

Abstract
Neuroendocrine to non-neuroendocrine plasticity supports small cell lung cancer (SCLC) tumorigenesis and promotes immunogenicity. Approximately 20–25% of SCLCs harbor loss of function (LOF) NOTCH mutations. Previous studies demonstrated that NOTCH functions as a SCLC tumor suppressor, but can also drive non-neuroendocrine plasticity to support SCLC growth. Given the dual functionality of NOTCH, it’s not understood why SCLCs select for LOF NOTCH mutations and how these mutations impact SCLC tumorigenesis. In a CRISPR-based genetically-engineered mouse model of SCLC, genetic loss of Notch1 or Notch2 modestly accelerated SCLC tumorigenesis. Interestingly, Notch-mutant SCLCs still formed non-neuroendocrine subpopulations, and these Notch-independent, non-neuroendocrine subpopulations were driven by Runx2-mediated regulation of Rest. Notch2-mutant non-neuroendocrine cells highly express innate immune signaling genes including STING and were sensitive to STING agonists. This work identifies a Notch-independent mechanism to promote non-neuroendocrine plasticity and suggests that therapeutic approaches to activate STING could be selectively beneficial for SCLCs with NOTCH2 mutations.
Keywords: NOTCH, RUNX, REST, STING, small cell lung cancer
Introduction
Small cell lung cancers (SCLCs) are high grade neuroendocrine tumors that account for ~15% of lung cancers(1–3). SCLCs are characterized by near universal loss of the RB1 and TP53 tumor suppressor genes(4–6), and ~70% of SCLCs highly express the neuroendocrine transcription factor ASCL1(4,7,8), which is required for SCLC tumorigenesis(9) and a dependency in a subset of SCLC cell lines(10–12). NOTCH receptors and target genes are repressed in nearly all ASCL1 positive SCLCs(4) suggesting that NOTCH and ASCL1 oppose each other during SCLC tumorigenesis(13,14). ~25% of SCLCs harbor mutually exclusive loss of function (LOF) mutations in NOTCH receptors (NOTCH1, NOTCH2, NOTCH3, and NOTCH4)(4) demonstrating that genetic loss of NOTCH is selected for during SCLC tumorigenesis and nominating NOTCH as a tumor suppressor gene in SCLC.
Functional studies in SCLC cell lines and SCLC genetically-engineered mouse models (GEMMs) support the role of NOTCH as a SCLC tumor suppressor(4,13). Re-expression of transcriptionally active NOTCH1 (NOTCH1-ICD) or NOTCH2 (NOTCH2-ICD) in the Rb1−/−, Trp53−/−, Rbl2−/− (RPP, also known as RPR2) SCLC GEMM, which normally forms SCLC tumors of the ASCL1 + subtype, dramatically inhibits SCLC tumor formation demonstrating that low NOTCH activity is required for SCLC tumor initiation(4). The SCLCs that escape tumor suppression by NOTCH and form with high NOTCH activity lose ASCL1 expression and neuroendocrine differentiation demonstrating that NOTCH functionally opposes the ASCL1 driven neuroendocrine phenotype in SCLC. In support of these in vivo findings, re-expression of NOTCH in SCLC cell lines blocks cellular proliferation and neuroendocrine differentiation(4,13,15,16). These data, together with selection for LOF NOTCH mutations in human SCLC tumors, supports a tumor suppressive role for NOTCH during SCLC tumorigenesis.
More recent data suggests that NOTCH has a dual role in SCLC and can function as both a suppressor and promoter of SCLC tumorigenesis(15). In the RPP SCLC GEMM, NOTCH-active non-neuroendocrine subpopulations of cells highly express the canonical NOTCH target gene HES1. These HES1-positive cells have decreased expression of ASCL1 and other neuroendocrine markers, can be induced by canonical DLL4-NOTCH signaling, and the NOTCH target gene REST is necessary and sufficient for their formation(15). Functionally, their presence promotes the growth of the bulk ASCL1 positive neuroendocrine cells in the tumor. These HES1-positive non-neuroendocrine cells are more resistant to standard chemotherapies compared to their neuroendocrine counterparts, which is supported by human data showing increased NOTCH expression in CDX-models from SCLC patients after chemotherapy resistance(17). Additionally, c-MYC can drive plasticity towards a non-neuroendocrine state during tumor evolution with high NOTCH and REST expression(18). Together, these data suggest that NOTCH also has a tumor promoting function during SCLC tumor maintenance.
Several studies have now demonstrated that SCLCs are highly plastic and can adopt distinct neuroendocrine and non-neuroendocrine fates(15,16,18–23). Non-neuroendocrine adherent/mesenchymal subpopulations within SCLC tumors promote SCLC tumorigenesis and metastasis(19) and share many common features with HES1-positive non-neuroendocrine cells(15). These subpopulations are present in SCLC GEMMs, spontaneously form in culture from primary cells derived from SCLC GEMM tumors, display loss of the oncogenic neuroendocrine regulators ASCL1 and MYCL, and can promote metastasis of their neuroendocrine counterparts(19). Moreover, two recent papers found that non-neuroendocrine SCLCs have restored antigen presentation and are enriched for anti-tumor immune responses with PD-1/PD-L1 immunotherapy(21,22).
Our laboratory recently developed a CRISPR-based SCLC GEMM using an adenovirus encoding Cre-recombinase and sgRNAs targeting Rb1, Trp53, and Rbl2 (RPP) to intratracheally inject into lox-stop-lox (LSL)-Cas9 mice. Somatic CRISPR/Cas9 editing of RPP in the lung results in SCLC tumors (referred to hereafter as CRISPR RPP GEMM)(16). Our model has the capability to inactivate a candidate target gene of interest at tumor initiation and study its role during SCLC tumorigenesis. The selection for LOF NOTCH mutations in human SCLC is paradoxical given data demonstrating that NOTCH functions both as a tumor suppressor and tumor promoter. We therefore used our CRISPR RPP GEMM to generate Notch1-Mutant or Notch2-mutant SCLCs and study the functional role and consequences of LOF Notch1 or Notch2 mutations during SCLC tumorigenesis.
Materials and Methods
Cell Lines and Cell Culture
NCI-H1694 (obtained 11/2018) were obtained from American Type Culture Collection (ATCC). CORL47 were obtained from Sigma (11/2018). 293FT were from Dr. Myles Brown laboratory at DFCI. NCI-H1694 cells were maintained in DMEM/F12 media supplemented with HITES [10 nM hydrocortisone (Sigma Aldrich # H0135), Insulin-Transferrin-Selenium (Gemini #400–145), and 10 nM beta-estradiol (Sigma Aldrich# E2257)]. CORL47 cells were maintained in RPMI medium. 293FT cells were maintained in DMEM media. The mouse cell lines derived from genetically-engineered small cell lung cancer (SCLC) mouse tumors (see below for description of cell line generation) were maintained in RPMI-1640 media supplemented with HITES. All media was supplemented with 10% fetal bovine serum (FBS, Gemini), 100 U/mL penicillin, and 100 μg/mL streptomycin except for NCI-H1694 cells, where the media was supplemented with 5% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Early passage cells of all cell lines listed above were tested for Mycoplasma (Lonza #LT07–218) and then were frozen using Bambanker’s freezing media (Bulldog Bio). The cells were last tested for mycoplasma in 06/2021. Cells were then maintained in culture for <3 months at which point new early passage vials were thawed. Nearly all experiments were performed in cells within the first 6 weeks after thawing. Where indicated, the following chemicals (stored at −20°C) were also added to the media as indicated in the text: Doxycycline (Sigma, stock 1 mg/mL in H20), Compound E (EMD Millipore # 565790, stock 2 mM in DMSO), ADU-S100 (ChemieTek # CT-ADU-S100, stock 50 mM in H20), and 2’3’-cGAMP (InvivoGen # tlrl-nacga23, stock 10 mg/ml in H20).
Adenovirus Production and Purification
5 μg of the adenovirus vector (pAd/PL Invitrogen #V494-20) containing the desired sgRNA sequences and Cre recombinase expression cassette (see Adenoviral Cloning section in Supplementary Methods) was digested with PacI (New England Biolabs) for 2 hours at 37°C according to the manufacturer’s instructions and column purified using Qiagen’s gel extraction kit. 1 μg of PacI-digested pAd/PL was transfected into 1.5 × 106 293AD cells plated on a 6 cm tissue-culture dish using Lipofectamine 2000. The following day, the media was exchanged, and subsequently every 48 hours thereafter. Once 293AD cells showed evidence of adenovirus production (determined by comet formation with lysis), the cells and supernatant were harvested, which were then subjected to 4 freeze-thaw cycles by alternating between an ethanol dry ice bath and 37°C. Cell debris was removed by centrifugation and the supernatant was collected, passed through a 0.45 μm filter, aliquoted, and frozen at −80 °C until use.
To generate high titer adenovirus for in vivo experiments, adenovirus was generated as described above. 50 μl of the adenovirus stock was added to each 10 cm tissue-culture dish of 293FT cells plated at 3 × 106 cells per dish (4 10 cm dishes in total for each purification). When 293FT cells showed evidence of adenovirus production, as determined by cell rounding and partial detachment (~48–72 hours after addition of adenoviral stock), the cells were collected, and adenovirus was purified using Virabind Adenovirus Purification Kit (VPK-100). The purified adenovirus was then dialyzed into PBS at 4° C overnight. Adenovirus was titered using QuickTiter Adenovirus Quantitation Kit (Cell Biolabs ##VPK106) according to the manufacturer’s instructions.
Mouse Experiments
All mouse experiments complied with National Institutes of Health guidelines and were approved by Dana-Farber Cancer Institute Animal Care and Use Committee (DFCI, protocol 19–009).
Intratracheal Injections
Intratracheal injections were performed as described previously(24). Briefly, mice were anesthetized with ketamine and xylazine and pedal reflexes were monitored to ensure adequate anesthesia. Mice were maintained on a heated stage at 37° C while anesthetized. Mice were hung on stage with their top incisors and intubated with a 22-gauge 1 inch catheter (ThermoFisher Scientific #1484120). Once intubated, adenovirus (4 × 108 VP/mouse) in a total volume of 75 μl (diluted in PBS) was added to the catheter and subsequently inhaled by the mice.
