

Abstract
Objectives
Aberrant NLRP3 inflammasome activation has been demonstrated in rheumatoid arthritis (RA), which may contribute to debilitating inflammation and bone destruction. Here, we explored the efficacy of the potent TGF‐β‐activated kinase‐1 (TAK1) inhibitor LL‐Z1640‐2 (LLZ) on joint inflammation and bone destruction in collagen‐induced arthritis (CIA).
Methods
LL‐Z1640‐2 was administered every other day in CIA mice. Clinical and histological evaluation was performed. Priming and activation of NLRP3 inflammasome and osteoclastogenic activity were assessed.
Results
NLRP3 inflammasome formation was observed in synovial macrophages and osteoclasts (OCs) in CIA mice. TACE and RANKL were also overexpressed in synovial macrophages and fibroblasts, respectively, in the CIA joints. Treatment with LLZ mitigated all the above changes. As a result, LLZ markedly suppressed synovial hypertrophy and pannus formation to alleviate pain and inflammation in CIA mice. LLZ could block the priming and activation of NLRP3 inflammasome in RAW264.7 macrophage cell line, primary bone marrow macrophages and OCs upon treatment with LPS followed by ATP, thereby suppressing their IL‐1β production. LLZ also suppressed LPS‐induced production of TACE and TNF‐α in bone marrow macrophages and abolished IL‐1β‐induced production of MMP‐3, IL‐6 and RANKL in synovial fibroblasts. In addition, LLZ directly inhibits RANKL‐mediated OC formation and activation.
Conclusion
TAK1 inhibition with LLZ may become a novel treatment strategy to effectively alleviate inflammasome‐mediated inflammation and RANKL‐induced osteoclastic bone destruction in joints alongside its potent suppression of TNF‐α and IL‐6 production and proteinase‐mediated pathological processes in RA.
Keywords: rheumatoid arthritis, NLRP3 inflammasome, TAK1, LL‐Z1640‐2, synovial fibroblasts, macrophage, osteoclast
In this study, we found that TGF‐β activated kinase‐1 (TAK1) mediates NLRP3 inflammasome‐induced inflammation and RANKL‐induced osteoclastic bone destruction alongside of TNF‐α and IL‐6 production and proteinase‐induced pathological processes in rheumatoid arthritis. The TAK1 inhibitor LL‐Z1640‐2 effectively alleviates joint inflammation and bone destruction in collagen‐induced arthritis.
Introduction
Rheumatoid arthritis (RA) is the most common autoimmune disorder in joints. Clinical application of biologics, such as IL‐6 or TNF‐α inhibitors, and small compounds to inhibit the intracellular signalling mediator Janus kinase (JAK) has significantly improved the therapeutic outcome of RA. However, a portion of patients are still refractory to currently available biologics. The diversity of activated cells and overproduced cytokines may limit their clinical efficacy. Inflammasomes are formed as innate defence against infection or tissue damage. NLRP3 inflammasome activation causes an inflammation and is actively involved in the pathogenesis of inflammatory disorders. 1 , 2 Aberrant NLRP3 inflammasome activation has also been reported in patients with RA. 3 , 4 The priming and activation of NLRP3 inflammasome convert pro‐IL‐1β into mature and biologically active IL‐1β. 1 , 2 , 5 Excessive IL‐1β production also contributes to the induction of various pathological conditions in RA. 6 , 7 The treatment with the selective NLRP3 inhibitor MCC950 has been reported to result in significantly less severe joint inflammation and bone destruction in type II collagen‐induced arthritis (CIA). 8 Therefore, activation of NLRP3 inflammasome appears to provide as a novel treatment target of RA.
Synovial fibroblasts (SFs) are activated and proliferated in inflamed joints in RA and play a critical role in joint inflammation and destruction. 9 , 10 SFs produce various pro‐inflammatory cytokines and proteinases, including TNF‐α, IL‐6 and alongside metalloproteinases (MMPs) and a disintegrin and metalloproteinase 17 (ADAM17)/TNF‐α‐converting enzyme (TACE). 9 , 10 , 11 , 12 These pro‐inflammatory cytokines and proteinases in turn activate SFs themselves and shift them towards an activated phenotype, which further potentiates inflammatory responses with inflamed synovium to cause joint damage. Jones et al. thoroughly screened kinase inhibitors and drugs available to block SF activation in multiple samples from patients with RA, and demonstrated that (5z)‐7‐oxozeaenol (LL‐Z1640‐2; LLZ), a resorcylic acid lactone derived from fungus, was the most effective in terms of blocking SF activation across all contexts they studied, including the aberrant production of TNF‐α and IL‐6. 13
LL‐Z1640‐2 is a preclinical drug whose primary target is transforming growth factor β‐associated kinase‐1 (TAK1). TAK1 is a member of the mitogen‐activated protein kinase kinase kinase (MAP3K), also known as MAP3K7. 14 It was originally identified as a key kinase to transduce the TGF‐β signalling down to p38 mitogen‐activated protein kinase (MAPK), c‐Jun and N‐terminal kinase (JNK). 15 , 16 Subsequently, TAK1 has been demonstrated to be associated with a wide range of intracellular signalling pathways important for various cellular functions, including those for the activation of nuclear factor‐κB (NF‐κB) and extracellular signal‐regulated kinase (ERK). 15 , 17 Therefore, TAK1 appears to be a gatekeeper to regulate multiple important intracellular signalling pathways. Besides, TAK1 activation strongly elicits cellular production of pro‐inflammatory cytokines, chemokines, adhesion molecules and the critical bone‐resorbing factor receptor activator of NF‐κB ligand (RANKL). 18 , 19
Pharmacological inhibition of TAK1 with the selective inhibitor takinib has been recently reported to alleviate clinical manifestation of inflammation, cartilage damage, pannus formation and bone resorption in CIA mouse models, further posing TAK1 as a therapeutic target to control inflammatory signatures of RA and reduce disease activity. 20 Given the most superb therapeutic efficacy with LLZ against SFs from patients with RA, 13 the objectives of the present study are to clarify the therapeutic efficacy of LLZ on pathological manifestation in RA, especially on NLRP3 inflammasome activation and osteoclastic bone destruction, and to delineate pathognomonic roles of the TAK1‐NLRP3 inflammasome pathway in RA. Here, we demonstrate that NLRP3 inflammasome is formed in synovial macrophages and osteoclasts (OCs) in CIA mouse models and that treatment with LLZ can block the priming and activation of NLRP3 inflammasome in RAW264.7 macrophage cell line, primary bone marrow macrophages (BMMs) and OCs upon LPS and subsequent ATP, thereby suppressing their IL‐1β production. LLZ also directly inhibits RANKL‐mediated OC formation and activation. Moreover, LLZ suppresses LPS‐induced production of TACE and TNF‐α in BMMs and abolishes IL‐1β‐induced production of MMP‐3, IL‐6 and RANKL in SFs. Therefore, TAK1 inhibition with LLZ may become a novel treatment strategy to effectively alleviate inflammasome‐mediated inflammation and RANKL‐induced osteoclastic bone destruction in joints alongside its potent suppression of TNF‐α, IL‐6 and proteinase‐mediated pathological processes in RA.
Results
The TAK1 inhibitor LLZ alleviates inflammation and bone destruction in CIA joints
To investigate the therapeutic effects of the TAK1 inhibitor LLZ on RA, type II collagen‐induced arthritis (CIA) was induced in DBA1/J mice, which is generally accepted as an RA animal model. 21 The second injection of type II collagen induced swelling in paws (Figure 1a). The TAK1 inhibitor LLZ was administrated by intraperitoneal injection once per every 2 days from the second injection. Treatment with LLZ suppressed the paw swelling (Figure 1a), and improved arthritis severity (Figure 1b left) and incidence (Figure 1b right), and also increased the withdrawal threshold of paws by pain in plantar tests (Figure 1c) in the CIA mice. Hypertrophy of synovial tissues and joint destruction was observed in joints of CIA mice in histopathological analysis; however, the treatment with LLZ suppressed the hypertrophy of synovial tissues and joint destruction (Figure 1d) along with the disappearance of rheumatoid pannus formation (Figure 1e). Along with joint destruction, OCs appeared on the surface of eroded bones in joints of CIA mice, which was reduced by the treatment with LLZ (Figure 1f). These results collectively suggest that treatment with LLZ effectively suppresses inflammation and bone destruction in RA. Although inflammation of synovial tissues is vital in RA progression, 10 , 11 the role of NLRP3 inflammasome in synovial tissues is not fully understood. Therefore, we immunohistochemically investigated the expression of NLRP3 inflammasome in joints in CIA mice. F4/80‐expressing macrophages markedly increased in number in the synovial tissues and expressed NLRP3 (Figure 1g). However, treatment with LLZ almost completely suppressed the emergence of NLRP3‐expressing synovial macrophages (Figure 1g) and reduced IL‐1β secretion in sera of CIA mice (Figure 1h). These observations suggest the role of TAK1 activation in NLRP3 inflammasome‐mediated inflammation in CIA mice.
Figure 1.
