

Abstract
Introduction:
The efficacy of pharmacotherapy and deep brain stimulation of the subthalamic nucleus in treating Parkinson’s disease motor symptoms is highly variable and may be influenced by patient genotype. The relatively common (prevalence about one in three) and protein-altering rs6265 single nucleotide polymorphism (C>T) in the gene BDNF has been associated with different clinical outcomes with levodopa.
Objective:
We sought to replicate this reported association in early-stage Parkinson’s disease subjects and to examine if a difference in clinical outcomes was present with subthalamic nucleus deep brain stimulation.
Materials and Methods:
Fifteen deep brain stimulation and thirteen medical therapy subjects were followed for 24 months as part of the Vanderbilt DBS in Early Stage PD clinical trial (NCT00282152, FDA IDE #G050016). Primary outcome measures were the Unified Parkinson’s Disease Rating Scale (UPDRS) and Parkinson’s Disease Questionnaire-39.
Results:
Outcomes with drug therapy in subjects carrying the rs6265 T allele were significantly worse following twelve months of treatment compared to C/C subjects (UPDRS: +20 points, p = 0.019; PDQ-39: +16 points, p = 0.018). In contrast, rs6265 genotype had no effect on overall motor response to subthalamic nucleus deep brain stimulation at any time point; further, rs6265 C/C subjects treated with stimulation were associated with worse UPDRS part II scores at 24 months compared to medical therapy.
Conclusions:
Genotyping for the rs6265 polymorphism may be useful for predicting long-term response to drug therapy and counseling Parkinson’s disease patients regarding whether to consider earlier subthalamic nucleus deep brain stimulation. Validation in a larger cohort of early-stage Parkinson’s disease subjects is warranted.
Keywords: Parkinson disease, brain-derived neurotrophic factor, levodopa, subthalamic nucleus deep brain stimulation, rs6265
Introduction
Oral levodopa and deep brain stimulation of the subthalamic nucleus (STN DBS) are the mainstay pharmacologic and surgical therapies, respectively, for Parkinson’s disease (PD). The introduction of levodopa often leads to a brief period of dramatic efficacy known as the ‘honeymoon’ phase. This honeymoon rapidly ends with increasing medication requirements and the onset of motor fluctuations, beginning after four to six years of levodopa treatment for about 40% of patients.1 During mid- and late-stage PD, some patients elect to undergo STN DBS and often experience a marked improvement in motor function, quality of life and motor fluctuations referred to by some as a ‘second honeymoon’.2 Although generally effective in treating PD motor symptoms, clinical response is highly variable for both of these therapies. For example, early-stage PD subjects receiving equivalent levodopa dosages experience a magnitude of response ranging from a 100% improvement to a 242% worsening as assessed using the Unified Parkinson’s Disease Rating Scale part III (UPDRS-III, motor score).3 In one of the largest clinical trials of STN DBS, late-stage PD subjects experienced improvements in UPDRS-III that ranged from 3% to 63% improvement.4 In addition, in a clinical trial of mid-stage PD subjects comparing STN DBS to medical therapy, UPDRS-III scores in the STN DBS treatment group ranged from an 83% improvement to a 7% worsening, whereas in the medication arm UPDRS-III scores ranged from a 50% improvement to a 42% worsening.5 These reports underscore the heterogeneity in clinical outcomes in association with different therapies across disease stages.
Variability in therapeutic outcomes may be explained from specific genetic variants,6, 7 like the rs6265 single nucleotide polymorphism (SNP) in the gene BDNF. A Val66Met substitution (C>T) in the prodomain of brain-derived neurotrophic factor (BDNF) decreases its activity-dependent release.8 T allele carriers (C/T or T/T) comprise approximately one third of the general population9 and have reduced plasticity and volume of the motor cortex and the caudate nucleus.10-12 The rs6265 SNP is associated with a milder early clinical phenotype in the unmedicated state of PD but not with increased incidence or rate of progression.13, 14 Of importance, the rs6265 SNP may have pharmacogenetic effects on PD clinical outcomes across disease stages. A recent report of early-stage PD subjects demonstrated worse clinical outcomes when rs6265 T allele carriers were treated with levodopa monotherapy compared to other dopamine replacement medications (e.g., dopamine agonists).15 Further, levodopa-induced dyskinesias develop earlier in T allele carriers in some studies16, 17 but not all.18 Beyond pharmacologic effects, BDNF signaling may also contribute to the efficacy of STN DBS.19, 20 Indeed, preclinical studies show that both chronic levodopa administration and STN DBS result in alterations in brain-derived neurotrophic factor (BDNF) levels.21-23 Therefore, we hypothesized the BDNF rs6265 T allele may alter antiparkinsonian treatment outcomes, including pharmacologic therapy with levodopa and surgical therapy with STN DBS. Retrospective examination of the completed Vanderbilt DBS in Early Stage PD clinical trial, which enrolled early-stage PD subjects who were randomized to medical therapy or STN DBS, provides an opportunity to test this hypothesis.24
Materials and Methods
We collected and genotyped DNA samples and retrospectively analyzed available clinical data from the Vanderbilt DBS in Early Stage PD trial. This study was approved by the Institutional Review Board of Vanderbilt University (#040797).