Generation of Genetically-Engineered Mouse Models of SCLC using CRISPR/Cas9
For all experiments, Lox-stop-lox (LSL) Cas9 BL6NJ mice (Jackson No. 026556) were crossed with lox-stop-lox (LSL) FLUC mice (Jackson No. 005125) yielding progeny that were heterozygous for LSL-Cas9/+, LSL-FLUC/+, whose genotypes were confirmed (Transnetyx). Of note, the intention was to use the firefly luciferase for bioluminescence imaging (BLI) to track tumor burden over time. However, at early timepoints comparing BLI to MRI imaging, we found that BLI imaging yielded several false negatives for unclear reasons. Therefore, we carried out the study with lung MRI imaging only. 3–4 month-old transgenic LSL-Cas9/+, LSL-FLUC/+ were intratracheally injected with adenovirus (4 × 108 VP/mouse) encoding effective sgRNAs targeting Rb1, Trp53, and Rbl2 (RPP, also known as RPR2) and CMV-Cre recombinase that also encoded either an effective sgRNA targeting Notch1#1 (sgNotch1 RPP), Notch2#1 (sgNotch2 RPP), Ascl1#4 (sgAscl1 RPP) or a non-targeting sgRNA as a control (sgControl RPP). MRI of the lungs were performed on mice beginning 8 months after intratracheal injection and were performed monthly thereafter until the mice became symptomatic defined as weight loss >15%, dyspnea, or had lung tumor volumes >200 mm3, and were then euthanized. Tumor volumes were calculated by lung MRIs (see Mouse MRI Imaging in Supplementary Methods).
Upon euthanization, ~1/3 of the lung tumor was immediately flash frozen in dry ice for subsequent DNA and RNA analysis, ~1/3 was fixed in 10% formalin for 24 hours and then stored in 70% ethanol, and the remaining 1/3 was used to establish cell lines (see Method section below). Liver was also harvested and fixed as described above to determine metastatic burden. The tissues were then embedded in paraffin. Slides were made for hematoxylin and Eosin (H&E) and immunohistochemistry (IHC) staining and H&E slides were analyzed by Dr. Rod Bronson for diagnosis.
For experiments in Figure S2F only, LSL-Cas9 BL6J mice homozygous mice were used (Jackson No. 026175).
Generation of cell lines from mouse SCLC tumors
To generate cell lines from tumors, LSL-Cas9/+, LSL-FLUC/+ mice (as described above) that grew tumors and were at their endpoint as defined above were euthanized and their tumors were quickly extracted, washed in ice cold PBS, and minced several times using an ethanol sterilized razor blade. 3 mLs of collagenase/hyaluronidase (Stem cell biology #07912) diluted 1:10 in complete RPMI (10% FBS, P/S, and HITES), and 1 mL dispase (Corning # 354235) was added to the tumor and incubated at 37°C for 20–40 minutes (until most tumor cells were dissociated). The cells were then collected, centrifuged at 1000 rpm for 5 minutes, resuspended in RPMI HITES media, filtered through a 70 μm cell strainer (BD #352350), centrifuged again at 1000 rpm for 5 minutes, resuspended in fresh complete RPMI HITES media and placed in ultra-low adherence tissue culture dishes (Corning #3471). Media was subsequently replaced every 3 days.
SCLC adherent cells derivatives of the parental cell line was established by plating cells on tissue culture plates at 200,000 cells/mL for 4 days and the supernatant containing suspension cells was removed and replacing with fresh complete media every ~3 days until the adherent cell lines were established.
RNA-Sequencing
For the RNA sequencing (seq) experiment using the SCLC tumors derived from the CRISPR-based SCLC GEMM (Fig. 3), tumors were harvested at necropsy and were flash-frozen. RNA was extracted using RNeasy mini kit including a DNase digestion step according to the manufacturer’s instructions and RNA sequencing was performed as described below.
Figure 3. Notch Inactivation Drives SCLCs with High L-Myc Expression.

(A) Hierarchical clustering heatmap from RNA-seq data of SCLC tumors generated with the indicated RPP adenoviruses. Red denotes genes with high expression levels, and blue denotes genes with low expression levels. The color ranging from red to blue indicates log10(FPKM+1) value from large to small. n=4 sgControl RPP lung tumors, n=4 sgNotch1 loss of function (LOF) RPP lung tumors, n=3 sgNotch1 heterozygous mutant (Hets) RPP lung tumors, n=5 lung tumors for sgNotch2 RPP lung tumors, and n=2 sgAscl1 RPP lung tumors. (B) Ascl1, Insm1, Neurod1, and Pou2f3 mRNA expression from the RNA-seq experiment in A. (C & D) Gene set enrichment analysis (GSEA) from the RNA-seq experiment in A using the hallmarks gene sets comparing sgNotch1 LOF RPP vs. sgControl RPP (C), or sgNotch2 RPP vs. sgControl RPP (D). q-values are indicated. (E) Immunoblot analysis using SCLC cell lines that are Notch-WT (from sgControl RPP tumors), Notch1-Mutant (from sgNotch1 RPP tumors), or Notch2-Mutant (from sgNotch2 RPP tumors), derived from SCLC tumors generated in our CRISPR-based SCLC GEMM. Human SCLC cell lines that are ASCL1 positive (CORL47) or ASCL1 negative/NOTCH1 positive (NCI-H1694) are included as controls. (F) Mycl, Myc, and Mycn mRNA expression relative to Actb, as determined by RT-qPCR, in the primary cell lines shown in E. n=3 biological replicates. (G) Mycl mRNA expression from F normalized to Actb. (H) Mycl mRNA data from G comparing all Notch-Mutant primary SCLC cell lines to all Notch-WT primary cell lines. (I, J, K), MYCL1 (I), MYC (J) and MYCN (K) mRNA expression of human SCLC tumors that lacked copy number alterations in MYC-family members and were either NOTCH-WT (n=18) or NOTCH-Mutant (n=8). p-values are indicated. (L) Immunoblot analysis of Notch-WT 703 and K93 adherent subpopulations of SCLC primary cell lines treated with the gamma secretase inhibitor Compound E at 1 μM for 4 days. (M) Mycl mRNA expression as determined by RT-qPCR of the cells treated as in L with Compound E. n=5 biological replicates. (N) Immunoblot analysis of the human CORL47 SCLC cell line first infected with a DOX-ON NOTCH1-ICD (N1-ICD) then grown in the presence or absence of DOX, as indicated, for 15 days. (O) MYCL mRNA expression determined by RT-qPCR of the cells from N. n =3 biological replicates. For all other panels, *=p<0.05, **=p<0.01.
For the RNA-seq experiment in mouse SCLC adherent cells (Fig. 5), cells were counted using Vi-Cell XR Cell Counter and plated at 200,000 cells/mL in one well of 6-well plate. Cells were harvested 72 hours later and RNA was extracted as above.
Figure 5. Notch2-Mutant Adherent Subpopulations Have Enriched Interferon and STING Signaling Compared to Notch-WT Adherent Subpopulations.

(A) Principal component analysis (PCA) of gene expression from RNA-seq data of 3 Notch-WT (K47, K93, 703 from sgControl RPP) established SCLC adherent cell lines and 3 Notch-Mutant [1(188–2) from sgNotch1 RPP and 2(K60, K62) sgNotch2 RPP] established SCLC adherent cell lines. n=2 biological replicates of each cell line. (B) Unsupervised hierarchical clustering heat map of top 500 upregulated and top 500 downregulated genes in Notch-WT vs. Notch-Mutant adherent cells from the RNA-seq experiment in A. (C) Heat map showing expression of canonical Notch target genes in Notch-WT Adherent vs Notch-Mutant Adherent cells. For B&C, the red to blue color scale indicates FPKM values from large to small. (D-G) GSEA comparing Notch-Mutant adherent cells vs. Notch-WT adherent cells using the cell type signature gene sets (D, E), top 100 neuroendocrine marker gene set (F), and interferon gamma response gene set (G). For D-G, FDR q-values are indicated. (H) Immunoblot analysis of established Notch-Mutant adherent cells and Notch-WT adherent cells with the antibodies indicated. The established suspension counterpart for each cell line is included on the right for comparison. (I) STING IHC of human NOTCH-WT or NOTCH2-Mutant SCLC tumors. Scale Bar=50 μM. (J) Quantification of CXCL10 secretion in the media by ELISA of established adherent SCLC cell lines treated with the STING agonist ADU-S100 or ddH20 for 24 hours. IFN-β was used as a positive control. n=3 biological replicates. (K) Cell counts of established adherent cells treated with ADU-S100 for 3 days normalized to the ddH20 control. n=4 biological replicates. (L) Quantification of CXCL10 secretion in the media by ELISA of K47 Notch-WT adherent SCLC cell line pre-treated with Compound E (1 μM) or DMSO for 24 hours and then ADU-S100 or ddH20 for an additional 24 hours. n=3 biological replicates. For all panels, *=p < 0.05, **=p<0.01, ****=p<0.0001 where indicated.
Total RNA samples in each experiment were submitted to Novogene Inc. The libraries for RNA-seq are prepared using NEBNext Ultra II non-stranded kit. Paired end 150bp sequencing was performed on Novaseq6000 sequencer using S4 flow cell. Sequencing reads were mapped to the mm10 genome by STAR. Statistics for differentially expressed genes were calculated by DESeq2.
RNA-Seq Analysis
For Gene Set Enrichment Analysis (GSEA), software was downloaded from the Gene Set Enrichment Analysis website [http://www.broad.mit.edu/gsea/downloads.jsp]. GSEA was performed using the ‘Hallmark’, cell type signature gene sets, and neuroendocrine gene sets. Gene Sets with an FDR<0.25 and a nominal p-value of <0.05 were considered significant.
For principal component analysis and cluster analysis, top 500 genes in terms of the largest standard deviation were subjected to principal component analysis using the prcomp function of R software. Clustering and heatmap were generated using the heatmap.2 function in the gplots package of R software.