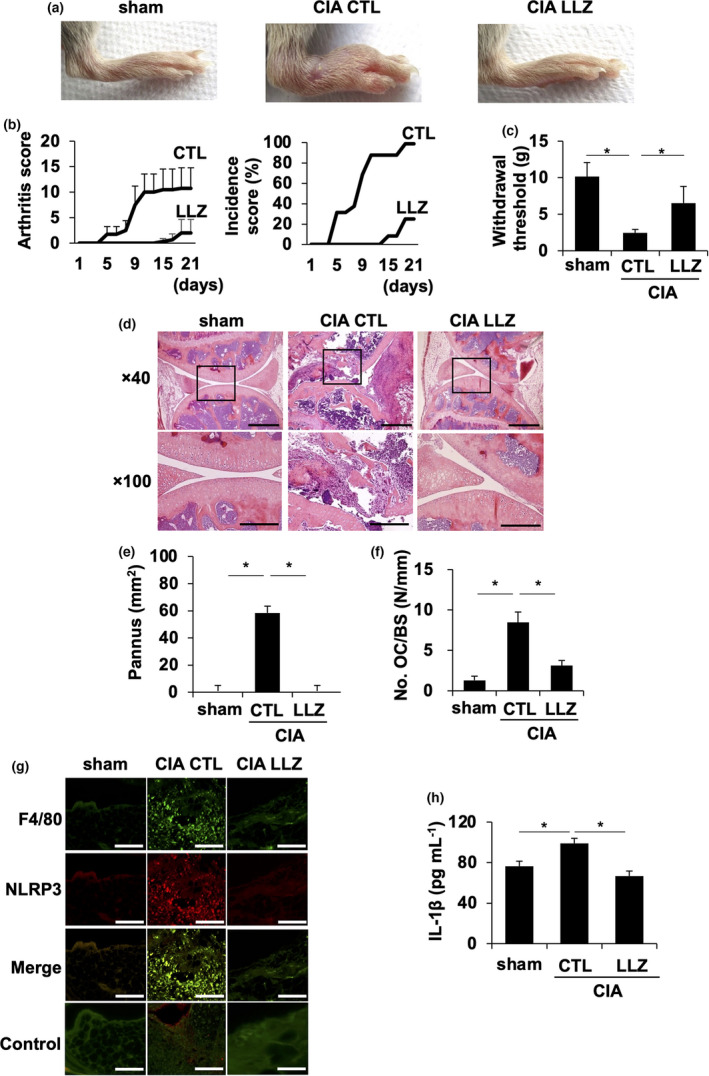
LLZ effectively alleviates inflammation and bone destruction in CIA mice. (a) CIA mice were treated every other day with vehicle control (CIA CTL) or LLZ (CIA LLZ) at 20 mg kg−1. Representative photographs of hind paws at day 21 from the second injection of the type II collagen are shown. (b) The inflammation of each paw was scored. The sum of scores for all 4 paws was expressed as an arthritis score (n = 4 for each group). The incidence of arthritis was presented as a percentage of swollen paws in each group (16 paws, n = 4 for each group). Data are expressed as mean ± SD. *P < 0.05 by two‐way ANOVA for arthritis scores and a log‐rank test for incidence scores. (c) Withdrawal threshold (g) was analysed with the plantar test at day 21 from the second injection of type II collagen. Data are expressed as mean ± SD (n = 4). *P < 0.05 by one‐way ANOVA with Tukey’s test. (d) HE staining of knee joints in sham and CIA mice is shown. Scale bars represent 200 μm. Original magnification ×40 and ×100 as indicated. (e, f) Pannus area (e) and numbers of osteoclasts over bone surface (No. OC/BS) (f) were measured. Data are expressed as mean ± SD (n = 4). *P < 0.05 by one‐way ANOVA with Tukey’s test. (g) Expression of NLRP3 in F4/80‐positive macrophages in synovial tissue sections of resected hindlegs in sham and CIA mice. The samples were stained with antibodies against F4/80 and NLRP3. The first antibodies were detected with the fluorescence‐conjugated secondary antibodies as described in the Methods. Scale bars represent 100 μm. Original magnification ×400. (h) Concentration of IL‐1β in sera from mice was quantified by ELISA. Data are expressed as mean ± SD (n = 4). *P < 0.05 by one‐way ANOVA with Tukey’s test.
LLZ suppresses the priming and activation of NLRP3 inflammasome in bone marrow macrophages
Inflammasomes are multiprotein signal platforms to elicit the innate immune system and inflammation in response to pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs). 22 , 23 , 24 Because LLZ potently suppressed NLRP3 expression in synovial macrophages in CIA mice (Figure 1g), we next looked at the effects of LLZ on in vitro NLRP3 inflammasome formation and activation. TLR4‐mediated activation has been well demonstrated to induce NLRP3 and pro‐IL‐1β expression as ‘a priming phase’. 1 , 25 The addition of the Toll‐like receptor 4 (TLR4) agonist LPS induced TAK1 phosphorylation, followed by the phosphorylation and disappearance of IκBα in the murine macrophage cell line RAW264.7 (Figure 2a). Furthermore, LPS induced the protein levels of NLRP3 and pro‐IL‐1β in RAW264.7 cells and primary BMMs, which was abolished upon treatment with LLZ (Figure 2b). TAK1 gene silencing by siRNA also reduced NLRP3 and pro‐IL‐1β levels in RAW264.7 cells (Figure 2c). After the priming phase, the protein complex, named NLRP3 inflammasome, is formed by factors released by tissue damage such as ATP to cause caspase‐1 activation, called as ‘an activation phase’. 1 , 25 Caspase‐1 then cleaves pro‐IL‐1β to generate biologically active IL‐1β. 1 , 25 After the priming by LPS, the subsequent addition of ATP activated caspase‐1 in the BMMs and increased IL‐1β levels in their culture supernatants, indicating the activation of NLRP3 inflammasome (Figure 2d). However, the addition of LLZ suppressed these changes by ATP. Interestingly, the addition of ATP induced TAK1 phosphorylation in RAW264.7 cells (Figure 2e); however, LLZ almost completely suppressed the TAK1 phosphorylation induced by ATP. Moreover, the gene expression of the ATP receptor P2rx7 was decreased by LLZ (Figure 2f). These results suggest that LLZ effectively suppresses the priming and activation of NLRP3 inflammasome and thereby IL‐1β production in BMMs.
Figure 2.
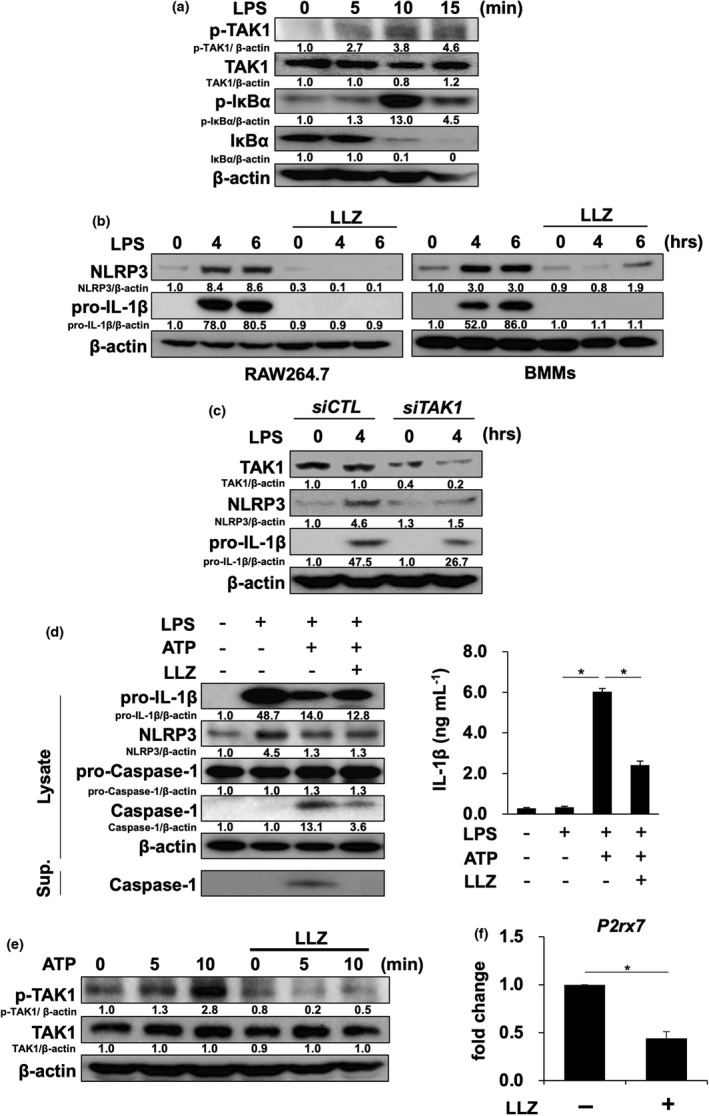
LLZ suppresses the priming and activation of NLRP3 inflammasome. (a) The murine macrophage cell line RAW264.7 was starved with 1% FBS for 12 h and then incubated with or without the TAK1 inhibitor LLZ at 500 nm, followed by the addition of LPS (100 ng mL−1) for the indicated time periods. Protein levels of phosphorylated TAK1 (p‐TAK1), TAK1 and phosphorylated IκBα (p‐IκBα) and IκBα were detected by Western blotting. β‐Actin was used as a protein loading control. Relative changes in the band intensities standardised by respective loading controls are indicated. Representative data of 2 independent experiments are shown. (b) RAW264.7 cells and mouse bone marrow macrophages (BMMs) were incubated with LPS (100 ng mL−1) in the presence or absence of LLZ (500 nm). Protein levels of NLRP3, pro‐IL‐1β and β‐actin were detected by Western blotting. Representative data of 2 independent experiments are shown. (c) TAK1 siRNA or control siRNA was transfected into RAW264.7 cells. After the transfection, the cells were treated with LPS (100 ng mL−1) for 4 h. Protein levels of NLRP3, pro‐IL‐1β, TAK1 and β‐actin were detected by Western blotting. Representative data of 2 independent experiments are shown. (d) BMMs were primed with LPS (100 ng mL−1) for 6 h, and then, ATP (2 mm) was added onto the BMMs with or without LLZ (500 nm). After incubating for 30 minutes, the cells were lysed and their supernatants (Sup.) were collected. Western blotting was performed to detect the protein levels of NLRP3, pro‐IL‐1β, pro‐caspase‐1, caspase‐1 and β‐actin in the cell lysates and caspase‐1 in their supernatants (left). IL‐1β production from BMMs was assessed by ELISA (right). Data are expressed as mean ± SD (n = 4). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative data of 4 independent experiments are shown. (e) RAW264.7 cells were starved with 1% FBS for 12 h and then incubated for 1 h with or without LLZ at 500 nm, followed by the addition of ATP at 2 mm for the indicated time periods. Protein levels of phosphorylated TAK1 (p‐TAK1) and TAK1 were analysed by Western blotting. β‐Actin was used as a protein loading control. Representative data of 2 independent experiments are shown. (f) RAW264.7 cells were treated with or without LLZ (500 nm) for 3 h. RNA was collected, and the expression of ATP receptor, P2rx7, was detected by real‐time PCR. Gapdh served as an endogenous control to normalise each sample. Real‐time PCR was performed with 3 independent samples. Data are expressed as mean ± SD. *P < 0.05 by the Student’s t‐test.
