

Abstract
BACKGROUND
Autosomal recessive spinocerebellar ataxia type 4 (SCAR4) is a type of SCA that is a group of hereditary diseases characterized by gait ataxia. The main clinical features of SCAR4 are progressive cerebellar ataxia, pyramidal signs, neuropathy, and macrosaccadic intrusions. To date, many gene dysfunctions have been reported to be associated with SCAR4.
CASE SUMMARY
Here, we report a novel compound heterozygous mutation, c.3288delA (p.Asp1097ThrfsTer6), in the VPS13D gene in a young female Chinese patient. The patient found something wrong with her legs about 10 years ago and presented with the typical characteristics of SCAR4 when she came to the hospital, including ataxia, neuropathy, and positive pyramidal signs. She was then diagnosed with SCAR4 and went home with symptomatic schemes.
CONCLUSION
SCAR4 is a hereditary disease characterized by ataxia, pyramidal signs, neuropathy, and macrosaccadic intrusions. We report a novel compound heterozygous mutation, c.3288delA (p.Asp1097ThrfsTer6), in the VPS13D gene, which enriches the gene mutation spectrum and provides additional information about SCAR4.
Keywords: Spinocerebellar ataxia, Recessive, VPS13D gene, Compound heterozygous mutation, Case report
Core Tip: We report a female Chinese patient diagnosed with autosomal recessive spinocerebellar ataxia type 4 (SCAR4) with a compound heterozygous mutation, c.3288delA (p.Asp1097ThrfsTer6), in the VPS13D gene. By reviewing the literature, we speculate that the mutation may cause SCAR4 by affecting mitochondrial function. However, there is currently no specific treatment for SCAR4.
INTRODUCTION
Spinocerebellar ataxia (SCA) is a group of hereditary diseases characterized by progressive gait ataxia, dysarthria, and oculomotor disorders[1-4], which can be caused by autosomal dominant, autosomal recessive, or X-linked mutations[5]. Autosomal recessive SCA (SCAR) represents the type of SCA caused by autosomal recessive mutations. To date, 13 types of SCAR (named SCAR1-13) have been reported[6], and we focused on SCAR4 in the present study. SCAR4 is characterized by cerebellar ataxia, pyramidal signs, neuropathy, and macrosaccadic intrusions, generally developing in early adulthood[7]. SCAR4 has been reported to be caused by many gene mutations, and mutations in the vacuolar protein sorting–associated protein 13D isoform 1 (VPS13D) gene represent the essential component[7]. However, the details of the mechanism have not been explored thoroughly. Here, we report a case of SCAR4 with a novel compound heterozygous mutation in the VPS13D gene.
CASE PRESENTATION
Chief complaints
A 33-year-old Chinese woman came to our department for not being able to run for 10 mo.
History of present illness
The patient felt tired when standing up from squatting and walking downstairs 10 years ago, but daily life was not affected at that time. After that, she had increasingly more difficulties in these actions in the following days. Five years ago, she could not walk steadily even on a level road. Ten months ago, she realized that she could not run anymore. No muscle atrophy or fasciculation was found.
History of past illness
The patient had no previous history of neurological disorders, and she did not suffer any significant injuries in these years.
Personal and family history
The patient was born in Beijing and had no remarkable family history. Her parents and sister were clinically healthy (Figure 1).
Figure 1.
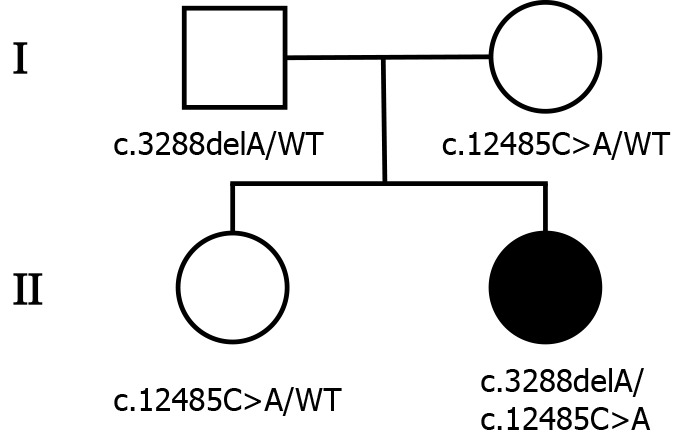
Pedigree of the patient’s family. The patient’s parents and sister were all healthy. Males and females are represented as squares and circles, respectively. The patient is indicated by black-filled symbols, and unfilled symbols indicate the unaffected individuals. The mutation state is given below.
