

Abstract
Hepatitis B virus (HBV)/hepatitis C virus (HCV) coinfection accelerates liver fibrosis progression compared to HBV or HCV mono-infection. The transcription factors octamer binding protein 4 (OCT4) and Nanog are direct targets of the profibrogenic transforming growth factor beta-1 (TGF-β1) signaling cascade. We leveraged a coculture model to monitor the effects of HBV and HCV coinfection on fibrogenesis in both NTCP-transfected Huh7.5.1 hepatoma cells and LX2 hepatic stellate cells (HSCs). We used CRISPR/Cas9 to knockout OCT4 and Nanog to evaluate their effects on HBV-, HCV-, or TGF-β1-induced liver fibrogenesis. HBV/HCV coinfection and HBx, HBV preS2, HCV core and HCV NS2/3 over-expression increased TGF-β1 mRNA levels in NTCP-Huh7.5.1 cells compared to controls. HBV/HCV coinfection further enhanced profibrogenic gene expression relative to HBV or HCV mono-infection. Coculture of HBV and HCV mono-infected or HBV/HCV coinfected hepatocytes with LX2 cells significantly increased profibrotic gene expression and LX2 cell invasion and migration. OCT4 and Nanog gRNA independently suppressed HBV-, HCV-, HBV/HCV-, and TGF-β1-induced α-SMA, TIMP-1 and Col1A1 expression and reduced Huh7.5.1, LX2, primary hepatocyte and pHSC migratory capacity. OCT4/Nanog protein expression also correlated positively with fibrosis stage in liver biopsies from patients with chronic HBV or HCV infection. In conclusion, HBV and HCV independently and cooperatively promote liver fibrogenesis through a TGF-β1-induced OCT4/Nanog-dependent pathway.
Keywords: HBV, HCV, immune response, liver fibrosis, OCT4/Nanog, TGF-β1
Introduction
Liver fibrosis is an immune-mediated wound-healing response to recurrent liver injury, marked by excessive deposition of extracellular matrix (ECM) proteins in the perisinusoidal space (1, 2). Hepatic stellate cells (HSCs) are the main source of ECM and the key drivers of the profibrotic process. Chronic liver injury enhances production of transforming growth factor beta-1 (TGF-β1), a canonical profibrogenic cytokine, in both hepatocytes and HSCs, that in turn activates HSCs to produce ECM proteins, such as collagen, fibronectin, and proteoglycans (1–5). Globally, approximately 240 million people suffer from chronic hepatitis B virus (HBV) infection while 71 million people have chronic hepatitis C virus (HCV) infection (6–8). HBV and HCV are primary causes of progressive liver fibrosis and hepatocellular carcinoma (HCC) worldwide, and thus are associated with significant morbidity and mortality (8–14). In contrast to mono-infection with HBV or HCV, HBV/HCV-coinfected patients have a higher risk of accelerated progression to cirrhosis, end-stage liver disease, and HCC (15–19). Interestingly, HBV and HCV actually inhibit each other’s replication in hepatocytes but cooperate in deteriorating normal hepatocellular processes and enhancing cytopathic effects (20). However, the precise mechanisms involved in HBV/HCV-induced liver disease remain unclear. In prior studies, we have demonstrated that HCV induces liver fibrosis through TGF-β1 production in HSCs and hepatocytes (21). Moreover, HBV infection has also been reported to increase hepatic TGF-β1 mRNA and protein levels (22). Therefore, both HBV and HCV appear to act through the TGF-β1 signaling pathway in the promotion of liver fibrosis (3, 23). Octamer-binding transcription factor 4 (OCT4) and Nanog, two important pluripotent markers that play significant roles in stem cell regulation and self-renewal (24), transduce TGF-β1 signaling in physiologic and pathologic processes (25, 26). Notably, TGF-β1-induced OCT4/Nanog activity is involved in both HBV replication and liver cancer (27–29). Huang et al found that HCC patients with elevated hepatic OCT4/Nanog mRNA and protein levels had a higher rate of HCC recurrence (30). Furthermore, HBV-associated HCC is associated with increased hepatic OCT4/Nanog expression relative to HCV-associated HCC (30). The HBV protein HBx has also been reported to promote Nanog expression in liver stem cells leading to increased self-renewal, tumorigenicity, chemoresistance and migration—properties associated with liver cancer progression. (31, 32).
In this study, we used JFH1 HCV-infected and HBV-infected NTCP (sodium taurocholate cotransporting polypeptide)-transfected Huh7.5.1 hepatoma cells (33–39) to support HBV and HCV infection in vitro. We also established HBV/HCV coinfection in NTCP-transfected Huh7.5.1 cells and primary human hepatocytes (PHHs) to explore the effects of HBV/HCV coinfection on liver fibrogenesis in a coculture model with LX2 hepatic stellate cells (HSCs) or primary human HSCs (pHSCs). We used gRNAs to knockdown OCT4 and Nanog in hepatocytes and HSCs to investigate the regulatory roles of TGF-β1/OCT4/Nanog in mediating HBV/HCV-induced liver fibrogenesis. We found that HBV and HCV infection enhance TGF-β1 production and hepatic fibrogenesis in hepatocytes and HSCs through OCT4 and Nanog.
Materials and methods
Cell culture
Huh7.5.1, LX2 HSCs, and HepAD38 cells (40) (which stably generate infectious HBV virions) were grown in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, Waltham, MA) with 10% FBS (GIBCO, Waltham, MA), 100 IU/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, Waltham, MA). We transfected Huh7.5.1 cells with an NTCP expression plasmid to generate NTCP-Huh7.5.1 stable cell lines using Lipofectamine LTX (Life Technologies, Carlsbad, USA) with G418 (Life Technologies) selection as previously described (39). pHSCs and PHHs were purchased from TRIANGLE Research Lab (Durham, NC, USA) and cultured according to the manufacturer’s protocol. In brief, PHHs or pHSCs were seeded onto matrix-coated plates and cultured with Hepatocyte Plating Medium (Durham, NC, USA). Cell viability was determined using the Cell Titer-Glo luminescent cell viability assay kit (Promega, Madison, WI) according to the manufacturer’s instructions and as previously described (1, 3).
Plasmids and transfection
HBV sub-genomic constructs including HBV P, X, C, pre C, S, preS1, and preS2 were synthesized and cloned into a pcDNA3.1(+) vector, carrying a neomycin resistance gene (GenScript corporation, Piscataway, NJ, USA). A full-length HBV construct pAAV-HBV1.2 was obtained from Dr. Pei-Jer Chen (National Taiwan University) (41). A pCMV-NTCP DNA construct was obtained from OriGene (Rockville, Maryland, USA). We used CRISPR/Cas9 guide RNAs (gRNAs) (pSpCas9 BB-2A-Puro PX459 all-in-one system, GenScript Corporation, NJ, USA) to generate OCT4 gRNA and Nanog gRNA. HCV sub-genomic constructs including HCV E1, Core, NS2/3, NS4A, NS4B, NS5A, NS5B and the pcDNA3.1(+) empty vector were purchased from Sino Biological (Beijing, China).
Viral stocks
Infectious HBV was derived from the culture supernatant of HepAD38 cells as previously described (37–39). In brief, the supernatant of HepAD38 was filtered through a 0.45 mm filter. HBV was precipitated using 2.3% NaCl and 10% PEG8000. The precipitates were washed with phosphate-buffered saline (PBS) and resuspended at approximately 200-fold concentration. HBV DNA was quantified by quantitative polymerase chain reaction (qPCR) (38, 39). Infectious HCV was collected from JFH1-infected Huh7.5.1 supernatant as previously described (39, 42, 43). The JFH1 crude virus supernatant (200 mL) was concentrated by centrifugation in a Centricon centrifugal device with a 100-kDa cutoff (Millipore, Temecula, CA), washed with PBS and resuspended to 1 mL as previously described (44). The purified HBV and HCV particles are referred to as HBVvp and HCVvp to distinguish from HBVs, HCVs, (HBV/HCV)s from the supernatants of HBV, HCV, or HBV/HCV co-infected NTCP-Huh7.5.1 cell.
Small molecular inhibitors
HCV NS5A inhibitor daclatasvir (Dac, BMS-790052), TGF-β1 receptor inhibitor (SB525334) (Shelleckchem, Houston, TX, USA) and entecavir (ETV) (Sigma, St. Louis, MO, USA) were dissolved in DMSO and further diluted with DMEM to the working concentrations.
TGF-β1 signaling-luciferase reporter assay.
TGF-β1 signaling activity in NTCP-Huh7.5.1 and LX2 cells was monitored by the Cignal SMAD Response Luciferase Reporters assay (SABiosciences, Frederick, MD, USA). pSMAD-luc (expressing firefly luciferase) and pRL-TK (expressing renilla luciferase) were co-transfected into the cells. The luciferase lysates were harvested at 72-hours post vector transfection and 24-hours post TGF-β1 treatment. TGF-β1-induced SMAD luciferase levels were normalized to RL-TK luciferase levels yielding the relative luciferase unit (RLU).
Enzyme-linked immunosorbent assay (ELISA)
TGF-β1 ELISA kits (no. DY240-05; R&D Systems) were used to measure the concentrations of the active form of TGF-β1 in cell culture supernatant and purified virus according to the manufacturers’ instructions.
Wound-healing scratch assay
The wound-healing scratch assay was performed in a six-well plate. NTCP-Huh7.5.1 or LX2 cells were transfected with Neg gRNA, OCT4 gRNA or Nanog gRNA for overnight. The cell monolayer was scratched using a sterilized P200 pipette tip. HBVvp, HCVvp, HBVs, HCVs, uninfected supernatant or TGF-β1 were added to the appropriate well for for 48 hours. Cell was captured using a phase-contrast microscope (200 X amplification, EVOS XL Core Cell Imaging System, Life Technologies, Carlsbad, CA, USA) at 0 and 48 hours.
Cell migration and invasion assays
The cell migration and invasion assay was performed in an 8 μm pore size transwell membrane (Corning Incorporated, NY, USA). The treated cells were placed in the upper chamber of the transwell for 48 hours. Cells that migrated from the upper chamber to the lower chamber were washed with PBS, fixed with 4% paraformaldehyde and stained with 0.05% crystal violet. Migrating or invading cells in the lower chamber were then counted using an inverted light microscope (200 X).
Transwell coculture of hepatocytes and hepatic stellate cells
To assess HBV/HCV-induced liver fibrogenesis in both hepatocyte and hepatic stellate cells, we employed a transwell coculture system as previously reported (45). Briefly, NTCP-Huh7.5.1 cells or PHHs were seeded onto the lower chamber of 12-well plates, and LX2 cells or pHSCs were plated on the upper chamber of a 0.4 μm transwell membrane. HBVvp, HCVvp, HBVs, HCVs, uninfected supernatant or TGF-β1 was added to the appropriate well. After coculture for 72 hours, cells were washed with PBS and each cell type was harvested in bulk for mRNA or protein studies.
DNA, RNA extraction and qPCR
HBV DNA was isolated from cells or supernatants by use of the QIAamp mini kit as previously described (39). Total cellular RNA was harvested from cells using the QIAshredder and RNeasy mini kits (QIAGEN, Valencia, CA, USA) as reported previously (35, 39, 42). RNA concentrations were determined using the NanoDrop ND-1000 spectrophotometer (NanoDrop Inc., DE, USA). cDNA was synthesized by reverse transcription using the High-Capacity cDNA Kit (Thermo Fisher Scientific). qPCR was performed using the Power Up SYBR Green Master Mix (Thermo Fisher Scientific) using the Quant Studio 3 platform qPCR machine (Thermo Fisher Scientific, Waltham, MA). The target mRNA expression was normalized to GAPDH using the 2-ΔΔCt method to obtain mRNA arbitrary units (fold-change). Primer sequences are listed in Table 1.
Table 1.