LLZ suppresses TNF‐α converting enzyme (TACE) expression and TNF‐α production by BMMs
Synovial macrophages produce TNF‐α to stimulate the inflammation and bone destruction in RA. 26 TACE, a sheddase for TNF‐like ligands/receptors, is known to be overexpressed in RA and modulate the biological activities of critical inflammatory mediators such as TNF‐α. 27 TACE was overexpressed in synovial tissues in CIA mice; interestingly, treatment with LLZ suppressed the expression of TACE in synovial tissues (Figure 3a). Furthermore, LPS induced TACE expression and TNF‐α production by BMMs, which was abrogated by LLZ (Figure 3b, c). Therefore, LLZ is suggested to suppress TACE‐mediated TNF‐α maturation and production in RA.
Figure 3.
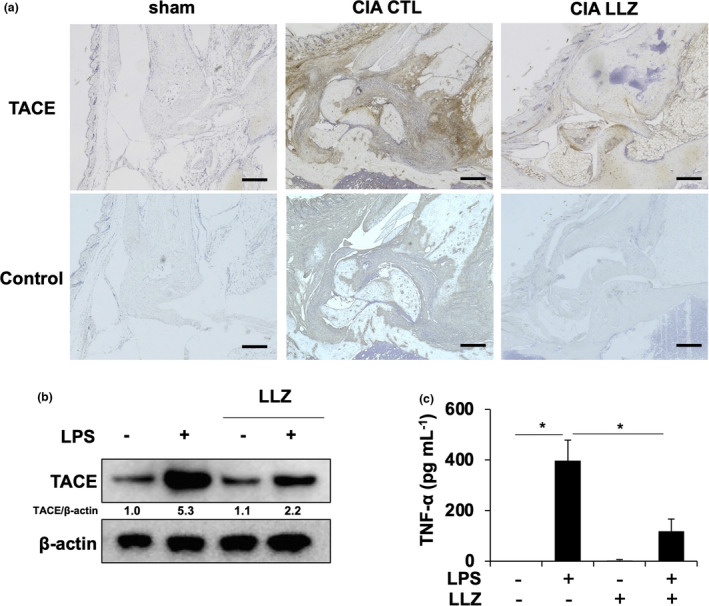
LLZ suppresses TACE expression and TNF‐α production in macrophages. (a) TACE immunoreactivity in synovial tissues in hindlegs of sham and CIA mice with or without LLZ treatment. Scale bars represent 200 μm. Original magnification ×100. (b) BMMs were treated with LPS (100 ng mL−1) in the presence or absence of LLZ (500 nm) for 24 h. Protein expression of TACE was detected by Western blotting. β‐actin served as a loading control. Relative changes in the band intensities standardised by respective loading controls are indicated. (c) BMMs were incubated with LPS (100 ng mL−1) in the presence or absence of LLZ (500 nm) for 12 h. Concentrations of TNF‐α in culture supernatants were measured by ELISA. Data are expressed as mean ± SD (n = 3). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative data of 2 independent experiments are shown.
LLZ suppresses NLRP3 formation in OCs in CIA mice
We next investigated NLRP3 inflammasome formation in OCs in inflamed joints in CIA mice. CTSK‐positive OCs appeared on the surface of eroded bone; NLRP3 was found to be expressed in large CTSK‐positive OCs (Figure 4a). To further clarify the activation of NLRP3 inflammasome in OCs, CTSK‐positive mature OCs were generated in vitro from mouse BMM by M‐CSF and RANKL. Treatment with LPS induced NLRP3 expression in the OCs; however, LLZ suppressed the induction of NLRP3 expression by LPS (Figure 4b), suggesting the priming of NLRP3 inflammasome in OCs. ATP addition following LPS triggered caspase‐1 activation and IL‐1β production in OCs (Figure 4c), indicating activation of NLRP3 inflammasome in OCs. However, LLZ reduced the caspase‐1 activation and IL‐1β production. To estimate the proportion of OCs and BMMs remained undifferentiated into OCs, we examined the expression of the OC marker Acp5 by comparing with the macrophage marker Adgre1 and confirmed the dominance of OCs (Supplementary figure 1). These results suggest that OCs are not only bone‐resorbing cells but also inducers of inflammasome‐mediated inflammation in CIA mice and that LLZ can suppress the priming and activation of NLRP3 inflammasome in OCs.
Figure 4.
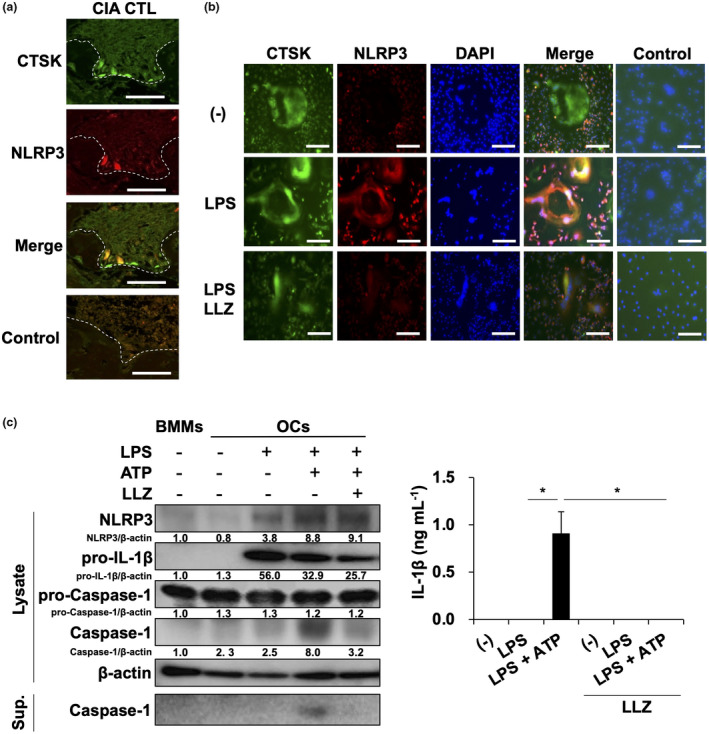
LLZ suppresses NLRP3 formation in OCs. (a) Coexpression of cathepsin K (CTSK), an osteoclastic marker, and NLRP3 in tissue sections of hind paws in CIA mice. Scale bars represent 50 μm. Original magnification ×400. (b) Osteoclastogenesis was induced from BMMs upon treatment with M‐CSF and RANKL. After non‐adherent cells were washed out, adherent osteoclastic lineage cells were cultured with LPS (100 ng mL−1) in the presence or absence of LLZ (500 nm) for 24 h. After fixation, the cells were stained with antibodies against CTSK and NLRP3. Nuclei were detected with DAPI staining. Scale bars represent 100 μm. Original magnification ×100. (c) OCs were prepared from BMMs as described in the Methods. OCs were cultured with LPS (100 ng mL−1) for 6 h. After washing, the OCs were then incubated with ATP (2 mm) for 30 min. Cell lysates and culture supernatants were collected. Western blotting was performed to assess the protein levels of NLRP3, pro‐IL‐1β, pro‐caspase‐1, caspase‐1 and β‐actin in the cell lysates and caspase‐1 in the culture supernatants (left). Relative changes in the band intensities standardised by respective loading controls are indicated. IL‐1β production from BMMs was assessed by ELISA (right). Data are expressed as mean ± SD (n = 4). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative data of 3 independent experiments are shown.