Physical examination
On physical and neurological examination, the patient had an unsteady walk and an ataxic gait. Other cerebellar signs, such as nystagmus, nose-finger test, and heel-shin slide, were normal. Additionally, she could not stand up when squatting. Her tendon reflexes were hyperactive in the lower limbs. Bilateral Babinski signs, Hoffmann signs, and a Rossolimo sign in the left hand were also observed. There was no problem with muscle strength or sensory examination.
Laboratory examinations
There were increases in the levels of anti-CCP, anti-Ro52, and ANA. However, we could not determine what type of immune system disease that the patient had even with the help of physicians in the Department of Rheumatology because she did not have any related symptoms. Cerebrospinal fluid analysis was normal.
Imaging examinations
Electroneuronography showed reduced amplitude of sensory potentials in the right median nerve. Magnetic resonance imaging of the brain (Figure 2) showed no obvious abnormality, and lumbar MRI showed only mild hyperostosis.
Figure 2.

Brain magnetic resonance imaging. The image on the left is the T1-weighted axial image, the image in the middle is the T2-weighted axial image, and the image on the right is the T2-weighted sagittal image. No cerebellar atrophy was observed.
Gene sequence analysis
To explore the underlying genetic patterns, we communicated with the patient and obtained informed consent for whole exon sequencing. We collected venous blood samples from the patient and her family at Peking University Third Hospital. We identified a novel compound heterozygous pathogenic mutation, c.3288delA (p.Asp1097ThrfsTer6; RefSeq NM_015378)/c.12485C>A (p.Thr4162Asn; RefSeq NM_015378), in the VPS13D gene in this patient. Her father was found to be heterozygous for the c.3288delA frameshift mutation, and her mother was found to be heterozygous for the c.12485C>A missense mutation in the VPS13D gene (Figure 3). Her parents reported no symptoms, which suggested that the disease was inherited in an autosomal recessive mode. Although there was also a c.6575C>T (p.Thr2192Ile; RefSeq NM_001376) missense variant in the DYNC1H1 gene in this patient, but this variant may be clinically irrelevant.
Figure 3.

Genomic sequence electropherograms. The patient carried a novel c.3288delA (p.Asp1097ThrfsTer6) frameshift mutation of the VPS13D gene, which was not detected in Chinese databases. Only the patient’s father was heterozygous for this mutation among her parents and sister, and her mother was heterozygous for c.12485C>A (p.Thr4162Asn). Pedigree analysis suggested that the disease was consistent with autosomal recessive inheritance.
FINAL DIAGNOSIS
The final diagnosis of this patient was SCAR4.
TREATMENT
The treatment of SCA has always been a difficult problem worldwide. However, there have been several clinical trials in recent years. There is no specific treatment for SCA to date. Currently, the treatment for the patient is mainly rehabilitation therapy.
OUTCOME AND FOLLOW-UP
The patient returned to the neurologic clinic regularly. She complained of worsening ataxia last time she returned in July 2021. We plan to continue the follow-up in the following years.
DISCUSSION
The clinical manifestations of SCAR are varied, including slowly progressive gait disorder, hypotonia, excessive clumsiness, etc.[8]. Similarly, SCAR mostly occur before the age of 30 years[2-9]. SCAR4, one type of SCAR, mainly presents with cerebellar ataxia, neuropathy, pyramidal signs, and macrosaccadic intrusions[7]. The patient's clinical presentation was consistent with the diagnosis of SCAR4. First, the patient developed symptoms early as her symptoms appeared at 22 years old and became evident at 32 years old. The main symptoms were unsteady walking and clumsiness when walking. In addition, the patient felt tired quickly before the unsteady walk. Consistently, the phenomena of pre-ataxia were confirmed by two earlier observational studies, which found other symptoms occurring several years before ataxia[10]. Pyramidal signs, as identified by physical examination, and neuropathy, as observed by electroneuronography, provided evidence consistent with the diagnosis. Because we did not perform electronystagmograms, we could not confirm the saccadic intrusions. Overall, the patient’s clinical manifestations were in accordance with SCAR4.