Transcriptional regulator and motif enrichment analyses were performed using epigenetic Landscape in Silico deletion Analysis (LISA), 200 top up-regulated and 200 top down-regulated genes from the Notch-Mutant vs. Notch-WT adherent cell RNA-seq experiment were used as the input for Fig. 6A, our previous 631RPP adherent vs. 631RPP suspension RNA-Seq dataset was used for Fig. 6B(22), and Notch-Mutant adherent cell line vs. Notch-WT SCLC tumors were used for Fig. S11C.
Figure 6. Runx2 Promotes Rest expression and is Necessary for Neuroendocrine to Non-Neuroendocrine Plasticity.
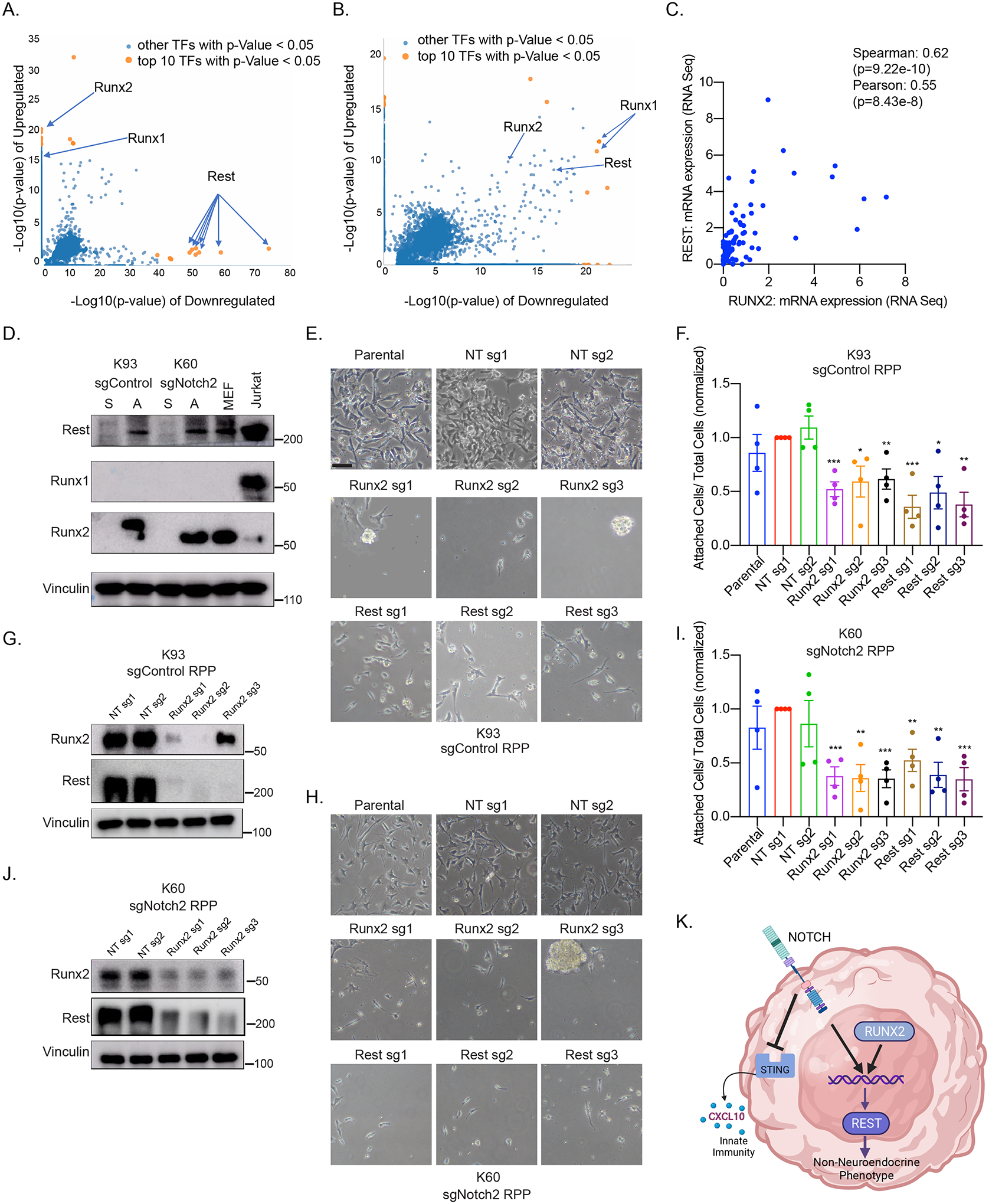
(A, B) Epigenetic Landscape In Silico deletion Analysis (LISA) using the top 200 upregulated and top 200 downregulated genes from our RNA-seq data comparing Notch-Mutant adherent cells vs. Notch-WT adherent cells (A) or comparing matched suspension 631RPP adherent cells vs. 631RPP suspension cells (B). (C) Correlation analysis RUNX2 vs. REST mRNA expression using publicly available RNA-seq data (4) from 81 human SCLC tumor samples. Spearman and Pearson correlation coefficients are indicated. (D) Immunoblot analysis of both suspension (S) and newly formed adherent (A) cells of the cell lines indicated. MEFs and Jurkat cells are included for benchmark controls for Runx expression. (E, F, H, I) Representative brightfield images (E&H) and quantitation of cell counts (F&I) of adherent cells formed from neuroendocrine suspension K93 Notch-WT (E, F) and K60 Notch2-Mutant (H, I) cells first infected with the indicated sgRNAs for CRISPR inactivation of Runx2 or Rest and then plated on tissue-culture treated plastic for 4 days to allow adherent cell formation. n=4 biological replicates. Scale Bar=100 μM. (G & J) Immunoblot analysis of newly formed K93 Notch-WT adherent cells (G) or K60 Notch2-Mutant adherent cells (J) derived from suspension cultures where Runx2 was CRISPR inactivated. NT=non-targeting sgRNA. (K) Model summarizing our findings. For all panels, *=p < 0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001 where indicated.
Analysis of publicly available cohort data
For the expression of MYC, MYCL and MYCN, the processed RNA-Seq, CNV, and SNV data was previously published(4). The samples were divided into two groups of NOTCH-Mutant when there was at least one short variation observed in any NOTCH family genes. Otherwise the sample was called NOTCH WT. For the analysis in Fig. 3I–K, only samples with copy number information were included (n=29 total) and any samples with copy number amplifications of MYC family members were excluded (n=3) and therefore 26 samples were analyzed. The gene expression of MYC, MYCL and MYCN between two groups was represented in the bar plot. Mann-Whitney test was used to calculate the p-value between two distributions. The analysis in Supplementary Fig. S4A–C comparing NOTCH-Mutant vs. NOTCH-WT included all 81 SCLC human samples with RNA-sequencing data(4).
For the co-expression of RUNX1, RUNX2, REST, AXL, TAP1, POU2F3, ASCL1, NEUROD1, and INSM1 in SCLC tumors, the processed RNA-Seq data was previously published(4) and data were downloaded from cBioPortal. All 81 samples were included in the analysis.
For the expression of STAT1, the RNA-seq of SCLC cell lines was from CCLE(25). The samples were divided into two groups of NOTCH-Mutant and NOTCH-WT. The gene expression of STAT1 between two groups was represented in bar plot. Mann-Whitney test was used to calculate the p-value between two distributions.
Cistrome Data Browser was used to identify potential regulators of REST and the transcription factors and chromatin regulators binding to REST. For potential Rest regulators, 1kb to mouse Rest transcriptional start site (chr5:77265493:77283696:NM_011263) was analyzed. For the transcription factors and chromatin regulators binding to REST, 1kb to human REST transcriptional start site (chr4:56906899–56907899) was analyzed.
Mouse SCLC Attachment Assays
To determine which primary mouse SCLC cells could form attached non-neuroendocrine subpopulations, mouse SCLC suspension cells that were previously only grown in ultra-low attachment flasks were counted on day 0 using a Vi-Cell XR Cell Counter and plated in tissue culture-treated 6 well plates at 200,000 cells/mL in 2 mL of complete media. After 3 days, all cells were collected, centrifuged, and replated in fresh media into the same well. After 6 days, representative images were acquired using brightfield microscopy with a 10X objective and then stained with crystal violet for visualization of the entire well.
For the attachment experiments with Compound E, mouse SCLC suspension cells that were previously only grown in ultra-low attachment flasks were counted on day 0 using a Vi-Cell XR Cell Counter and plated in tissue culture-treated 6-well plates at 100,000 cells/mL in 2 mLs with 1 μM Compound E or DMSO. 4 days later representative images were acquired using brightfield microscopy with a 10X objective and then stained with crystal violet for visualization of the entire well. Attached cells were counted using a Vi-Cell XR Cell Counter and harvested for immunoblot analysis.
STING Agonist Experiments
For the CXCL10 ELISA experiments, mouse SCLC adherent cells were counted on day 0 using a Vi-Cell XR Cell Counter and plated in tissue culture-treated 12-well plates at 300,000 cells/mL in 1 mL of complete media. Once the cells attached, they were then treated with STING Agonist ADU-S100 (10 or 50 μM), 2’3’-cGAMP (10 μg/ml) or ddH20. The concentrations of ADU-S100 of 10 or 50 μM were chosen based on the original publication from Aduro BioTech Inc. describing these cyclic dinucleotide derivatives(26). 24 hours later, conditioned media from the cells was collected. Mouse CXCL10 ELISA (#DY466, R&D Systems, Minneapolis, MN) was performed according to manufacturer’s instructions. The cell counting assays were performed as described above except that the cells were plated at 50,000 cells/mL in 1 mL of complete media and counted 72 hours later using a Vi-Cell XR Cell Counter. The combination Compound E and ADU-S100 CXCL10 ELISAs were performed as above with the following modification: K47 SCLC adherent cells were pretreated with 1 μM Compound E or DMSO for 24 hours before the ADU-S100 was added.