LLZ attenuates RANKL expression in SFs in CIA joints and SF‐mediated osteoclastogenesis by IL‐1β
Synovial fibroblasts robustly produce various pro‐inflammatory cytokines and catabolic factors such as MMP‐3 and the critical osteoclastogenic factor RANKL and play a critical role in joint inflammation and destruction in RA. 9 , 28 Immunohistochemical study on CIA joints revealed TAK1 phosphorylation and RANKL expression in SFs, whereas LLZ markedly suppressed the TAK1 phosphorylation and RANKL expression (Figure 5a). Because NLRP3 inflammasome‐mediated inflammation appears to be involved in CIA mice, we next investigated the effects of IL‐1β on RANKL expression and osteoclastogenic activity by SFs isolated from CIA mice. IL‐1β induced TAK1 phosphorylation, followed by the phosphorylation of IκBα and the reduction in IκBα protein in SFs (Figure 5b). However, LLZ suppressed the phosphorylation of TAK1 in the presence of IL‐1β in SFs. The addition of IL‐1β and TNF‐α induced RANKL expression at mRNA (Figure 5c) and protein (Supplementary figure 2) levels in SFs isolated from CIA mice and could induce osteoclastogenesis from pre‐osteoclasts in the presence of the SFs (Figure 5d). However, LLZ suppressed the upregulation of RANKL expression and osteoclastogenesis by IL‐1β or TNF‐α (Figure 5c and d). Therefore, IL‐1β and TNF‐α produced by synovial macrophages are suggested to induce RANKL expression in SFs and thereby osteoclastogenesis in CIA joints in an LLZ‐inhibitable manner. Besides, SFs are major cells to overproduce MMP‐3, a cartilage‐degrading enzyme, and IL‐6 in RA joints. 29 , 30 IL‐1β also enhanced MMP‐3 expression (Figure 5e) and IL‐6 production (Figure 5f) by SFs. However, LLZ abolished the upregulation of MMP‐3 and IL‐6 by IL‐1β.
Figure 5.
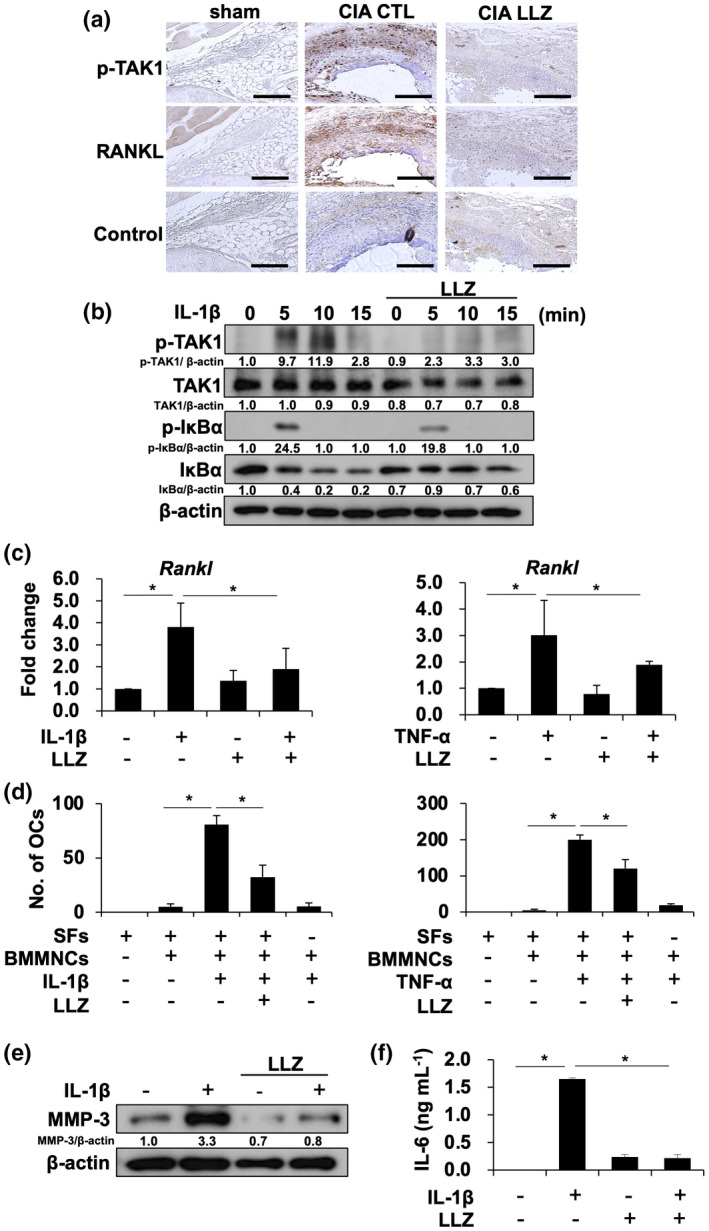
LLZ attenuates RANKL expression in synovial fibroblasts, while directly inhibiting OC differentiation. (a) Phosphorylated TAK1 (p‐TAK1) and RANKL expression in synovial tissues in knee joints of sham and CIA mice. Scale bars represent 200 μm Original magnification ×100. (b) Synovial fibroblasts isolated from CIA mice were starved for 12 h with 1% FBS, and then incubated for 1 h with or without LLZ at 500 nm, followed by the addition of IL‐1β at 20 ng mL−1 for the indicated time periods. Protein levels of p‐TAK1, TAK1, p‐IκBα and IκBα were detected by Western blotting. β‐Actin was used as a protein loading control. Relative changes in the band intensities standardised by respective loading controls are indicated. (c) Synovial fibroblasts were cultured for 8 h with IL‐1β (10 ng mL−1) (left) or TNF‐α (10 ng mL−1) (right) in the presence or absence of LLZ at 250 nm. Rankl mRNA expression was determined using real‐time PCR. Gapdh served as an endogenous control to normalise each sample. Data are expressed as mean ± SD (n = 4). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative data of 3 independent experiments are shown. (d) Murine bone marrow mononuclear cells (BMMNC) (2 × 105 cells/well) were seeded onto synovial fibroblasts (SFs) (5 × 103 cells/well in 24‐well culture plates). The cells were treated with M‐CSF (10 ng mL−1) for 3 days to generate pre‐osteoclastic cells, followed by the addition of IL‐1β (20 ng mL−1) (left) or TNF‐α (25 ng mL−1) (right) with or without LLZ (250 nm). TRAP‐positive cells containing three or more nuclei were counted as mature OCs after culturing for 14 days. Data are expressed as mean ± SD (n = 4). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative data of 3 independent experiments are shown. (e) Synovial fibroblasts were cultured for 24 h with or without IL‐1β (20 ng mL−1) in the presence or absence of LLZ at 500 nm. Protein levels of MMP‐3 and β‐actin were analysed by Western blotting. Representative data of 3 independent experiments are shown. (f) Synovial fibroblasts were cultured for 24 h with or without IL‐1β (20 ng mL−1) in the presence or absence of LLZ at 500 nm. IL‐6 concentrations in culture supernatants were determined by ELISA. Data are expressed as mean ± SD (n = 3). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative data of 3 independent experiments are shown.
LLZ impairs RANKL‐induced osteoclastogenesis
Because RANKL expression was upregulated in SFs in CIA joints (Figure 5a), we next investigated the direct effects of LLZ on RANKL‐induced osteoclastogenesis. BMMs were primed with M‐CSF and then cultured with RANKL. RANKL substantially induced multinucleated OC formation (Figure 6a) and activity (Figure 6b); however, LLZ dose‐dependently suppressed the OC formation and activity induced by RANKL. The time‐dependent induction in BMMs of NFATc1 and c‐fos, critical transcription factors for osteoclastogenesis, and CTSK was abolished by LLZ (Figure 6c). In parallel with RANKL upregulation, IL‐1β, TNF‐α and IL‐6 are overproduced in RA. Therefore, we next investigated the effects of LLZ on RANKL‐induced osteoclastogenesis in the presence of these pro‐inflammatory cytokines. IL‐1β, TNF‐α and IL‐6 cooperatively enhanced RANKL‐induced osteoclastogenesis from BMMs (Figure 6d). However, LLZ could mitigate the RANKL‐mediated osteoclastogenesis enhanced by these pro‐inflammatory cytokines. These results suggest that LLZ suppresses not only RANKL induction in inflamed joints but also RANKL‐mediated signalling in osteoclastic lineage cells to effectively prevent osteoclastic joint destruction under inflammation in RA.
Figure 6.