Currently, the diagnosis of SCAR4 relies on genetic testing[11]. SCAR4 is mapped to chromosome 16q22.1, and the VPS13D gene has been reported to be closely related to SCAR4[7]. In this case, we found a novel compound heterozygous mutation in the VPS13D gene, c.3288delA (p.Asp1097ThrfsTer6; RefSeq NM_015378)/c.12485C>A (p.Thr4162Asn; RefSeq NM_015378). Pedigree analysis suggested that the disease was autosomal recessive inherited. This mutation has not been described in the previous literature. Furthermore, the new mutation is located in a relatively conserved domain, suggesting that the variant may be a pathogenic mutation (Figure 4).
Figure 4.
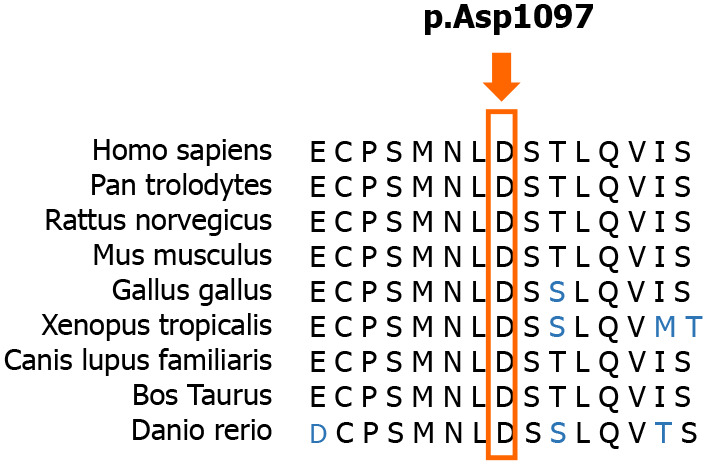
Conservation of amino acids in the context of the frameshift mutation. Amino acid sequence alignments of some of the amino acids affected by the mutation are shown for selected species. The red rectangle highlights the first amino acid affected by the frameshift mutation. This novel mutation is located in a relatively conserved domain.
VPS13D (NM_015378.3) consists of 69 exons spanning approximately 281000 nucleotides, and it encodes a 4388 amino acid protein (NP_056193.2)[12,13]. Although large, VPS13D is intolerant to variations[14]. Only 27 individuals from 15 families have been identified with VPS13D mutations (Figure 5)[7,14-18]. Previous studies have reported that VPS13D gene variants could lead to normal growth and development but with cerebellar ataxia, pyramidal signs, and extrapyramidal signs[7-14,19].
Figure 5.

Schematic representation of the VPS13D protein (RefSeq NM_015378). The novel p.Asp1097ThrfsTer6 mutation is marked red. The others are mutations that have since been found in other studies: p.Ser405Arg, p.Thr1118Met, p.Arg3141*, and p.Thr2945Ala (Koh et al[13], 2020); p.Gly1190Asp, p.Ala4210Val, p.Met1307Leu, p.Gly4149Ser, p.Asn4107Ile, p.Gln1106Ter, p.Tyr1803Ter, p.Gln662Ter, p.Gln2572Ter, p.Val2987Glyfs*14, and p.Leu2277Ter (Seong et al[7], 2018); p.Val2445Glufs*16, p.Leu2900Ser, p.Asn3521Ser, p.Glu3573Aspfs*3, p.Arg4228Gln, p.Arg3253Gln, p.Leu2900Ser, p.Thr865Ala, p.Arg674Gly, and p.Gly1200Asp (Gauthier et al[12], 2018); p.Arg260Trp (McCarthy et al[14], 2014); p.Arg674Gly (Shamseldin HE et al[16], 2017); and p.Leu2587Pro (Lee JS et al[15], 2020). N1: VPS13 1st N-terminal domain (aa 2-115); N2: VPS13 2nd N-terminal domain (aa 137-356); U: Ubiquitin-associated (UBA)-like domain (aa 2627-2679); SHR: VPS13 SHORT ROOT transcription factor-binding domain (aa 3276-3558); C: VPS13 C-terminal domain (aa 3983-4129).