Vanderbilt Subjects and Trial Design
We genotyped subjects enrolled in the previously published Vanderbilt DBS in Early Stage PD trial.24 Twenty-nine, early-stage PD subjects prospectively treated for two years with bilateral STN DBS plus medication (“DBS”) or medical therapy (MT) completed the two-year trial. Subjects in the MT arm were treated primarily with levodopa and catechol-O-methyltransferase (COMT) inhibition or a dopamine agonist.25 Twenty-eight subjects (15 DBS, 13 MT) consented for genetic testing. Subjects were age 50-75 years, diagnosed with idiopathic PD, Hoehn and Yahr Stage II when off medication, treated with antiparkinsonian medications for >6 months but ≤4 years and with no history of dyskinesia or other motor fluctuations (Table 1). All subjects randomized to DBS were implanted with bilateral STN leads. Stimulation was optimized throughout the trial to maximize clinical benefit while minimizing adverse effects. Medications were optimized by each subject’s original treating neurologist to reduce investigator bias. Subjects were followed for 24 months. Assessments included UPDRS at baseline and six-month intervals. UPDRS-III (excluding rigidity) was videotaped in the ON therapy state and was scored by an independent, blinded PD expert certified in scoring the UPDRS. Levodopa-equivalent daily doses (LEDDs) were calculated as described previously.26
Table 1.
Vanderbilt Cohort Baseline Demographic Characteristics by rs6265 Genotype.
| Baseline Characteristics | All C/C (n=17) |
All C/T or T/T (n=11) |
DBS C/C (n=10) |
DBS C/T or T/T (n=5) |
MT C/C (n=7) |
MT C/T or T/T (n=6) |
|---|---|---|---|---|---|---|
| Gender | ||||||
| Men | 14 | 11 | 9 | 5 | 5 | 6 |
| Women | 3 | 0 | 1 | 0 | 2 | 0 |
| Age (years) at enrollment | ||||||
| Mean | 61.2 (1.8) |
59.7 (1.5) |
60.4 (2.4) |
61.1 (1.9) |
62.2 (2.8) |
58.5 (2.4) |
| Range | 52.2 - 73.9 | 50.2 - 67.9 | 52.2 - 73.9 | 55.4 - 67.0 | 52.4 - 69.6 | 50.2 - 67.9 |
| PD Duration (years) | 2.2 (0.5) |
2.1 (0.6) |
2.6 (0.7) |
1.4 (0.7) |
1.7 (0.5) |
2.7 (0.9) |
| Medicine Use | ||||||
| Mean Duration (years) | 2.3 (0.4) |
1.8 (0.4) |
2.7 (0.7) |
1.5 (0.7) |
1.8 (0.3) |
2.1 (0.6) |
| Mean Levodopa equivalents, Total (mg/day) | 472.1 (73.2) |
427.6 (62.0) |
405.9 (100.5) |
469.7 (130.8) |
566.6 (102.6) |
392.5 (46.1) |
| From Levodopa and COMT inhibitors (mg/day) | 338.5 | 140.8 | 242.9 | 259.7 | 475.2 | 41.7 |
| From Dopamine agonists (mg/day) | 132.1 | 221.4 | 160.5 | 190.0 | 91.4 | 300.8 |
| From MAOB Inhibitors or antivirals (mg/day) | 1.5 | 65.4 | 2.5 | 20.0 | 0.0 | 50.0 |
| UPDRS Scores | ||||||
| Mean Total (ON) | 33.0 (3.9) |
37.7 (3.8) |
36.1 (5.4) |
34.2 (6.6) |
27.8 (5.1) |
40.6 (4.6) |
| Mean UPDRS I | 2.1 (0.4) |
1.5 (0.3) |
1.6 (0.5) |
1.8 (0.6) |
1.3 (0.4) |
2.3 (0.6) |
| Mean UPDRS II (OFF) | 11.2 (1.2) |
10.1 (1.3) |
13.1 (1.5) |
8.2 (1.5) |
8.4 (1.3) |
11.7 (1.5) |
| Mean UPDRS II (ON) | 8.2 (1.5) |
8.1 (1.1) |
9.3 (1.5) |
5.4 (1.9) |
6.3 (1.6) |
10.5 (1.8) |
| Mean UPDRS III (OFF) | 28.3 (2.3) |
30.6 (2.6) |
26.8 (3.3) |
30.2 (4.60 |
30.3 (3.3) |
30.8 (3.3) |
| Mean UPDRS III (ON) | 21.1 (2.9) |
25.2 (2.9) |
24.6 (6.0) |
23.2 (4.0) |
17.5 (4.2) |
25.7 (2.5) |
| Mean UPDRS IV | 2.0 (0.4) |
2.3 (0.8) |
2.0 (0.5) |
2.4 (1.3) |
2.0 (0.6) |
2.2 (1.0) |
MT = medical therapy, DBS = deep brain stimulation, COMT = catechol-O-methyltransferase, MAOB = monoamine oxidase B. Values represent the mean (SEM).