Runx2 Rest sgRNA Attachment Experiments
K93 Notch-WT and K60 Notch-Mutant mouse SCLC suspension cells were first infected with lentiviruses encoding an sgRNA targeting Runx2 (sgRunx2 #1,#2,#3), Rest (sgRest #1,#2,#3) or a nontargeting sgRNAs (non-targeting sgRNA #1,#2). Puromycin-resistant cells, indicative of successful infection with the sgRNA lentiviruses, were counted using the Vi-Cell XR Cell Counter and plated in tissue culture-treated 6-well plates at 100,000 cells/mL in 2 mLs in complete media. After 4 days, representative images were acquired using brightfield microscopy with a 10X objective. Both the attached population and suspension populations in the same well were counted independently and an identical plate was used to count the total cells (both suspension and adherent) using a Vi-Cell XR Cell Counter. The fraction of attached cells relative to the total number of cells (adherent + suspension) is plotted to account for any differences in proliferation during the 4-day assay. The data was then normalized to the non-targeting sgRNA #1. Isolated attached cells (from a separate plating experiment with identical design) were harvested for immunoblot analysis.
Statistical Analysis
For the RNA-seq experiments and GSEA analysis in Figs. 3 and 5, and Supplemental Fig. S11, statistical significance was calculated using FDR corrected for multiple hypothesis testing where q-value of <0.25 is considered statistically significant.
For the in vivo studies using sgNotch1 RPP, sgNotch2 RPP, sgAscl1 RPP, and sgControl RPP in Fig. 1, Chi-Square test for trend was used to determine the p-value of % of mice with lung lesions in Fig. 1E, Gehan-Breslow-Wilcoxon test was used to determine the p-value of the Kaplan-Meier survival analysis in Fig. 1G.
Figure 1. Loss of Notch1 or Notch2 Accelerates SCLC Tumorigenesis.

(A) Schematic of the adenovirus used to infect lox-stop-lox (LSL)-Cas9 mice. RPP = sgRb1, sgTrp53, sgRbl2. sg “T”= sgNotch1, sgNotch2, sgAscl1 or sgControl (nontargeting sgRNA). (B) Immunoblot analysis of MEFs expressing Cas9 infected with the sgControl RPP, sgNotch1 RPP, or sgNotch2 RPP adenoviruses as indicated. (C) Immunoblot analysis of sgControl RPP 631 cells (top) or MEFs expressing Cas9 (bottom) infected with the sgControl RPP and sgAscl1 RPP adenoviruses as indicated. (D) Schematic of the intratracheal injection (IT) approach to deliver adenoviruses to the lungs of mice. (E) Quantification of the percent of mice with lung lesions determined by monthly lung MRIs up until 12 months. For sgNotch2 RPP or sgAscl1 RPP vs. sgControl RPP, p<0.0001; for sgNotch1 RPP vs sgControl RPP p<0.05 determined by Chi-Square Test for Trend. (F) Tumor volume measurements of individual mice determined by lung MRIs at the times indicated (in months) after IT injection of the adenovirus indicated into LSL-Cas9 mice. (G) Median overall survival up until 18 months of LSL-Cas9 mice IT injected with the indicated adenoviruses. p=0.1579 for sgNotch1 RPP vs. sgControl RPP, p=0.1860 for sgNotch2 RPP vs. sgControl RPP, p=0.024 for sgAscl1 RPP vs. sgControl RPP. For E-G, n=10 mice (sgNotch1 RPP), n=10 mice (sgNotch2 RPP), n=7 mice (sgAscl1 RPP) or n=8 mice (sgControl RPP). (H) Representative lung MRIs of mice with lung lesions at the times indicated (in months) after IT injection of the indicated adenoviruses.
For all other experiments, statistical significance was calculated using unpaired, two-tailed Students t-test. p-values were considered statistically significant if the p-value was <0.05. For all figures, * indicates p-value <0.05, ** indicates p-value <0.01, *** indicates p-value <0.001, and **** indicates p-value <0.0001. Error bars represent SEM unless otherwise indicated.
Data and Materials Availability
Data from RNA-seq experiments will be deposited in the GEO database prior to publication. All FPKM RNA-seq data is included in the supplementary tables. All other data and materials can be requested from the corresponding author upon reasonable request.
Results
Genetic Inactivation of Notch1 or Notch2 Promotes SCLC Tumorigenesis In Vivo
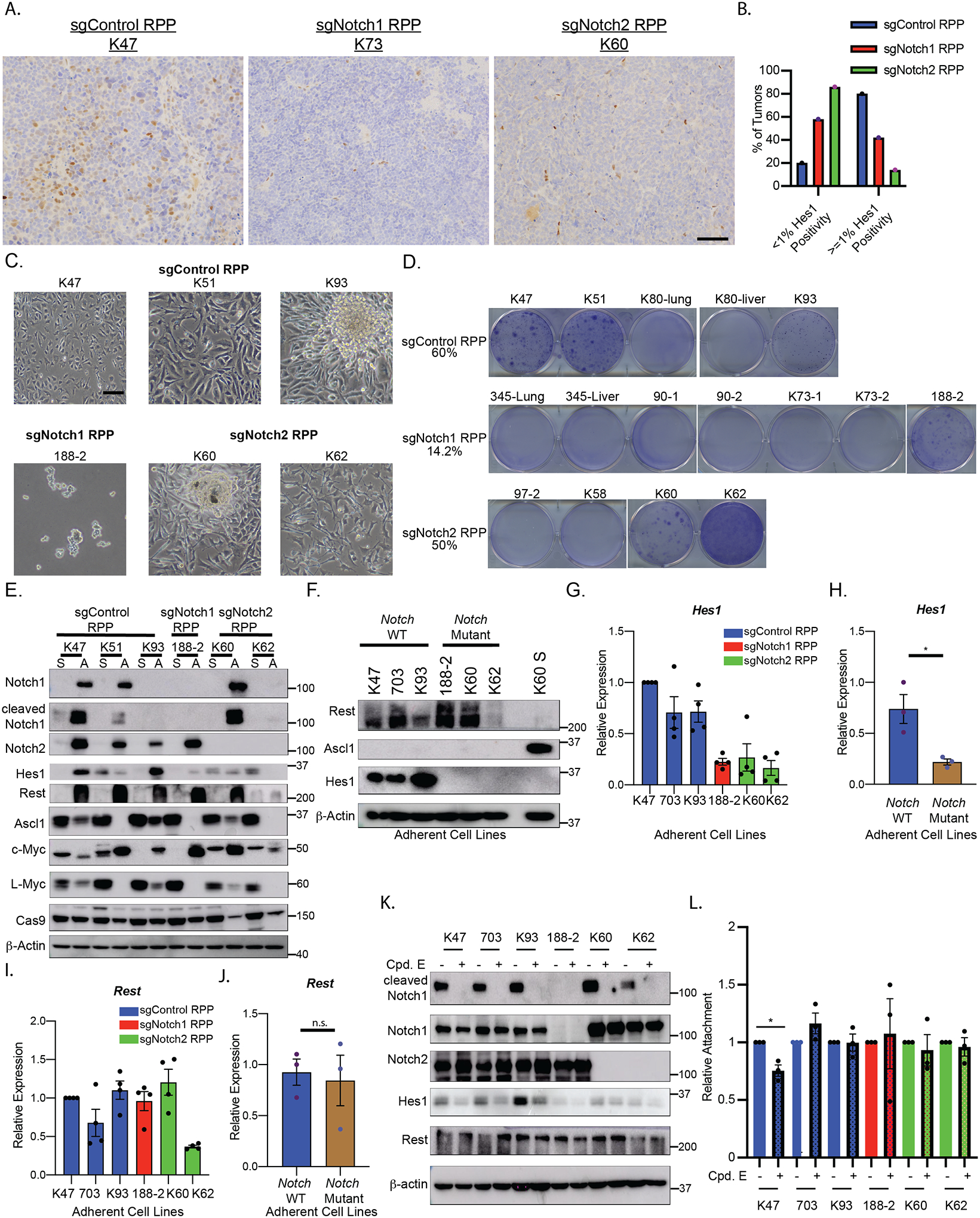
Since LOF NOTCH mutations are found in ~25% of human SCLCs, we hypothesized that genetic inactivation of NOTCH1 or NOTCH2 would accelerate SCLC tumorigenesis. To test this, we used our SCLC CRISPR RPP (also known as RPR2) GEMM(16) to generate autochthonous SCLCs that were Notch1-Mutant, Notch2-Mutant, or Notch-WT (Fig. 1A). We also used the same approach to generate autochthonous SCLCs with Ascl1 deletion (Fig. 1A), as we hypothesized that Ascl1 loss would have the opposite effect of Notch inactivation, and also to compare negative selection in our CRISPR RPP GEMM with the traditional RPP GEMM where Ascl1 is required for SCLC tumorigenesis(9). We first validated that individual sgRNAs targeting Notch1, Notch2, or Ascl1 effectively knocked out their intended target genes. We then generated adenoviruses that encoded Cre recombinase and sgRNAs targeting Rb1, Trp53, and Rbl2 (RPP) and Notch1 (sgNotch1 RPP), Notch2 (sgNotch2 RPP), Ascl1 (sgAscl1 RPP), or a non-targeting control (sgControl RPP). We confirmed that adenoviruses transduced into MEFs could simultaneously knock out Notch1, Notch2, or Ascl1 along with RPP (Fig. 1B and 1C).
These sgNotch1 RPP, sgNotch2 RPP, sgAscl1 RPP, or sgControl RPP adenoviruses were then introduced into the lungs of lox-stop-lox (LSL)-Cas9 mice by intratracheal injection (IT) (Fig. 1D). Beginning 8 months after injection, we performed monthly lung MRIs to monitor lung tumors. Lung tumors were detected earlier (Fig. 1E–F, 1H; Supplementary Fig. S1A) in mice injected with sgNotch1 RPP or sgNotch2 RPP compared to mice that received sgControl RPP. Eventually ~100% of mice injected with sgNotch1 RPP, sgNotch2 RPP, and sgControl RPP developed lung tumors. Although not statistically significant by Kaplan-Meier survival estimate, there was a trend toward decreased overall survival in sgNotch1 RPP and sgNotch2 RPP mice compared to sgControl RPP mice (Fig. 1G; Supplementary Fig. S1B). Consistent with a previous study in the traditional RPP GEMM(9), tumor onset was dramatically delayed in sgAscl1 RPP mice, which translated to an increased overall survival compared to sgControl RPP mice (Fig. 1E–H). Only 42.8% (3/7 mice) of the mice injected with sgAscl1 RPP adenovirus developed SCLC lung tumors (Fig. 1E–F; Supplementary Fig. S2D). 0/15 mice in an additional cohort injected with 3 different sgRNAs targeting ASCL1 formed SCLCs demonstrating that Ascl1 is required for SCLC tumorigenesis and that escape after Ascl1 inactivation is a rare event (Fig. 1E; Supplementary Fig. S2E). Collectively, these data demonstrate that loss of Notch1 or Notch2 modestly promotes SCLC tumorigenesis, while loss of Ascl1 inhibits SCLC tumorigenesis.
