

Neurodegenerative disorders are characterized by disruptions to neuronal function and circuitry, leading to a variety of clinical syndromes depending on the affected neuroanatomic regions (Geula, 1998). Many proteinopathies implicated in neurodegenerative diseases are characterized by the pathologic accumulation of proteins into inclusions that are initially deposited in specific areas of the brain and spread widely with disease progression, leading to significant neuronal loss and gliosis (Brettschneider et al., 2013, 2014; Josephs et al., 2016; Jamshidi et al., 2020). There is substantial evidence that amyloid-β, phosphorylated tau, and α-synuclein spread through cell-to-cell propagation of pathological seeds in a prion-like manner, most likely involving trans-synaptic spread of pathology (Brettschneider et al., 2013). In recent years, research has begun to address the mechanism of propagation underlying the transactive response DNA-binding protein 43-kDa (TDP-43) abnormal species of frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) (Brettschneider et al., 2013). In a recent study, our laboratory produced evidence suggesting that TDP-43 inclusions spread throughout the hippocampus in a manner that supports transsynaptic propagation along axonal pathways (Jamshidi et al., 2020). Here, we briefly review prior research on the propagation of TDP-43 pathology and suggest putative mechanisms.
TDP-43 in neurodegenerative disorders: TDP-43 is an RNA/DNA-binding protein that is located primarily within the nucleus in non-pathological states and is an essential part of messenger ribonucleoprotein particles involved in RNA processing (Wong et al., 2021). In contrast, pathological TDP-43 is largely phosphorylated and mislocalized in the cytoplasm and processes of neurons where its clearance is disrupted, and accumulation of TDP-43 in the cytoplasm, nuclei, and neurites contributes to neurodegeneration (Ding et al., 2015; Iguchi et al., 2016; Wong et al., 2021). TDP-43 inclusions are commonly found at autopsy in a large proportion of the brains of individuals diagnosed with frontotemporal lobar degeneration (FTLD-TDP), which accounts for a substantial share of dementia diagnoses under age 65 (Brettschneider et al., 2014). Although five subtypes of TDP-43 inclusions have been described (types A-E), types A, B, and C account for the majority of FTLD-TDP cases (Mackenzie et al., 2011). Briefly, TDP-43 type A is characterized by the presence of neuronal cytoplasmic inclusions, neuronal intranuclear inclusions, and short dystrophic neurites; type B by the predominance of cortical neuronal cytoplasmic inclusions; and type C by the predominance of long, thick cortical dystrophic neurites (Mackenzie et al., 2011).
Clinical syndromes associated with TDP-43 proteinopathy: FTLD-TDP may present as a variety of clinical dementia syndromes, including behavioral variant frontotemporal dementia (bvFTD), primary progressive aphasia (PPA), and ALS (Mackenzie et al., 2011). Patients with bvFTD typically demonstrate changes in personality and comportment with or without executive functioning deficits (Rascovsky et al., 2011). In contrast, PPA is characterized by a salient progression in language dysfunction, with primary deficits in grammaticality (agrammatic PPA), word retrieval (logopenic PPA), and single-word comprehension (semantic PPA) (Mesulam et al., 2014). In ALS, pathologic accumulation and spread of TDP-43 inclusions in the brain and spinal cord contributes to a significant loss of motor neurons, ultimately leading to paralysis and death, with a subset of patients also showing deficits characteristic of bvFTD (Brettschneider et al., 2013, 2014). TDP-43 types A and B are implicated in bvFTD and ALS, whereas type B is frequently found in agrammatic PPA, and type C is the most common cause of semantic PPA (Mackenzie et al., 2011). TDP-43 precipitates that are considered “non-FTLD” can also accumulate in medial temporal areas, often in the presence of comorbid hippocampal sclerosis and/or Alzheimer's disease pathology, and contribute to an amnestic phenotype (Josephs et al., 2016).