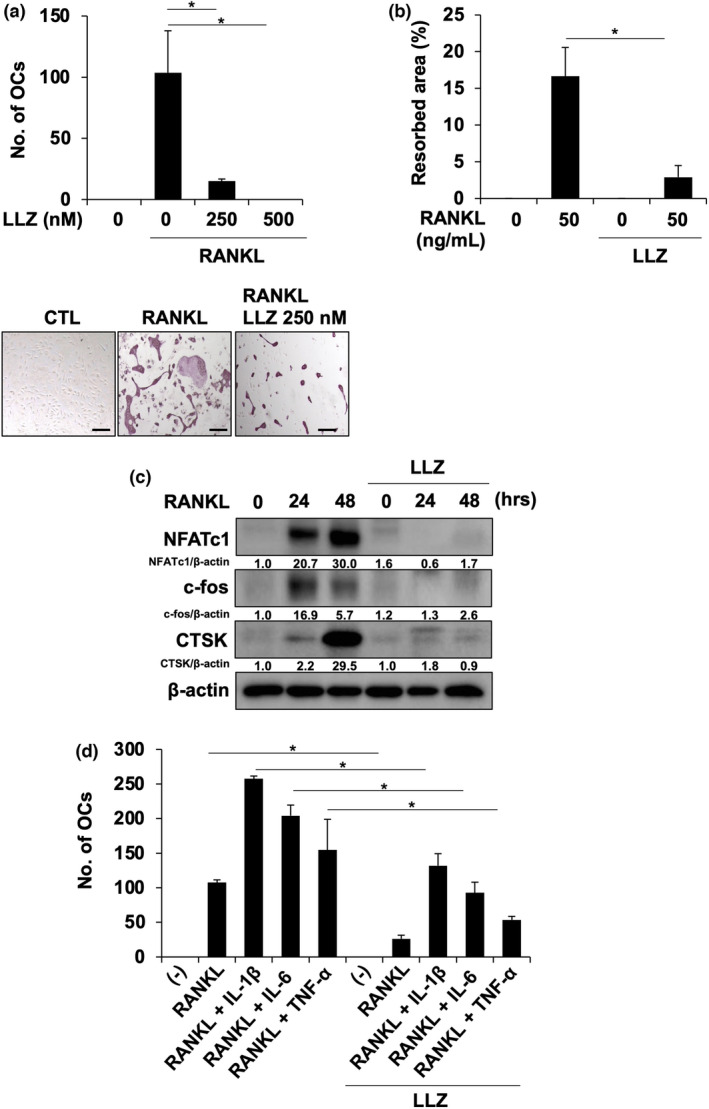
LLZ impairs RANKL‐induced osteoclastogenesis. (a) BMMs were cultured with M‐CSF (10 ng mL−1) and RANKL (50 ng mL−1) for 10 days in the presence or absence of LLZ at the indicated concentrations. TRAP‐positive cells containing three or more nuclei were counted as osteoclasts (OCs). Data are expressed as mean ± SD (n = 3). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative photographs are shown. Scale bars represent 100 μm. Original magnification ×200. Representative data of 3 independent experiments are shown. (b) BMMs were cultured with M‐CSF (10 ng mL−1) and RANKL (50 ng mL−1) in the presence or absence of LLZ at 250 nm for 10 days on Corning Osteo‐Assay Surface 96‐well plates. Resorption area was visualised by von Kossa staining and analysed using a light microscope. The results are expressed as % area of bone resorption and expressed as mean ± SD (n = 3). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative data of 3 independent experiments are shown. (c) BMMs were treated with M‐CSF (10 ng mL−1) for 3 days. After washing, RANKL (50 ng mL−1) was added for the indicated time periods in the presence or absence of LLZ at 500 nm. Cell lysates were collected, and protein levels of NFATc1, CTSK and c‐Fos were analysed by Western blotting. β‐Actin served as a loading control. Relative changes in the band intensities standardised by respective loading controls are indicated. Representative data of 3 independent experiments are shown. (d) BMMs were cultured with M‐CSF (10 ng mL−1) and RANKL (50 ng mL−1) for 10 days. IL‐1β (10 ng mL−1), TNF‐α (10 ng mL−1) and IL‐6 (10 ng mL−1) were added as indicated in the presence or absence of LLZ (250 nm). TRAP‐positive cells containing three or more nuclei were counted as osteoclasts (OCs). Data are expressed as mean ± SD (n = 3). *P < 0.05 by one‐way ANOVA with Tukey’s test. Representative data of 3 independent experiments are shown.
Discussion
TGF‐β‐activated kinase‐1 activation is vital in the production of various pro‐inflammatory cytokines and inflammatory mediators and potentiation of intracellular signalling pathways driven by these cytokines and mediators in RA. Therefore, therapeutic strategies to inhibit TAK1 activation appear to be essential in the treatment of RA. In accordance with the previous report on in vitro efficacy of LLZ for primary SFs, 13 LLZ exerted superb therapeutic efficacy for joint swelling and pain, pannus formation and osteoclastic bone destruction in RA animal models.
NLRP3 inflammasome formation was observed in synovial macrophages and OCs in CIA mice. NLRP3 inflammasome formation and activation have been studied in synovial macrophages but not in OCs in RA, although stimulation by LPS, an inducer of NLRP3 inflammasome, can enhance osteoclastogenesis. 31 , 32 The present study demonstrates that NLRP3 inflammasome can be activated downstream of TAK1 in macrophages and OCs to shift SFs towards an activated phenotype through elaborating IL‐1β, orchestrating inflammatory responses with inflamed synovium and joint destruction. The induction of IL‐1β is suggested to result in overproduction of MMP‐3, IL‐6 and RANKL in SFs. In addition, TAK1 activation in inflamed joints upregulated the expression of TACE, a critical modulator of TNF‐like ligands/receptors, to release and activate pro‐inflammatory mediators in RA, including TNF‐α. Treatment with LLZ abrogated NLRP3 expression in synovial macrophages and OCs and TACE in synovial macrophages, as well as TAK1 phosphorylation and RANKL overexpression in SFs in CIA joints. LLZ can abolish the priming and activation phases of NLRP3 inflammasome along with suppression of TACE induction in cultured BMMs. LLZ also reduced the IL‐1β‐induced production of MMP‐3, IL‐6 and RANKL in primary SFs. LLZ has been demonstrated to inhibit MEK1/2 and MKK3/6. 13 The p38 MAPK signal transduction is a key pathway of IL‐1β and TNF‐α production in RA. The MAP kinase kinases (MAPKKs) MKK3 and MKK6, upstream regulators of p38 MAPK, are activated in RA synovium. 33 In addition, MEK1/2 inhibition has been demonstrated to exert potent anti‐arthritic effects in RA animal models. 34 Therefore, suppression of MKK3/6 and MEL1/2 together with TAK1 by LLZ may contribute to potent anti‐RA activity exerted by LLZ. Furthermore, LLZ suppressed not only RANKL induction in SFs but also directly inhibited RANKL‐mediated signalling in osteoclastic lineage cells to abolish OC formation and function. To further confirm the efficacy of LLZ for RA, we looked at the effects of LLZ on Th17 cells in CIA models. Th17 cells were substantially infiltrated in synovial tissues in CIA models, which was abrogated by treatment with LLZ. Furthermore, serum levels of IL‐17A and anti‐type II collagen IgG antibody were also increased in CIA models; however, treatment with LLZ reduced these levels (Supplementary figure 3). These results suggest therapeutic efficacy of LLZ in terms of suppression of Th17 cells and humoral responses in RA as well. We also looked at the effects of LLZ in another arthritis model and obtain improvement in arthritis scores in SKG mice upon stimulation with mannan (Supplementary figure 4). These results collectively suggest beneficial mechanisms of action of LLZ as an anti‐RA agent.
The role of TAK1 in promoting receptor‐induced NF‐κB and MAPK signalling events has been well established in various inflammation and immune activation. In line with the conventional role of TAK1, we studied the role of TLR‐mediated TAK1 activation by exogenous stimuli in macrophages and OCs in RA pathogenesis. In contrast, the absence of basal TAK1 expression in macrophages has been reported to induce spontaneous activation of the NLRP3 inflammasome without the requirement of exogenous priming and activation signals. 35 TAK1 deficiency leads to basal activation of IKKα/β to promote TNF release and thereby spontaneous inflammasome activation without requiring the priming and activating signals. Therefore, TAK1 inhibition has two different aspects depending on the presence or absence of exogenous priming and activating signals for the NLRP3 inflammasome. Alleviation of aberrant activation of TAK1 appears to be beneficial in pathological conditions with inflammation, immune activation and cancers; however, we should be careful about dysregulation of NLRP3 inflammasome homeostasis in a quiescent state with continuous potent TAK1 inhibition, and need to clarify the best way to deliver TAK1 inhibitors against TAK1 overactivation in RA. Intermittent inhibition of TAK1 overactivation is suggested to therapeutically work well in this regard.