VPS13D is an important protein involved in mitochondrial metabolism, including autophagy (mitophagy), fission, and clearance in Drosophila[18,19]. VPS13D is also essential in human cells. Researchers have observed enlarged mitochondria in human HeLa cells with VPS13D knockout[20]. Gauthier et al[12] reported that T2 hyperintensities in the basal ganglia and/or white matter could be observed on brain MRI in VPS13D-caused movement disorders. Considering that mitochondrial leukodystrophies also show a pattern of diffuse subcortical white matter and bilateral basal ganglia involvement, we suggest that the impact of VPS13D mutation on mitochondrial function may be part of the pathophysiological mechanisms of these diseases[14].
CONCLUSION
To date, studies on SCAR4 are insufficient with only a few reported cases. Nonetheless, SCAR4 shows genetic heterogeneity and the pathogenesis and treatment are far from clear at present. It is necessary to collect data on mutations in the VPS13D gene and to further explore the correlations between genotype and phenotype. We report a female Chinese patient diagnosed with SCAR4 with a compound heterozygous mutation, c.3288delA (p.Asp1097ThrfsTer6), in the VPS13D gene, which enriches the gene mutation spectrum and is valuable information for SCAR4. The physiopathological mechanism of the gene variant requires further investigation.
Footnotes
Informed consent statement: Informed written consent was obtained from the patient for publication of this report and any accompanying images.
Conflict-of-interest statement: There are no conflicts of interest to disclose.
CARE Checklist (2016) statement: The authors have read the CARE Checklist (2016), and the manuscript was prepared and revised according to the CARE Checklist (2016).
Provenance and peer review: Unsolicited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: July 10, 2021
First decision: October 22, 2021
Article in press: December 3, 2021
Specialty type: Neurosciences
Country/Territory of origin: China
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Faruk M S-Editor: Ma YJ L-Editor: Wang TQ P-Editor: Ma YJ
Contributor Information
Xin Huang, Department of Neurology, Peking University Third Hospital, Beijing 100191, China; Municipal Key Laboratory of Biomarker and Translational Research in Neurodegenerative Diseases, Beijing 100191, China.
Dong-Sheng Fan, Department of Neurology, Peking University Third Hospital, Beijing 100191, China; Municipal Key Laboratory of Biomarker and Translational Research in Neurodegenerative Diseases, Beijing 100191, China. dsfan2010@aliyun.com.
References
- 1.Sullivan R, Yau WY, O'Connor E, Houlden H. Spinocerebellar ataxia: an update. J Neurol. 2019;266:533–544. doi: 10.1007/s00415-018-9076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hetzelt KLML, Kraus C, Kusnik S, Thiel CT, Uebe S, Ekici AB, Trollmann R, Reis A, Zweier C. A case of severe autosomal recessive spinocerebellar ataxia type 18 with a novel nonsense variant in GRID2. Eur J Med Genet. 2020;63:103998. doi: 10.1016/j.ejmg.2020.103998. [DOI] [PubMed] [Google Scholar]
- 3.Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- 4.Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019;5:24. doi: 10.1038/s41572-019-0074-3. [DOI] [PubMed] [Google Scholar]
- 5.Palau F, Espinós C. Autosomal recessive cerebellar ataxias. Orphanet J Rare Dis. 2006;1:47. doi: 10.1186/1750-1172-1-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morino H, Miyamoto R, Ohnishi S, Maruyama H, Kawakami H. Exome sequencing reveals a novel TTC19 mutation in an autosomal recessive spinocerebellar ataxia patient. BMC Neurol. 2014;14:5. doi: 10.1186/1471-2377-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seong E, Insolera R, Dulovic M, Kamsteeg EJ, Trinh J, Brüggemann N, Sandford E, Li S, Ozel AB, Li JZ, Jewett T, Kievit AJA, Münchau A, Shakkottai V, Klein C, Collins CA, Lohmann K, van de Warrenburg BP, Burmeister M. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann Neurol. 2018;83:1075–1088. doi: 10.1002/ana.25220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med. 2012;366:636–646. doi: 10.1056/NEJMra1006610. [DOI] [PubMed] [Google Scholar]