SNP Genotyping
Vanderbilt subjects’ blood or saliva samples were collected, and high-quality genomic DNA was isolated using the prepIT-L2P (DNA Genotek) reagent. Samples were genotyped for the BDNF rs6265 SNP using the 5’ exonuclease allelic discrimination Taqman assay. Genotyping was performed with a minimum 20 ng of genomic DNA in a 25 μl reaction volume in duplicate using Taqman Genotyping Mastermix (Applied Biosystems, 4371353). Reactions were run using a Real-Time PCR instrument (Viia 7, Applied Biosystems). Data were analyzed for genotype determination calls made by Taqman Genotyper software (Applied Biosystems).
Vanderbilt Analysis and Statistics
We compared the outcomes of DBS and MT patients with and without rs6265 T alleles (i.e., C/T and T/T vs. C/C) using a mixed effects model that included the fixed effects (genotype, treatment, time) as well as their pairwise and three-way interactions and an autoregressive covariance structure to account for repeated measures at baseline, 6, 12, 18, and 24 months. Parameters were estimated from this model and used to compare group differences in terms of average scores over time. UPDRS and its four subparts were analyzed. Statistical significance was set at p < 0.05. All tests were two-tailed. These analyses were performed using SAS (v9.3; Cary, NC) and R (http://www.r-project.org/).
Results
We genotyped subjects enrolled in the Vanderbilt trial in which bilateral STN DBS plus medication (“DBS”) was compared prospectively to medical therapy (MT) over two years in early-stage PD.24 Five of fifteen subjects (33%) in the DBS arm and six of thirteen subjects (46%) in the MT arm carried at least one rs6265 T allele (C/T or T/T; Table 1).
At baseline, all clinical endpoints were similar across rs6265 genotypes (Table 1). However, over time, T allele carriers on MT exhibited higher (worse) average ON UPDRS scores compared to C/C MT subjects. Specifically, at 18 (p = 0.017) and 24 months (p = 0.019) UPDRS scores were higher in T allele carriers compared to C/C MT subjects (Figure 1A). At 24 months, the average UPDRS score of T allele carriers was ≈20 points higher (C/C MT = 33.1 (SE 4.3); C/T or T/T MT = 53.2 (6.3)). Similarly, T allele carriers in the MT arm displayed significantly higher (≈16 points) PDQ-39 scores at 12 (p = 0.033) and 24 months (31.80 (8.01), p = 0.018) compared to C/C subjects in the MT arm (14.97 (5.62), Figure 1B).
Figure 1. Impact of the BDNF rs6265 SNP on UPDRS and PDQ-39 over 2 years after initiation of DBS or with Medical Therapy.
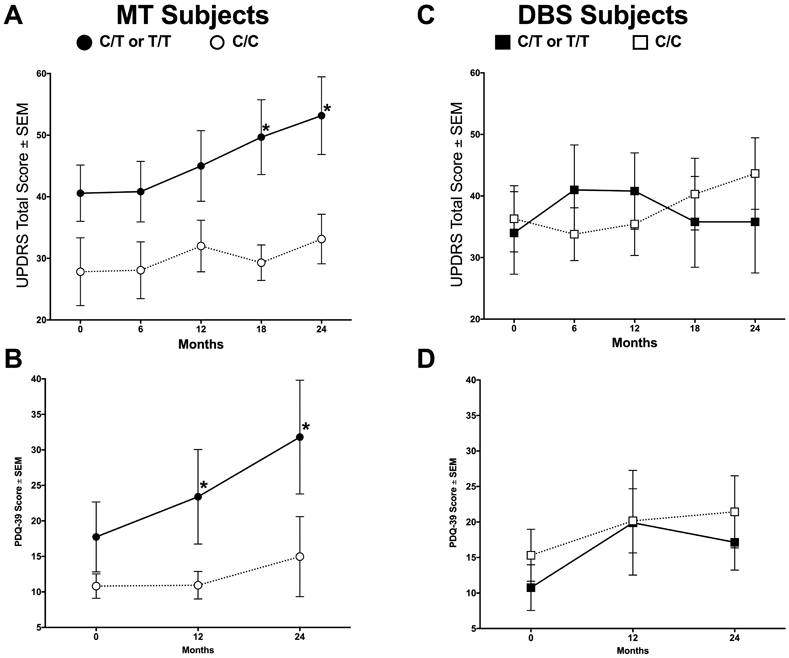
T allele carriers in the medical therapy (MT) treatment arm exhibited significantly higher (worse) ON total UPDRS at 18 and 24 months compared to C/C MT subjects (A, *, p = 0.017 and p = 0.019, respectively). T allele carriers also displayed higher (worse) PDQ-39 scores at 12 and 24 months compared to C/C subjects on MT (B, *, p = 0.033 and p = 0.018, respectively). No significant differences were observed between T allele carriers and C/C subjects receiving deep brain stimulation (DBS, ON medication and ON stimulation) at any time point (C, D, p > 0.05). Values represent the mean ± SEM.