SCLCs Select for LOF Notch Mutations and In-Frame Deletions in Ascl1 that Restore Partial Ascl1 Expression
We hypothesized that LOF Notch1 and Notch2 mutations were positively selected and LOF Ascl1 mutations were negatively selected during SCLC tumorigenesis. To test this, we performed CRISPR amplicon sequencing on SCLC tumors at the genomic loci targeted by Notch1, Notch2, and Ascl1 sgRNAs. Consistent with a tumor suppressive role for Notch2, all sgNotch2 RPP tumors evaluated had insertions or deletions (indels) leading to LOF frame-shift mutations in Notch2 (Fig. 2A, 2E–F; Supplementary Fig. S2C). Interestingly, all 3 tumors that formed in mice injected with the sgAscl1 RPP harbored large in-frame indels (24, 48, and 57 nucleotides) in roughly half of the Ascl1 sequencing reads, while the remaining sequencing reads had a LOF indel (Fig. 2B, 2E–F; Supplementary Fig. S2D). Large in-frame indels are a rare event during CRISPR/Cas9 editing and did not occur in MEFs infected with the Ascl1 sgRNA (Supplementary Fig. S2F) demonstrating strong selective pressure to retain 1 functional copy of Ascl1 during SCLC tumorigenesis. Finally, 11 of 15 sgNotch1 RPP tumors examined had indels that led to complete LOF frame-shift mutations in Notch1 (Fig. 2C, 2E–F; Supplementary Fig. S2B). 27% (4 of 15) of the sgNotch1 RPP lung tumors examined had larger in-frame deletions in roughly half of the sequencing reads, while the remaining sequencing reads harbored a LOF indel (Fig. 2D–F; Supplementary Fig. S2B). This was unexpected given that the survival of the mice with these tumors were the same as the tumors with complete LOF Notch1 frameshift mutations (Supplementary Fig. S1B and S1C), and shorter than the sgControl RPP tumors (Supplementary Fig. S1B) suggesting that in some mice there was selective pressure for SCLCs to have the capability to restore Notch1 expression.
Figure 2. SCLC Tumors Select for LOF Notch Mutations and In-Frame Deletions to Restore Ascl1 Expression.

(A-D) Representative examples of CRISPR amplicon sequencing of lung tumors that developed in LSL-Cas9 mice injected with the sgNotch2 RPP (A), sgAscl1 RPP (B) or sgNotch1 RPP (C, D) adenoviruses. sgRNA sequences are labeled in blue within the consensus sequence. Gene editing is highlighted in red. (E, F) CRISPR amplicon sequencing of Notch1, Notch2 or Ascl1 genomic loci from tumors isolated from LSL-Cas9 mice infected with the respective adenoviruses, showing the proportion of mutant and wild-type sequencing reads (E), and the proportion of each type of indel for each tumor (F). FS: frame shift, NFS: non-frame shift. (G) Representative H&E and IHC for Ascl1 in SCLC lung tumors from LSL-Cas9 mice injected with the indicated RPP adenoviruses. Scale Bar=50 μM. (H) Representative IHC for Notch1 and Notch2 from SCLC lung tumors formed in LSL-Cas9 mice injected with the indicated RPP adenoviruses. Scale Bar=50 μM.
We next asked whether the tumors that formed in sgNotch1 RPP, sgNotch2 RPP, and sgAscl1 RPP mice had histological and neuroendocrine features of SCLC. Similar to sgControl RPP tumors, sgNotch1 RPP and sgNotch2 RPP tumors had histological features of SCLC and high Ascl1 expression (Fig. 2G; Supplementary Fig. S2A–C). Immunohistochemistry (IHC) for Notch1 or Notch2 showed that sgControl RPP tumors had subpopulations of Notch1 or Notch2 positive cells, while sgNotch1 RPP or sgNotch2 RPP tumors did not (Fig. 2H) consistent with previous work demonstrating that Notch is expressed in only a small fraction of cells within SCLC RPP GEMMs(15,16). Consistent with the presence of in-frame Ascl1 indels, the tumors that formed in the sgAscl1 RPP mice had histological features of SCLC, and retained some Ascl1 expression (Fig. 2B and 2G; Supplementary Fig. S2D).
Loss of NOTCH Drives MYCL Expression in SCLC
To understand the transcriptional changes in tumors inactivated for Notch or Ascl1, we performed bulk RNA-seq on sgNotch1 RPP, sgNotch2 RPP, sgAscl1 RPP, and sgControl RPP lung tumors. Based on our Notch1 CRISPR amplicon sequencing results, we segregated sgNotch1 RPP tumors into 2 groups: 1. sgNotch1 RPP LOF tumors, where both copies of Notch1 harbored indels that resulted in LOF or 2. sgNotch1 RPP heterozygous (Het) tumors where 1 copy of Notch1 harbored an in-frame indel. Hierarchical clustering showed that sgNotch1 RPP LOF tumors were transcriptionally distinct from sgNotch1 RPP Het tumors suggesting that this selection had functional consequences on transcription (Fig. 3A; Supplementary Table S1). Consistent with the dramatic effect of Ascl1 deletion on SCLC tumorigenesis, tumors that formed in the sgAscl1 RPP mice were most distinct from all other tumor genotypes (Fig. 3A). Similar to sgControl RPP and previous studies with the RPP GEMM(9,27–29), the sgNotch1 RPP and sgNotch2 RPP tumors had high expression of Ascl1 and Insm1, but not Neurod1 or Pou2f3 (Fig. 3B).
Gene-Set Enrichment Analysis (GSEA) of the RNA-seq data revealed that MYC and E2F target gene signatures were highly enriched in both sgNotch2 RPP and sgNotch1 LOF RPP tumors compared to sgControl RPP tumors, with a greater enrichment in sgNotch2 RPP tumors (Fig. 3C and 3D; Supplementary Table S2) suggesting that MYC pathway activation is linked to Notch inactivation in SCLC. To study this and other mechanisms by which loss of Notch drives SCLC tumorigenesis, we generated SCLC cell lines from sgNotch1 RPP, sgNotch2 RPP, and sgControl RPP tumors. Similar to ASCL1+ human SCLC cell lines, cells derived from sgNotch1 RPP, sgNotch2 RPP, and sgControl RPP tumors grew as clusters of cells in suspension when plated on ultra-low attachment dishes and highly expressed Ascl1 (Fig. 3E; Supplementary Fig. S3A). As expected, Notch1 or Notch2 expression was absent in sgNotch1 RPP and sgNotch2 RPP tumors, respectively. Expression of Hes1, a canonical Notch target gene, was nearly completely lost in all cell lines derived from sgNotch1 RPP or sgNotch2 RPP tumors demonstrating that loss of a single Notch receptor paralog markedly decreases Notch target gene expression. Pan-Myc expression was increased in nearly all sgNotch1 RPP and sgNotch2 RPP cell lines compared the sgControl RPP cell lines (Fig. 3E). RT-qPCR for specific Myc paralogs demonstrated that Mycl was highly upregulated in 4 of the 6 Notch-Mutant primary cell lines compared to sgControl RPP cell lines (Fig. 3F–H; Supplementary Fig. S3B–E) consistent with previous studies showing SCLC RPP GEMM models highly express L-Myc and not the other Myc paralogs(9,27–29).
To further explore the link between Notch mutations and L-Myc expression, we analyzed data of 29 SCLC human tumors where both RNA-seq data and copy number alteration data were available(4). 3 of these tumors had MYC paralog amplifications and were excluded. Consistent with our data in mouse tumors, we found that human NOTCH-Mutant SCLCs had higher expression of MYCL1, but not MYC and MYCN paralogs, compared to NOTCH-WT tumors (Fig. 3I–K). Analysis of the RNA-seq data from all 81 human SCLC tumors(4) showed a similar trend (Supplementary Fig. S4A–C).
We then asked whether Notch activity regulates L-Myc expression. To test this, we isolated subpopulations of adherent Notch-WT mouse SCLC derived from sgControl RPP tumors that had high levels of active Notch (Fig. 3L). Blocking NOTCH activity with the gamma secretase inhibitor Compound E (Fig. 3L; Supplementary Fig. S4D) increased L-Myc expression (Fig. 3M). Conversely, expression of a DOX-ON NOTCH1-ICD (transcriptionally active NOTCH) to induce NOTCH activity (Supplementary Fig. S4E) in the CORL47 human SCLC cell line, which has repressed NOTCH1 expression and activity at baseline (Fig. 3N), decreased MYCL and ASCL1 expression (Fig. 3N and 3O; Supplementary Fig. S4F). Together, these data demonstrate that low NOTCH activity is required for high MYCL expression in SCLC and suggest that NOTCH inactivation is a potential mechanism (and likely indirect given NOTCH canonically functions as a transcriptional activator) to increase MYCL1 expression in SCLC in the absence of a MYCL1 copy number amplification.
Non-Neuroendocrine Adherent SCLC Subpopulations Can Form in the Absence of NOTCH Activity.
NOTCH activity promotes non-neuroendocrine plasticity within SCLC tumors. These cells are identified by HES1-positivity and their formation is dependent on the NOTCH target gene REST (15). We performed HES1 IHC to ask whether these HES1-positive cells were reduced or absent in the sgNotch1 RPP or sgNotch2 RPP tumors. Consistent with previous observations, sgControl RPP tumors contained HES1-positive subpopulations (Fig. 4A and 4B). In contrast, HES1-positive cells were either markedly reduced or completely absent in the majority of sgNotch1 RPP and almost all sgNotch2 RPP tumors (Fig. 4A and 4B), consistent with our findings in Notch-Mutant primary cell lines (Fig. 3E). This was paradoxical given previous data demonstrating a role for these HES1-positive subpopulations in promoting SCLC tumor growth(15) and our data demonstrating that inactivation of Notch, which ablates these HES1-positive subpopulations, accelerates SCLC tumorigenesis (Fig. 1). One explanation to reconcile these observations was that Notch-Mutant tumors selected for Notch-independent mechanisms to promote the formation of non-neuroendocrine adherent cells. Alternatively, it is possible that non-neuroendocrine subpopulations were dispensable for Notch-Mutant SCLC tumorigenesis.