- 9.Guan RY, Wu JJ, Ding ZT, Wang J, Sun YM. Clinical and genetic findings in a cohort of Chinese patients with autosomal recessive spinocerebellar ataxia. Clin Genet. 2020;97:532–535. doi: 10.1111/cge.13669. [DOI] [PubMed] [Google Scholar]
- 10.Meijer IA. VPS13D Movement Disorder. 2019 Feb 21. In: GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 1993-2022. [PubMed] [Google Scholar]
- 11.Velayos-Baeza A, Vettori A, Copley RR, Dobson-Stone C, Monaco AP. Analysis of the human VPS13 gene family. Genomics. 2004;84:536–549. doi: 10.1016/j.ygeno.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 12.Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, Tsiakas K, Prokisch H, Rossignol E, Helm MH, Rodan LH, Karamchandani J, Carecchio M, Lubbe SJ, Telegrafi A, Henderson LB, Lorenzo K, Wallace SE, Glass IA, Hamdan FF, Michaud JL, Rouleau GA, Campeau PM. Recessive mutations in VPS13D cause childhood onset movement disorders. Ann Neurol. 2018;83:1089–1095. doi: 10.1002/ana.25204. [DOI] [PubMed] [Google Scholar]
- 13.Koh K, Ishiura H, Shimazaki H, Tsutsumiuchi M, Ichinose Y, Nan H, Hamada S, Ohtsuka T, Tsuji S, Takiyama Y. VPS13D-related disorders presenting as a pure and complicated form of hereditary spastic paraplegia. Mol Genet Genomic Med. 2020;8:e1108. doi: 10.1002/mgg3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, Mistry M, Pavlidis P, Solomon R, Ghiban E, Antoniou E, Kelleher E, O'Brien C, Donohoe G, Gill M, Morris DW, McCombie WR, Corvin A. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry. 2014;19:652–658. doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JS, Yoo T, Lee M, Lee Y, Jeon E, Kim SY, Lim BC, Kim KJ, Choi M, Chae JH. Genetic heterogeneity in Leigh syndrome: Highlighting treatable and novel genetic causes. Clin Genet. 2020;97:586–594. doi: 10.1111/cge.13713. [DOI] [PubMed] [Google Scholar]
- 16.Shamseldin HE, Kurdi W, Almusafri F, Alnemer M, Alkaff A, Babay Z, Alhashem A, Tulbah M, Alsahan N, Khan R, Sallout B, Al Mardawi E, Seidahmed MZ, Meriki N, Alsaber Y, Qari A, Khalifa O, Eyaid W, Rahbeeni Z, Kurdi A, Hashem M, Alshidi T, Al-Obeid E, Abdulwahab F, Ibrahim N, Ewida N, El-Akouri K, Al Mulla M, Ben-Omran T, Pergande M, Cirak S, Al Tala S, Shaheen R, Faqeih E, Alkuraya FS. Molecular autopsy in maternal-fetal medicine. Genet Med. 2018;20:420–427. doi: 10.1038/gim.2017.111. [DOI] [PubMed] [Google Scholar]
- 17.Steel D, Kurian MA. Recent genetic advances in early-onset dystonia. Curr Opin Neurol. 2020;33:500–507. doi: 10.1097/WCO.0000000000000831. [DOI] [PubMed] [Google Scholar]
- 18.Andersen JN, Sathyanarayanan S, Di Bacco A, Chi A, Zhang T, Chen AH, Dolinski B, Kraus M, Roberts B, Arthur W, Klinghoffer RA, Gargano D, Li L, Feldman I, Lynch B, Rush J, Hendrickson RC, Blume-Jensen P, Paweletz CP. Pathway-based identification of biomarkers for targeted therapeutics: personalized oncology with PI3K pathway inhibitors. Sci Transl Med. 2010;2:43ra55. doi: 10.1126/scitranslmed.3001065. [DOI] [PubMed] [Google Scholar]
- 19.Fogel BL. Collaborative science unites researchers and a novel spastic ataxia gene. Ann Neurol. 2018;83:1072–1074. doi: 10.1002/ana.25262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anding AL, Wang C, Chang TK, Sliter DA, Powers CM, Hofmann K, Youle RJ, Baehrecke EH. Vps13D Encodes a Ubiquitin-Binding Protein that Is Required for the Regulation of Mitochondrial Size and Clearance. Curr Biol. 2018;28:287–295.e6. doi: 10.1016/j.cub.2017.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]