The differences in UPDRS scores were examined further by its parts I-IV. T allele carriers on MT had higher UPDRS-II scores than C/C subjects at 18 months (13 (2.1) versus 6.6 (1.2), respectively; p = 0.03) and at 24 months (14.5 (2.3) versus 7.4 (1.5), respectively; p = 0.017, Figure 2B) with large numerical differences in UPDRS-III. In contrast, no differences due to genotype were observed between T allele carriers and C/C subjects receiving DBS at any time with any metric (Figure 1C,D and Figure 2E-H, p > 0.05). UPDRS-IV scores were low overall (Figure 2D,H).
Figure 2. Impact of the BDNF rs6265 SNP on UPDRS parts I-IV over 2 years.
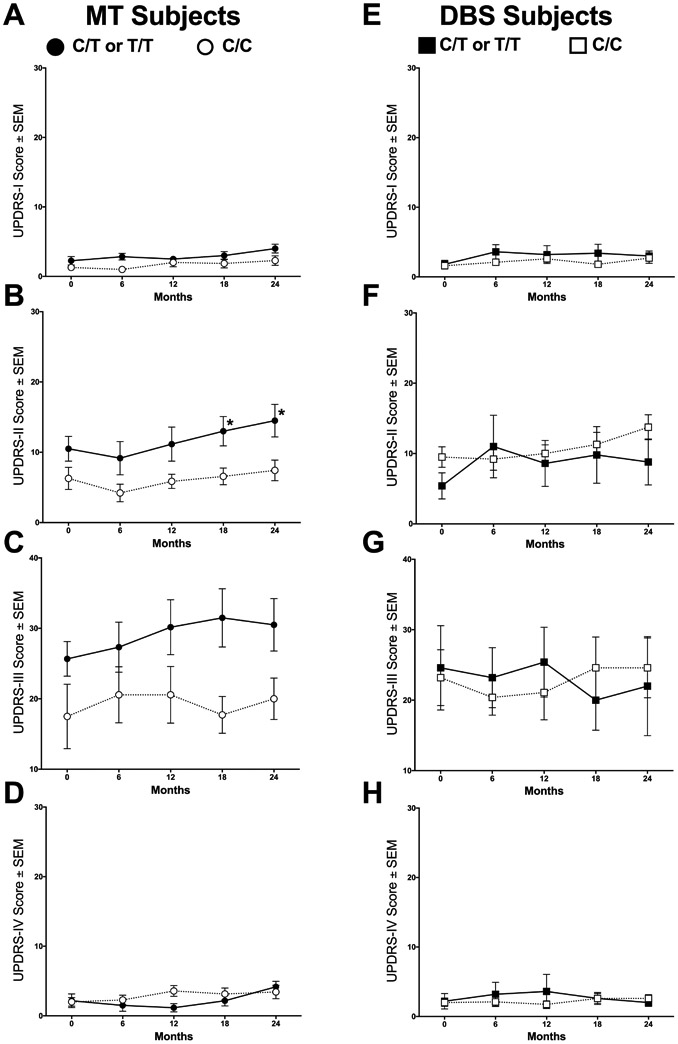
T allele carriers in the MT arm displayed significantly higher (worse) UPDRS-II scores at 24 months compared to C/C subjects (B, *, p = 0.017). In the other UPDRS parts, no significant differences were observed due to either rs6265 genotype or treatment (A, C-H, p > 0.05). Values represent the mean ± SEM.