Stages of TDP-43 proteinopathy progression: Over the past decade, researchers have characterized the stages by which TDP-43 inclusions spread throughout the brain, varying according to the specific clinical dementia syndrome or the presence of multiple co-occurring pathologies (Josephs et al., 2016). For example, in bvFTD due to TDP-43 type A or B, TDP-43 deposition begins in the amygdala, orbital gyri, and gyrus rectus (stage I); spreads to the middle frontal gyrus, anterior cingulate gyrus, anteromedial temporal lobe regions, superior and medial temporal gyri, striatum, red nucleus, thalamus, and precerebellar nuclei (stage II); progresses to the motor cortex, bulbar somatomotor neurons, and the spinal cord anterior horn (stage III); and finally reaches the visual cortex (stage IV) (Brettschneider et al., 2014). This hierarchical spread of disease corresponds to the emergence of clinical symptoms. In contrast, in ALS, TDP-43 is initially restricted to cortical upper motor neurons, brainstem lower motor nuclei, and spinal cord lower motor neurons followed by progression to neocortical and other subcortical regions (Brettschneider et al., 2013). Relative to TDP-43 types A and B in bvFTD and ALS, less is known about the stages of TDP-43 progression in PPA, particularly TDP-43 type C in semantic PPA. Based on substantially higher densities of TDP-43 inclusions in cortical areas affiliated with language function, it is reasonable to assume that TDP-43 inclusions first appear in language regions in PPA, and gradually spread to other cortical areas (Kim et al., 2019). In the presence of co-occurring Alzheimer's disease pathology, TDP-43 appears to have a greater initial predilection for limbic regions, similar to the spreading of tau pathology in Alzheimer's disease according to Braak staging (Josephs et al., 2016).
Mechanism of TDP-43 propagation: The systematic spread of TDP-43 inclusions to functionally connected brain regions with disease progression suggests the possibility of transsynaptic spread, or propagation along axonal pathways. Transsynaptic spread has been commonly implicated in other neurodegenerative diseases, with Braak staging in Alzheimer's disease serving as one of the most widely regarded examples (Brettschneider et al., 2013, 2014). In a recently published study (Jamshidi et al., 2020), our group sought to determine whether quantification of TDP-43 inclusions and pre-inclusions could provide evidence for the transsynaptic spread of TDP-43 pathology along with monosynaptically-connected subregions of the hippocampus.
The hippocampus is a medial temporal lobe structure primarily involved in learning and memory; it has been highly studied in the context of normal human cognition as well as neurodegenerative diseases, and consequently its circuitry has been charted in great detail (Geula, 1998). Briefly, the hippocampus is comprised of the dentate gyrus (DG), the Ammon's horn, and the subiculum (Figure 1). Ammon's horn, or cornu Ammonis (CA) is made up of four distinct subfields, CA1-CA4, comprised of a major pyramidal cell layer among other layers of axons and dendrites. Three distinct layers make up the DG, including the inner polymorphic layer, granule cell layer, and neuron-free molecular layer. The granule cell layer of the DG is highly vulnerable to TDP-43 pathology (Jamshidi et al., 2020) and is the first relay station in the hippocampus, receiving input from the entorhinal cortex via the perforant pathway (Geula, 1998). Information from the DG granule cell layer is relayed to the DG polymorphic layer, CA4, and CA3. Axons from CA3 and the DG polymorphic layer then project back to the DG molecular layer. Projections from CA3 and CA2 terminate locally in CA1, and finally, axons from CA1 carry information to the subiculum.
Figure 1.
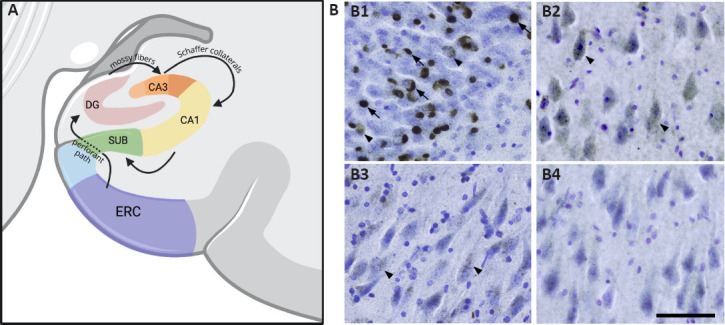
(A) Information from layer II neurons in the entorhinal cortex (ERC) travels to the granule cells of the dentate gyrus (DG) via the perforant pathway (Geula, 1998).
Mossy fiber axons from the DG granule cells then carry information to CA3, and Schaffer axon collaterals project to the CA1 (Jamshidi et al., 2020). Finally, information from CA1 travels to the subiculum. (B) Hippocampal distribution of TDP-43 mature inclusions and pre-inclusions. (B1) Granule cells of the dentate gyrus show a high density of both mature inclusions (arrows) and pre-inclusions (arrowheads). (B2) CA3 pyramidal cells demonstrate a high abundance of pre-inclusions (arrows) but scarce mature inclusions. (B3) The CA2 subfield shows relatively fewer pre-inclusions compared to the CA3 subfield and a similar absence of mature inclusions. (B4) In the CA1 subfield, pre-inclusions and mature inclusions are virtually absent. Photomicrographs were acquired using 40 μm hippocampal sections stained with a human phosphorylated TDP-43 antibody and counterstained for Nissl using Cresyl violet. Scale bar: 100 μm.