Figure 4. Non-Neuroendocrine Adherent SCLC Subpopulations Can Form in the Absence of NOTCH.
(A) Representative IHC for Hes1 in SCLC lung tumors from LSL-Cas9 mice injected with the indicated RPP adenoviruses. Scale Bar=50 μM. (B) Quantification of Hes1-positive cells in sgControl RPP tumors (n=5), sgNotch1 RPP tumors (n=12) and sgNotch2 RPP tumors (n=7). p=<0.0001 of sgNotch1 RPP or sgNotch2 RPP vs. sgControl RPP determined by Chi-Square Test. (C) Representative brightfield images and (D) crystal violet staining of primary neuroendocrine suspension cell lines plated on tissue culture plastic for 6 days. Scale Bar=100 μM. (E) Immunoblot analysis of both suspension (S) and newly formed adherent (A) SCLC cell lines 6 days after plating. (F) Immunoblot analysis of established adherent SCLC cell lines that are either Notch-WT or Notch-Mutant. (G-J) RT-qPCR for Hes1 (G,H) and Rest (I,J) in the indicated Notch-WT and Notch-Mutant established adherent SCLC cell lines. n = 4 biological replicates. (K) Immunoblot analysis of newly formed adherent SCLC cell lines from suspension cells that were plated on tissue culture plastic and treated with or without Compound E at 1 μM for 4 days. (L) Quantification of the number of attached cells from K that formed when each cell line was plate with Compound E normalized to the same cell line treated with DMSO. n=3 biological replicates. For all panels, *=p < 0.05 where indicated.
To investigate whether non-neuroendocrine subpopulations exist in Notch-Mutant tumors, we plated sgNotch1 RPP, sgNotch2 RPP, and sgControl RPP primary cell lines on tissue culture plastic. 4 days later, some primary cell lines spontaneously adhered to tissue culture (TC) plastic and exhibited a mesenchymal-like phenotype (Fig. 4C and 4D), which has been observed previously(19). 60% of cell lines derived from sgControl RPP tumors and 50% of sgNotch2 RPP tumors formed these adherent subpopulations, while only 1 out of 7 of the sgNotch1 RPP cell lines formed adherent cells (Fig. 4C and 4D). We then established these adherent cells as distinct cell lines from their suspension counterparts. CRISPR-amplicon sequencing for Rb1, Notch1, Notch2 of neuroendocrine suspension cells and non-neuroendocrine adherent cells pairs confirmed that the adherent cell subpopulations harbored identical CRISPR-mediated indels as their suspension neuroendocrine counterparts confirming that they were tumor cells derived from neuroendocrine suspension cells and not contaminating cells from the tumor microenvironment (Supplementary Fig. S5 and S6). Although these data suggest that Notch1 is important for the formation of these adherent cell subpopulations, it is surprising that the Notch-Mutant tumors formed adherent subpopulations at all; particularly the sgNotch2 RPP tumors, which formed adherent subpopulations at the same frequency as the sgControl RPP tumors but had near-absence of HES1-positive subpopulations in tumors (Fig. 4A and 4B).
Adherent cell lines derived from sgControl RPP, sgNotch1 RPP, and sgNotch2 RPP tumors had reduced expression of Ascl1 and Mycl compared to their suspension counterparts (Fig. 4E; Supplementary Fig. S7A–C). In sgControl RPP adherent cell lines, Notch, Hes1, and Rest expression was increased relative to their neuroendocrine suspension counterparts (Fig. 4E; Supplementary Fig. S7D and S7E), which was observed previously(15). Consistent with our data in Notch-Mutant SCLC tumors, the sgNotch1 RPP and sgNotch2 RPP adherent subpopulations had markedly reduced expression of Hes1 demonstrating that canonical Notch signaling was decreased in Notch-Mutant adherent cells compared to Notch-WT adherent cells (Fig. 4E–H; Supplementary Fig. S7D). Surprisingly, Rest mRNA expression was increased in Notch-Mutant adherent cells compared to their suspension counterparts, and expressed at similar levels when compared to sgControl RPP adherent cell lines despite Notch inactivation and their reduction in Hes1 expression (Fig. 4F–J; Supplementary Fig. S7E) suggesting selection for Notch-independent mechanisms to activate Rest in sgNotch1 RPP and sgNotch2 RPP tumors. Loss of Ascl1 and gain of Rest persisted after replating adherent subpopulations on ultra-low attachment dishes suggesting that gene expression changes in adherent cells were not simply a consequence of plating the neuroendocrine cultures on TC plastic (Supplementary Fig. S7F and S7G).
One trivial explanation for this would be that Rest expression is regulated by residual Notch activity in the Notch-Mutant adherent cells. To test this, we used Compound E to block any residual Notch activity in the Notch-Mutant suspension cells (Fig. 4K). Compound E treatment did not block the formation of adherent subpopulations in all Notch-Mutant cell lines and only partially blocked the formation of adherent subpopulations in 1 of the 3 Notch-WT cell lines (Fig. 4L; Supplementary Fig. S8A and S8B) demonstrating that Notch activity is not required for adherent cell formation in all Notch-Mutant cell lines and some Notch-WT cell lines. Furthermore, Rest expression in newly formed adherent cells was not significantly altered by Compound E, which effectively blocked all Notch activity and decreased Hes1 levels (Fig. 4K), consistent with a recent publication demonstrating that Notch activity is not required for Rest expression(30). Consistent with prior work(15), re-expression of transcriptionally active NOTCH markedly induced REST expression and promoted an adherent cell phenotype in the CORL47 cells (Supplementary Fig. S4G and S4H) demonstrating that increasing NOTCH activity is sufficient to induce REST expression and non-neuroendocrine plasticity. However, our data clearly suggest that additional NOTCH independent mechanisms are capable of inducing REST and promoting non-neuroendocrine plasticity in SCLC.
Notch2-Mutant Adherent Subpopulations Have Enriched Interferon and STING Signaling Compared Notch-WT Adherent Subpopulations
To examine whether loss of NOTCH might alter the non-neuroendocrine phenotype, we performed RNA-seq of adherent cell lines derived from 3 Notch-WT tumors compared to 3 Notch-Mutant tumors (Fig. 4F). Principal component analysis (PCA) and unsupervised hierarchical clustering analysis revealed significant differences between the Notch-Mutant adherent cells compared to the Notch-WT adherent cells (Fig. 5A and 5B; Supplementary Table S3). Consistent with their genotype, Notch-Mutant adherent cells had lower expression of canonical Notch target genes demonstrating that inactivation of a single Notch receptor paralog is sufficient to decrease canonical Notch target gene expression (Fig. 5C), with the exception being REST, which we found is regulated in a Notch-independent manner (Fig. 4K and 4L).
To better understand the differences between Notch-Mutant and Notch-WT adherent cells, we performed GSEA analysis using cell type signature gene sets. Notch-WT adherent cells were more similar to neurons and enteroendocrine cells (Fig. 5D; Supplementary Fig. S9A; Supplementary Table S4), while Notch-Mutant adherent cells were most similar to mesenchymal and fibroblast cells (Fig. 5E; Supplementary Fig. S9B) suggesting that Notch confines the plasticity of SCLC within the neuroendocrine state, while loss of Notch selects for fibroblastic non-neuroendocrine plasticity. Consistent with this, the gene set for top 100 neuroendocrine markers(31) was decreased in the Notch-Mutant adherent cells compared to Notch-WT adherent cells (Fig. 5F; Supplementary Fig. S9C).
GSEA for hallmarks gene sets revealed that Notch-Mutant adherent cells were significantly enriched in interferon signaling, the inflammatory response, and JAK-STAT signaling (Fig. 5G; Supplementary Fig. S9D–F). Immunoblot analysis revealed that phospho-STAT1, total STAT1, and stimulator of interferon genes (STING) protein levels were selectively increased in the Notch2-Mutant adherent cells compared to the Notch-WT adherent cells (Fig. 5H). IHC for STING expression in the limited set of NOTCH2-mutant human SCLCs that were available revealed that all NOTCH2-mutant tumors examined had high STING expression while none of the NOTCH-WT tumors expressed STING (Fig. 5I). Although a limited sample size, these findings were intriguing as STING protein expression is largely absent in human SCLCs(20,22). Also consistent with these findings, total STAT1 mRNA expression was modestly increased in NOTCH-Mutant SCLC cell lines compared to NOTCH-WT SCLC cell lines (Supplementary Fig. S9G). Treatment of adherent cells with the STING agonist ADU-S100 dramatically induced CXCL10 production (a cytokine used to measure output of the STING pathway) in Notch2-Mutant adherent cells compared to Notch-WT adherent cells to similar levels induced by the interferon-β positive control (Fig. 5J). Consistent with this, ADU-S100 inhibited proliferation in Notch2-Mutant adherent cells, but not Notch-WT adherent cells (Fig. 5K). Similarly, 2’3’-cGAMP, the endogenous second messenger that binds and stimulates STING(32) also selectively inhibited proliferation in Notch2-Mutant adherent cells (Supplementary Fig. S10A), which was likely due to its ability to induce apoptosis in these cells (Supplementary Fig. S10B–G). CXCL10 production induced by ADU-S100 was significantly potentiated by blocking Notch activity with Compound E in K47 Notch-WT adherent cells demonstrating that Notch activity represses STING pathway activation (Fig. 5L). Together, these data show that Notch2-Mutant adherent cells are unique from Notch-WT adherent cells with high interferon signaling, high expression of components of the STING pathway, and are selectively vulnerable to STING agonists.