Primers used for quantitative reverse transcription-PCR
| Gene | Forward Primer (5’-3’) | Reverse Primer (5’-3’) |
|---|---|---|
| TGF-β1 | CTCTCCGACCTGCCACAGA | AACCTAGATGGGCGCGATCT |
|
| ||
| OCT4 | GTCCGAGTGTGGTTCTGTA | CTCAGTTTGAATGCATGGGA |
| NANOG | CTAAGAGGTGGCAGAAAAACA | CTGGTGGTAGGAAGAGTAAAGG |
| α-SMA | AAAAGACAGCTACGTGGGTGA | GCCATGTTCTATCGGGTACTTC |
| Col1A1 | CAGCCGCTTCACCTACAGC | TCAATCACTGTCTTGCCCCA |
| TIMP-1 | ACTTCCACAGGTCCCACAAC | GCTAAGCTCAGGCTGTTCCA |
| cccDNA | GGAAAGAAGTCAGAAGGCAA | GTGCCTTCTCATCTGCCGG |
| HBV DNA | GGAAAGAAGTCAGAAGGCAA | CACCTCTGCCTAATCATC |
| JFH1 | TCTGCGGAACCGGTGAGTA | TCAGGCAGTACCACAAGGC |
| GAPDH | ACCTTCCCCATGGTGTCTGA | GCTCCTCCTGTTCGACAGTCA |
Human liver specimens.
Liver sections from patients with HBV mono-infection (n=4), HCV mono-infection (n=4), HBV/HCV coinfection (n=4) and uninfected controls (n=4), were obtained from Anhui Provincial Hospital and The Affiliated Infectious Diseases Hospital of Soochow University and immediately stored in RNALater (Ambion, TX, USA) or fixed in formalin. H&E staining and Masson’s trichrome staining were used to score hepatic inflammation and fibrosis according to the Ishak scoring system. Immunohistochemical staining was performed on 4 μm thickness paraffin-embedded liver sections to determine the expression and distribution of OCT4 and Nanog. We determined OCT4 and Nanog protein expression in human liver samples using primary antibodies, rabbit anti-OCT4 (Cat# ab134218, Abcam, Cambridge, MA, USA) and rabbit anti-Nanog (Cat# ab109250, Abcam). Liver sections were scored by two independent expert pathologists blinded to sample study groups. Protein expression of OCT4 and Nanog was evaluated by immunoreactive score (IRS), assessing the staining intensity as follows: 0=negative, 1=weak intensity, 2=moderate intensity, and 3=strong intensity.
This study was approved by the Medical Ethics Committee of the First Affiliated Hospital of University of Science and Technology of China (Anhui Provincial Hospital), China and The Affiliated Infectious Diseases Hospital of Soochow University, China. Written informed consent was obtained from each patient, and all ex vivo human experiments were carried in accordance with the ethical standards of the institutional and/or national research committee, the Helsinki declaration and its later amendments, or comparable ethical standards.
Western blot assays
Cells were washed with PBS and lysed using protein RIPA Lysis Buffer containing protease inhibitor cocktail (Sigma Life Science and Biochemicals, St. Louis, MO, USA). Equal quantities of protein (20 μg) were loaded into each lane. Protein samples were separated by SDS-PAGE gel electrophoresis with NuPAGE Novex pre-cast 4-12% Bis-Tris gradient gels (Thermo Fisher Scientific) and blotted onto nitrocellulose membranes. The membranes were blocked with 5% bovine serum albumin (BSA). Targeted proteins were detected with specific primary antibodies including mouse anti-HCV core C7-50, rabbit anti-Col1A1 (Thermo Fisher Scientific), rabbit anti-α-SMA, rabbit anti-TIMP-1 (Cell Signaling Technology, Danvers, MA, USA), rabbit anti-OCT4, and rabbit anti-Nanog (Abcam). The secondary antibodies used were horseradish peroxidase (HRP)-conjugated ECL donkey anti-rabbit IgG and HRP-conjugated ECL sheep anti-mouse IgG (GE Healthcare Biosciences, Pittsburgh, PA, USA). The blots were detected by chemiluminescence using the Amersham ECL Western blotting detection kit (GE Healthcare Biosciences).
Statistical analysis
Statistical differences between groups were assessed by one-way analysis of variance (ANOVA) and SNK-q test using GraphPad Prism v6 (GraphPad, La Jolla, CA, USA). Data are expressed as mean ± standard deviation (error bar) of at least three sample replicates, unless stated otherwise. In all analyses, * P < 0.05; ** P < 0.01; *** P < 0.001 indicates statistically significant differences between indicated experimental comparisons.
Results
HBV infection increases TGF-β1/OCT4/Nanog gene expression in hepatocytes.
To explore the effects of HBV and HCV on the TGF-β1/OCT4/Nanog pathway, we used the HBV inhibitor entecavir (ETV, 20 μM) and the HCV NS5A inhibitor daclatasvir (Dac, 10 pM). Treatment with 20 μM ETV significantly reduced HBV DNA levels in both HepAD38 cells and supernatant in a time-dependent manner (Fig. 1A–B). Furthermore, HBV-induced TGF-β1/OCT4/Nanog upregulation in HepAD38 cells (Fig. 1C–D) was markedly suppressed by ETV treatment (Fig. 1C–D). Based on previous studies, we also used Huh7.5.1 cells overexpressing NTCP to investigate HBV pathogenesis (37–39). HBV increased OCT4 and Nanog expression in NTCP-Huh7.5.1 cells while ETV treatment significantly reduced HBV DNA levels and TGF-β1/OCT4/Nanog mRNA levels in HBV-infected NTCP-Huh7.5.1 cells (Fig.1E–H). In PHHs that supported HBV replication, ETV treatment significantly reduced HBV DNA levels in both cells and supernatant in a time-dependent fashion(Fig. 1I–J). As in the hepatoma cell lines, HBV-induced TGF-β1/OCT4/Nanog upregulation could be suppressed by ETV treatment (Fig. 1K).
Figure 1. HBV replication enhances TGF-β1, OCT4 and Nanog expression in NTCP-Huh7.5.1 and PHHs.
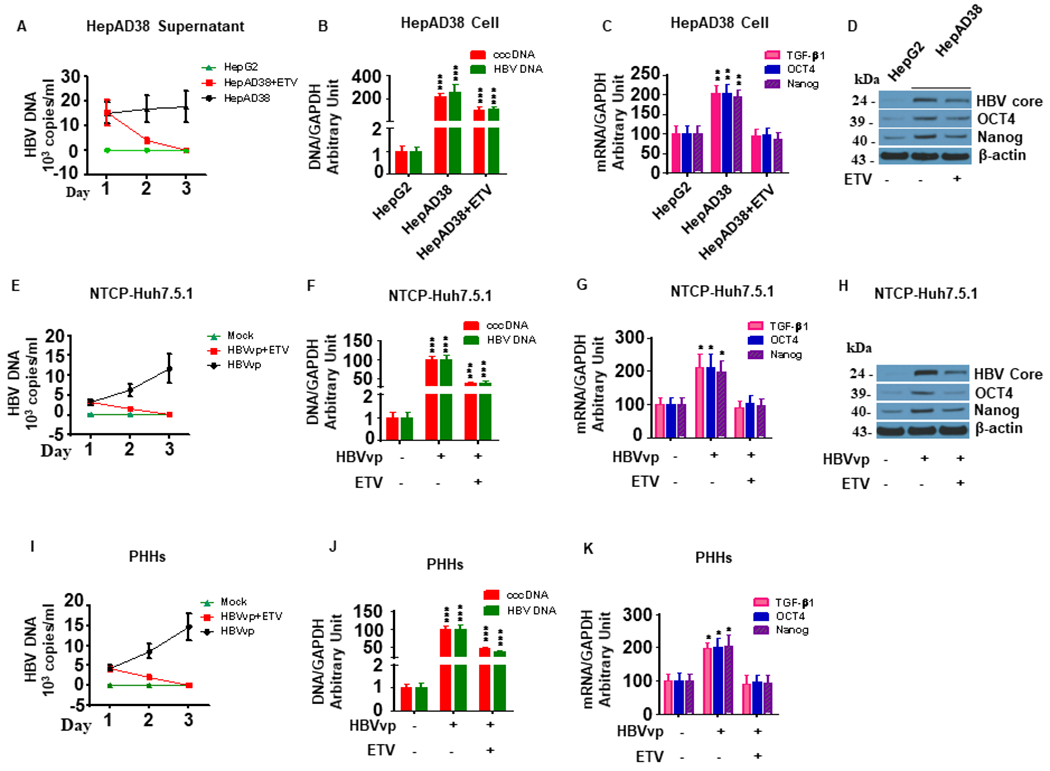
HepAD38 cells (30,000 cell/well in 1 mL) were seeded into 24-well plates and incubated overnight.NTCP-Huh7.5.1 (30,000 cell/well) and PHHs (30,000 cell/well) were seeded into 24-well plates (1 mL) and infected with 100 μL HBVvp (0.2 MOI) overnight. Culture supernatant was aspirated, and the cells were washed with PBS and maintained with fresh 10% FBS DMEM. Entecavir (ETV) (20 μM final concentration) was added to the appropriate wells. The culture supernatant and cellular mRNA, DNA and protein were harvested at 24, 48 and 72 hours post ETV treatment, respectively.
(A). ETV treatment significantly reduced HBV DNA levels in the supernatant of HepAD38 cells in a time-dependent manner.
(B). ETV significantly inhibited HBV DNA and cccDNA expression in HepAD38 cells.
(C). HBV replication in HepAD38 cells significantly increased TGF-β1, OCT and Nanog mRNA levels compared to HepG2 cells. ETV treatment blocked HBV replication-induced TGF-β1, OCT4 and Nanog upregulation in HepAD38cells.
(D). HBV replication in HepAD38 cells significantly increased HBV core, OCT and Nanog protein levels compared to HepG2 cells. ETV treatment blocked HBV replication-induced OCT4 and Nanog upregulation and reduced HBcAg levels in HepAD38cells.
(E). ETV treatment significantly reduced HBV DNA levels in the supernatants of NTCP-Huh7.5.1 in a time-dependent manner.
(F). ETV significantly inhibited HBV DNA and cccDNA expression in HBV infected NTCP-Huh7.5.1 cells.
(G). ETV treatment leads to a significant decrease in mRNA expression of TGF-β1, OCT4, Nanog in NTCP-Huh 7.5.1 cells.
(H). ETV treatment leads to a significant decrease in protein expression of OCT4, Nanog, HBcAg in NTCP-Huh 7.5.1 cells.
(I). ETV treatment significantly reduced HBV DNA levels in the supernatants of PHHs cells in a time-dependent manner.
(J) ETV significantly inhibited HBV DNA and cccDNA expression in HBV infected PHHs.
(K). ETV treatment leads to a significant decrease in mRNA expression of TGF-β1, OCT4, Nanog in PHHs.
Data are representative of 3 independent experiments with similar results. Bars represent means ± SD of 3 biological repeats. *, p < 0.05. **, p < 0.01. ***, p < 0.001.
HBV/HCV coinfection further enhances TGF-β1/OCT4/Nanog expression in hepatocytes relative to HBV or HCV monoinfection.
HBV and JFH1-HCV mono-infection significantly increased TGF-β1/OCT4/Nanog expression compared to uninfected NTCP-Huh7.5.1 cells (Fig. 2A). HBV/JFH1-HCV coinfection in NTCP-Huh7.5.1 cells further increased TGF-β1/OCT4/Nanog expression relative to either HBV or JFH1-HCV mono-infection (Fig. 2A). In addition, HCV RNA levels were lower in HBV/JFH1-HCV coinfected cells than in JFH1-HCV mono-infected cells (Table 2), which is consistent with prior reports on the inhibition of HCV replication in the context of HBV coinfection (20). Treatment with either ETV or Dac significantly reduced HBV/JFH1-HCV-induced TGF-β1/OCT4/Nanog upregulation (Fig. 2A, 2B). Combination therapy of ETV and Dac further reduced TGF-β1/OCT4/Nanog expression to levels similar to those observed in uninfected controls (Fig. 2A–B). Western blot analyses confirmed that treatment with ETV and Dac inhibited HBV and HCV core protein in HBV and JFH1-HCV-infected NTCP-Huh7.5.1 cells, respectively (Fig. 2B). These experiments were repeated in PHHs, where HBV and JFH1-HCV mono-infection also resulted in TGF-β1/OCT4/Nanog upregulation, while HBV/JFH1-HCV coinfection further enhanced TGF-β1/OCT4/Nanog activity relative to controls, suggesting an additive or synergistic effect of the two viruses in activation of the TGF-β1/OCT4/Nanog pathway (Fig. 2C). HBV/HCV infection, ETV and Dac treatment did not significantly affect cell viability (Fig. 2D–F).
Figure 2. HBV/JFH1-HCV coinfection/coexposure additively promotes TGF-β1 and OCT4/Nanog activity in NTCP-Huh7.5.1, PHHs and human liver tissue.
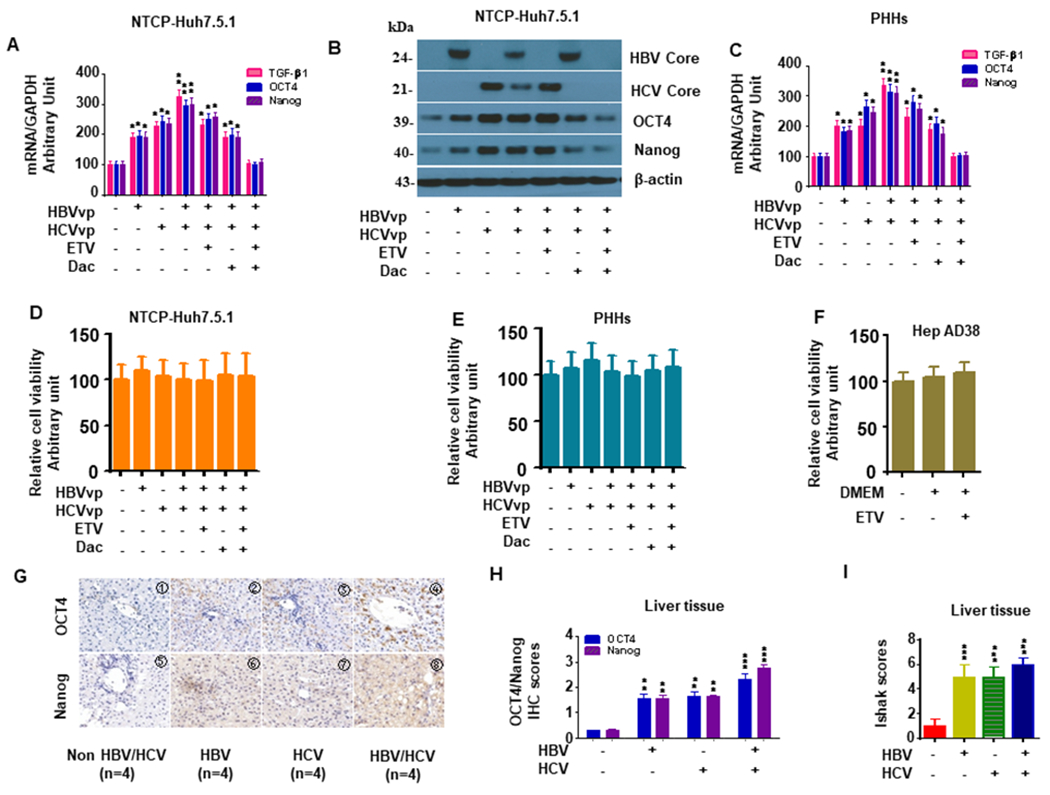
NTCP-Huh7.5.1 (30,000 cell/well) and PHHs (30,000 cell/well) in 1 mL were seeded into 24-well plates and infected with 100 μL HBVvp (0.2 MOI) overnight. Cells were washed and maintained with fresh media with 20 μM ETV or 10 pM Dac for 72 hrs. Cellular mRNA and protein were harvested for qPCR or western blotting, respectively. Cell viability was determined using the Cell Titer-Glo luminescent cell viability assay kit (Promega, Madison, WI) according to the manufacturer’s instructions.
(A). ETV and Dac treatment blocked HBV/JFH1-HCV coinfection enhanced TGF-β1/OCT4/Nanog mRNA levels in NTCP-Huh7.5.1 cells.
(B). ETV and Dac treatment blocked HBV/JFH1-HCV coinfection enhanced TGF-β1/OCT4/Nanog protein levels in NTCP-Huh7.5.1 cells.
(C). ETV and Dac treatment blocked HBV/JFH1-HCV coinfection enhanced TGF-β1/OCT4/Nanog mRNA levels in PHHs.
(D). HBV/HCV infection and ETV or Dac treatment did not significantly affect cell viability in NTCP-Huh7.5.1 cells.
(E). HBV/HCV infection and ETV or Dac treatment did not significantly affect cell viability in PHHs.
(F). HBV infection and ETV treatment did not significantly affect cell viability of HepAD38.
(G). Representative Immunohistochemistry images of OCT4 and Nanog (200 X) in liver specimens from non-HBV/HCV (NBNC), HBV-infected, HCV-infected and HBV/HCV coinfected patients.
(H). Quantification of OCT4 and Nanog in HBV-infected, HCV-infected or HBV/HCV coinfected liver samples.
(I). Ishak liver fibrosis scores.
Data are representative of 3 independent experiments with similar results. Bars represent means ± SD of 3 biological repeats. *, p < 0.05. **, p < 0.01. ***, p < 0.001.
Table 2.
JFH1 replication in NTCP-Huh7.5.1 cells with antiviral treatments
| Cell | Treatment | JFH1/GAPDH in cells |
|---|---|---|
| NTCP-Huh 7.5.1 | NA | 1.00 |
| NTCP-Huh 7.5.1+HBV | NA | 1.12 |
| NTCP-Huh 7.5.1+JFH1 | NA | 942,156.23 |
| NTCP-Huh 7.5.1+HBV+JFH1 | NA | 86,105.11 |
| NTCP-Huh 7.5.1+HBV+JFH1 | ETV | 808,241.42 |
| NTCP-Huh 7.5.1+HBV+JFH1 | Dac | 10.33 |
| NTCP-Huh 7.5.1+HBV+JFH1 | ETV+Dac | 11.02 |
HBV or HCV infection is associated with increased OCT4/Nanog expression in human liver samples.
We performed immunohistochemistry (IHC) to assess OCT4/Nanog protein expression in liver tissue from HBV-, HCV- and HBV/HCV-infected patients (Table 3). HBV/HCV coinfected patients had the highest expression levels compared to either HCV or HBV mono-infected patients, while uninfected controls had the lowest OCT4 and Nanog protein expression (Fig. 2G–I). Furthermore, patients with HBV/HCV coinfection had higher Ishak fibrosis scores compared to liver samples from patients with HCV or HBV mono-infection (Fig. 2G–I).
Table 3.
Patient demographics
| Characterize | NBNC | HBV | HCV | HBV/HCV | P |
|---|---|---|---|---|---|
| Gender (M/F) | 1/3 | 2/2 | 3/1 | 1/3 | >0.05 |
| Age (years) | 29.2±9.9 | 25.3±7.6 | 31.3±8.2 | 27.1±11.2 | >0.05 |
| ALT(IU/L) | 24.5±9.6 | 45.3±11.4 | 53.1±10.7 | 63±13.4 | <0.05 |
| AST(IU/L) | 22.8±9.6 | 42.3±10.9 | 52.3±12.2 | 59.5±9.9 | <0.05 |
| Total bilirubin(umol/L) | 8.0±3.2 | 11.3±6.4 | 13.2±35.7 | 25.9±9.3 | <0.05 |
| Albumin (g/L) | 48.1±15.3 | 35.4±10.2 | 37.5±12.6 | 33.1±10.9 | <0.05 |
| HBV DNA (log copy/ml) | 0 | 6.5±1.1 | 0 | 4.7±1.5 | <0.05 |
| HCV RNA (log copy/ml) | 0 | 0 | 6.8±1.7 | 3.5±1.0 | <0.05 |
| Ishak Score | 0.50±0.28 | 4.75±0.48 | 5.00±0.41 | 5.75±0.25 |
Knockdown of either OCT4 or Nanog in NTCP-Huh7.5.1 and LX2 cells inhibits TGF-β1-induced hepatic fibrogenesis.
To determine how HBV and HCV infection affect signaling downstream of TGF-β1 ligation, we utilized a SMAD luciferase reporter assay. HBV and JFH1-HCV infection increased TGF-β1 signaling by 3.8 ± 0.4 and 5.5 ± 0.6-fold, respectively, when compared to uninfected controls (Fig. 3A). In NTCP-Huh7.5.1 cells, HBV/JFH1-HCV coinfection and TGF-β1 (10 ng/mL) further enhanced luciferase activity to 6.2 ± 0.7 and 7.0 ± 0.6-fold, respectively (Fig. 3A). We observed similar effects of HBV and JFH1-HCV exposure on TGF-β1 signaling in LX2 cells (Fig. 3A). We next measured secreted TGF-β1 by ELISA and found that HCV and HBV monoculture and HCV/HBV coculture significantly increased TGF-β1 production compared to uninfected Huh7.5.1 cells (Figure 3B). Furthermore, OCT4/Nanog was significantly upregulated in LX2 cells exposed to HBV, HCV, and HBV+HCV supernatant relative to cells exposed to uninfected supernatant. Notably, the highest expression levels of OCT4/Nanog were observed in cells incubated in supernatant from both viruses (HBVs/HCVs) (Fig. 3C–D). However, the purified virus (HBVvp and HCVvp without TGF-β1) did not upregulate profibrotic genes, TGF-β1, OCT4 and Nanog in HSCs (Fig. 3B–D).
Figure 3. OCT4 and Nanog gRNA decreases TGF-β1-induced profibrogenic gene expression in HBV/JFH1-HCV coinfection/coexposure in NTCP-Huh7.5.1 and LX2 cells.
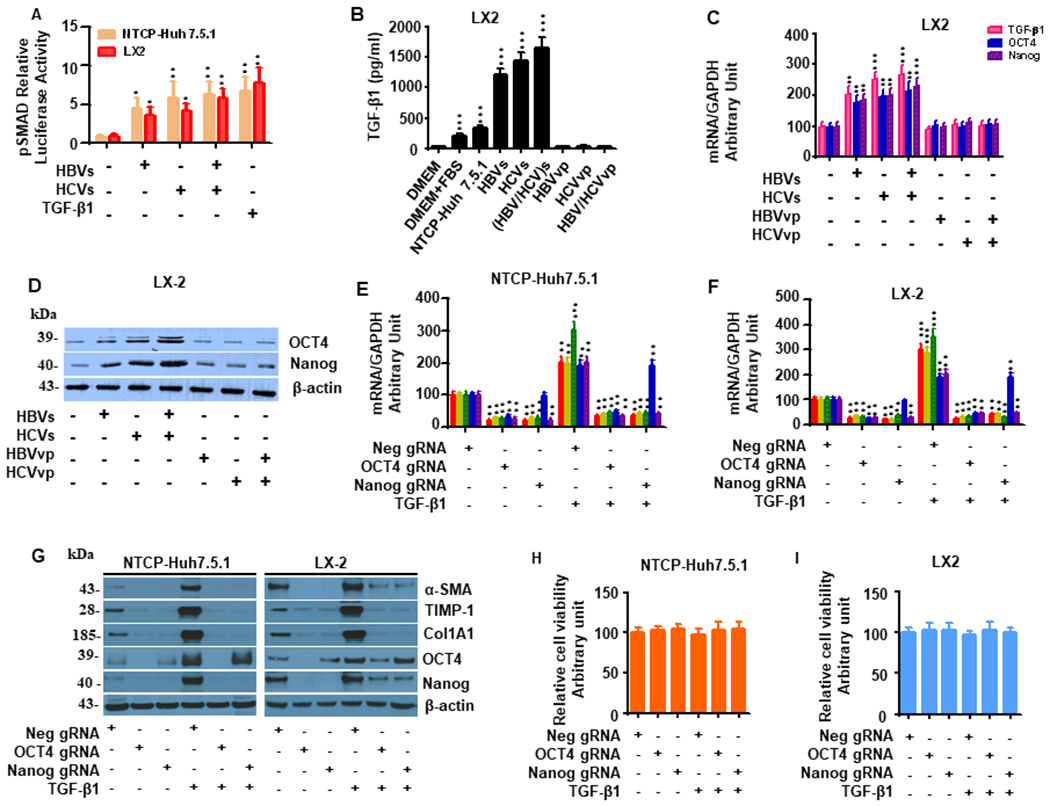
(A). HBVvp/HCVvp infection in NTCP-Huh7.5.1 cells or HBVs/HCVs (HBV/HCV)s exposure in LX2 cells increased pSMAD signaling. NTCP-Huh7.5.1 and LX2 cells (100,000 cell/well in 2 mL) were seeded into 6 well-plates and co-transfected with pSMAD-luc (expressing firefly luciferase) and pRL-TK (expressing renilla luciferase) overnight. After that, cells were harvested and seeded into 96 well plate (3000 cell/well in 100 μL). 10 μL of HBVvp or HCVvp was added to the appropriate well for 48 hours. 10 μL of 100 ng/mL TGF-β1 (10 ng/mL final concentration) was added to the appropriate well for another 24 hours. TGF-β1 signaling activity in these cells were tested by the dual-luciferase assay.
(B). Exposure of LX2 cells to HBVs, HCVs, or (HBV/HCV)s increased TGF-β1 protein levels. Levels of TGF-β1 in the supernatant or medium were examined by ELISA kits for the samples including DMEM, DMEM+10%FBS, HBVvp, HCVvp, and supernatants from LX2 cells exposure to HBVs, HCVs, (HBV/HCV)s or uninfected NTCP-Huh7.5.1 cell. We found that there is little TGF-β1 in DMEM, HBVvp, HCVvp. Exposure of LX2 cells to HBVs, HCVs, or (HBV/HCV)s increased TGF-β1 protein levels compared to the uninfected NTCP-Huh7.5.1 cell supernatant.
(C). Exposure of LX2 cells to HBVs and/or HCVs increased TGF-β1, OCT4 and Nanog mRNA. LX2 cells were seeded (30,000 cell/well in mL) into 24 well-plates and 100 μL of uninfected supernatant, HBVs, HCVs, (HBV/HCV)s, HBVvp, and/or HCVvp was added for 48 hours. Cellular mRNA was collected for qPCR. Exposure of LX2 cells to HBVs and/or HCVs increased OCT4 and Nanog mRNA levels compared to uninfected supernatant.
(D). Exposure of LX2 cells to HBVs and/or HCVs increased OCT4 and Nanog protein level. LX2 cells treatments were described in (C). Cellular protein lysates were collected for Western blot. Exposure of LX2 cells to HBVs and/or HCVs increased OCT4 and Nanog protein levels compared to uninfected supernatant.
(E). OCT4 gRNA and Nanog gRNA independently inhibited TGF-β1-induced mRNA enhancement of α-SMA, TIMP-1 and Col1A1 in NTCP-Huh7.5.1. NTCP-Huh7.5.1 (30,000 cell /well in 1 mL) cells were seeded into 24 well-plates and transfected with Neg gRNA, OCT4 gRNA or Nanog gRNA in combination with 10 ng/mL TGF-β1 and incubated for 72 hours. Cellular mRNA was collected for qPCR. TGF-β1 treatment increased mRNA level of α-SMA, TIMP-1 and CoL1A1 in NTCP-Huh7.5.1. OCT4 gRNA and Nanog gRNA independently blocked TGF-β1-stimulated mRNA level of α-SMA, TIMP-1 and Col1A1 in NTCP-Huh7.5.1 cells. OCT4 gRNA decreased OCT4 and Nanog mRNA levels.
(F). OCT4 gRNA and Nanog gRNA independently inhibited TGF-β1-induced mRNA enhancement of liver fibrosis related genes in LX2 cells. LX2 cells (30,000 cell /well in 1 mL) cells were seeded into 24 well-plates and transfected with Neg gRNA, OCT4 gRNA or Nanog gRNA in combination with 10 ng/mL TGF-β1 and incubated for 72 hours. Cellular mRNA was collected for qPCR. TGF-β1 treatment increased mRNA level of α-SMA, TIMP-1 and Col1A1 in LX2 cells. OCT4 gRNA and Nanog gRNA independently blocked TGF-β1-stimulated mRNA level of α-SMA, TIMP-1 and Col1A1 in LX2 cells. OCT4 gRNA decreased OCT4 and Nanog mRNA expression.
(G). OCT4 gRNA and Nanog gRNA independently inhibited TGF-β1-induced protein levels of α-SMA, TIMP-1 and Col1A1 in NTCP-Huh7.5.1 and LX2 cells. NTCP-Huh7.5.1 and LX2 cells treatments were described in (E) and (F). Cellular protein lysates were collected for Western blot. WB confirmed that TGF-β1 treatment increased protein level of α-SMA, TIMP-1 and Col1A1 in NTCP-Huh7.5.1 and LX2 cells. OCT4 gRNA and Nanog gRNA independently inhibited TGF-β1-induced protein levels of α-SMA, TIMP-1 and Col1A1 in NTCP-Huh7.5.1 and LX2 cells.
(H) OCT4 gRNA and Nanog gRNA did not significantly affect NTCP-Huh 7.5.1 cell viability.
(I) OCT4 gRNA and Nanog gRNA did not significantly affect LX2 cell viability.
Data are representative of 3 independent experiments with similar results. Bars represent means ± SD of 3 biological repeats. *, p < 0.05. **, p < 0.01. ***, p < 0.001.
We next sought to evaluate the role of OCT4 and Nanog in HBV- and HCV-induced TGF-β1 activation. TGF-β1 (10 ng/mL) exposure to NTCP-Huh7.5.1 and LX2 cells for 12-96 hours significantly increased profibrotic gene expression, including α-SMA, TIMP-1, and Col1A1, along with OCT4/Nanog (SFig. 1A,1B). OCT4 and Nanog gRNAs significantly reduced profibrotic gene expression and protein levels in NTCP-Huh7.5.1 and LX2 cells both in the presence and absence of TGF-β1 (Fig. 3E–G). Interestingly, both OCT4 and Nanog gRNA inhibited Nanog mRNA and protein expression in NTCP-Huh7.5.1 and LX2 cells, while Nanog gRNA did not affect OCT4 expression (Fig. 3E–G) suggesting that OCT4 may lie upstream of Nanog in its intersection with the TGF-β1 signaling pathway. OCT4 and Nanog gRNAs did not affect cell viability in NTCP-Huh7.5.1 or HSC LX2 cells (Fig. 3H–I).
Since HSC migration is important to the wound-healing process, we also performed scratch and invasion assays with NTCP-Huh7.5.1 and LX-2 cells. TGF-β1 (10 ng/mL), and HBV/HCV coinfection/coexposure increased cell migration and invasion compared to cells incubated with uninfected supernatant (Fig. 4A–D). Notably, both OCT4 and Nanog gRNAs inhibited TGF-β1- and HBV/HCV-induced cell migration in Huh7.5.1 cells and LX2 cells (Fig. 4A–D).
Figure 4. OCT4 and Nanog gRNA inhibit TGF-β1-induced NTCP-Huh7.5.1 and LX2 cell migratory capacity.
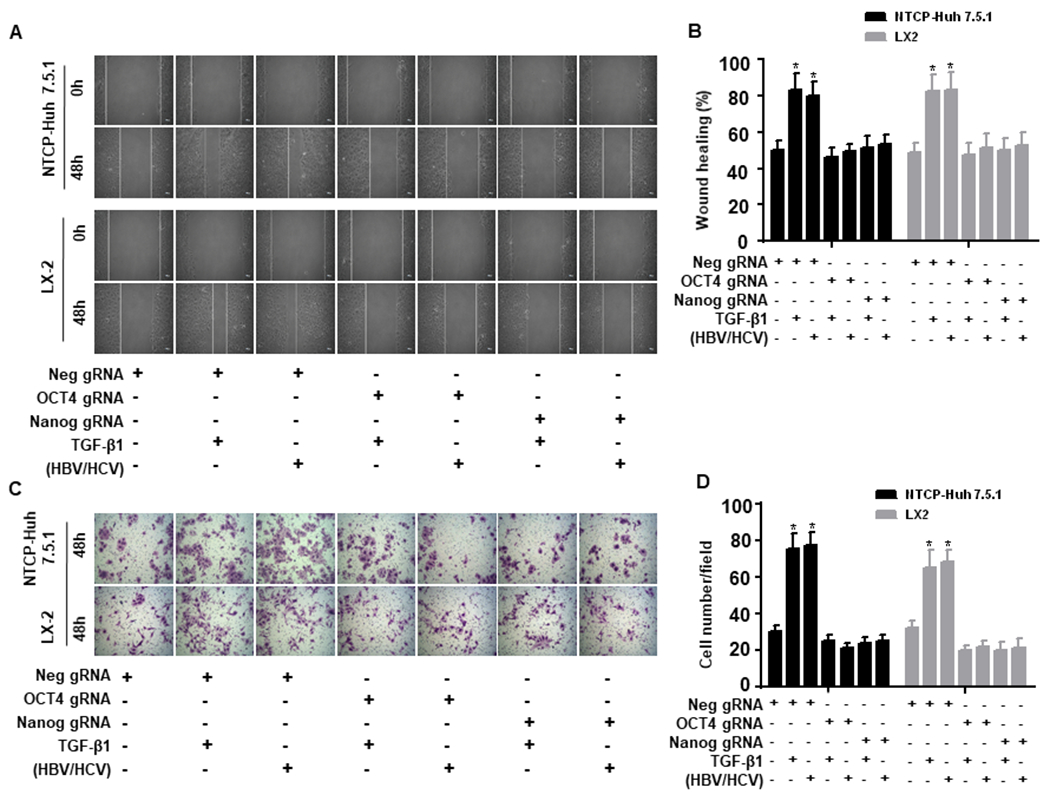
NTCP-Huh7.5.1 or LX2 (100,000 cell/well in 2 mL) cells were seeded into 6 well-plates and transfected with Neg gRNA, OCT4 gRNA or Nanog gRNA for overnight. The cell monolayer was scratched using a sterilized P200 pipette tip. Cells were washed with PBS and maintained in 2 mL fresh DMEM with 10% FBS. 100 μL of HBVvp and 100 μL HCVvp were added to the appropriate NTCP-Huh7.5.1 wells, 100 μL of HBVs and 100 μL HCVs were added to the appropriate LX2 wells, 200 μL of uninfected supernatant or TGF-β1(100 ng/mL) was added to the appropriate well for 48 hours.
(A). Images of NTCP-Huh7.5.1 (top) and LX-2 cell (bottom) scratch assays at 0 and 48 hours.
(B). Proportion of migrated NTCP-Huh7.5.1 or LX2 cells.
(C). NTCP-Huh7.5.1 or LX2 cells (100,000 cell/well in 2 mL) were seeded into 6 well-plates and transfected with empty gRNA, OCT4 gRNA or Nanog gRNA for overnight. The treated cells were harvested and seeded onto the up chamber (50,000 cell/well in 2 mL) of a 12 well Corning transwell plate (with 8 μm transmembrane pore). 100 μL of HBVvp and 100 μL of HCVvp were added to the appropriate NTCP-Huh7.5.1 cells, 100 μL of HBVs and 100 μL of HCVs were added to the appropriate LX2 cells, 200 μL of uninfected supernatant or TGF-β1(100 ng/mL) was added to the appropriate well for 48 hours. Cells that migrated to the bottom chamber were washed three times with PBS, fixed with 4% paraformaldehyde and stained with 0.05% crystal violet. Cells in the bottom chamber were counted using an inverted light microscope (200 X). (D) Number of migrated NTCP-Huh7.5.1 or LX2 cells. Data are representative of 3 independent experiments with similar results. Bars represent means ± SD of 3 biological repeats. *, p < 0.05. **, p < 0.01.
HBV/HCV-induced fibrogenesis through OCT4/Nanog is dependent on the surface TGF-β1 receptor
We used a TGF-β1 receptor inhibitor (SB525334) to determine whether HBVs- and HCVs-induced HSC activation through OCT4/Nanog occurs through TGF-β1 receptor activation (46). In LX2 cells, HBVs, HCVs and (HBV/HCV)s exposure significantly increased expression of TGF-β1, OCT4/Nanog and fibrosis-related genes (SFig. 2), while SB525334 treatment attenuated HBV- and HCV-induced fibrogenesis (SFig. 2). These findings suggest HBV and/or HCV replication in hepatocytes results in the release of TGF-β1 that activates HSCs through TGF-β1 surface receptor ligation.
HBV preS2 and HBx proteins promote liver fibrosis via activation of the OCT4/Nanog pathway.
To determine which HBV proteins induce hepatic fibrogenesis and whether they act through the OCT4/Nanog pathway, we overexpressed HBV genome proteins including HBV P, X, C, preC, S, preS1, preS2 as well as full-length HBV construct pAAV-HBV1.2 in Huh7.5.1 cells. pcDNA 3.1vector (pEmpty) was transfected as a negative control. We found that only overexpression of HBV preS2, HBx and full-length HBV genes significantly up-regulated TGF-β1 production (Fig. 5A) and enhanced mRNA and protein expression of OCT4/Nanog and the profibrotic genes α-SMA, TIMP-1 and Col1A1 in Huh7.5.1 cells compared to cells transfected with pEmpty (Fig. 5B–D). Western blot analyses confirmed overexpression of HBV P, X, C, preC, S, preS1, preS2, and full-length HBV in Huh7.5.1 cells (Fig. 5E). Overexpression of HBV proteins did not significantly affect cell viability in Huh7.5.1 cells (Fig. 5F). Importantly, HBV preS2- and HBX-induced hepatic fibrogenesis could be abrogated by OCT4 or Nanog knockdown via gRNA (Fig. 5G–I).
Figure 5. OCT4 and Nanog gRNA attenuate HBV-induced liver fibrosis in NTCP-Huh7.5.1 cells.
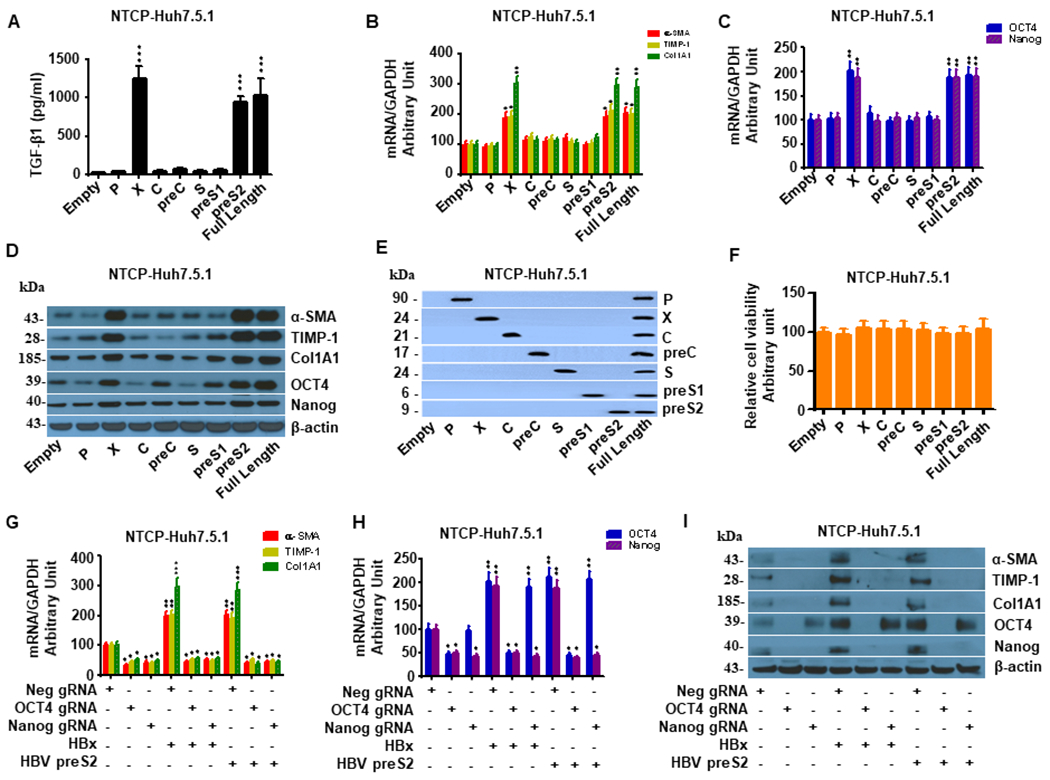
NTCP-Huh7.5.1 cells (30,000 cells/well in 1 mL) were seeded into a 24-well plate and incubated overnight. The cells were transfected with HBV full length and sub-genomic constructs including HBV P, X, C, preC, S, preS1, preS2 or pcDNA3.1(+) empty vector for 72 hr. Cell lysates were harvested for mRNA quantification or protein analysis by western blot.
(A). Overexpression of HBV full length and HBV X and preS2 significantly increased TGF-β1 levels in supernatant compared to the empty vector in NTCP-Huh7.5.1 cells. In contrast, overexpression of HBV P, C, PreC, S, preS1 or Empty vector did not significantly affect TGF-β1 levels in supernatant.
(B). Overexpression of HBV X, preS2 and the full-length HBV genome significantly increased mRNA of liver fibrosis-related genes (α-SMA, TIMP-1, and Col1A1) in NTCP-Huh7.5.1 cells. In contrast, HBV P, C, PreC, S, preS1 and empty vector did not have significant effects on these profibrotic genes.
(C). Overexpression of HBV X, preS2 and the full-length HBV genome significantly increased OCT4/Nanog mRNA levels in NTCP-Huh7.5.1 cells. In contrast, HBV P, C, PreC, S, preS1 and empty vector did not have significant effects on Oct-4/Nanog mRNA levels.
(D). Overexpression of HBV X, preS2 and the full-length HBV genome significantly increased protein levels of α-SMA, TIMP-1, Col1A1 and Oct-4/Nanog in NTCP-Huh7.5.1 cells. In contrast, HBV P, C, PreC, S, preS1 and empty vector did not have significant effects on these gene protein expressions.
(E). Confirmation of HBV full length and subgenomic overexpression in NTCP-Huh7.5.1 cells. The antibodies used for western blotting included anti-HBV P (Affinity, DF13563, USA), anti-HBX (Abcam, ab2741), anti-HBV Core (Abcam, ab8637), anti-HBV preC (Abcam, ab228709), anti-HBV S (Abcam, ab8636), anti-HBV preS1(Abm59501-4, Beijing, China), and anti-preS2 (Abcam, ab8635).
(F). Overexpression of HBV full length and sub-genomic constructs including HBV P, X, C, preC, S, preS1, preS2 did not significantly affect cell viability in NTCP-Huh7.5.1 cells.
(G) OCT4 and Nanog gRNA attenuate HBV-induced liver fibrosis related gene mRNA enhancement in NTCP-Huh7.5.1 cells. NTCP-Huh7.5.1 cells (30,000 cell /well in 1 mL) were seeded into a 24-well plate and incubated overnight. The cells were transfected with HBX, preS2 and/or OCT4 gRNA, Nanog gRNA, or Neg gRNA for 72 hours. Cell lysates were harvested for mRNA quantification by qRT-PCR. Both OCT4 gRNA and Nanog gRNA reduced HBx- or HBV preS2-induced α-SMA, TIMP-1 and Col1A1 mRNA expressions.
(H) OCT4 and Nanog gRNA attenuate HBV-stimulated OCT4/Nanog mRNA levels in NTCP-Huh7.5.1 cells. NTCP-Huh7.5.1 cells treatments were described in (G). We found that both OCT4 gRNA and Nanog gRNA blocked HBx- or HBV preS2-induced OCT4/Nanog mRNA levels.
(I) OCT4 and Nanog gRNA blocked HBV-stimulated liver fibrosis related gene and Oct-4/Nanog protein levels in NTCP-Huh7.5.1 cells. NTCP-Huh7.5.1 cells treatments were described in (G). Cell lysates were harvested for protein analysis by western blot. We found that both OCT4 gRNA and Nanog gRNA attenated HBx- or HBV preS2-induced α-SMA, TIMP-1, Col1A1 and OCT4/Nanog protein levels.
Data are representative of 3 independent experiments with similar results. Bars represent means ± SD of 3 biological repeats. *, p < 0.05. **, p < 0.01. ***, p < 0.001.
HCV Core and NS2/3 proteins promote liver fibrosis via activation of the OCT4/Nanog pathway.
We next examined HCV sub-genomic constructs including HCV E1, Core, NS2/3, NS4A, NS4B, NS5A and NS5B to determine which specific HCV proteins promote fibrogenesis through OCT4/Nanog. HCV Core and NS2/3 were the only proteins that significantly up-regulated TGF-β1 production (Fig. 6A) and enhanced mRNA and protein expression of OCT4/Nanog and the profibrotic genes TGF-β1, α-SMA, TIMP-1 and Col1A1 in Huh7.5.1 cells compared to cells transfected with pEmpty (Fig. 6 B–D). The aforementioned constructs were overexpressed in NTCP-Huh7.5.1 cells and confirmed by western blot analyses (Fig. 6E). Overexpression of HCV proteins did not significantly affect cell viability in Huh7.5.1 cells (Fig. 6F). As with the HBV proteins, OCT4 and Nanog silencing attenuated HCV Core- and NS2/3-induced liver fibrogenesis (Fig. 6G–I).
Figure 6. OCT4 and Nanog gRNAs reduce HCV-induced liver fibrogenesis in NTCP-Huh7.5.1 cells.
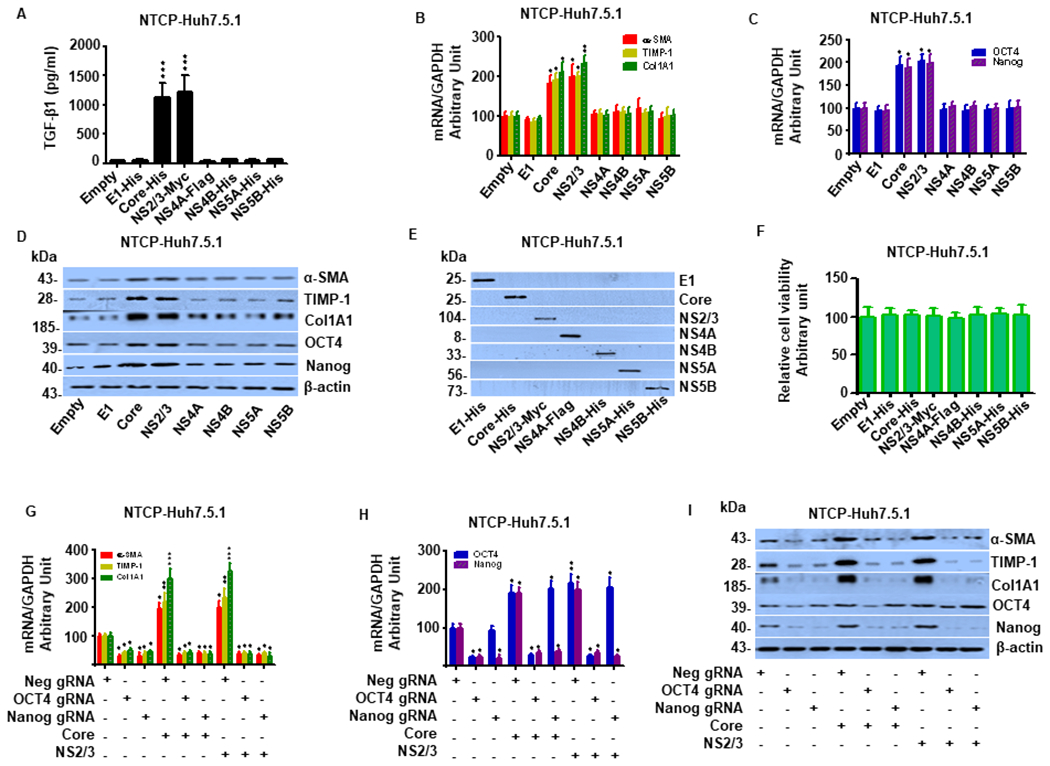
(A). Overexpression of HCV Core or NS2/3 significantly increased TGF-β1 levels in supernatant compared to the Empty vector in NTCP-Huh7.5.1 cells. In contrast, Overexpression HCV E1, NS4A, NS4B, NS5A, NS5B or Empty vector did not significantly affect TGF-β1 levels in supernatant.
(B). Overexpression of HCV Core or NS2/3 significantly increased mRNA expression of liver fibrosis-related genes (α-SMA, TIMP-1 and Col1A1). NTCP-Huh7.5.1 cells (30,000 cell /well in 1 mL) were seeded into a 24-well plate and incubated overnight. The cells were transfected with HCV sub-genomic constructs including HCV E1, core, NS2-3, NS4A, NS4B, NS5A, NS5B or the pcDNA3.1(+) empty vector for 72 hours. Cell lysates were harvested for mRNA quantification by qRT-PCR. We found that overexpression of HCV Core or NS2/3 significantly increased mRNA expression of liver fibrosis-related genes α-SMA, TIMP-1 and Col1A1. In contrast, HCV E1, NS4A, NS4B, NS5A, NS5B and empty vector did not have significant effects on profibrotic gene expression.
(C). Overexpression of HCV Core or NS2/3 significantly increased OCT4/Nanog mRNA levels. NTCP-Huh7.5.1 cells treatments were described in (B). We found that overexpression of HCV Core or NS2/3 significantly increased OCT4/Nanog mRNA expression. In contrast, HCV E1, NS4A, NS4B, NS5A, NS5B and empty vector did not have significant effects on OCT4/Nanog mRNA levels.
(D). Overexpression of HCV Core or NS2/3 significantly increased α-SMA, TIMP-1, Col1A1 and OCT4/Nanog protein levels. NTCP-Huh7.5.1 cells treatments were described in (B). Cell lysates were harvested for protein analysis by western blot. We found that overexpression of HCV Core or NS2/3 significantly increased α-SMA, TIMP-1, Col1A1 and OCT4/Nanog protein levels. In contrast, HCV E1, NS4A, NS4B, NS5A, NS5B and empty vector did not have significant effects on these gene protein levels.
(E). Confirmation of HCV sub-genomic construct overexpression, including HCV E1, Core, NS2-3, NS4A, NS4B, NS5A, NS5B or pcDNA3.1(+) Empty vector in NTCP-Huh7.5.1 cells. The antibodies used included anti-His (Biovision, 3646-100), anti-Myc (Abcam, ab32072), or anti-Flag (Abcam, ab205606). (HRP)-conjugated ECL donkey anti-rabbit IgG (GE Healthcare Biosciences, Pittsburgh, PA, USA) was used as a secondary antibody.
(F). Overexpression HCV sub-genomic constructs including HCV E1, Core, NS2-3, NS4A, NS4B, NS5A, NS5B or Empty vector in NTCP-Huh7.5.1 cells did not significantly affect cell viability.
(G) OCT4 and Nanog gRNAs blocked HCV-stimulated liver fibrosis related gene mRNA in NTCP-Huh7.5.1 cells. NTCP-Huh7.5.1 cells (30,000 cell /well in 1 mL) were seeded into a 24-well plate and incubated overnight. The cells were transfected with HCV Core, NS2/3 and/or OCT4 gRNA, Nanog gRNA, or Neg gRNA for 72 hours. Cell lysates were harvested for mRNA quantification by qRT-PCR. Both OCT4 gRNA and Nanog gRNA suppressed HCV Core- or NS2/3-induced mRNA expression of α-SMA, TIMP-1, and Col1A1.
(H) OCT4 and Nanog gRNAs reduced HCV-stimulated OCT4/Nanog mRNA in NTCP-Huh7.5.1 cells. NTCP-Huh7.5.1 cells treatments were described in (G). Both OCT4 gRNA and Nanog gRNA suppressed HCV Core- or NS2/3-induced OCT4/Nanog mRNA enhancements.
(I) OCT4 and Nanog gRNAs attenuated HCV-stimulated liver fibrosis related gene and OCT4/Nanog protein expression in NTCP-Huh7.5.1 cells. NTCP-Huh7.5.1 cells treatments were described in (G). Cell lysates were harvested for protein analysis by western blot. Both OCT4 gRNA and Nanog gRNA suppressed overexpression of HCV Core- or NS2/3-induced of α-SMA, TIMP-1, Col1A1 and OCT4/Nanog mRNA expressions.
Data are representative of 3 independent experiments with similar results. Bars represent means ± SD of 3 biological repeats. *, p < 0.05. **, p < 0.01. ***, p < 0.001.
OCT4 and Nanog gRNA inhibit HBV/JFH1-HCV-induced fibrogenesis in hepatocytes and hepatic stellate cells in a coculture system.
We previously demonstrated that HCV induces fibrogenesis in HSC monoculture, with additive induction observed in HSC and hepatocyte coculture, thereby demonstrating the importance of cooperative interactions between cell types in the promotion of liver fibrosis. To determine whether HBV- and JFH1-HCV-induced fibrosis depends on synergistic interactions between liver cell types, we performed coculture studies using hepatocytes and hepatic stellate cells in a transwell system. In this model, the 0.4 μm pore membrane allowed cellular communication between compartments via soluble mediators.
We found that JFH1-HCV and HBV mono-infection in NTCP-Huh7.5.1 cells significantly enhanced expression of Col1A1 and TIMP-1 as well as OCT4/Nanog in co-cultured NTCP-Huh7.5.1 and LX2 cells (Fig. 7A–C; SFig. 3A–B), which was potentiated in the setting of HBV/JFH1-HCV coinfection (Fig. 7A–C; SFig. 3A–B). In this coculture system, TGF-β1 levels in the supernatant from HBV/HCV monoinfected hepatocytes were significantly higher than mock infection while lower than HBV/HCV coinfected hepatocytes (SFig. 3C). Interestingly, HBV/HCV coinfetion in NTCP-Huh7.5.1 cells inhibited HBV or HCV replication (SFig. 3D). HBV/HCV exposure or OCT4 gRNA and Nanog gRNA did not affect LX2 cell viability (SFig. 3E).
Figure 7. OCT4 and Nanog gRNA attenuate enhanced profibrogenic gene expression induced by HBV/JFH1-HCV in hepatocytes and hepatic stellate cells in coculture.
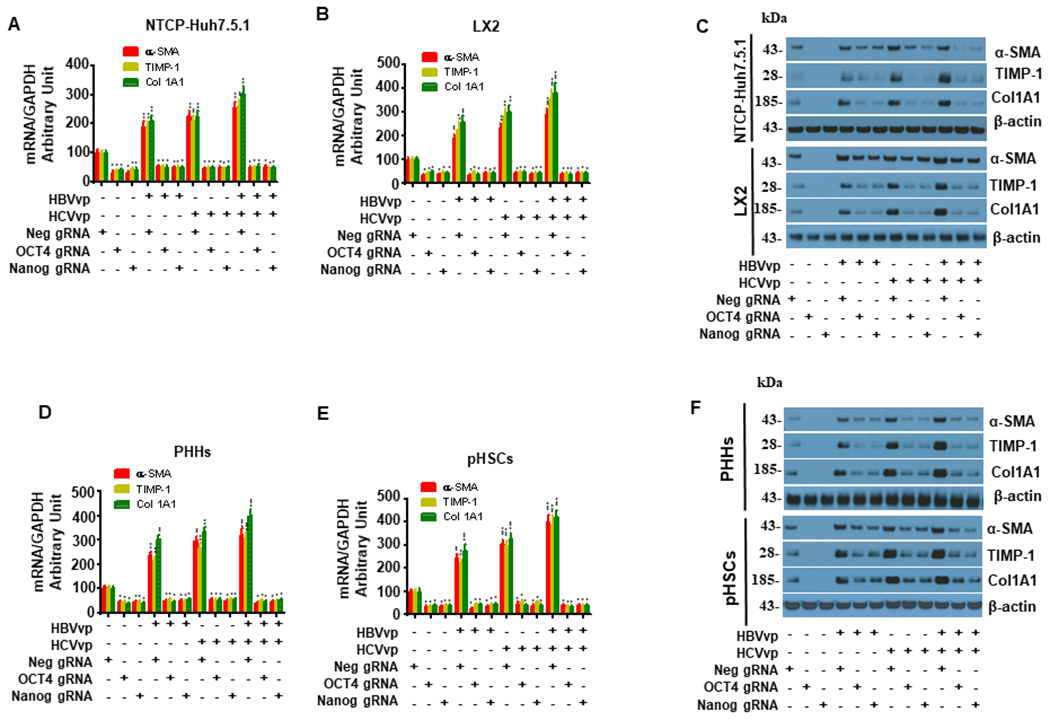
NTCP-Huh7.5.1 or PHHs (50,000 cell/well) were seeded onto the lower chamber of 12-well transwell plates. LX2 cells or pHSCs (50,000 cell/well) were plated in 2 mL 10%FBS DMEM on the upper chamber with a 0.4 micrometer (μm) membrane. NTCP-Huh7.5.1/LX2 cells were in one chamber while PHHs/pHSCs were in another chamber. 200 μL HBVvp or HCVvp were added to the appropriate well for 4 hours. Cells in the corresponding wells were transfected with OCT4, Nanog, or Neg gRNA for 72 hours. Cells were washed with PBS and cell lysates were harvested for mRNA quantification by qRT-PCR or protein analysis by western blot.
(A). OCT4 and Nanog gRNA inhibited HBV-, JFH1 HCV- or HBV+HCV-induced α-SMA, TIMP-1 and Col1A1 mRNA upregulation in NTCP-Huh7.5.1 in NTCP-Huh7.5.1/LX2 coculture.
(B). OCT4 and Nanog gRNA inhibited HBV-, JFH1 HCV- or HBV+HCV-induced α-SMA, TIMP-1 and Col1A1 mRNA level in LX2 in NTCP-Huh7.5.1/LX2 coculture.
(C). OCT4 and Nanog gRNA blocked HBV-, JFH1 HCV- or HBV+HCV-stimulated α-SMA, TIMP-1 and Col1A1 protein upregulation in NTCP-Huh7.5.1 and LX2 cells in NTCP-Huh7.5.1/LX2 coculture.
(D). OCT4 and Nanog gRNA reduced HBV-, JFH1 HCV- or HBV+HCV-induced α-SMA, TIMP-1 and Col1A1 mRNA upregulation in PHHs in PHHs/pHSCs coculture.
(E). OCT4 and Nanog gRNA inhibited HBV-, JFH1 HCV- or HBV+HCV-induced α-SMA, TIMP-1 and Col1A1 mRNA level in pHSCs in PHHs/pHSCs coculture.
(F). OCT4 and Nanog gRNA blocked HBV-, JFH1 HCV- or HBV+HCV-stimulated α-SMA, TIMP-1 and Col1A1 protein upregulation in PHHs and pHSCs cells in PHHs/pHSCs coculture.
Data are representative of 3 independent experiments with similar results. Bars represent means ± SD of 3 biological repeats. *, p < 0.05. **, p < 0.01. ***, p < 0.001.
We then confirmed these findings in PHHs and pHSCs with HBV and JFH1-HCV in coculture(Fig. 7D–F; SFig. 4A–F). Our data shows that JFH1-HCV and HBV replication in PHHs significantly upregulated Col1A1, TIMP-1, and OCT4/Nanog in cocultured PHHS and pHSCs (Fig. 7D–F, SFig. 4A–B). Moreover, HBV/HCV coinfection produced the highest levels of TGF-β1 compared to uninfected mock and HBV and HCV mono-infection (SFig. 4C). Similarily, HBV/HCV coinfetion in PHHs inhibited HBV or HCV replication (SFig. 4D). As in prior experiments, OCT4 and Nanog knockdown via gRNA (Fig. 7D–F, SFig. 4A–B) suppressed HBV- and JFH1-HCV-induced fibrogenesis in the coculture model without any effect on cell viability (SFig. 4E–F).
Discussion
HBV and HCV infection remain major public health concerns worldwide (6, 7) and , unfortunately due to shared routes of transmission, HBV/HCV coinfection is common (15, 47). Several studies have shown that coinfection is associated with accelerated progression to cirrhosis, end-stage liver disease and HCC (8, 15, 48, 49). Currently, there are no approved antifibrotic therapies and treating the underlying virus may not fully reverse the liver damage that has accumulated over years of infection prior to initiation of antiviral therapy. It is therefore important to understand the mechanisms through which HBV/HCV coinfection results in progressive liver fibrosis, to identify novel targets and strategies that may ameliorate or reverse the hepatic profibrogenic state.
TGF-β1 is perhaps the most important cytokine involved in liver fibrosis as binding to its cell surface receptor activates SMAD proteins that promote profibrotic gene transcription (50). The transcription factors OCT4 and Nanog are also regulated by TGF-beta signaling (27, 51), activated by HBV replication in hepatocytes (28, 52), and associated with liver fibrosis and liver cancer (30). We previously determined that HCV initiates liver fibrosis through the TGF-β1 signaling pathway (1, 4), however, the role of the TGF-β1/OCT4/Nanog pathway in HBV/HCV coinfection is not well understood. Using various cell culture models that support HBV infection, including HepAD38 cells, NTCP-Huh 7.5.1 cells and PHHs, we have shown that HBV replication potentiates TGF-β1/OCT4/Nanog activity, and that OCT4/Nanog upregulation is inhiibited by anti-HBV treatment, suggesting that HBV-induced fibrogenesis occurs through the TGF-β1/OCT4/Nanog pathway.
In accordance with prior studies, we also found that JFH1-HCV infection inhibits HBV replication (and vice versa) in HBV/JFH1-HCV coinfected NTCP-Huh 7.5.1 cells and PHHs. However, in the context of HBV/JFH1-HCV coinfection, there was upregulation of TGF-β1, OCT4, and Nanog beyond what was observed in HBV mono-infection in both NTCP-Huh 7.5.1 cells and PHHs. Furthermore, only concurrent inhibition of both HBV and JFH1-HCV replication could decrease TGF-β1/OCT4/Nanog activity in HBV/JFH1-HCV coinfected NTCP-Huh 7.5.1 and PHHs. More importantly, HBV/HCV coinfected patients had higher fibrosis scores and hepatic OCT4/Nanog protein levels than mono-infected patients. These findings suggest that the additive or synergistic effect of HBV and HCV on hepatic fibrogenesis is dependent on OCT4 and Nanog (Figure 8). In addition, we demonstrated that TGF-β1 increased profibrotic gene expression in both hepatocytes and HSCs, through an OCT4 or Nanog-dependent pathway. Silencing of OCT4 or Nanog also inhibited TGF-β1-induced Huh7.5.1 and LX-2 cell migratory capacity. It was previously shown that the HBx protein is directly involved in the pathogenesis of liver fibrosis and liver cancer (32, 52). Our data further demonstrate that the HBx and preS2 viral proteins increase profibrogenic gene expression in hepatocytes in an OCT4 or Nanog-dependent manner. Furthermore, we found that HCV Core and NS2/3 proteins also induced fibrogenesis through the TGF-β1/OCT4/Nanog pathway (53, 54).
Figure 8. Proposed pathway of hepatic fibrogenesis in HBV/HCV coinfection.
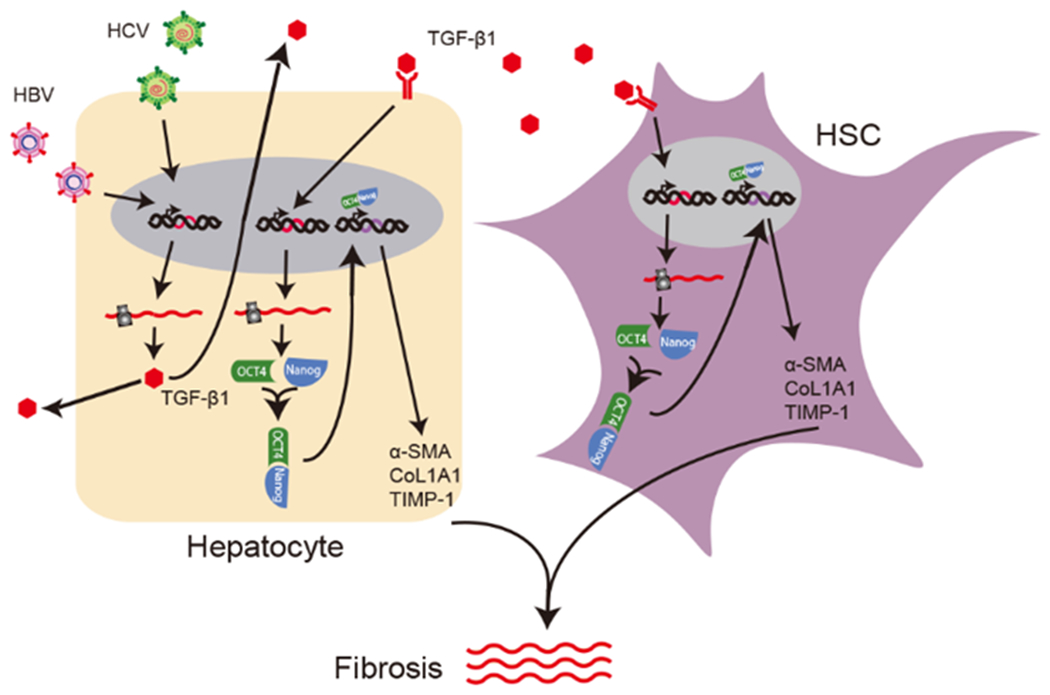
HBV and HCV stimulate TGF-β1 production in hepatocytes, which activates OCT4/Nanog in both hepatocytes and hepatic stellate cells. OCT4/Nanog then translocate to the nucleus, where they potentiate the expression of profibrogenic genes such as α-SMA, TIMP-1 and Col1A1.
In this study, we applied a multicellular coculture system to simulate the more complex in vivo liver microenvironment where cell-cell communication is important for maximal induction of a profibrogenic program (45). This model consisted of hepatocytes with or without HBV/JFH1-HCV infection and HSCs. The expression of profibrotic genes, such as Col1A1 and TIMP-1, in LX2 cells was significantly greater in the coculture system compared to LX2 cells in monoculture. Furthermore, dual exposure to supernatants from both HBVs and HCVs further augmented HSC activation. Using this model, we then depleted OCT4/Nanog in HSCs, which precluded HBVs- and HCVs-induced fibrogenesis in HSCs. Notably, purified HCV or HBV (without TGF-β1) did not induce a profibrotic response in HSCs, indicating that the cytokines and chemokines produced by hepatocytes infected with HBV and HCV are responsible for HSC activation.
The novel aspect of this study is the elucidation of a mechanism of HBV- and HCV-induced liver fibrosis through OCT4 and Nanog that is mediated by TGF-β-1 signaling. Furthermore, this pathway may be involved in hastened fibrosis progression seen in HBV-HCV coinfected patients and may serve as a new therapeutic target. Although this study did not involve all relevant cell types, a major strength of our study is the use of primary hepatic cells including PHHs and pHSCs, which allowed us to more aptly mimic in vivo conditions. These cells were used to confirm the results obtained in immortalized hepatic cell lines, specifically that HBV/HCV coinfection in hepatocytes induced liver fibrogenesis via the OCT4/Nanog pathway. Furthermore, PHH exposure to HBV/HCV increased TGF-β1 cytokine production.
In conclusion, HBV/HCV coinfection in hepatocytes and HBVs/HCVs coexposure to HSCs enhanced fibrogenesis relative to HBV or HCV mono-infection/exposure. TGF-β1 receptor blockade prevented HBVs and HCVs-induced liver fibrosis through the TGF-β1-OCT4/Nanog pathway. In HBV protein overexpression studies, we identified HBx and preS2 as the two primary triggers of HBV-related fibrogenesis, while the HCV core and NS2/3 proteins were the two main inducers of HCV-related liver fibrosis through the TGF-β1/OCT4/Nanog pathway. These experiments demonstrate that HBV, HCV, HBV/HCV coinfection/coexposure in hepatocytes and HSCs induces fibrogenesis through the TGF-β1-OCT4/Nanog pathway. Taken together, these findings provide new and exciting evidence that OCT4/Nanog may be a novel target(s) for antifibrotic therapeutic development.
Supplementary Material
Key Points:
HBV/HCV coinfection enhanced profibrogenic compared to HBV or HCV mono-infection.
HBV, HCV, and HBV/HCV induced liver fibrogenesis through the TGF-β1-OCT4/Nanog pathway.
Acknowledgments:
We are grateful to these investigators for supplying the reagents listed here: Dr. Takaji Wakita (National Institute of Infectious Disease, Japan) for providing JFH1 HCV DNA construct; Dr. Francis Chisari (The Scripps Research Institute, CA) for providing Huh7.5.1 cells; Dr. Ai-long Huang (Chongqing Medical University, China) for providing HepAD38 cells.
Funding:
This work was supported by grants from Natural Science Foundation of China (NSFC 82102383 to X. Duan, NSFC 81871661 to W. Lin, NSFC 81770591 to C. Zhu), the Fundamental Research Funds for the Central Universities of China (WK9110000048) to W. Li, Key project Foundation of Anhui Province, China (S202104j07020097) to W. Li, Project Foundation of academic leader Anhui Province, China (2018H178) to W. Li, Project supported by Hainan Province Clinical Medical Center to W. Li, NIH/NIAID R01AI155140 (RTC and WLin), NIH AI069939 (RTC), AI082630 (RTC), DK098079 (RTC), DK108370 (RTC).
Abbreviations
- HBV
hepatitis B virus
- HBcAg
hepatitis B core antigen
- HCV
hepatitis C virus
- HCC
hepatocellular carcinoma
- NTCP
sodium taurocholate cotransporting polypeptide
- PHHs
primary human hepatocytes
- Dac
daclatasvir
- ETV
entecavir
Footnotes
Conflicts of interest: The authors disclose no conflicts of interest.
References
- 1.Lin W, Wu G, Li S, Weinberg EM, Kumthip K, Peng LF, Mendez-Navarro J, Chen WC, Jilg N, Zhao H, Goto K, Zhang L, Brockman MA, Schuppan D, and Chung RT. 2011. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFkappaB. J Biol Chem 286: 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin W, Weinberg EM, and Chung RT. 2013. Pathogenesis of accelerated fibrosis in HIV/HCV co-infection. J Infect Dis 207 Suppl 1: S13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin W, Tsai WL, Shao RX, Wu G, Peng LF, Barlow LL, Chung WJ, Zhang L, Zhao H, Jang JY, and Chung RT. 2010. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 138: 2509–2518, 2518.e2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin W, Weinberg EM, Tai AW, Peng LF, Brockman MA, Kim KA, Kim SS, Borges CB, Shao RX, and Chung RT. 2008. HIV increases HCV replication in a TGF-beta1-dependent manner. Gastroenterology 134: 803–811. [DOI] [PubMed] [Google Scholar]
- 5.Imai Y, Yoshida O, Watanabe T, Yukimoto A, Koizumi Y, Ikeda Y, Tokumoto Y, Hirooka M, Abe M, and Hiasa Y. 2019. Stimulated hepatic stellate cell promotes progression of hepatocellular carcinoma due to protein kinase R activation. PLoS One 14: e0212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortiz E, Scanlon B, Mullens A, and Durham J. 2020. Effectiveness of Interventions for Hepatitis B and C: A Systematic Review of Vaccination, Screening, Health Promotion and Linkage to Care Within Higher Income Countries. J Community Health 45: 201–218. [DOI] [PubMed] [Google Scholar]
- 7.Krarup HB, Rex KF, and Andersen S. 2020. Risk of hepatitis B when migrating from low to high endemic areas. Int J Circumpolar Health 79: 1817274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu G, Chi X, Wu R, Wang X, Gao X, Kong F, Feng X, Gao Y, Huang X, Jin J, Qi Y, Tu Z, Sun B, Zhong J, Pan Y, and Niu J. 2015. Replication Inhibition of Hepatitis B Virus and Hepatitis C Virus in Co-Infected Patients in Chinese Population. PLoS One 10: e0139015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu D, and Ning Q. 2017. Toward a Cure for Hepatitis B Virus Infection: Combination Therapy Involving Viral Suppression and Immune Modulation and Long-term Outcome. J Infect Dis 216: S771–s777. [DOI] [PubMed] [Google Scholar]
- 10.Lanini S, Pisapia R, Capobianchi MR, and Ippolito G. 2018. Global epidemiology of viral hepatitis and national needs for complete control. Expert Rev Anti Infect Ther 16: 625–639. [DOI] [PubMed] [Google Scholar]
- 11.Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C, Wang X, Ryffel B, and Sun B. 2013. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. Journal of immunology (Baltimore, Md. : 1950) 191: 1835–1844. [DOI] [PubMed] [Google Scholar]
- 12.Wu Q, and Liu Q. 2012. Do hepatitis B virus and hepatitis C virus co-infections increase hepatocellular carcinoma occurrence through synergistically modulating lipogenic gene expression? Hepatol Res 42: 733–740. [DOI] [PubMed] [Google Scholar]
- 13.Wu Q, Li Z, and Liu Q. 2018. An important role of SREBP-1 in HBV and HCV co-replication inhibition by PTEN. Virology 520: 94–102. [DOI] [PubMed] [Google Scholar]
- 14.Xu L, Yu D, Yao YL, Gu T, Zheng X, Wu Y, Luo RH, Zheng YT, Zhong J, and Yao YG. 2020. Tupaia MAVS Is a Dual Target during Hepatitis C Virus Infection for Innate Immune Evasion and Viral Replication via NF-kappaB. Journal of immunology (Baltimore, Md. : 1950) 205: 2091–2099. [DOI] [PubMed] [Google Scholar]
- 15.Konstantinou D, and Deutsch M. 2015. The spectrum of HBV/HCV coinfection: epidemiology, clinical characteristics, viralinteractions and management. Ann Gastroenterol 28: 221–228. [PMC free article] [PubMed] [Google Scholar]
- 16.Sagnelli E, Coppola N, Pisaturo M, Masiello A, Tonziello G, Sagnelli C, Messina V, and Filippini P. 2009. HBV superinfection in HCV chronic carriers: a disease that is frequently severe but associated with the eradication of HCV. Hepatology 49: 1090–1097. [DOI] [PubMed] [Google Scholar]
- 17.Lee LP, Dai CY, Chuang WL, Chang WY, Hou NJ, Hsieh MY, Lin ZY, Chen SC, Hsieh MY, Wang LY, Chen TJ, and Yu ML. 2007. Comparison of liver histopathology between chronic hepatitis C patients and chronic hepatitis B and C-coinfected patients. J Gastroenterol Hepatol 22: 515–517. [DOI] [PubMed] [Google Scholar]
- 18.Shi J, Zhu L, Liu S, and Xie WF. 2005. A meta-analysis of case-control studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma in China. Br J Cancer 92: 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donato F, Boffetta P, and Puoti M. 1998. A meta-analysis of epidemiological studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma. Int J Cancer 75: 347–354. [DOI] [PubMed] [Google Scholar]
- 20.Grewal US, Walia G, Bakshi R, and Chopra S. 2018. Hepatitis B and C Viruses, Their Coinfection and Correlations in Chronic Liver Disease Patients: A Tertiary Care Hospital Study. Int J Appl Basic Med Res 8: 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chusri P, Kumthip K, Hong J, Zhu C, Duan X, Jilg N, Fusco DN, Brisac C, Schaefer EA, Cai D, Peng LF, Maneekarn N, Lin W, and Chung RT. 2016. HCV induces transforming growth factor beta1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci Rep 6: 22487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feili X, Wu S, Ye W, Tu J, and Lou L. 2018. MicroRNA-34a-5p inhibits liver fibrosis by regulating TGF-beta1/Smad3 pathway in hepatic stellate cells. Cell Biol Int 42: 1370–1376. [DOI] [PubMed] [Google Scholar]
- 23.Guo P, Sun X, Feng X, and Zhang C. 2018. Transforming growth factor-β1 gene polymorphisms with liver cirrhosis risk: A meta-analysis. Infect Genet Evol 58: 164–170. [DOI] [PubMed] [Google Scholar]
- 24.Hoseini SM, Montazeri F, Bahrami AR, Kalantar SM, Rahmani S, Zarein F, and Matin MM. 2020. Investigating the expression of pluripotency-related genes in human amniotic fluid cells: A semi-quantitative comparison between different subpopulations, from primary to cultured amniocytes. Reprod Biol. [DOI] [PubMed] [Google Scholar]
- 25.Alemohammad H, Asadzadeh Z, Motafakker Azad R, Hemmat N, Najafzadeh B, Vasefifar P, Najafi S, and Baradaran B. 2020. Signaling pathways and microRNAs, the orchestrators of NANOG activity during cancer induction. Life Sci 260: 118337. [DOI] [PubMed] [Google Scholar]
- 26.Sharma P, Gupta S, Chaudhary M, Mitra S, Chawla B, Khursheed MA, and Ramachandran R. 2019. Oct4 mediates Müller glia reprogramming and cell cycle exit during retina regeneration in zebrafish. Life Science Alliance 2: e201900548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Au HK, Chang JH, Wu YC, Kuo YC, Chen YH, Lee WC, Chang TS, Lan PC, Kuo HC, Lee KL, Lee MT, Tzeng CR, and Huang YH. 2015. TGF-betaI Regulates Cell Migration through Pluripotent Transcription Factor OCT4 in Endometriosis. PLoS One 10: e0145256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mani SK, Zhang H, Diab A, Pascuzzi PE, Lefrancois L, Fares N, Bancel B, Merle P, and Andrisani O. 2016. EpCAM-regulated intramembrane proteolysis induces a cancer stem cell-like gene signature in hepatitis B virus-infected hepatocytes. J Hepatol 65: 888–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang C, Xu Y, Ge H, Li G, and Wu J. 2018. Clinicopathological and prognostic significance of OCT4 in patients with hepatocellular carcinoma: a meta-analysis. Onco Targets Ther 11: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang TS, Wu YC, Chi CC, Su WC, Chang PJ, Lee KF, Tung TH, Wang J, Liu JJ, Tung SY, Kuo LM, Ho HN, Ling TY, and Huang YH. 2015. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin Cancer Res 21: 201–210. [DOI] [PubMed] [Google Scholar]
- 31.Ching RHH, Sze KMF, Lau EYT, Chiu YT, Lee JMF, Ng IOL, and Lee TKW. 2017. C-terminal truncated hepatitis B virus X protein regulates tumorigenicity, self-renewal and drug resistance via STAT3/Nanog signaling pathway. Oncotarget 8: 23507–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Oishi N, Shimakami T, Yamashita T, Honda M, Murakami S, and Kaneko S. 2017. Hepatitis B virus X protein induces hepatic stem cell-like features in hepatocellular carcinoma by activating KDM5B. World J Gastroenterol 23: 3252–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, and Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11: 791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H, Lin W, Kumthip K, Cheng D, Fusco DN, Hofmann O, Jilg N, Tai AW, Goto K, Zhang L, Hide W, Jang JY, Peng LF, and Chung RT. 2012. A functional genomic screen reveals novel host genes that mediate interferon-alpha’s effects against hepatitis C virus. J Hepatol 56: 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Duan X, Holmes JA, Li W, Lee SH, Tu Z, Zhu C, Salloum S, Lidofsky A, Schaefer EA, Cai D, Li S, Wang H, Huang Y, Zhao Y, Yu ML, Xu Z, Chen L, Hong J, Lin W, and Chung RT. 2019. A Long Noncoding RNA Regulates Hepatitis C Virus Infection Through Interferon Alpha-Inducible Protein 6. Hepatology 69: 1004–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin W, Zhu C, Hong J, Zhao L, Jilg N, Fusco DN, Schaefer EA, Brisac C, Liu X, Peng LF, Xu Q, and Chung RT. 2015. The spliceosome factor SART1 exerts its anti-HCV action through mRNA splicing. J Hepatol 62: 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, and Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 3. [DOI] [PubMed] [Google Scholar]
- 38.Watashi K, Sluder A, Daito T, Matsunaga S, Ryo A, Nagamori S, Iwamoto M, Nakajima S, Tsukuda S, Borroto-Esoda K, Sugiyama M, Tanaka Y, Kanai Y, Kusuhara H, Mizokami M, and Wakita T. 2014. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 59: 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duan X, Li S, Holmes JA, Tu Z, Li Y, Cai D, Liu X, Li W, Yang C, Jiao B, Schaefer EA, Fusco DN, Salloum S, Chen L, Lin W, and Chung RT. 2018. MicroRNA 130a Regulates both Hepatitis C Virus and Hepatitis B Virus Replication through a Central Metabolic Pathway. J Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, and King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41: 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin YJ, Huang LR, Yang HC, Tzeng HT, Hsu PN, Wu HL, Chen PJ, and Chen DS. 2010. Hepatitis B virus core antigen determines viral persistence in a C57BL/6 mouse model. Proc Natl Acad Sci U S A 107: 9340–9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.W L, Z. C, H. J, Z L, J N, F DN, S EA, B C, L X, P LF, X Q, and C RT. 2015. The spliceosome factor SART1 exerts its anti-HCV action through mRNA splicing. Journal of hepatology 62: 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, and Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102: 9294–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, and Chisari FV. 2010. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol 84: 10999–11009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salloum S, Holmes JA, Jindal R, Bale SS, Brisac C, Alatrakchi N, Lidofsky A, Kruger AJ, Fusco DN, Luther J, Schaefer EA, Lin W, Yarmush ML, and Chung RT. 2016. Exposure to human immunodeficiency virus/hepatitis C virus in hepatic and stellate cell lines reveals cooperative profibrotic transcriptional activation between viruses and cell types. Hepatology 64: 1951–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dewidar B, Meyer C, Dooley S, and Meindl-Beinker AN. 2019. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shih YF, and Liu CJ. 2020. Hepatitis C Virus and Hepatitis B Virus Co-Infection. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pol S, Haour G, Fontaine H, Dorival C, Petrov-Sanchez V, Bourliere M, Capeau J, Carrieri P, Larrey D, Larsen C, Marcellin P, Pawlostky JM, Nahon P, Zoulim F, Cacoub P, de Ledinghen V, Mathurin P, Negro F, Pageaux GP, Yazdanpanah Y, Wittkop L, Zarski JP, Carrat F, and C. French Anrs Co22 Hepather. 2017. The negative impact of HBV/HCV coinfection on cirrhosis and its consequences. Aliment Pharmacol Ther 46: 1054–1060. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen LH, and Nguyen MH. 2013. Systematic review: Asian patients with chronic hepatitis C infection. Aliment Pharmacol Ther 37: 921–936. [DOI] [PubMed] [Google Scholar]
- 50.Yang F, Wang N, Wang Y, Yu T, and Wang H. 2017. Activin-SMAD signaling is required for maintenance of porcine iPS cell self-renewal through upregulation of NANOG and OCT4 expression. J Cell Physiol 232: 2253–2262. [DOI] [PubMed] [Google Scholar]
- 51.Itoh F, Watabe T, and Miyazono K. 2014. Roles of TGF-beta family signals in the fate determination of pluripotent stem cells. Semin Cell Dev Biol 32: 98–106. [DOI] [PubMed] [Google Scholar]
- 52.Zhu M, Li W, Lu Y, Dong X, Lin B, Chen Y, Zhang X, Guo J, and Li M. 2017. HBx drives alpha fetoprotein expression to promote initiation of liver cancer stem cells through activating PI3K/AKT signal pathway. Int J Cancer 140: 1346–1355. [DOI] [PubMed] [Google Scholar]
- 53.Goto K, Roca Suarez AA, Wrensch F, Baumert TF, and Lupberger J. 2020. Hepatitis C Virus and Hepatocellular Carcinoma: When the Host Loses Its Grip. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakata K, Hara M, Terada T, Watanabe N, Takaya D, Yaguchi S, Matsumoto T, Matsuura T, Shirouzu M, Yokoyama S, Yamaguchi T, Miyazawa K, Aizaki H, Suzuki T, Wakita T, Imoto M, and Kojima S. 2013. HCV NS3 protease enhances liver fibrosis via binding to and activating TGF-beta type I receptor. Sci Rep 3: 3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.