In our study (Jamshidi et al., 2020), we quantified TDP-43 pre-inclusions and mature inclusions using unbiased stereological counting methods in the granule cell layer of the DG and pyramidal cell layer of CA1-3. Hippocampal sections were cut at a thickness of 40 μm, stained with an antibody recognizing human TDP-43 phosphorylated (p) at Ser409/410, and counterstained for Nissl using Cresyl violet. TDP-43 pre-inclusions were identified by the appearance of smooth, granular, or fibrillary accumulation of phosphorylated TDP-43 protein in the cytoplasm or nucleus, and mature inclusions were identified as neuronal intranuclear inclusions, neuronal cytoplasmic inclusions, or dystrophic neurites, all showing pSer409/410 immunoreactivity (Kim et al., 2019). All participants in our study (Jamshidi et al., 2020) carried clinical diagnoses of PPA (n = 5) or bvFTD (n = 2) and were found to have TDP-43 pathology at autopsy (n = 4 type A and n = 3 type B). Our findings revealed that the density of mature TDP-43 inclusions was significantly higher in the DG, bilaterally, relative to other hippocampal subregions (Figure 1B). Both the DG and CA3 had higher densities of pre-inclusions than CA1 and CA2, and within CA3, the density of pre-inclusions was significantly higher than the density of mature inclusions (Figure 1B). Moreover, across the CA subfields, we observed mostly TDP-43 pre-inclusions and very few mature inclusions (Figure 1B). We have previously shown that in PPA due to underlying TDP-43 types A and B, pre-inclusions may be found in the absence of mature inclusions, but the reverse is not observed (Kim et al., 2019). In this earlier study, a significantly greater density of mature inclusions was seen in the language-dominant hemisphere, while the reverse was observed in the language non-dominant hemisphere, suggesting earlier inclusion formation in the former. Taken together, these findings (Kim et al., 2019; Jamshidi et al., 2020) suggest propagation of TDP-43 pathology, first as pre-inclusions followed by mature inclusions, along monosynaptically connected regions of the hippocampus. Within the hippocampus specifically, it appears that TDP-43 pathology is first deposited in the DG, followed by CA3, and finally by CA1-2. Importantly, differences in packing densities of neurons between the subfields did not account for these findings. Given that CA1-3, comprised of similar groups of pyramidal neurons, showed highly different levels of pre-inclusions, it was also unlikely that findings simply reflected differences in the selective vulnerability of neurons to TDP-43.
In a recent study, Wong et al. (2021) used far-field super-resolution microscopy to visualize TDP-43 in the postsynaptic density of dendritic spines. Additional experiments using cellular and animal models as well as the brains of FTLD-TDP patients suggested that messenger ribonucleoprotein particles containing pathological TDP-43 may act as seeds promoting TDP-43 aggregation. Using similar methods, visualization of TDP-43 within synapses may permit future studies to determine whether this occurs transsynaptically.
In addition to trans-synaptic spread, there is evidence that pathologic TDP-43 may also propagate in a seeded manner through mechanisms previously implicated in other proteinopathies (Brettschneider et al., 2013, 2014). Studies have demonstrated that exosomes isolated from human ALS-FTLD brains contain elevated levels of full-length TDP-43 and TDP-43 C-terminal fragments, and exposure of neuron-like or glioblastoma cells in culture to these exosomes facilitates mislocalization of TDP-43 to the cytoplasm and transcellular propagation of TDP-43 aggregates (Ding et al., 2015; Iguchi et al., 2016). The exact mechanism by which this occurs is unclear, as there is conflicting evidence as to whether exosomal secretion of TDP-43 occurs in both neurons and glial cells, and inhibition of exosomal secretion does not necessarily attenuate aggregation and disease phenotype (Ding et al., 2015; Iguchi et al., 2016). It is possible that exosomes containing TDP-43 contribute to both clearances of TDP-43 from neurons and its propagation across brain regions. Propagation of TDP-43 may also occur through the formation of tunneling nanotubes, long fine F-actin-containing plasma membrane extensions that permit direct transmission between the cytoplasm of neighboring cells, and this process may be facilitated by exosomes (Ding et al., 2015). This observation suggests that exosomes containing pathological TDP-43 may act as ‘seeds’, promoting the formation of TDP-43 aggregates that are then propagated via tunneling nanotubes. Overall, these findings highlight the need for future research to clarify the nature of transcellular TDP-43 propagation, particularly as it relates to the role of exosomes.