Runx2 Regulates Rest and is necessary for Neuroendocrine to Non-Neuroendocrine Plasticity
Given that non-neuroendocrine adherent cells form in the absence of Notch activity (Fig. 4L), we performed Epigenetic Landscape In Silico deletion Analysis (LISA)(33) using SCLC RNA-seq data sets to identify candidate master transcription factors that drive non-neuroendocrine plasticity in the absence of Notch. Rest was identified as a top regulator of downregulated genes when comparing Notch-Mutant adherent cells to Notch-WT adherent cells and comparing Notch-Mutant adherent cells to SCLC mouse tumors (Fig. 6A; Supplementary Fig. S11A and S11C; Supplementary Table S5) supporting our findings that Rest remains at least as active, if not more active, in Notch-Mutant adherent cells compared to Notch-WT adherent cells (Fig. 4F, 4I, 4J; Supplementary Fig. S7E). Rest was also one of the top regulators of differentially expressed genes in Notch-WT non-neuroendocrine adherent cells compared to their neuroendocrine suspension counterpart (Fig. 6B). Together, these data nominate Rest as a master repressor that drives non-neuroendocrine plasticity in both Notch-Mutant and Notch-WT SCLC, which is consistent with previous findings showing that Rest is necessary and sufficient to drive non-neuroendocrine plasticity in SCLC(15).
We then used both LISA analysis of RNA-seq data and public ChIP-seq datasets on Cistrome.Db(33–35) to identify candidate transcriptional activators that upregulate Rest expression in a Notch-independent manner. LISA analysis of Notch-Mutant vs. Notch-WT adherent cells and Notch-WT adherent vs. Notch-WT suspension cells both showed that the Runx family of transcription factors are master transcriptional regulators of upregulated genes in adherent non-neuroendocrine SCLC cells with some selectivity in Notch-Mutant adherent cells compared to Notch-WT adherent cells (Fig. 6A and 6B; Supplementary Fig. S11B). Regulatory potential analysis of ChIP-seq data from Cistrome.Db(34,35) 1 kilobase (kb) upstream of the REST transcription start site showed that RUNX2 is one of the top regulators of REST, and RUNX1 is one of the top transcription factors that bind to REST (Supplementary Fig. S11F and S11G). Publicly available ChIP-seq data showed that RUNX1 directly binds to the REST promoter in bone marrow-derived mast cells and RUNX2 directly binds to the REST promoter in preosteoblast cells (Supplementary Fig. S11J). Among the top transcription factors that bind REST, only RUNX and MYC were highly and selectively expressed in Notch-WT adherent cells compared to Notch-WT suspension cells (Supplementary Fig. S11G and S11H). Additionally, RUNX (both RUNX1 and RUNX2) and REST mRNA expression is significantly correlated in human SCLC samples (Fig. 6C; Supplementary Fig. S11I). Interestingly, RUNX2 mRNA expression highly positively correlates with expression of genes that are enriched in the non-neuroendocrine subtypes of SCLC including AXL and TAP1 (Supplementary Fig. S12A and S12B) which are markers of the “inflammatory” subtype of SCLC(21,36) and POU2F3(37) (Supplementary Fig. S12C), and negatively correlates with the neuroendocrine subtype markers ASCL1, NEUROD1, and INSM1 (Supplementary Fig. S12D–F).
Together these findings nominate Runx1 and Runx2 as candidate activators of Rest expression to drive non-neuroendocrine SCLC plasticity. Immunoblot analysis of paired neuroendocrine suspension cells and non-neuroendocrine adherent cells showed that Runx2 and Rest were highly upregulated in adherent non-neuroendocrine subpopulations compared to their neuroendocrine counterparts and that Runx1 was not expressed in either subpopulation (Fig. 6D). To test whether Runx2 and Rest were required for neuroendocrine to non-neuroendocrine plasticity, CRISPR/Cas9 was used to inactivate Runx2 and Rest using multiple independent sgRNAs in the K60 Notch-Mutant and K93 Notch-WT suspension SCLC cell lines. Consistent with previous data, CRISPR inactivation of Rest decreased non-neuroendocrine adherent cell formation (Fig. 6E, 6F, 6H, 6I; Supplementary Fig. S11K, and S11L). Similarly, CRISPR inactivation of Runx2 decreased the formation of non-neuroendocrine adherent cells and also dramatically downregulated REST expression in both Notch-WT and Notch-Mutant cell lines demonstrating that Runx2 is necessary for the transition from neuroendocrine to non-neuroendocrine adherent cells in both Notch-WT and Notch-Mutant SCLC (Fig. 6E–J). Collectively, our data demonstrate that Notch-Mutant SCLCs can still adopt plasticity states in the absence of canonical Notch signaling and use Runx2 activity to drive Rest and non-neuroendocrine SCLC phenotypes.
Discussion
20–25% of SCLCs select for loss of function (LOF) mutations in the NOTCH receptors with the most frequent mutations found in NOTCH1 or NOTCH2(4). Restoration of NOTCH has potent tumor suppressive effects in SCLC tumors demonstrating that NOTCH has tumor suppressor activity in SCLC(4). Our data showing that genetic inactivation of Notch1 or Notch2 accelerates SCLC tumorigenesis suggests a dominant tumor suppressive role for Notch in SCLC tumorigenesis, which is supported by recent studies demonstrating that inhibition of NOTCH can cooperate with RB1 and TP53 loss to sustain neuroendocrine differentiation of pulmonary neuroendocrine cells during early events of SCLC initiation(38,39) and is consistent with studies in cell lines where reactivation of NOTCH blocks ASCL1 expression and cellular proliferation(13,14,16,23).
Notch also possesses tumor promoting functions in SCLC through its ability to drive non-neuroendocrine subpopulations in vitro and within SCLC tumors in vivo marked by HES1 positivity(15). Our data are consistent with these findings in SCLC tumors and cell lines that are Notch-WT. We find that Notch is sufficient to drive the formation of non-neuroendocrine adherent cells in culture. Notch1 CRISPR-mediated genetic inactivation harbored in-frame insertions/deletions (indels) in 1 Notch1 allele in ~25% of tumors, leaving open the possibility that these tumors selected for the ability to partially restore Notch1 expression. Alternatively, these in-frame indels could partially attenuate Notch1 function given the tumor onset was similar to Notch1-Mutant tumors where both alleles were deleted and slightly accelerated compared to Notch-WT tumors. All Notch2-Mutant tumors had complete loss of Notch2 without any tumors harboring in-frame indels. Both Notch1-Mutant and Notch2-Mutant tumors had near complete depletion of Hes1-positive subpopulations demonstrating that loss of a single Notch receptor, which is analogous to what has been observed in human SCLCs(4), was sufficient to significantly reduce Notch activity. This suggests that HES1-positive non-neuroendocrine subpopulations were not supporting tumorigenesis in these Notch-Mutant mouse models. Notch has recently been shown to have a tumor promoting function in ASCL1 negative SCLC GEMM tumors that are driven by MYC(18). Collectively, these data are consistent with Notch serving a tumor promoting function in established SCLC tumors that are genetically NOTCH-WT or are ASCL1 negative SCLCs driven by MYC, but suggests that NOTCH dominantly functions as a tumor suppressor at earlier stages of SCLC tumorigenesis in the ASCL1 + SCLC subtype(4,23,38,39). It remains an open question whether the tumors in our study that selected for the ability to restore partial Notch1 expression had a selective functional advantage.
This led us to ask the question why would ~25% of human SCLCs select for LOF NOTCH mutations if NOTCH is required to drive non-neuroendocrine subpopulations that support SCLC tumorigenesis? Consistent with previous findings(15), primary cells isolated from Notch-WT tumors spontaneously formed adherent non-neuroendocrine cells that highly express Notch, Hes1, and Rest, and NOTCH was sufficient to drive the formation of HES1-positive, REST-positive non-neuroendocrine cells. Surprisingly, we observed that some primary cells from the Notch-Mutant tumors, primarily Notch2-mutant tumors, were able to form non-neuroendocrine subpopulations with exceedingly low Hes1 levels and still formed when Notch activity was completely blocked with the gamma-secretase inhibitor Compound E demonstrating that these adherent non-neuroendocrine cells formed by Notch-independent mechanisms. Interestingly, all non-neuroendocrine subpopulations from both Notch-Mutant and Notch-WT highly expressed the neuronal transcriptional repressor Rest, which drives non-neuroendocrine differentiation in SCLC(15,18,30,40). We found that Runx2 promotes Rest expression and is necessary for the formation of adherent non-neuroendocrine cells in both Notch-Mutant and Notch-WT SCLC models uncovering a Notch-independent mechanism to drive non-neuroendocrine plasticity in SCLC that converges on Rest (Fig. 6K). Consistent with our study, Runx2 has also recently been identified as an escape mechanism driving osteogenic non-neuroendocrine differentiation in MYC-driven SCLC tumors that are deleted for ASCL1(41). RUNX1 and NOTCH often bind DNA at nearby sites and coordinately regulate target genes in T-ALL(42) and Runx1 rescues defective hematopoiesis in Notch1-null cells(43) providing evidence for their overlapping functions in different cancer types. In neuroendocrine SCLCs, RUNX1 and RUNX2 are largely repressed while RUNX1T1, a fusion protein that contains the RUNX DNA-binding domain at its N-terminus, is highly expressed and can function to promote SCLC proliferation(44). An “inflammatory” subtype of human SCLC has recently been identified and these SCLCs are non-neuroendocrine, have restored antigen presentation, and have prolonged responses to immune checkpoint blockade(21,22). Our data shows that RUNX2 expression is enriched in this subset of tumors and also in the non-neuroendocrine POU2F3 subtype(37). Future studies will focus on more detailed mechanisms for how RUNX2 regulates REST and promotes non-neuroendocrine differentiation in SCLC and whether RUNX2 could be used as a biomarker to identify non-neuroendocrine SCLCs.
Another recent study found that REST is regulated by MYC in a NOTCH-independent manner(30), which is supported by our LISA analysis which also identified MYC as a candidate regulator of the transcriptional program in Notch-Mutant adherent non-neuroendocrine subpopulations. Together these studies demonstrate that plasticity mechanisms to promote non-neuroendocrine differentiation converge on REST, which can be regulated by at least 3 mechanisms including NOTCH, RUNX2, and MYC(15,18,30). It will be interesting to determine whether there are additional positive upstream regulators of REST and to uncover the functional consequences of inactivating REST at SCLC tumor initiation in vivo to better understand the function of non-neuroendocrine subpopulations in promoting SCLC tumorigenesis and therapeutic resistance. Alternatively, this could uncover additional escape mechanisms that drive non-neuroendocrine plasticity.