Treatments were compared within genotype from a precision medicine perspective. At 24 months, T allele carriers on DBS had lower UPDRS scores at 24 months versus those on MT, although this difference (≈17 points) was not statistically significant in this small study (Figure 3B, p = 0.06, β = 0.714). Further, at 24 months T allele carriers treated with DBS had lower numerical PDQ-39 scores compared to subjects on MT, but this difference (≈15 points) was not significant statistically as well (Figure 3C, p = 0.08, β = 0.748). Of note, C/C subjects on MT had better UPDRS-II scores at 24 months (7.43 (1.58)) compared to those on DBS (13.75 (1.77), Figure 3A, p = 0.016).
Figure 3. BDNF rs6265 genotype as a surgicogenetic approach.
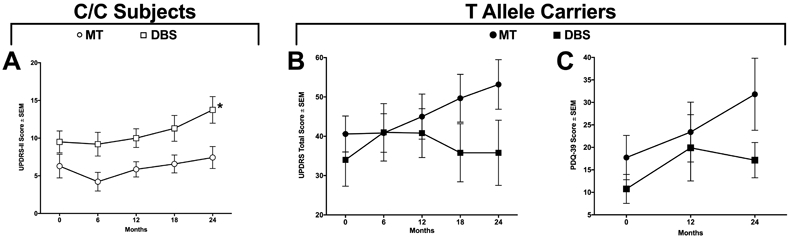
C/C subjects are associated with higher (worse) UPDRS-II scores at 24 months with DBS compared to MT (A, p = 0.016). T allele carriers in the MT arm displayed higher (worse) UPDRS (B) and PDQ-39 (C) scores at 24 months compared to T allele carriers in the DBS arm, though these trends did not reach statistical significance (UPDRS: p = 0.06; PDQ-39: p = 0.08). Values represent the mean ± SEM.
To determine if drug prescribing practices and dosages contributed to observed differences, LEDDs were compared between genotype-therapy groups. LEDDs (mg/day) at 24 months were not different between DBS T allele carriers (491 (213)), DBS C/C subjects (526 (65)), MT T allele carriers (537 (110)) and MT C/C subjects (715 (101)).
Discussion
Using a hypothesis-driven approach, three potential conclusions can be made about disease outcomes for PD subjects and the BDNF rs6265 SNP. First, these data replicate in an independent cohort that the rs6265 SNP contributes to the heterogeneous outcomes of the treatment strategies commonly employed in early-stage PD, particularly for levodopa.15 Second, there is no difference between T allele carriers and C/C subjects within the DBS arm. Third, C/C subjects with early-stage PD are associated with improved activities of daily living, as measured by UDPRS-II, when treated with pharmacotherapy over STN DBS.
A recent retrospective analysis of a large sample of early-stage PD subjects over three years was the first to demonstrate an association with worse clinical outcomes in rs6265 T allele carriers, specifically with levodopa monotherapy compared to a non-levodopa treatment strategy.15 The present study replicates this finding since within the MT arm T allele carriers were associated with numerically worse UPDRS scores throughout the study and significantly diverging at 18 and 24 months. Of note, the scores at 24 months are about 20 points higher, a large clinically important difference.27 The UPDRS parts II and III, measures in activities of daily living and motor outcomes, respectively, contributed to this spread. Of note, while the rs6265 SNP has been associated with earlier development of levodopa-induced dyskinesia,16, 17 these were not significantly present in this study of early-stage subjects based on the overall very low UPDRS part IV scores that measure this and other complications of therapy. Conversely, in the present study the rs6265 T allele was not associated with worse clinical outcomes with STN DBS over two years. Of importance, whereas the rs6265 SNP did not account for heterogeneity in DBS outcomes, the T allele may be a biomarker for considering early treatment with STN DBS over continuing standard pharmacotherapeutic strategies. Therefore, to understand this interaction better, future clinical trials exploring STN DBS in early-stage PD should consider including genotyping for rs6265.
Preclinical work has implicated BDNF-tropomyosin-related kinase type 2 (trkB) signaling in the antiparkinsonian efficacy of both levodopa and STN DBS.21-23 Specifically, levodopa administration to unilaterally lesioned rats increased transcript expression of BDNF in the frontal cortex and trkB in the striatum21; further, a progressive, levodopa-induced increase in corticostriatal BDNF release was critical to enhanced responsiveness to levodopa and its beneficial effects on motor performance.21 In a similar PD rat model, STN DBS significantly increased BDNF in the nigrostriatal system and primary motor cortex,22 and pharmacologic blockade of trkB attenuated stimulation efficacy.23 The decrease in activity-dependent BDNF release resulting from possession of the T allele28 may confer a suboptimal, striatal dopaminergic tone that is insufficient to maintain a prolonged response to dopaminergic medication. Indeed, healthy control subjects with the T allele display altered striatal dopamine signaling.29 Conversely, the lack of impact we observed of the T allele on DBS outcomes may be due to the limited involvement of dopaminergic signaling in DBS therapeutic efficacy.30, 31 Future preclinical studies will examine the potential mechanisms driving our clinical observations.