Conclusions and future directions: TDP-43 is a common underlying pathology in FTLD, leading to a variety of young-onset clinical dementia syndromes, including bvFTD, PPA, and ALS, which are characterized by devastating cognitive, behavioral, and motor symptoms. More recently, TDP-43 has also been characterized as an etiology of amnestic dementia syndromes (Josephs et al., 2016). Topographic staging schemes have been suggested to evaluate the spread of pathology across regions selectively vulnerable to TDP-43. Evidence of propagation along synaptic connections in the human brain had largely been limited to qualitative and semiquantitative observations. Here, we reviewed our prior findings using hippocampi from individuals with non-amnestic clinical dementia syndromes to provide quantitative evidence in support of a sequential transsynaptic propagation mechanism of TDP-43 aggregates. There are other putative mechanisms to consider, and exosomes and/or tunneling nanotubes have also been suggested as possible vehicles for spread. Currently, there are no treatments capable of reversing or hampering the progression of TDP-43 proteinopathies, and patients with TDP-43-associated dementia syndromes are underserved, partially due to the complex heterogeneity of clinical syndromes. A nuanced understanding of TDP-43 propagation across selectively vulnerable brain regions, in a variety of clinical syndromes, will be crucial for developing effective treatments and identifying therapeutic targets.
This work was supported by grants from the National Institute of Neurological Disorders and Stroke, No. NS085770; National Institute on Deafness and Other Communication Disorders No. DC008552;and National Institute on Aging, No. AG062566 and AG065463; by an institutional training grant from the National Institute of Neurological Disorders and Stroke, No. NS047987; and by an Alzheimer's Disease Center Grant from the National Institute on Aging, No. AG013854.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Liu WJ, Li JY; T-Editor: Jia Y.
References
- 1.Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB, Fang L, Van Deerlin VM, Ludolph AC, Lee VMY. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD) Acta Neuropathol. 2014;127:423–439. doi: 10.1007/s00401-013-1238-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding X, Ma M, Teng J, Teng RK, Zhou S, Yin J, Fonkem E, Huang JH, Wu E, Wang X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget. 2015;6:24178. doi: 10.18632/oncotarget.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geula C. Abnormalities of neural circuitry in Alzheimer's disease: hippocampus and cortical cholinergic innervation. Neurology. 1998;51:18–29. doi: 10.1212/wnl.51.1_suppl_1.s18. [DOI] [PubMed] [Google Scholar]
- 5.Iguchi Y, Eid L, Parent M, Soucy G, Bareil C, Riku Y, Kawai K, Takagi S, Yoshida M, Katsuno M. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain. 2016;139:3187–3201. doi: 10.1093/brain/aww237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jamshidi P, Kim G, Shahidehpour RK, Bolbolan K, Gefen T, Bigio EH, Mesulam MM, Geula C. Distribution of TDP-43 pathology in hippocampal synaptic relays suggests transsynaptic propagation in frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2020;79:585–591. doi: 10.1093/jnen/nlaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, Liesinger AM, Petersen RC, Parisi JE, Dickson DW. Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol. 2016;131:571–585. doi: 10.1007/s00401-016-1537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim G, Bolbolan K, Shahidehpour R, Jamshidi P, Gefen T, Ayala IA, Weintraub S, Bigio EH, Mesulam MM, Geula C. Morphology and distribution of TDP-43 pre-inclusions in primary progressive aphasia. J Neuropathol Exp Neurol. 2019;78:229–237. doi: 10.1093/jnen/nlz005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mesulam MM, Rogalski EJ, Wieneke C, Hurley RS, Geula C, Bigio EH, Thompson CK, Weintraub S. Primary progressive aphasia and the evolving neurology of the language network. Nat Rev Neurol. 2014;10:554. doi: 10.1038/nrneurol.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, Van Swieten JC, Seelaar H, Dopper EG, Onyike CU. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong CE, Jin LW, Chu YP, Wei WY, Ho PC, Tsai KJ. TDP-43 proteinopathy impairs mRNP granule mediated postsynaptic translation and mRNA metabolism. Theranostics. 2021;11:330–345. doi: 10.7150/thno.51004. [DOI] [PMC free article] [PubMed] [Google Scholar]