Therapeutic efficacy of TAK1 inhibition has also been studied in preclinical models of various cancers, including mantle cell lymphoma, 36 breast cancer 37 and ovarian cancer. 38 We reported the therapeutic impacts of LLZ on tumor progression and bone destruction in animal models of the bone residing tumor multiple myeloma (MM). 19 MM cells perturb bone metabolism to cause bone destruction and thereby form crosstalk between MM cells and the microenvironment in bone lesions, which leads to a progressive vicious cycle phase of tumor growth and bone destruction. 39 , 40 , 41 TAK1 is activated in MM cells and their environmental cells in bone marrow, including OCs and bone marrow stromal cells. Treatment with LLZ effectively disrupts the key signal transduction pathways responsible for tumor progression and bone destruction in MM, including those activated by IL‐6 and TNF‐α. 19 , 42 Moreover, TAK1 inhibition clearly blocked the upregulation of RANKL and IL‐6 production by bone marrow stromal cells by MM cells. 19 The present results suggest that TAK1 inhibition with LLZ may provide a therapeutic approach towards treating autoimmune and inflammatory diseases, such as RA, besides cancers. Cancer development and/or lymphoproliferative disorders emerge as a serious concern in the treatment of RA with immunosuppressive agents currently available in clinic. LLZ’s antitumor potential may be beneficial in this regard. We also reported that LLZ (1) suppressed osteoclastogenesis from monocytes, (2) suppressed TNF‐α‐induced VCAM1 and RANKL expression in bone marrow stromal cells and (3) abrogated TNF‐α‐mediated suppression of osteoblastogenesis in the presence of BMP‐2. 19 In addition, although LLZ did not impair monocyte differentiation into dendritic cells with GM‐CSF and IL‐4 in combination, LLZ blunted the upregulation of CD80 and CD86 in the dendritic cells by LPS (Supplementary figure 5). Much broader functions of TAK1 remain to be clarified in other immune and non‐immune cell types.
LL‐Z1640‐2 is a resorcyclic acid lactone‐related compound, was discovered in fungi and has been shown to be an irreversible inhibitor of TAK1. 43 The irreversibility by the formation of a covalent bond and relatively low metabolic stability of LLZ limit its clinical application. Despite the known limitations of LLZ, 44 the preclinical efficacy of LLZ is superb. 13 E6201, a closely related analogue of LLZ, was generated with improved stability, although its activity is milder than that of LLZ. 45 Precise dosing schedules to maximise their clinical efficacy and tolerability should be examined to make the best use of these inhibitors. Development of new compounds retaining active motifs of LLZ is underway in our university to maintain therapeutic efficacy of LLZ with less toxicity and improved stability. Novel ways of delivering these TAK1 inhibitors should also be investigated to increase their bioavailability and reduce their toxicity. In this regard, we are also exploring the liposome‐mediated drug delivery system with LLZ.
Methods
Reagents
The following reagents were purchased from the indicated manufacturers: rabbit polyclonal antibodies against TAK1, IκBα and TACE, rabbit monoclonal antibodies against phosphorylated IκBα, c‐fos and β‐actin, mouse monoclonal anti‐IL‐1β, horseradish peroxidase (HRP) anti‐rabbit IgG and anti‐mouse IgG from Cell Signaling Technology (Beverly, MA, USA); rabbit polyclonal anti‐IL‐17A and rabbit monoclonal anti‐caspase‐1 from Abcam (Cambridge, UK); mouse monoclonal antibodies against NFATc1, RANKL, cathepsin K (CTSK), MMP‐3 and F4/80 from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA); rabbit polyclonal anti‐NLRP3 antibody from Novus Biologicals (Centennial, CO, USA); rabbit polyclonal anti‐phosphorylated TAK1 from Cusabio (Houston, TX, USA); rabbit polyclonal anti‐RANKL from GeneTex (Irvine, CA, USA); TAK1 inhibitor, LLZ, from BioAustralis (Smithfield, NSW, Australia); fluorescein isothiocyanate (FITC)‐conjugated mouse anti‐human CD1a and phycoerythrin (PE)‐conjugated mouse anti‐human CD14 from BD Biosciences Pharmingen (San Jose, CA, USA); PE‐conjugated mouse anti‐human CD80, PE‐conjugated mouse anti‐human CD86 and PE‐conjugated mouse IgG1‐κ isotype control from BioLegend (San Diego, CA, USA); recombinant human IL‐1β, M‐CSF, IL‐6, IL‐4, GM‐CSF, lipopolysaccharide (LPS) (E. coli O111) and mouse IgG and rabbit IgG from Wako (Osaka, Japan); recombinant human TNF‐α from R&D Systems (Minneapolis, MN, USA); human‐soluble RANKL from Oriental Yeast Co., Ltd (Shiga, Japan); and adenosine 5'‐triphosphate (ATP) from Tokyo Chemistry Industry (Tokyo, Japan).
Cells
RAW264.7 cell line was obtained from ATCC (Rockville, MD, USA). BMMs were prepared from mouse bone marrow cells as previously described. 42 Briefly, whole bone marrow cells were harvested from the femurs of DBA1/J mice (SLC, Tokyo, Japan), and non‐adherent cells were cultured with M‐CSF (10 ng mL−1) for 3 days to generate mouse BMMs. The isolation of SFs was performed following the previous report. 46 SFs were isolated as previously reported. 46 The hindlegs were collected from CIA mice, and synovial tissues of knee joints were exposed after removing muscles and tendons. The synovial tissues were incubated in 10 mL of culture media containing 1.0 mg mL−1 of collagenase type 4 (Worthington Biochemical Corp., Lakewood, NJ, USA) and 0.1 mg mL−1 of deoxyribonuclease I (Sigma‐Aldrich, St. Louis, MO, USA) for 1 h at 37°C. The cells were collected and cultured, and adherent cells were used as SFs after 2–4 passages. The cells were cultured in Eagle's Minimal Essential Medium Alpha Modification (α‐MEM) (Sigma‐Aldrich) supplemented with 10% FBS, l‐glutamine and 50 mg mL−1 penicillin/streptomycin.
CIA models
Animal experiments were performed under the regulation and permission of the Animal Care and Use Committee of the Tokushima University, Tokushima, Japan (T27‐73). CIA was induced following previous studies. 47 , 48 Female DBA1/J mice (5 weeks old; SLC, Tokyo, Japan) were immunised by intradermal injection of bovine type II collagen (100 μL per mouse; Chondrex, Inc., Redmond, WA, USA) in mixture of incomplete adjuvant and M. tuberculosis H37 RA (Difco Laboratories, Detroit, MI, USA). The second injection was performed 21 days after the first injection. The TAK1 inhibitor LLZ (20 mg kg−1) or PBS as a vehicle control was intraperitoneally administered every other day from the second injection of type II collagen. Arthritis and incidence scores were monitored every day until day 42 after the second injection as previously described. 49 Incidence scores were expressed as the percentage of swollen paws in each group (16 paws, 4 mice in each group). To evaluate pain in CIA mice, a plantar test was performed with the Plantar Test Instrument (Ugo Basile, Varese, Italy) at the same time of the sacrifice as previously described. 50
SKG models
Eight‐week‐old female SKG mice were purchased from CLEA Japan (Tokyo, Japan) and stimulated with mannan (10 mg/mouse, Sigma‐Aldrich) to induce arthritis. Two weeks later, the mice were divided into two groups. LLZ (20 mg kg−1) or PBS as a vehicle control was intraperitoneally administered every other day for additional 2 weeks. Joint swelling was monitored by inspection and scored as previously described 51 : 0, no joint swelling; 0.1, swelling of one finger joint; 0.5, mild swelling of one wrist or ankle; and 1.0, severe swelling of one wrist or ankle. Scores for all fingers of forepaws and hind paws, wrists and ankles were totalled in each mouse.
Osteoclast formation and activity
The generation of OCs from mouse BMMs was performed as previously described. 52 BMMs were cultured for 10–14 days with M‐CSF (10 ng mL−1) and RANKL (50 ng mL−1) to generate OCs. To investigate the effects of SFs on OC differentiation, murine non‐adherent bone marrow mononuclear cells (2 × 105 cells/well) were plated onto SFs (5 × 103 cells). TRAP‐positive cells were detected with a Leukocyte Acid Phosphatase Assay Kit (Sigma‐Aldrich), and TRAP‐positive cells containing three or more nuclei were counted as mature OCs under a light microscope (BX50; Olympus, Tokyo, Japan). Bone resorption assay was performed using Corning Osteo‐Assay Surface 96‐well plates (Corning, Lowell, MA, USA), as described previously. 42 Resorbed area was determined using image analysis techniques (NIH Image J System; http://imagej.nih.gov/ij/), after the cells were removed from the slides using 6% sodium hypochlorite.
Dendritic cell differentiation
Human CD14+ cells were isolated from peripheral blood mononuclear cells (PBMCs) of healthy donors using Human Pan Monocyte Isolation Kit (Miltenyi Biotec, Sunnyvale, CA, USA). The CD14+ cells were cultured with IL‐4 (50 ng mL−1) and GM‐CSF (25 ng mL−1) in combination for 5 days to differentiate into dendritic cells in RPMI 1640 medium (Sigma‐Aldrich) supplemented with 10% FBS, penicillin G at 50 mg mL−1 and streptomycin at 50 mg mL−1. The differentiation into CD14− CD1a+ dendritic cells and their expression of CD80 and CD86 were analysed using flow cytometry using Gallios (Beckman Coulter, CA, USA). Data were edited using FlowJo software (BD Biosciences). All procedures involving human specimens were performed with written informed consent according to the Declaration of Helsinki and using a protocol approved by the Institutional Review Board for human protection in Tokushima University Hospital (#240‐2).