The present study applies a precision medicine approach to the approximate one third of the PD population carrying at least one rs6265 T allele9 to improve clinical decisions. This study replicates in early-stage PD subjects that rs6265 T allele carriers have worse outcomes with a primarily levodopa pharmacotherapeutic strategy. Pharmacogenetic considerations for PD have previously focused on genes that alter drug metabolism or dopamine transmission.32, 33 Surgicogenetic studies for stimulation procedures for PD have yielded mixed results and without a pharmacotherapy control group.34-37 The present results suggest that the brain environment created by possession of the T allele shapes the response to antiparkinsonian pharmacotherapy. This study is consistent with prior work demonstrating T allele carriers are associated with better clinical outcomes from avoiding levodopa especially as a monotherapy.15 Whether early STN DBS is another approach is unknown, as it was not compared directly to a levodopa-sparing strategy (e.g., dopamine agonist monotherapy). Further, there was not adequate power to avoid a false-negative result, as evidenced by the large β errors in comparing T allele carriers randomized to MT versus DBS. Of note, a previous analysis of this same clinical trial demonstrated MT subjects were treated with polypharmacy more often over time compared to DBS subjects,25 and this partly may be due to the T allele carriers treated with levodopa monotherapy are not converting to polypharmacy in the presence of stimulation. If these results are replicated in a prospective clinical trial, rs6265 SNP genotyping would be the first precision medicine approach that alters a pharmacotherapy treatment decision and perhaps the recommended timing of a surgical treatment.
Since this study was a retrospective analysis, stratification by genotype could not be performed, so the data may be subject to bias not addressed through randomization. T allele carriers on MT show a lower average LEDD than C/C subjects on MT that could contribute to the different UPDRS-III scores at 24 months; however, the magnitude of difference in LEDDs between these groups was nearly identical at the start compared to the end of the study (144.5 vs. 148.4 mg/day, or expressed as percentages, T allele carriers were prescribed an average LEDD of 69% and 75% of the C/C subjects at 0 and 24 months, respectively). It is unlikely that underlying differences in prescribing practices of the treating neurologists account for all of the variance in the LEDDs between T allele carriers and C/C subjects in the MT arm. Differences in LEDDs between subgroups were not statistically significant at baseline or at the end of the study. In contrast, there was no difference in outcomes with MT between C/C subjects and T allele carriers at baseline, but over the course of 24 months these differences were magnified and became significant. Lastly, some group variability may be influenced by differences in electrode placement and were not assessed by an active electrode contact analysis; however, these likely are randomly distributed across genotypes within the DBS group. To strengthen further the evidence for this study’s conclusions, similar analyses on clinical outcomes with different pharmacotherapeutic strategies and with STN DBS in a prospective trial are the suggested next step.
In conclusion, the BDNF rs6265 T allele is associated with worse disease outcomes in those treated with pharmacotherapy but not STN DBS in early-stage PD patients. The approximate one-third of PD patients carrying a T allele may have improved early therapeutic outcomes when treated with STN DBS. If these results are replicated prospectively, accounting for the BDNF rs6265 SNP in clinical practice and in planning clinical trials would be warranted. Future investigation should include the interaction of the BDNF rs6265 SNP and antiparkinsonian treatment strategies as a potential avenue for personalized medicine and therapy development.
Funding Statement:
This research was supported by the Morris K. Udall Center of Excellence for Parkinson’s Disease Research at Michigan State University (P50 NS058830), Vanderbilt CTSA grant UL1TR000445 from the National Center for Advancing Translational Sciences (NCATS), NCATS/NIH award UL1TR000011, NIH EB006136 and the Saint Mary’s Foundation (Grand Rapids, MI, USA). Medtronic, Inc. provided funding for the original study and analysis of the Vanderbilt DBS in Early PD trial but not the analyses presented above.
Footnotes
Data Accessibility
The individual, de-identified subject data and related study documents are not being shared publicly at this time since they currently are being used for the development of a proprietary, multicenter, phase III, pivotal clinical trial (IDE G050016).
Author Disclosures:
Caryl E. Sortwell receives research funding from the NIH, Michael J. Fox Foundation, the Saint Mary’s Foundation and the U.S. Department of Defense. She also receives income for reviewing for the NIH, the Michael J. Fox Foundation, and the Weston Brain Institute.
Mallory L. Hacker receives research funding from the DoD, NIH, and the American Parkinson Disease Association.
Peter E. Konrad receives research funding from Medtronic and the NIH, is on the speaker’s bureau for Medtronic and FHC, and also holds a fiduciary position (Board of Directors) with Neurotargeting, the American Society for Stereotactic and Functional Neurosurgery, and the North American Neuromodulation Society.
Joseph S. Neimat has done consulting work for Medtronic, Monteris and Abbott; and has received research funding from Medtronic and the NIH.
Jack W. Lipton receives research funding from the NIH and the Michael J. Fox Foundation, and he also receives income for reviewing for the NIH.
P. David Charles receives income from USWorldMeds, Alliance for Patient Access and Revance for consulting services. Vanderbilt University Medical Center receives income from grants or contracts with Abbott, Abbvie, Allergan, Boston Scientific, Intec, Ipsen, Lundbeck, Medtronic, Merz, Novartis, Pharma Two B, USWorldmeds, and Voyager for research or educational programs led by David Charles.
D. Luke Fischer, Allyson Cole-Strauss, Thomas L. Davis, Lily Wang, Yanna Song and Zach R. Mattingly have no reportable conflicts of interest.
References
- 1.Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. May 2001;16(3):448–458. [DOI] [PubMed] [Google Scholar]
- 2.Tanner CM. A second honeymoon for Parkinson's disease? N Engl J Med. Feb 14 2013;368(7):675–676. [DOI] [PubMed] [Google Scholar]
- 3.Hauser RA, Auinger P, Oakes D, Parkinson Study G. Levodopa response in early Parkinson's disease. Mov Disord. Dec 15 2009;24(16):2328–2336. [DOI] [PubMed] [Google Scholar]
- 4.Weaver FM, Follett KA, Stern M, et al. Randomized trial of deep brain stimulation for Parkinson disease: thirty-six-month outcomes. Neurology. Jul 3 2012;79(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuepbach WM, Rau J, Knudsen K, et al. Neurostimulation for Parkinson's disease with early motor complications. N Engl J Med. Feb 14 2013;368(7):610–622. [DOI] [PubMed] [Google Scholar]
- 6.Espay AJ, Schwarzschild MA, Tanner CM, et al. Biomarker-driven phenotyping in Parkinson's disease: A translational missing link in disease-modifying clinical trials. Mov Disord. Mar 2017;32(3):319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurzawski M, Bialecka M, Drozdzik M. Pharmacogenetic considerations in the treatment of Parkinson's disease. Neurodegener Dis Manag. Feb 2015;5(1):27–35. [DOI] [PubMed] [Google Scholar]
- 8.Chen ZY, Patel PD, Sant G, et al. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. May 5 2004;24(18):4401–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egan MF, Kojima M, Callicott JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. Jan 24 2003;112(2):257–269. [DOI] [PubMed] [Google Scholar]
- 10.Kleim JA, Chan S, Pringle E, et al. BDNF val66met polymorphism is associated with modified experience-dependent plasticity in human motor cortex. Nat Neurosci. Jun 2006;9(6):735–737. [DOI] [PubMed] [Google Scholar]
- 11.McHughen SA, Rodriguez PF, Kleim JA, et al. BDNF val66met polymorphism influences motor system function in the human brain. Cereb Cortex. May 2010;20(5):1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pezawas L, Verchinski BA, Mattay VS, et al. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci. Nov 10 2004;24(45):10099–10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karamohamed S, Latourelle JC, Racette BA, et al. BDNF genetic variants are associated with onset age of familial Parkinson disease: GenePD Study. Neurology. Dec 13 2005;65(11):1823–1825. [DOI] [PubMed] [Google Scholar]
- 14.Fischer DL, Auinger P, Goudreau JL, et al. Bdnf variant is associated with milder motor symptom severity in early-stage Parkinson's disease. Parkinsonism Relat Disord. Aug 2018;53:70–75. [DOI] [PubMed] [Google Scholar]
- 15.Fischer DL, Auinger P, Goudreau JL, et al. BDNF rs6265 Variant Alters Outcomes with Levodopa in Early-Stage Parkinson's Disease. Neurotherapeutics. Oct 2020;17(4):1785–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foltynie T, Cheeran B, Williams-Gray CH, et al. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson's disease. J Neurol Neurosurg Psychiatry. Feb 2009;80(2):141–144. [DOI] [PubMed] [Google Scholar]
