 58.Palmieri D, Tessari A, and Coppola V 2018. Scorpins in the DNA Damage Response. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagel AK, and Ball LE 2015. Intracellular protein O-GlcNAc modification integrates nutrient status with transcriptional and metabolic regulation. Adv. Cancer Res 126: 137–166. [DOI] [PubMed] [Google Scholar]
- 60.Wu JL, Chiang MF, Hsu PH, Tsai DY, Hung KH, Wang YH, Angata T, and Lin KI 2017. O-GlcNAcylation is required for B cell homeostasis and antibody responses. Nat Commun 8: 1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Golks A, Tran TT, Goetschy JF, and Guerini D 2007. Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. EMBO J 26: 4368–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lund PJ, Elias JE, and Davis MM 2016. Global Analysis of O-GlcNAc Glycoproteins in Activated Human T Cells. J. Immunol 197: 3086–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y, Xie M, Men L, and Du J 2019. O-GlcNAcylation in immunity and inflammation: An intricate system (Review). Int. J. Mol. Med 44: 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soliman H, Mediavilla-Varela M, and Antonia S 2010. Indoleamine 2,3-dioxygenase: is it an immune suppressor? Cancer journal (Sudbury, Mass 16: 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soliman HH, A. S., Sullivan D, Vanahanian N, and Link C 2009. Overcoming tumor antigen anergy in human malignancies using the novel indoleamine 2,3-dioxygenase (IDO) enzyme inhibitor, 1-methyl-D-tryptophan (1MT). J. Clin. Oncol 27: Abstract 3004. [Google Scholar]
- 66.Soliman HH, Minton SE, Han HS, Ismail-Khan R, Mahipal A, Janssen W, Streicher H, Vahanian NN, Link CJ, Ramsey WJ, Antonia SJ, and Sullivan D 2013. A phase I study of ad.p53 DC vaccine in combination with indoximod in metastatic solid tumors. J. Clin. Oncol: suppl abstract 3069.23918942 [Google Scholar]
- 67.Soliman HH, Jackson E, Neuger T, Dees EC, Harvey RD, Han H, Ismail-Khan R, Minton S, Vahanian NN, Link C, Sullivan DM, and Antonia S 2014. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget 5: 8136–8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yue EW, Sparks R, Polam P, Modi D, Douty B, Wayland B, Glass B, Takvorian A, Glenn J, Zhu W, Bower M, Liu X, Leffet L, Wang Q, Bowman KJ, Hansbury MJ, Wei M, Li Y, Wynn R, Burn TC, Koblish HK, Fridman JS, Emm T, Scherle PA, Metcalf B, and Combs AP 2017. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med Chem Lett 8: 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Criado G, Simelyte E, Inglis JJ, Essex D, and Williams RO 2009. Indoleamine 2,3 dioxygenase-mediated tryptophan catabolism regulates accumulation of Th1/Th17 cells in the joint in collagen-induced arthritis. Arthritis Rheum 60: 1342–1351. [DOI] [PubMed] [Google Scholar]
- 70.Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, Munn DH, Mellor AL, Karlsson MC, and McGaha TL 2012. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc. Natl. Acad. Sci. U. S. A 109: 3909–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Merlo LM, Bowers J, Stefanoni T, Getts R, and Mandik-Nayak L 2020. B-Cell-Targeted 3DNA Nanotherapy Against Indoleamine 2,3-Dioxygenase 2 (IDO2) Ameliorates Autoimmune Arthritis in a Preclinical Model. Clin Pathol 13: 2632010X20951812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merlo LM, Grabler S, DuHadaway JB, Pigott E, Manley K, Prendergast GC, Laury-Kleintop LD, and Mandik-Nayak L 2017. Therapeutic antibody targeting of indoleamine-2,3-dioxygenase (IDO2) inhibits autoimmune arthritis. Clin. Immunol 179: 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]