Immunohistochemistry
Hindlegs of each mouse were collected and fixed in 70% ethanol. The ethanol was changed to flesh one every 3 days. After decalcification, samples were embedded in paraffin. Tissue sections (5 μm) of knee joints were stained with haematoxylin and eosin, and immunohistochemistry was performed on continuous tissue sections. After deparaffinisation, endogenous peroxidase was blocked using 3% H2O2 for 10 min and microwave treatment with citric acid at 500 W for 5 min was performed. The sections were treated with anti‐RANKL (1:200), anti‐phosphorylated TAK1 (1:100), anti‐TACE (1:100), anti‐IL‐17A (1:100) or control IgG overnight at 4°C, followed by incubation with peroxidase‐labelled anti‐rabbit and mouse antibodies using Dako EnVision Systems (Dako Corporation, Hamburg, Germany). Colour was developed with diaminobenzidine, and tissues were counterstained with haematoxylin.
Bone histomorphometry and histological evaluation of arthritis
For both bone histomorphometry and histological evaluation of arthritis, the knee joint (from the distal one‐third of the femur to one‐third of the proximal tibia), dissected at death, was fixed in 70% alcohol. The knee joints were then stained with Villanueva bone stain and embedded in methyl methacrylate resin without decalcification. The resulting blocks of knee joint specimens were sectioned in the frontal plane at a thickness of 5 μm with a microtome (RM2255; Leica Biosystems, Nussloch, Germany). Both bone histomorphometric analysis and histological evaluation were performed using semiautomatic image analysis software (System Supply, Nagano, Japan) and an optical fluorescence microscope (BX51; Olympus). The osteoclast number/bone surface (No.Oc/BS, mm−1) was measured in the secondary spongiosa, extending 1.3–3.9 mm distally from the proximal growth cartilage of the tibia. The pannus area (in mm2) was measured in the medial compartment at the tibial and femoral sides using a microscope with a magnifying power of 40. These parameters were measured by an investigator blinded to the experimental groups.
Immunofluorescence staining
For immunofluorescence staining, BMMs were cultured with M‐CSF (10 ng mL−1) and RANKL (50 ng mL−1) for 10 days on glass‐bottom dishes to generate OCs. OCs were fixed for 10 min with 10% neutral buffer formalin and permeabilised with 0.1% Triton X‐100 in PBS. After blocking with 3% bovine serum for 1 h, OCs were incubated with antibodies against NLRP3 (1:50) or CTSK (1:100), or control IgG overnight at 4°C, followed by staining for 1 h with secondary antibodies, anti‐rabbit IgG conjugated with Alexa Fluor® 594 and anti‐mouse IgG conjugated with Alexa Fluor® 488 (1:100 in PBS; Thermo Fisher Scientific, Waltham, MA, USA), respectively. To analyse the NLRP3 expression in CIA mice, the tissue sections (5 μm) of knee joints were incubated with anti‐NLRP3 (1:50), anti‐F4/80 (1:50), anti‐CTSK (1/100) or control IgG overnight at 4°C after deparaffinisation and blocking (3% bovine serum, 1 h). Secondary antibodies were used as described above. The nuclei were stained with 4′,6‐diamino‐2‐phenylindole (DAPI) (Thermo Fisher Scientific). Wide field‐of‐view fluorescence images were examined using a fluorescence microscope (BZ‐X800; Keyence, Osaka, Japan).
Western blot analysis
Cells were collected and lysed in RIPA buffer supplemented with protease inhibitor at 10 μL, phosphatase inhibitor at 10 μL, dithiothreitol (DTT) at 1.0 μL and phenylmethylsulphonyl fluoride (PMSF) at 10 μL (Sigma‐Aldrich). To detect IL‐1β in culture supernatants, culture supernatants were precipitated with cold acetone overnight and dissolved with RIPA buffer. The lysates were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) on a 10% polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were then blocked in 3% skimmed milk and incubated with a primary antibody overnight, followed by an appropriate secondary antibody. The membranes were then examined using Amersham Imager 600 (GE Life Sciences, Little Chalfont, England) in a fluorescence mode. The band intensities were quantitated using ImageJ Gel Analysis program (NIH Image J System).
Small interfering RNA (siRNA) transfection
RAW264.7 cells were seeded at a density of 2 × 105 cells per well on 6‐well plates in α‐MEM at 37°C with 5% CO2 overnight. TAK1 siRNA was purchased from Santa Cruz and transfected into RAW264.7 cells using RNAi MAX Transfection Reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer's protocol.
Real‐time PCR
Total RNA was extracted using TRIzol reagent (Gibco BRL, Rockville, MD, USA). Two μg of total RNA was reverse‐transcribed with PrimeScript RT (Takara Bio Inc., Shiga, Japan) in a 20 μL reaction solution. To perform the real‐time PCR, each cDNA sample was amplified using TB Green (Takara Bio Inc.) on the 7300 Real‐time PCR System (Thermo Fisher Scientific). Briefly, the reaction conditions consisted of 2 μL of cDNA and 0.4 μm primers in a total volume of 20 μL. Gapdh was used as an endogenous control to normalise each sample. The following primer sequences were used:
mouse Rankl F: ATGATGGAAGGCTCATGGT and R: CCAAGAGGACAGAGTGACTTT;
mouse Adgre1 F: CTTTGGCTATGGGCTTCCAGTC, R: GCAAGGAGGACAGAGTTTATCGTG;
mouse Acp5 F: ACTTCCCCAGCCCTTACTACCG, R: TCAGCACATAGCCCACACCG;
mouse Gapdh F: GTCTTCCTGGGCAAGCAGTA, R: CTGGACAGAAACCCCACTTC; and
mouse P2rx7 F: ATATCCACTTCCCCGGCCAC, R: TCGGCAGTGATGGGACCAG.
ELISA
Concentrations of IL‐1β, IL‐6, TNF‐α and IL‐17A in sera or culture supernatants were measured using an ELISA kit from R&D Systems. Serum concentration of anti‐type II collagen IgG in mice was assessed using mouse anti‐bovine type II collagen IgG antibody assay kit (Chondrex). The absorbance of each well was measured with an iMark microplate reader (Bio‐Rad Laboratories, Hercules, CA, USA).
Statistical analysis
Statistical analysis was performed using the Student's t‐test for two groups. For multiple groups, statistical differences were assessed by one‐way analysis of variance (ANOVA) with Tukey’s test. In the animal study, two‐way ANOVA and a log‐rank test were used to evaluate statistical differences. P ≤ 0.05 was considered as a significant difference. In this study, all statistical analyses were performed using Statcel 4 Software (OMS Publishing, Saitama, Japan).
CONFLICT OF INTEREST
Masahiro Abe received research funding from Chugai Pharmaceutical, Sanofi KK, Pfizer Seiyaku KK, Kyowa Hakko Kirin, MSD KK, Astellas Pharma, Takeda Pharmaceutical, Teijin Pharma and Ono Pharmaceutical, and honoraria from the Daiichi Sankyo Company. The other authors declare no competing financial interests.
Author contribution
Hirofumi Tenshin: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing – original draft. Jumpei Teramachi: Conceptualization; Formal analysis; Investigation; Methodology; Writing – original draft. Mohannad Ashtar: Formal analysis; Investigation. Masahiro Hiasa: Formal analysis; Investigation; Methodology. Yusuke Inoue: Formal analysis; Investigation; Methodology. Asuka Oda: Investigation; Methodology. Kotaro Tanimoto: Formal analysis; Investigation. So Shimizu: Formal analysis; Investigation. Yoshiki Higa: Formal analysis; Investigation. Takeshi Harada: Formal analysis; Investigation; Methodology. Masahiro Oura: Formal analysis; Investigation; Validation. Kimiko Sogabe: Formal analysis; Investigation; Validation. Tomoyo Hara: Formal analysis; Investigation. Ryohei Sumitani: Formal analysis; Investigation. Tomoko Maruhashi: Formal analysis; Investigation. Mayu Sebe: Formal analysis; Investigation. Rie Tsutsumi: Formal analysis; Investigation; Methodology. Hiroshi Sakaue: Methodology; Project administration; Supervision. Itsuro Endo: Methodology; Supervision. Toshio Matsumoto: Methodology; Project administration; Supervision. Eiji Tanaka: Methodology; Project administration; Supervision. Masahiro Abe: Conceptualization; Methodology; Project administration; Supervision; Writing – review & editing.