- 17.Kusters CDJ, Paul KC, Guella I, et al. Dopamine receptors and BDNF-haplotypes predict dyskinesia in Parkinson's disease. Parkinsonism Relat Disord. Feb 2018;47:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplan N, Vituri A, Korczyn AD, et al. Sequence variants in SLC6A3, DRD2, and BDNF genes and time to levodopa-induced dyskinesias in Parkinson's disease. J Mol Neurosci. Jun 2014;53(2):183–188. [DOI] [PubMed] [Google Scholar]
- 19.Fischer DL, Kemp CJ, Cole-Strauss A, et al. Subthalamic Nucleus Deep Brain Stimulation Employs trkB Signaling for Neuroprotection and Functional Restoration. J Neurosci. Jul 12 2017;37(28):6786–6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer DL, Sortwell CE. BDNF provides many routes toward STN DBS-mediated disease modification. Mov Disord. Jan 2019;34(1):22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature. May 3 2001;411(6833):86–89. [DOI] [PubMed] [Google Scholar]
- 22.Spieles-Engemann AL, Steece-Collier K, Behbehani MM, et al. Subthalamic Nucleus Stimulation Increases Brain Derived Neurotrophic Factor in the Nigrostriatal System and Primary Motor Cortex. J Parkinsons Dis. May 31 2011;1(1):123–136. [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer DL, Polinski NK, Kemp CJ, et al. trkB signaling mediates neuroprotective and behavioral effects of long-term, high-frequency subthalamic nucleus deep brain stimulation. Movement Disorders. May 2014;29:S438–S439. [Google Scholar]
- 24.Charles D, Konrad PE, Neimat JS, et al. Subthalamic nucleus deep brain stimulation in early stage Parkinson's disease. Parkinsonism Relat Disord. Jul 2014;20(7):731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hacker ML, Currie AD, Molinari AL, et al. Subthalamic Nucleus Deep Brain Stimulation May Reduce Medication Costs in Early Stage Parkinson's Disease. J Parkinsons Dis. 2016;6(1):125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov Disord. Nov 15 2010;25(15):2649–2653. [DOI] [PubMed] [Google Scholar]
- 27.Shulman LM, Gruber-Baldini AL, Anderson KE, Fishman PS, Reich SG, Weiner WJ. The clinically important difference on the unified Parkinson's disease rating scale. Arch Neurol. Jan 2010;67(1):64–70. [DOI] [PubMed] [Google Scholar]
- 28.Chen ZY, Jing D, Bath KG, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. Oct 6 2006;314(5796):140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pecina M, Martinez-Jauand M, Love T, et al. Valence-specific effects of BDNF Val66Met polymorphism on dopaminergic stress and reward processing in humans. J Neurosci. Apr 23 2014;34(17):5874–5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muthuraman M, Koirala N, Ciolac D, et al. Deep Brain Stimulation and L-DOPA Therapy: Concepts of Action and Clinical Applications in Parkinson's Disease. Front Neurol. 2018;9:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stefani A, Cerroni R, Mazzone P, et al. Mechanisms of action underlying the efficacy of deep brain stimulation of the subthalamic nucleus in Parkinson's disease: central role of disease severity. Eur J Neurosci. Mar 2019;49(6):805–816. [DOI] [PubMed] [Google Scholar]
- 32.Agundez JA, Garcia-Martin E, Alonso-Navarro H, Jimenez-Jimenez FJ. Anti-Parkinson's disease drugs and pharmacogenetic considerations. Expert Opin Drug Metab Toxicol. Jul 2013;9(7):859–874. [DOI] [PubMed] [Google Scholar]
- 33.Kalinderi K, Fidani L, Katsarou Z, Bostantjopoulou S. Pharmacological treatment and the prospect of pharmacogenetics in Parkinson's disease. Int J Clin Pract. Dec 2011;65(12):1289–1294. [DOI] [PubMed] [Google Scholar]
- 34.Schupbach M, Lohmann E, Anheim M, et al. Subthalamic nucleus stimulation is efficacious in patients with Parkinsonism and LRRK2 mutations. Mov Disord. Jan 2007;22(1):119–122. [DOI] [PubMed] [Google Scholar]
- 35.Johansen KK, Jorgensen JV, White LR, Farrer MJ, Aasly JO. Parkinson-related genetics in patients treated with deep brain stimulation. Acta Neurol Scand. Mar 2011;123(3):201–206. [DOI] [PubMed] [Google Scholar]
- 36.Greenbaum L, Israeli-Korn SD, Cohen OS, et al. The LRRK2 G2019S mutation status does not affect the outcome of subthalamic stimulation in patients with Parkinson's disease. Parkinsonism Relat Disord. Nov 2013;19(11):1053–1056. [DOI] [PubMed] [Google Scholar]
- 37.Sayad M, Zouambia M, Chaouch M, et al. Greater improvement in LRRK2 G2019S patients undergoing Subthalamic Nucleus Deep Brain Stimulation compared to non-mutation carriers. BMC Neurosci. Feb 1 2016;17:6. [DOI] [PMC free article] [PubMed] [Google Scholar]