We found that non-neuroendocrine subpopulations in Notch2-Mutant tumors highly express genes that promote innate immunity including STING and that Notch2-Mutant adherent cell lines are highly sensitive to STING agonists. This was surprising as we and others(20–22) have shown that non-neuroendocrine SCLCs, which overall have higher NOTCH expression and can be driven by NOTCH, have restored antigen presentation and increased sensitivity to STING agonists relative to ASCL1 positive neuroendocrine SCLCs. More recent data supports these findings showing that increased NOTCH signaling is a significant predictor of clinical benefit to immune checkpoint blockade(45). These studies together with our findings suggest that the non-neuroendocrine state, which can be driven in a NOTCH-dependent or NOTCH-independent manner, is enriched in genes that control antigen presentation and innate immune signaling. Therefore, a conserved gene set that controls the non-neuroendocrine state in both NOTCH-Mutant and NOTCH-WT SCLCs; including RUNX2 and REST, should also be explored as a signature to predict immunotherapy response.
Our data shows that inhibition of NOTCH activity augments the sensitivity to STING agonists in non-neuroendocrine SCLC cells that are Notch-WT consistent with a recent study showing that NOTCH-ICD directly inhibits STING activity(46). Together this suggests that NOTCH-independent REST-dependent causal mechanisms that drive the non-neuroendocrine cell state are primed for STING agonism, and that the presence of NOTCH dampens the ability to stimulate the STING pathway (Fig. 6K). NOTCH-Mutant NSCLCs, selectively NOTCH2-Mutant and NOTCH3-Mutant, show improved responses to immune checkpoint blockade compared to NOTCH-WT NSCLCs(47). Whether this observation is caused by NOTCH attenuating STING pathway activation or due to other mechanisms by which NOTCH opposes immunity, such as its ability to oppose JAK/STAT signaling(48,49), is not known. Together, this suggests the intriguing possibility that LOF NOTCH-mutations in lung cancer could enrich for responses to STING agonists, or that NOTCH inhibition could potentiate STING agonism in the NOTCH-WT setting. Additional work is needed to determine the mechanisms for how NOTCH inhibits STING and whether the link between NOTCH and STING has therapeutic utility in NOTCH-Mutant SCLC.
Supplementary Material
Statement of Significance.
A genetically engineered mouse model of NOTCH-mutant small cell lung cancer reveals that non-neuroendocrine plasticity persists in the absence of NOTCH, driven by a RUNX2-REST-dependent pathway and innate immune signaling.
Acknowledgements
M.G.O. is supported as a William Raveis Charitable Fund Clinical Investigator of the Damon Runyon Cancer Research Foundation (CI-101-19), by an NCI/NIH K08 grant (no. K08CA222657), the Kaplan Family Fund, and a developmental research project award from the DF/HCC Lung Cancer Program. E.H.K. is supported by the Gross-Loh research fellowship. Special thanks to Pingping Mao for help with GSEA analysis and Shengqing Stan Gu for help with LISA analysis, and members of the Oser, Barbie, and Janne laboratories for helpful discussions. We thank Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core, which provided histology and immunohistochemistry service. Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant # NIH 5 P30 CA06516.
Conflict of Interest
E.H. Knelson reports grants from Takeda outside the submitted work. D.A. Barbie reports personal fees from Qiagen; personal fees and other from Xsphera, grants from Gilead, grants from Novartis, Lilly/Loxo Oncology, and BMS, and personal fees from Tango Therapeutics outside the submitted work. M.G Oser reports grants from Takeda, Eli Lilly, Novartis, and BMS outside the submitted work.
References
- 1.Kalemkerian GP, Akerley W, Bogner P, Borghaei H, Chow LQ, Downey RJ, et al. Small cell lung cancer. J Natl Compr Canc Netw 2013;11:78–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers 2021;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet 2011;378:1741–55 [DOI] [PubMed] [Google Scholar]
- 4.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012;44:1104–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 2012;44:1111–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019;19:289–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baine MK, Hsieh MS, Lai WV, Egger JV, Jungbluth AA, Daneshbod Y, et al. SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J Thorac Oncol 2020;15:1823–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep 2016;16:1259–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Augustyn A, Borromeo M, Wang T, Fujimoto J, Shao C, Dospoy PD, et al. ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc Natl Acad Sci U S A 2014;111:14788–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell 2017;170:564–76 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang T, Collins BJ, Jin N, Watkins DN, Brock MV, Matsui W, et al. Achaete-scute complex homologue 1 regulates tumor-initiating capacity in human small cell lung cancer. Cancer Res 2009;69:845–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sriuranpong V, Borges MW, Ravi RK, Arnold DR, Nelkin BD, Baylin SB, et al. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res 2001;61:3200–5 [PubMed] [Google Scholar]
- 14.Sriuranpong V, Borges MW, Strock CL, Nakakura EK, Watkins DN, Blaumueller CM, et al. Notch signaling induces rapid degradation of achaete-scute homolog 1. Mol Cell Biol 2002;22:3129–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim JS, Ibaseta A, Fischer MM, Cancilla B, O’Young G, Cristea S, et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017;545:360–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oser MG, Sabet AH, Gao W, Chakraborty AA, Schinzel AC, Jennings RB, et al. The KDM5A/RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis. Genes Dev 2019;33:1718–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simpson KL, Stoney R, Frese KK, Simms N, Rowe W, Pearce SP, et al. A biobank of small cell lung cancer CDX models elucidates inter- and intratumoral phenotypic heterogeneity. Nature Cancer 2020;1:437–51 [DOI] [PubMed] [Google Scholar]
- 18.Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020;38:60–78 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calbo J, van Montfort E, Proost N, van Drunen E, Beverloo HB, Meuwissen R, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 2011;19:244–56 [DOI] [PubMed] [Google Scholar]
- 20.Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med 2018;24:1143–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021;39:346–60 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigi M, Campisi M, et al. Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Augert A, Eastwood E, Ibrahim AH, Wu N, Grunblatt E, Basom R, et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci Signal 2019;12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 2009;4:1064–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 2015;11:1018–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DW, Wu N, Kim YC, Cheng PF, Basom R, Kim D, et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev 2016;30:1289–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaffer BE, Park KS, Yiu G, Conklin JF, Lin C, Burkhart DL, et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res 2010;70:3877–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gazdar AF, Savage TK, Johnson JE, Berns A, Sage J, Linnoila RI, et al. The comparative pathology of genetically engineered mouse models for neuroendocrine carcinomas of the lung. J Thorac Oncol 2015;10:553–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel AS, Yoo S, Kong R, Sato T, Sinha A, Karam S, et al. Prototypical oncogene family Myc defines unappreciated distinct lineage states of small cell lung cancer. Sci Adv 2021;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balanis NG, Sheu KM, Esedebe FN, Patel SJ, Smith BA, Park JW, et al. Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer Cell 2019;36:17–34 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 2020;10:26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin Q, Fan J, Zheng R, Wan C, Mei S, Wu Q, et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol 2020;21:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mei S, Qin Q, Wu Q, Sun H, Zheng R, Zang C, et al. Cistrome Data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res 2017;45:D658–D62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res 2019;47:D729–D35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigi M, Campisi M, et al. Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov 2021;11:1952–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang YH, Klingbeil O, He XY, Wu XS, Arun G, Lu B, et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev 2018;32:915–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen HJ, Poran A, Unni AM, Huang SX, Elemento O, Snoeck HW, et al. Generation of pulmonary neuroendocrine cells and SCLC-like tumors from human embryonic stem cells. J Exp Med 2019;216:674–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouadah Y, Rojas ER, Riordan DP, Capostagno S, Kuo CS, Krasnow MA. Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch. Cell 2019;179:403–16 e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin Y, Ma D, Gramyk T, Guo C, Fang R, Ji H, et al. Kdm1a promotes SCLC progression by transcriptionally silencing the tumor suppressor Rest. Biochem Biophys Res Commun 2019;515:214–21 [DOI] [PubMed] [Google Scholar]
- 41.Olsen RR, Ireland AS, Kastner DW, Groves SM, Spainhower KB, Pozo K, et al. ASCL1 represses a SOX9(+) neural crest stem-like state in small cell lung cancer. Genes Dev 2021;35:847–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A 2014;111:705–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakagawa M, Ichikawa M, Kumano K, Goyama S, Kawazu M, Asai T, et al. AML1/Runx1 rescues Notch1-null mutation-induced deficiency of para-aortic splanchnopleural hematopoiesis. Blood 2006;108:3329–34 [DOI] [PubMed] [Google Scholar]
- 44.He T, Wildey G, McColl K, Savadelis A, Spainhower K, McColl C, et al. Identification of RUNX1T1 as a potential epigenetic modifier in small-cell lung cancer. Mol Oncol 2021;15:195–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roper N, Velez MJ, Chiappori A, Kim YS, Wei JS, Sindiri S, et al. Notch signaling and efficacy of PD-1/PD-L1 blockade in relapsed small cell lung cancer. Nat Commun 2021;12:3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Long J, Yang C, Zheng Y, Loughran P, Guang F, Li Y, et al. Notch signaling protects CD4 T cells from STING-mediated apoptosis during acute systemic inflammation. Sci Adv 2020;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang K, Hong X, Song Z, Xu Y, Li C, Wang G, et al. Identification of Deleterious NOTCH Mutation as Novel Predictor to Efficacious Immunotherapy in NSCLC. Clin Cancer Res 2020;26:3649–61 [DOI] [PubMed] [Google Scholar]
- 48.Assa-Kunik E, Torres IL, Schejter ED, Johnston DS, Shilo BZ. Drosophila follicle cells are patterned by multiple levels of Notch signaling and antagonism between the Notch and JAK/STAT pathways. Development 2007;134:1161–9 [DOI] [PubMed] [Google Scholar]
- 49.Liu W, Singh SR, Hou SX. JAK-STAT is restrained by Notch to control cell proliferation of the Drosophila intestinal stem cells. J Cell Biochem 2010;109:992–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.