Supporting information
Acknowledgments
This work was supported in part by the JSPS KAKENHI Grant Numbers JP18K08329, JP16K11504, JP17H05104, JP17KK0169, JP18H06294, JP19K21382, 19K22719, 19K08839, 20K18784 and 21H03111 and the Research Clusters Program of the Tokushima University. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
REFERENCES
- 1. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015; 21: 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murakami T, Ockinger J, Yu J, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA 2012; 109: 11282–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choulaki C, Papadaki G, Repa A, et al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther 2015; 17: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mathews RJ, Robinson JI, Battellino M, et al. Evidence of NLRP3‐inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3‐inflammasome complex in relation to susceptibility to RA and response to anti‐TNF treatment. Ann Rheum Dis 2014; 73: 1202–1210. [DOI] [PubMed] [Google Scholar]
- 5. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 2016; 41: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kay J, Calabrese L. The role of interleukin‐1 in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2004; 43(Suppl 3): iii2‐iii9. [DOI] [PubMed] [Google Scholar]
- 7. Dayer JM, Oliviero F, Punzi L. A brief history of IL‐1 and IL‐1 Ra in rheumatology. Front Pharmacol 2017; 8: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo C, Fu R, Wang S, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol 2018; 194: 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neumann E, Lefevre S, Zimmermann B, Gay S, Muller‐Ladner U. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol Med 2010; 16: 458–468. [DOI] [PubMed] [Google Scholar]
- 10. Okamoto K, Takayanagi H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther 2011; 13: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karmakar S, Kay J, Gravallese EM. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum Dis Clin N Am 2010; 36: 385–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J, Hsu HC, Mountz JD. Managing macrophages in rheumatoid arthritis by reform or removal. Curr Rheumatol Rep 2012; 14: 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones DS, Jenney AP, Swantek JL, et al. Profiling drugs for rheumatoid arthritis that inhibit synovial fibroblast activation. Nat Chem Biol 2017; 13: 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci 2012; 33: 522–530. [DOI] [PubMed] [Google Scholar]
- 15. Mihaly SR, Ninomiya‐Tsuji J, Morioka S. TAK1 control of cell death. Cell Death Differ 2014; 21: 1667–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huangfu WC, Omori E, Akira S, Matsumoto K, Ninomiya‐Tsuji J. Osmotic stress activates the TAK1‐JNK pathway while blocking TAK1‐mediated NF‐kappaB activation: TAO2 regulates TAK1 pathways. J Biol Chem 2006; 281: 28802–28810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sylvain‐Prévost S, Ear T, Simard FA, et al. Activation of TAK1 by chemotactic and growth factors, and its impact on human neutrophil signaling and functional responses. J Immunol 2015; 195: 5393–5403. [DOI] [PubMed] [Google Scholar]
- 18. Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG. TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol 2013; 33: 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Teramachi J, Tenshin H, Hiasa M, et al. TAK1 is a pivotal therapeutic target for tumor progression and bone destruction in myeloma. Haematologica 2021; 106: 1401–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scarneo SA, Eibschutz LS, Bendele PJ, et al. Pharmacological inhibition of TAK1, with the selective inhibitor takinib, alleviates clinical manifestation of arthritis in CIA mice. Arthritis Res Ther 2019; 21: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pietrosimone KM, Jin M, Poston B, Liu P. Collagen‐induced arthritis: a model for murine autoimmune arthritis. Bio Protoc 2015; 5: e1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takagi M. Toll‐like receptor–a potent driving force behind rheumatoid arthritis. J Clin Exp Hematop 2011; 51: 77–92. [DOI] [PubMed] [Google Scholar]
- 23. Chen Y, Sun W, Gao R, et al. The role of high mobility group box chromosomal protein 1 in rheumatoid arthritis. Rheumatology (Oxford) 2013; 52: 1739–1747. [DOI] [PubMed] [Google Scholar]
- 24. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010; 2010: e672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 2016; 41: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leirisalo‐Repo M, Paimela L, Jaattela M, Koskimies S, Repo H. Production of TNF by monocytes of patients with early rheumatoid arthritis is increased. Scand J Rheumatol 1995; 24: 366–371. [DOI] [PubMed] [Google Scholar]
- 27. Ohta S, Harigai M, Tanaka M, et al. Tumor necrosis factor‐α (TNF‐α) converting enzyme contributes to production of TNF‐α in synovial tissues from patients with rheumatoid arthritis. J Rheumatol 2001; 28: 1756–1763. [PubMed] [Google Scholar]
- 28. Takayanagi H, Iizuka H, Juji T, et al. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum 2000; 43: 259–269. [DOI] [PubMed] [Google Scholar]
- 29. Galil SM, El‐Shafey AM, Hagrass HA, Fawzy F, Sammak AE. Baseline serum level of matrix metalloproteinase‐3 as a biomarker of progressive joint damage in rheumatoid arthritis patients. Int J Rheum Dis 2016; 19: 377–384. [DOI] [PubMed] [Google Scholar]
- 30. Yoshida Y, Tanaka T. Interleukin 6 and rheumatoid arthritis. Biomed Res Int 2014; 2014: e698313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Teramachi J, Inagaki Y, Shinohara H, et al. PKR regulates LPS‐induced osteoclast formation and bone destruction in vitro and in vivo . Oral Dis 2017; 23: 181–188. [DOI] [PubMed] [Google Scholar]
- 32. Strålberg F, Kassem A, Kasprzykowski F, et al. Inhibition of lipopolysaccharide‐induced osteoclast formation and bone resorption in vitro and in vivo by cysteine proteinase inhibitors. J Leukoc Biol 2017; 101: 1233–1243. [DOI] [PubMed] [Google Scholar]
- 33. Inoue T, Hammaker D, Boyle DL, Firestein GS. Regulation of p38 MAPK by MAPK kinases 3 and 6 in fibroblast‐like synoviocytes. J Immunol 2005; 174: 4301–4306. [DOI] [PubMed] [Google Scholar]
- 34. Yamaguchi T, Kakefuda R, Tanimoto A, Watanabe Y, Tajima N. Suppressive effect of an orally active MEK1/2 inhibitor in two different animal models for rheumatoid arthritis: a comparison with leflunomide. Inflamm Res 2012; 61: 445–454. [DOI] [PubMed] [Google Scholar]
- 35. Malireddi RKS, Gurung P, Mavuluri J, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med 2018; 215: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buglio D, Palakurthi S, Byth K, et al. Essential role of TAK1 in regulating mantle cell lymphoma survival. Blood 2012; 120: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Safina A, Ren MQ, Vandette E, Bakin AV. TAK1 is required for TGF‐beta 1‐mediated regulation of matrix metalloproteinase‐9 and metastasis. Oncogene 2008; 27: 1198–1207. [DOI] [PubMed] [Google Scholar]
- 38. Cai PC, Shi L, Liu VW, et al. Elevated TAK1 augments tumor growth and metastatic capacities of ovarian cancer cells through activation of NF‐κB signaling. Oncotarget 2014; 5: 7549–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pearse RN, Sordillo EM, Yaccoby S, et al. Multiple myeloma disrupts the TRANCE/ osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci USA 2001; 98: 11581–11586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roodman GD, Dougall WC. RANK ligand as a therapeutic target for bone metastases and multiple myeloma. Cancer Treat Rev 2008; 34: 92–101. [DOI] [PubMed] [Google Scholar]
- 41. Abe M, Hiura K, Wilde J, et al. Osteoclasts enhance myeloma cell growth and survival via cell‐cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood 2004; 104: 2484–2491. [DOI] [PubMed] [Google Scholar]
- 42. Tenshin H, Teramachi J, Oda A, et al. TAK1 inhibition subverts the osteoclastogenic action of TRAIL while potentiating its antimyeloma effects. Blood Adv 2017; 1: 2124–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Z, Zhang H, Shi M, et al. TAK1 inhibitor NG25 enhances doxorubicin‐mediated apoptosis in breast cancer cells. Sci Rep 2016; 6: 32737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu J, Powell F, Larsen NA, et al. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)‐7‐Oxozeaenol. ACS Chem Biol 2013; 8: 643–650. [DOI] [PubMed] [Google Scholar]
- 45. Shen Y, Boivin R, Yoneda N, et al. Discovery of anti‐inflammatory clinical candidate E6201, inspired from resorcylic lactone LL‐Z1640‐2. III. Bioorg Med Chem Lett 2010; 20: 3155–3157. [DOI] [PubMed] [Google Scholar]
- 46. Hardy RS, Hulso C, Liu Y, et al. Characterisation of fibroblast‐like synoviocytes from a murine model of joint inflammation. Arthritis Res Ther 2013; 15: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kai H, Shibuya K, Wang Y, et al. Critical role of M. tuberculosis for dendritic cell maturation to induce collagen‐induced arthritis in H‐2b background of C57BL/6 mice. Immunology 2006; 118: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yi H, Kim J, Jung H, et al. Induced production of anti‐etanercept antibody in collagen‐induced arthritis. Mol Med Rep 2014; 9: 2301–2308. [DOI] [PubMed] [Google Scholar]
- 49. Brand DD, Latham KA, Rosloniec EF. Collagen‐induced arthritis. Nat Protoc 2007; 2: 1269–1275. [DOI] [PubMed] [Google Scholar]
- 50. Hiasa M, Okui T, Allette YM, et al. Bone pain induced by multiple myeloma is reduced by targeting V‐ATPase and ASIC3. Cancer Res 2017; 77: 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao M, Svensson MND, Venken K, et al. Altered thymic differentiation and modulation of arthritis by invariant NKT cells expressing mutant ZAP70. Nat Commun 2018; 9: 2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shinohara H, Teramachi J, Okamura H, et al. Double stranded RNA‐dependent protein kinase is necessary for TNF‐α‐induced osteoclast formation in vitro and in vivo. J Cell Biochem 2015; 116: 1957–1967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials