

Abstract
In Type 1 and advanced Type 2 diabetes mellitus, elevation of plasma epinephrine plays a key role in normalizing plasma glucose during hypoglycaemia. However, recurrent hypoglycaemia blunts this elevation of plasma epinephrine. To determine whether recurrent hypoglycaemia affects peripheral components of the sympatho-adrenal system responsible for epinephrine release, male rats were administered subcutaneous insulin daily for 3 days. These recurrent hypoglycaemic animals showed a smaller elevation of plasma epinephrine than saline-injected controls when subjected to insulin-induced hypoglycaemia. Electrical stimulation of an adrenal branch of the splanchnic nerve in recurrent hypoglycaemic animals elicited less release of epinephrine and norepinephrine than in controls, without a change in adrenal catecholamine content. Responsiveness of isolated, perfused adrenal glands to acetylcholine and other acetylcholine receptor agonists was also unchanged. These results indicate that recurrent hypoglycaemia compromised the efficacy with which peripheral neuronal activity stimulates adrenal catecholamine release and demonstrate that peripheral components of the sympatho-adrenal system were directly affected by recurrent hypoglycaemia.
Keywords: Adrenal gland, recurrent hypoglycaemia, insulin, diabetes, splanchnic nerve, catecholamines, rat
Introduction
Strict avoidance of hyperglycaemia has been shown to slow the progression of cardiovascular complications in diabetic patients.1,2 However, iatrogenic hypoglycaemia is the major barrier and limiting factor in the treatment of diabetes-induced hyperglycaemia.3–5 During hypoglycaemia, the sympatho-adrenal system is activated, resulting in catecholamine (CAT) release, primarily epinephrine (EPI), from the adrenal medulla.6,7 The benefit of increased plasma EPI is restoration of euglycaemia by inducing hepatic gluconeogenesis and glycogenolysis8 as well as neurogenic responses such as tremors and anxiety that trigger hypoglycaemia awareness.9,10 During hypoglycaemia, elevated EPI also increases cardiac work11 and promotes electrocardiogram (ECG) changes, for example, increased QT interval dispersion,12 that can be pro-arrhythmogenic, especially in the face of coronary artery disease that is another common complication of diabetes.13
Even a single episode of hypoglycaemia can reduce the release of EPI in response to a subsequent bout of hypoglycaemia.14,15 This reduced EPI release impairs recovery from hypoglycaemia and makes patients prone to hypoglycaemia unawareness and hypoglycaemia-associated autonomic failure (HAAF)16,17 but may also be cardioprotective.18 Thus, this adaptive change in EPI release with recurring episodes of hypoglycaemia can be both beneficial and deleterious to the patient. In either case, understanding how regulation of EPI release changes during recurrent episodes of iatrogenic hypoglycaemia is important to improve therapies whose goal is to achieve glycaemic control in diabetic patients.
Studies investigating the effects of recurrent hypoglycaemia (RH) on the sympatho-adrenal system have found numerous changes in brain loci known to regulate EPI secretion.15,19–25 Whether RH also directly affects peripheral components of the sympatho-adrenal system, separately from changes in the brain, has been minimally investigated. However, one observation of interest is that the increase in sympathetic activity in an adrenal branch of the splanchnic nerve recorded during hypoglycaemia in rats was found to be unaltered, but elevation of plasma EPI was reduced with RH.26,27 This finding raised the possibility that RH might directly alter adrenal medullary function, and that such alterations might contribute to reduced EPI secretion.
This study was undertaken to determine the effect of RH on adrenal CAT release evoked by splanchnic nerve stimulation and by direct application of acetylcholine receptor agonists. The data show that RH can reduce in vivo adrenal CAT release, independently of alterations in brain function, with no changes in adrenal CAT content or responsiveness of the adrenal gland to acetylcholine receptor agonists. These results suggest that signalling between the splanchnic nerve fibres and the adrenal medulla is compromised by RH.
Materials and methods
Subjects
Male Sprague Dawley (SD) rats (Charles River Laboratories, Wilmington, MA, USA) weighing 295–315 g were conventionally housed with standard chow and water available ad libitum on a 12:12-h light–dark cycle. Animals were acclimatized to handling for a period of 1 week prior to experimental procedures approved by the New Jersey Medical School Institutional Animal Care and Use Committee.
Right jugular vein catheterization
A sterile Silastic® catheter was implanted under pentobarbital anaesthesia as previously described.28 The catheter was externalized behind the neck of the animal and filled and flushed daily with 10 U/mL heparin in 0.9% NaCl (saline). Animals that underwent this procedure were allowed to recover from surgery for 5 days to regain their preoperative body weights before being subjected to the RH protocol below.
Recurrent hypoglycaemia
Hypoglycaemia was induced in the morning (10 a.m.) with a single subcutaneous (sc) injection of Humulin® regular insulin (Eli Lilly, Indianapolis, IN, USA) at a dose of 1 U/kg, once a day, for three consecutive days. Control animals were similarly injected with equivalent volumes of saline. Food was removed 1 h prior and returned 2 h after treatments. Glucose was measured in blood from tail nick every 30 min using a calibrated ACCU-CHECK® active glucometer (Roche, Basel, Switzerland). The average minimum blood glucose level attained was 46 ± 5 mg/dL (Figure S1). Animals whose blood glucose level decreased to less than 30 mg/dL were removed from the study to minimize the possibility that hypoglycaemia-induced convulsions would complicate interpretation of the data. In this study, two animals were excluded for this reason.
Blood collection during insulin-induced hypoglycaemia
The day after completion of the RH protocol (Day 4), RH and some control rats (acutely hypoglycaemic, Acute) were subjected to insulin-induced hypoglycaemia (IIH) produced by a single sc injection of Humulin, while collecting blood via the right jugular catheter every 30 min and measuring blood glucose levels as described above. The remaining control animals were injected with saline (Saline) and subjected to the same manipulations as RH and acute rats. Plasma was separated and stored for quantification of counter-regulatory hormones. Red blood cells were resuspended in equivalent volumes of saline and reinfused into the rats through the jugular catheter to prevent anaemia and hypovolaemia.
In vivo CAT release by electrical stimulation of splanchnic nerves innervating the left adrenal gland
Rats (non-catheterized) were subjected to the RH protocol. On Day 4, both RH and Saline animals were subjected to a nerve stimulation technique adapted from Vollmer et al.29 Stage 3 anaesthesia was induced with a single intraperitoneal injection of pentobarbital sodium (50 mg/kg). Catheters (polyethylene tubing, PE50) were inserted in the right femoral vein and artery and filled with 50 and 1500 U/mL heparin, respectively. The arterial catheter was connected to a pressure transducer linked to a Grass polygraph, and haemodynamic data were digitized with an Axon Digidata 1440 A at a sampling rate of 100 Hz and recorded using Clampex® 10.2 software (Molecular Devices; Sunnyvale, CA, USA). The venous catheter was initially used to infuse boluses of urethane (maximum dose of 1.2 g/kg) to maintain anaesthesia and then used to infuse donor blood during electrical stimulation of the splanchnic nerve and collection of adrenal outflow.
To enable mechanical ventilation, tubing (PE240) connected to a rodent ventilator (Harvard Apparatus, Holliston, MA, USA) was inserted in the trachea below the larynx. Air volumes of 2.5 cm3 were ventilated at a rate of 50 ± 5 strokes per minute. Animal body temperature was monitored and maintained at 37°C ± 1°C using an anal probe connected to a Physitemp TCAT-2AC temperature controller and a heating lamp.
The splanchnic nerve was exposed and ligated both rostral and caudal to the suprarenal ganglion without affecting the nerve fibres innervating the left adrenal gland (LAG). An electrode was placed on the isolated segment of the splanchnic nerve between the rostral ligature and the suprarenal ganglion (Figure S2(a)). Electrical stimulation was carried out at frequencies ranging from 1 to 100 Hz with a S44 stimulator (Grass Technologies, Warwick, RI, USA) linked to a stimulus isolation unit (Grass SIU 5). Stimulus voltage was monitored using an oscilloscope. The suprathreshold stimulus intensity (10 V) was chosen to produce maximal CAT release (Figure S3). Stimulus train duration was either 30 s (short) to determine the frequency and voltage dependence of CAT release or 5 min (sustained) to better mimic the prolonged elevation of splanchnic nerve spike frequency observed during hypoglycaemia.26,27
To collect blood flowing out of the LAG, the left renal vein was separated and ligated at its junctions with the vena cava and the kidney, without occluding outflow from the adrenal vein to the renal vein (Figure S2(a)). A cannula (PE190) was inserted in this ligated segment of the renal vein to collect the outflow into cold tubes containing 5 U of heparin and 180 μg of ascorbic acid in saline. Adrenal outflows were collected for 2 min before and during short simulations and for 5 min during sustained stimulations. Net CAT release was calculated by subtracting the amounts of CAT in the basal samples from those in samples collected during stimulation periods. Periods of electrical stimulation were separated by 10 min to allow release rate to return to its basal level.
During collection of adrenal outflow, donor blood, collected immediately before the experiment, was infused at an initial rate of 12 mL/h with the rate adjusted to be at least 20% greater than the collection rate. Donor blood was collected via catheters placed in the right jugular veins of donor rats after treatment with heparin (300 U). Donor blood was gently mixed, passed through a clean 200-μm polypropylene mesh and refrigerated until use. Plasma concentrations of EPI and norepinephrine (NE) in the donor blood were 200 and 300 pg/mL, respectively.
Direct stimulation of CAT release from the LAG
A day after evaluating the counter-regulatory hormonal responses to IIH and saline treatment (Day 5), animals were anaesthetized, and during Stage 3 anaesthesia, the right adrenal gland (RAG) of RH and Saline rats was harvested and stored at −70°C for further analysis. Some of these animals were then subjected to a retrograde perfusion technique of the LAG, adapted from Wakade.30 Briefly, collateral vessels branching off the left renal and adrenal veins were ligated. The left renal vein was also ligated at its junctions with the vena cava and the left kidney (Figure S2(b)). A cannula (PE190) was inserted to perfuse the adrenal gland in a retrograde fashion with Tyrode’s buffer (composition in mM: 10 dextrose, 145 NaCl, 5 mM KCl, 1.1 HEPES, 2 CaCl2, 1 MgCl2) pH 7.4 bubbled with 100% O2 at 37°C. Perfusion was maintained at a rate of 0.4 mL/min using a peristaltic Gilson Minipuls 2 pump. Perfusate was collected from a small incision made on the capsule of the adrenal gland. CAT release was evoked by bolus injection (100 μL) of acetylcholine receptor (AchR) agonist, acetylcholine (Ach), and with nicotine (Nico) and pilocarpine (Pilo) to selectively stimulate nicotinic and muscarinic AchR, respectively. Infusion of isotonic Tyrode’s buffer containing 100 mM KCl was used to evaluate non-receptor-mediated release. Basal and stimulated samples were collected for 5 min, and net stimulated release was calculated as described above. Applications of AchR agonists and KCl were separated by perfusion periods of 20 min with agonist-free Tyrode’s buffer to allow release rate to return to basal levels.
Homogenization of the RAG
RAG harvested from RH and Saline rats were thawed, cleaned of debris, weighed and homogenized in 1 mL of 0.1 N perchloric acid (PCA). Homogenates were then centrifuged for 5 min at 10,000g. Supernatants were collected for determination of CAT content.
Quantification of counter-regulatory hormones
CAT were separated and quantified in adrenal supernatants, perfusates and plasma by HPLC-ECD using an ESA pump 584 and a Coulochem III detector. Plasma samples were treated as described by Levin.31 Adrenal supernatants and perfusates were diluted with 0.1 N PCA, spiked with 3,4-dihydroxybenzylamine and filtered through 0.2-μm polyvinylidene fluoride syringe filters (Millex-GV; Millipore Corporation, Billerica, MA, USA) before chromatographic analysis. Adrenal perfusates and supernatant samples were also analysed using the fluorometric method of Anton and Sayre32 as modified by Wilke et al.33 to obtain estimates of total CAT concentration. Glucagon was determined in plasma samples using an 125I radioimmunoassay (Millipore Corporation). Radioactivity was measured in a Packard Cobra III Auto Gamma scintillation spectrometer.
Statistical analysis
RH effects were evaluated using SigmaPlot™ software with an a priori alpha level of 0.05. Animal group sizes for each experiment were estimated using a desired power of 0.9. Glycaemic levels, plasma concentrations of counter-regulatory hormones, Ach and frequency responses were analysed using repeated measures two-way analysis of variance (ANOVA). Basal and stimulated release of adrenal CAT evoked by repeated 100 mM KCl infusions and short stimulations at 10 Hz were analysed using repeated measures one-way ANOVA. Adrenal CAT content and release evoked by sustained electrical stimulation were analysed by t-test. Post hoc analyses were conducted using the Holm–Sidak test.
Results
Effects of RH on the concentrations of blood glucose and counter-regulatory hormones during IIH
Previous reports have shown that RH decreases EPI release in response to IIH.15,19,20,25,34,35 In this study, RH was produced with single daily sc injections of insulin (1 U/kg) for 3 days, while control animals were treated with saline. On the day after completion of the RH protocol, RH and control rats (Acute) were injected sc with insulin (1 U/kg) to induce hypoglycaemia, while other control animals were treated with saline (Saline). The time course of blood glucose decline and levels of hypoglycaemia in RH rats and those treated with insulin for the first time (Acute) were similar (Figure 1(a)). Furthermore, elevations of plasma glucagon concentration during hypoglycaemia were similar in Acute and RH rats (Figure 1(b)). The concentrations of glucose (Figure 1(a)) and glucagon (Figure 1(b)) measured in the Saline rats showed no significant changes.
Figure 1.
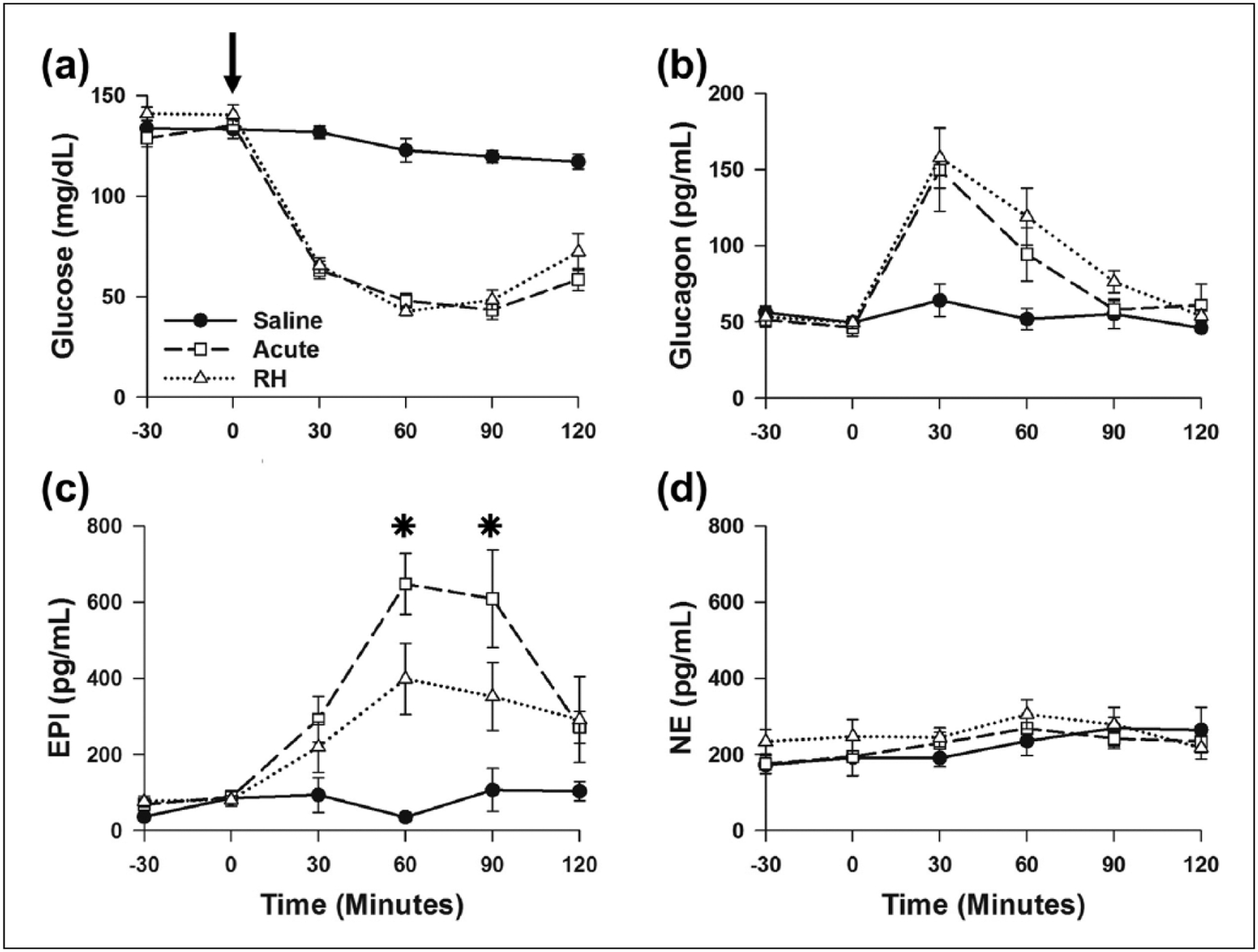
RH specifically decreased EPI response to a subsequent bout of IIH: (a) blood glucose, (b) plasma glucagon, (c) EPI and (d) NE concentrations. Saline (n = 6), Acute (n = 9) and RH (n = 9).
RH: recurrent hypoglycaemia; EPI: epinephrine; IIH: insulin-induced hypoglycaemia; NE: norepinephrine.
*P < 0.05, Acute versus RH. Arrow indicates time of saline (Saline) and insulin (Acute and RH) treatments.
To monitor activation of the sympatho-adrenal axis during hypoglycaemia, plasma CAT concentrations were measured before and after insulin or saline injection on Day 4. Elevations in plasma EPI concentration were significantly reduced 60 and 90 min after insulin treatment in RH when compared to Acute rats (Figure 1(c)). The maximal elevation of EPI in the RH group (458 ± 135 pg/mL, n = 9) was approximately 35% less than in the Acute rats (707 ± 89 pg/mL, n = 9). Saline treatment, blood collection and reinfusion of blood cells did not alter EPI concentrations in the Saline rats. Hypoglycaemia with this dose of insulin did not produce elevations in plasma NE levels (Figure 1(d)). No differences were observed in the plasma NE concentrations of RH, Acute and Saline animals. These data show that our RH protocol specifically blunted the increase in EPI during IIH. Thus, our RH protocol reduces the sympatho-adrenal response to IIH, similar to previous reports.15,19–21,23,25
Adrenal CAT release evoked by splanchnic nerve stimulation after RH
Most studies investigating the blunting of the EPI response produced by RH have focused on brain loci that regulate EPI secretion;15,19–25 however, much less is known about the effect of RH on the functional properties of peripheral components of the sympatho-adrenal axis. To address this issue, this study investigated adrenal CAT release in non-diabetic rats subjected to RH. The day after completion of the RH protocol (Day 4), Saline and RH animals were subjected to an in vivo electrical stimulation protocol to trigger adrenal CAT release. In this protocol, the splanchnic nerve was ligated rostral to the site of stimulation to prevent descending neuronal activity from triggering adrenal CAT release (Figure S2(a)). CAT release was evoked by suprathreshold stimuli (10 V; see Figure S3) delivered to the nerve fibres innervating the LAG to insure that all nerve fibres were activated during the protocol. Each stimulation episode was separated by 10 min to allow CAT release to return to basal levels. Adrenal CAT release was determined in blood samples collected via a catheter placed in the left renal vein to collect the outflow from the LAG.
To monitor adrenal gland stability during the experimental period, both basal CAT release and that evoked by short (30 s) 10 Hz stimulus trains were measured repeatedly (Figure 2(a)). In addition, measurements of arterial blood pressure and heart rate showed that during the experimental period, basal (Figure 2(b)) and stimulated adrenal CAT release (Figure 2(c)) as well as haemodynamic measurements (Figure S4(a)) were stable in Saline and RH animals.
Figure 2.
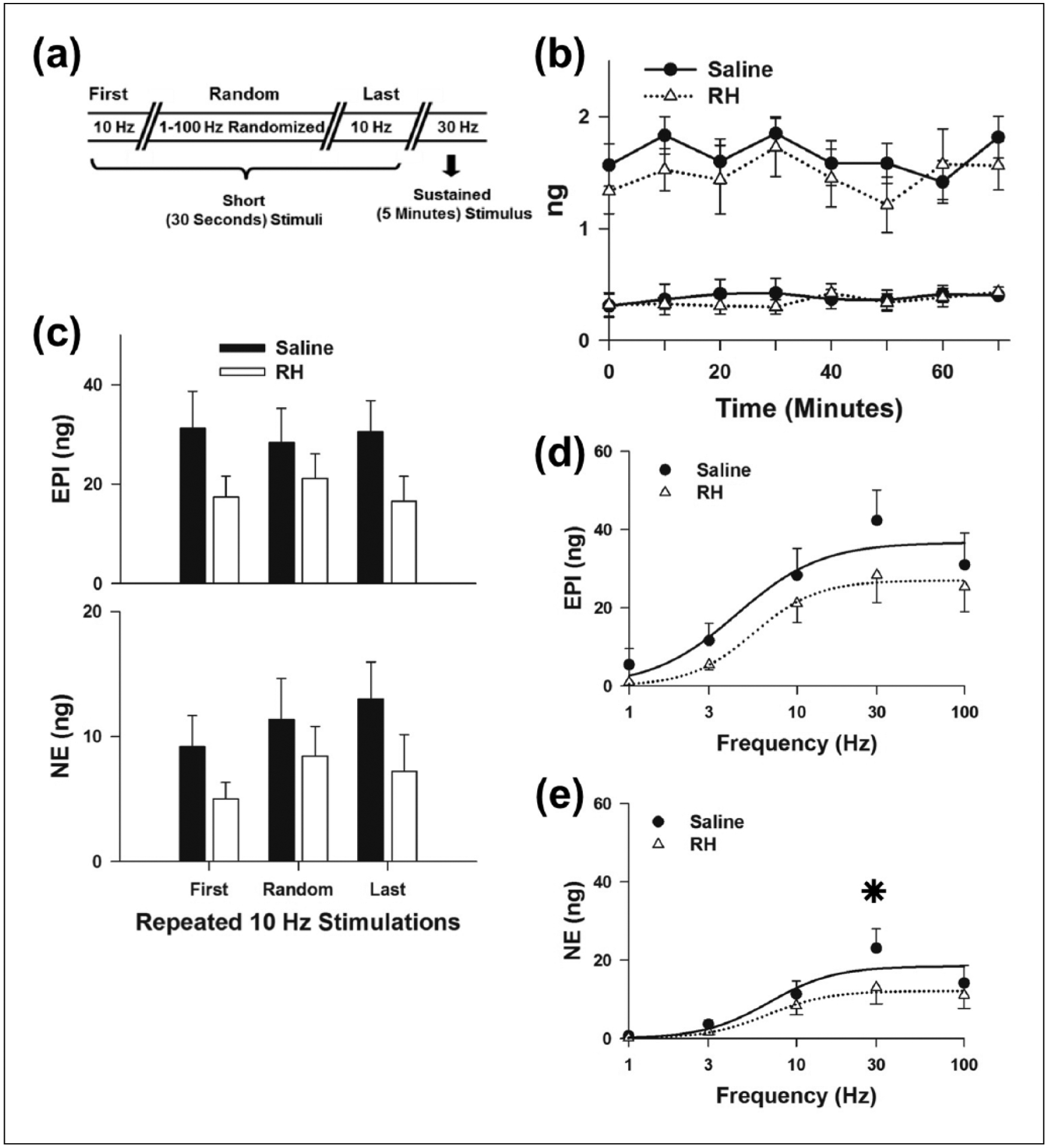
Characterization of CAT release evoked by in vivo electrical stimulation of the splanchnic nerves innervating the LAG: (a) sequence of electrical stimulations applied during the experimental period, (b) basal release of CAT before each electrical stimulation, (c) CAT release evoked by repeated short stimulations at 10 Hz, (d) EPI and (e) NE release evoked by randomized short stimulations (1–100 Hz). Sigmoidal curves were fitted to Saline (—) and RH data (····), respectively. Saline (n = 6) and RH (n = 8).
CAT: catecholamine; RH: recurrent hypoglycaemia; EPI: epinephrine; NE: norepinephrine; LAG: left adrenal gland.
*P < 0.05, Saline versus RH.
IIH is reported to produce a sustained and large increase (40–50 Hz) in the splanchnic nerve activity of rats.26 For this reason, the frequency dependence of CAT release was examined with short stimulus trains of 1–100 Hz, applied in a random order (Figure 2(a)). Release of EPI (Figure 2(d)) and NE (Figure 2(e)) both increased from 1 to 30 Hz, but 100 Hz stimulation yielded no greater release. These data could indicate that IIH in previous studies26,27 produced maximal activation of the adrenal CAT release. Thus, to simulate the sustained and large increase in splanchnic nerve activity produced by IIH, prolonged (5 min) 30 Hz stimulus trains were also applied. Under these conditions, EPI (76 ± 18 ng) and NE (30 ± 9 ng) release in RH animals was significantly less than EPI (163 ± 33 ng) and NE (83 ± 20 ng) release in controls (Figure 3). Since the rates of blood collection were similar in Saline and RH animals (Figure S4(b)), these reductions in stimulated CAT release represent a true decrease in adrenal CAT release rate following RH.
Figure 3.
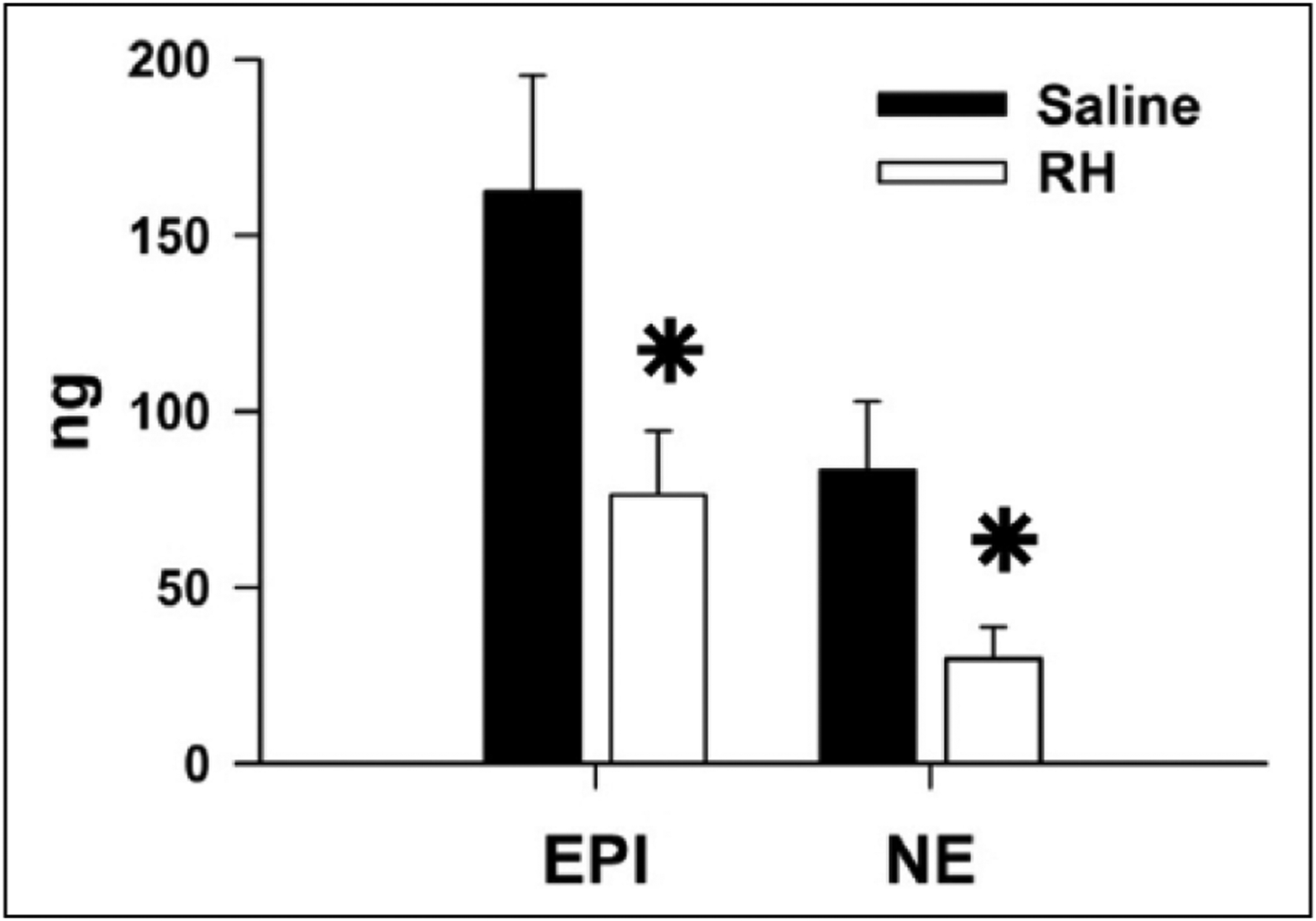
RH decreases adrenal CAT release evoked by sustained stimulation of the splanchnic nerve. The splanchnic nerve was electrically stimulated at 30 Hz for 5 min in Saline (n = 6) and RH animals (n = 8).
CAT: catecholamine; RH: recurrent hypoglycaemia; EPI: epinephrine; NE: norepinephrine.
*P < 0.05, Saline versus RH.
Effect of RH on the capacity of the adrenal medulla to release CAT
Previous reports have shown that RH can deplete adrenal CAT stores.27,36,37 However, in the present experiments, no differences in the adrenal EPI and NE content were observed between Saline and RH animals subjected to four episodes of IIH (Figure 4). In addition, the weight of the RAG did not differ between Saline (25 ± 1 mg) and RH animals (24 ± 1 mg). Similarly, one episode of IIH did not alter adrenal CAT contents measured in Acute rats (Figure S5(a)). These results show that our RH protocols did not produce a sustained depletion of adrenal CAT stores with up to four episodes of prior IIH.
Figure 4.
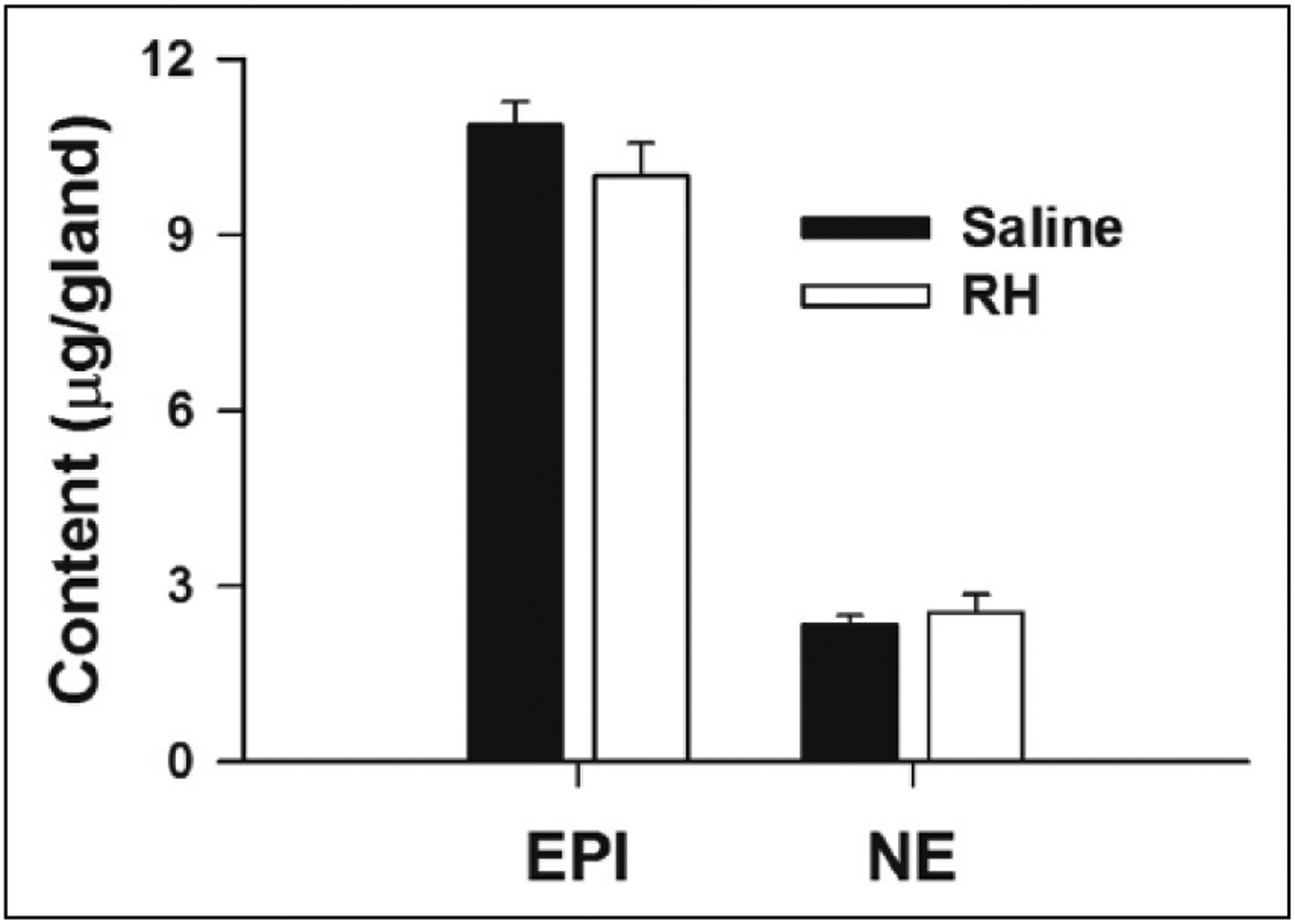
Effects of RH on adrenal CAT content. EPI and NE measured in the RAG of Saline (n = 6) and RH (n = 9) rats.
CAT: catecholamine; RH: recurrent hypoglycaemia; EPI: epinephrine; NE: norepinephrine; RAG: right adrenal gland.
Another possible explanation for the reduced release of CAT in RH rats is that adrenal medullary chromaffin cells are less responsive to stimuli that release CAT.38 To examine whether RH altered adrenal medullary responsiveness, CAT release induced by AchR agonists and membrane depolarization with KCl was evaluated in isolated, perfused LAG from animals in each group. In these experiments, the LAG was perfused with Tyrode’s buffer in a retrograde fashion. Perfusate was collected every 5 min and analysed for CAT content. CAT release was stimulated by direct bolus (100 μL) infusions of isotonic Tyrode’s buffer containing 100 mM KCl, randomized doses of Ach (0.15–400 μg) and maximal doses of the muscarinic and nicotinic AchR agonists, Pilo (200 μg) and Nico (20 μg), respectively (Figure 5(a)). Agonist application was bracketed with infusion of isotonic Tyrode’s buffer containing 100 mM KCl to evaluate receptor-independent CAT release. Each bolus infusion was separated by 20 min to allow release rate to return to basal levels.
Figure 5.
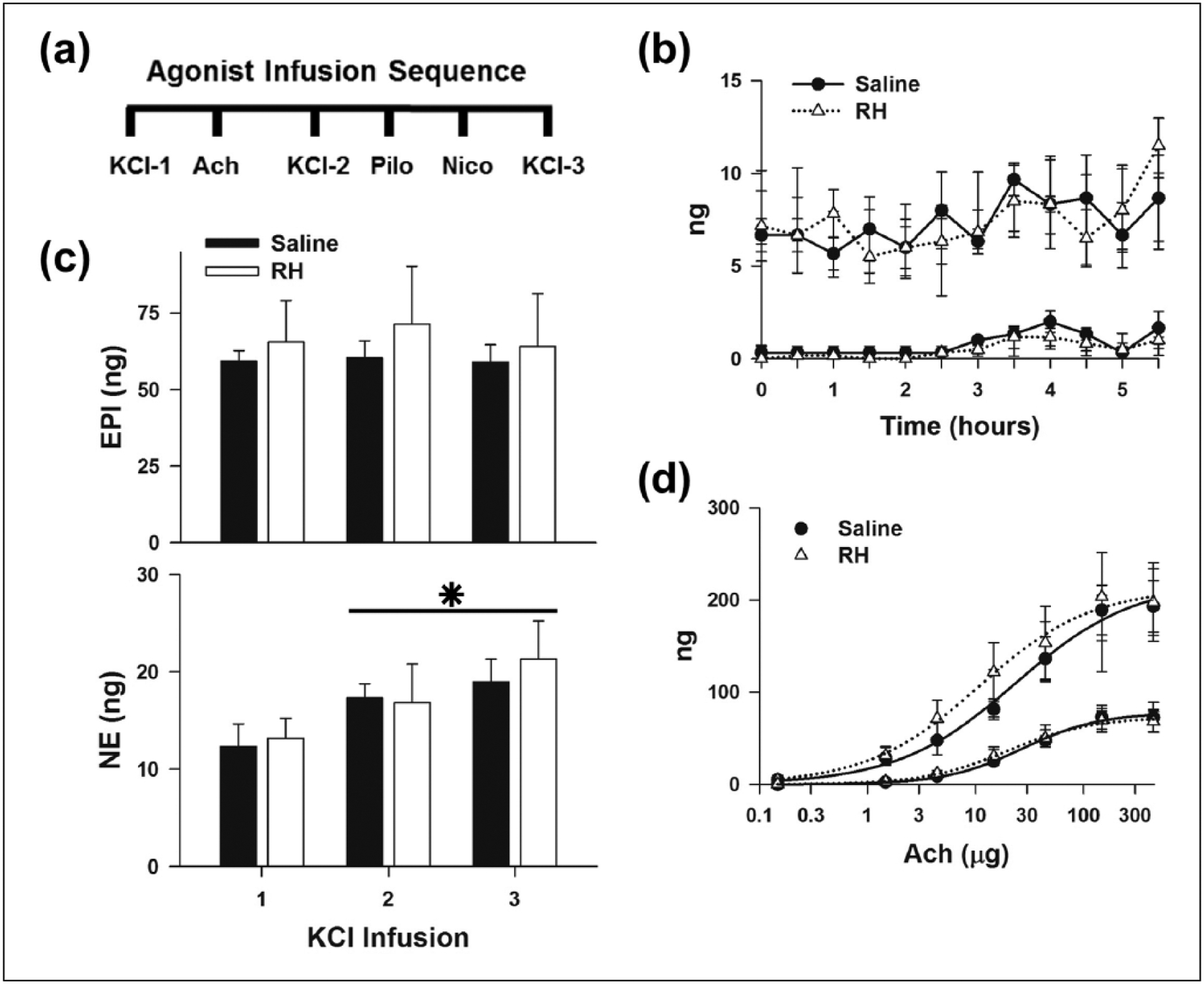
Effects of RH on adrenal CAT release evoked by membrane depolarization and activation of Ach receptors: (a) sequence of infusions applied to elicit CAT release from the LAG, (b) basal release of CAT before each agonist infusion, (c) CAT release evoked by repeated KCl infusions and (d) randomized Ach doses. Sigmoidal curves were fitted to Saline (——) and RH (······) data, respectively. Saline (n = 3) and RH (n = 6).
CAT: catecholamine; RH: recurrent hypoglycaemia; EPI: epinephrine; NE: norepinephrine; LAG: left adrenal gland; Ach: acetylcholine; Nico: nicotine; Pilo: pilocarpine.
*P < 0.05, NE release evoked by KCl infusion, 1 versus 2 and 3.
Basal release of EPI and NE was similar in both groups before and after direct infusion of agonists (Figure 5(b)). There were no differences in the release of adrenal EPI or NE produced by repeated KCl infusions between the Saline and RH animals (Figure 5(c)). However, NE release was significantly larger after the second and third KCl infusions compared to the first infusion in all cases. These results indicate that RH did not alter non-receptor-mediated release of CAT. However, the experimental protocol had some effect on the release of NE but not EPI.
To determine whether RH affected AchR-dependent release of CAT, Ach dose response relationships and the responses to AchR-type specific agonists were examined. Ach dose dependently increased the release of EPI and NE from the LAG of all animals (Figure 5(d)). No differences were observed in the calculated maximum releases of EPI (Saline = 220 ± 32 ng vs RH = 213 ± 40 ng) and NE (Saline = 78 ± 9 ng vs RH = 73 ± 9 ng) evoked by Ach, or the Ach dose that produced half maximal release of EPI (Saline = 24 ± 13 μg vs RH = 11 ± 8 μg), and NE (Saline = 27 ± 10 μg vs RH = 20 ± 8 μg). In addition, EPI and NE release evoked by maximal activation of muscarinic and nicotinic AchR with Pilo and Nico, respectively, were also unchanged (Figure S6(d)). Basal and stimulated release evoked by KCl, Ach, Nico and Pilo were unchanged in LAG from Acute animals (Figure S5). These results show that RH did not alter the responsiveness of the adrenal medulla to Ach nor nicotinic or muscarinic AchR activation. Thus, the reduction in CAT release with electrical stimulation of the splanchnic nerve after RH appears to be due to alterations in signalling between the adrenal medulla and splanchnic nerve.
Discussion
RH can reduce in vivo CAT release during hypoglycaemia by direct effects on adrenal gland function
The goal of this study was to determine whether RH per se can directly affect regulation of adrenomedullary CAT release, independently of its well-known effects in the central nervous system.15,19–25 The data show that RH decreases release of EPI and NE evoked by electrical stimulation of preganglionic splanchnic nerves innervating the LAG (Figure 3) without altering the adrenal CAT content (Figure 4) or release triggered directly by application of exogenous Ach (Figure 5(d)) and other AchR agonists (Figure S6(d)). The most straightforward interpretation of these data is that RH can affect adrenal CAT release by reducing the efficacy with which nerve stimulation triggers CAT release, without affecting the ability of Ach to trigger release of adrenal CAT. Thus, RH appears to alter the signalling between splanchnic nerve fibres and the adrenal medullary chromaffin cells. The implication of these data is that RH can impair adrenal CAT release independently of its effects on brain loci regulating sympathetic nervous system activation.
Some caution must be exercised in extrapolating the adrenal CAT release data to all conditions, because only a very limited set of stimulus conditions were examined, that is, prolonged nerve stimulation at a rate that produced maximal release. Nonetheless, based on in vivo recordings of splanchnic nerve activity,26,27 we believe that these stimulus conditions closely approximate those occurring during IIH.
The aforementioned in vivo splanchnic nerve activity measurements during IIH were conducted by Sivitz and colleagues.26,27 They also studied the effect of RH on the sympatho-adrenal axis activation by measuring plasma CAT levels and found that RH reduced the elevation of plasma EPI during IIH (and during nitroprusside-induced hypotension) in conscious rats, even though spike frequency in an adrenal branch of the nerve increased similarly in both control and RH animals. In their experiments, RH produced a depletion of adrenal EPI content.27 Since reduced adrenal CAT content can decrease stimulated CAT release,36,37,39 it is reasonable to conclude that increased splanchnic nerve activity during IIH produced less EPI release in RH animals, thereby contributing to the reduced elevation of plasma EPI, independently of the effects of RH in the brain. However, in the face of reduced adrenal CAT content, the possibility that RH also affected signalling between splanchnic nerve fibres and adrenal chromaffin cells could not be investigated.
It is worth noting that the results of Sivitz and colleagues26,27 differ from those of other studies measuring sympathetic nerve activity during IIH. For example, prior hypoglycaemia blunted the increase in muscle sympathetic nerve activity during IIH.40 The results of these studies might not be at odds since sympathetic pathways innervating different tissue beds can be regulated differently.41
In our experiments, adrenal CAT content was unchanged by RH. However, RH,26,27,36 and even one bout of severe hypoglycaemia,36,37,42 can produce a sustained depletion of adrenal CAT stores. This difference from our results is probably due to the higher doses of insulin used to induce hypoglycaemia in previous studies. Taken together with our results, we conclude that RH can directly affect adrenal EPI release by at least two mechanisms: (a) depletion of CAT stores and (b), as shown here for the first time, compromised signalling between splanchnic nerve fibres and adrenal medullary chromaffin cells during activation of the sympatho-adrenal system.
In this study, splanchnic nerve activation of adrenal gland CAT release was impaired after three antecedent episodes of IIH. A similar protocol has been used previously to show that the function of brain regions involved in glucosensing is affected by RH.21,24,25 Even a single episode of antecedent hypoglycaemia can also affect central23 and peripheral components36 of the sympatho-adrenal axis. Nonetheless, whether any temporal relationship exists between the development of defects in central and peripheral components of the sympathetic nervous system during RH is unknown.
Possible mechanisms by which RH decreases signalling between the splanchnic nerve fibres and adrenal chromaffin cells
The molecular mechanism by which RH compromises splanchnic nerve activation of adrenal chromaffin cell CAT release was not examined in this study. However, several mechanisms are plausible. For example, a prolonged elevation of splanchnic nerve activity26,27 that might deplete presynaptic Ach stores in the nerve fibre termini.43,44 In addition, presynaptic Ach release is modulated by a number of non-cholinergic hormones present in the nerve terminal,45–49 and the release of these neuromodulators might be expected to change with sustained nerve activity. RH has also been shown to increase acetylcholinesterase activity in rat cerebellum.50 A similar increase in acetylcholinesterase activity in the adrenal gland could decrease the effect of Ach released at nerve termini. Changes in any or all of these factors could affect signalling between nerve fibres and adrenal medullary chromaffin cells, so the mechanism underlying RH induced changes in splanchnic nerve signalling in the adrenal gland is a promising area for future investigation.
Effect of RH on elevation of counter-regulatory hormones in blood plasma during IIH
The decreased elevation in plasma EPI concentrations during IIH in RH animals observed in this study (Figure 1(c)) is consistent with previous reports.14,15,19,20,25,35 The time course and magnitude of IIH were similar with or without RH, and in this respect, our data differ from some reports in which the degree of hypoglycaemia becomes more severe with RH.19,23
The magnitude of the elevation of plasma EPI concentration produced by acute IIH in our study is also less pronounced than reported in the literature during similar levels of hypoglycaemia in non-diabetic rats.19,24,25,35,51–53 This difference may be explained by the lower dose of insulin used to attain hypoglycaemia in our studies. As illustrated in Figure S7(a), and reported from the Beverly laboratory,54 a similar time course of blood glucose decline can be attained using different doses of insulin, but with a lower dose, recovery towards euglycaemia begins sooner. The higher insulin dose does, however, cause a much larger increase in plasma EPI concentration and an increase in plasma NE (Figure S7(b)), similar to previous reports.55,56
In our hands, higher doses of insulin (2 and 4 U/kg) sometimes led to seizures during hypoglycaemia, while none were observed in animals treated with 1 U/kg insulin. Acute hypoglycaemia produced with high doses of insulin may also impair the glucagon response57 and hamper normalization of glycaemic levels.54 Thus, one advantage of our RH protocol is that it impairs the EPI response (Figure 1(c)) without altering the rise in plasma glucagon (Figure 2(b)) and without causing an increase in plasma NE (Figure 1(d)) during IIH. These data suggest that RH in our model specifically impairs activation of adrenal CAT release without more generalized effects on the peripheral sympathetic nervous system. In this regard, our RH protocol provides a specific model to study alterations in the sympatho-adrenal system that can contribute to the deficit of the EPI response induced by recurrent iatrogenic hypoglycaemia.
In comparing our in vivo measurements of plasma CAT during IIH versus the release of CAT triggered by electrical stimulation of the splanchnic nerve, it is notable that only EPI increased during IIH, while both EPI and NE increased with electrical stimulation. In fact, this difference between experimental protocols is expected because nerve pathways innervating EPI and NE containing adrenal chromaffin cells originate from different populations of cells in the hypothalamus and medulla.58 Hypoglycaemia appears to preferentially activate those areas controlling sympathetic outflow to EPI containing cells.6,7,59 In contrast, suprathreshold stimulation of the splanchnic nerve is expected to activate all efferent fibres irrespective of the type of innervated chromaffin cell.
Summary
RH reduced the sympatho-adrenal system response to IIH, as indicated by a decreased elevation of plasma EPI concentration. Adrenal CAT release evoked by electrical stimulation of the splanchnic nerve innervating the LAG was also reduced after RH. However, adrenal EPI levels and release evoked by Ach were unchanged in rats that had reduced in vivo EPI responses to IIH. Based on these data, we hypothesize that RH reduces CAT release from adrenal medullary chromaffin cells by altering synaptic signalling in the adrenal medulla. This change in function is independent of RH effects on the brain. Thus, the impaired EPI response to IIH may arise from RH induced changes in central and peripheral components of the sympatho-adrenal system. This impaired responsiveness of the sympatho-adrenal system may be beneficial or deleterious, depending on the severity of the hypoglycaemic episode and patient health. Reduced EPI release is detrimental to normalization of plasma glucose,9 especially in Type 1 and advanced Type 2 diabetes mellitus that are characterized by greatly reduced glucagon release during hypoglycaemia.8 Conversely, a less marked sympatho-adrenal response will attenuate the increased workload of the heart and lessen the arrhythmogenic actions of beta-adrenergic activation,13 both of which could be beneficial since the risk of cardiovascular disease is increased in Type 160 and Type 2 diabetes mellitus.2 Whether regulation of adrenal gland EPI release in diabetes is also directly impacted by RH, independently of its effects on brain loci regulating sympathetic nervous system activation, will nonetheless require additional experiments.
Supplementary Material
Acknowledgements
The authors thank Sunny Lee, Dr Xavier Fioramonti and Antoinette Moralishivili for technical assistance on catheter placement and catecholamine determinations, Drs Hreday Sapru and Vineet Chitravanshi for providing guidance in the isolation and stimulation of the splanchnic nerve, and Dr Walter N. Duran for assistance in vascular ligation and monitoring haemodynamics.
Funding
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-086681 (to B.O.O.) and the Alfred P. Sloan Foundation.
Footnotes
Declaration of conflicting interests
No conflicts of interest are declared by the authors.
References
- 1.The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin dependent diabetes mellitus. N Engl J Med 1993; 329: 977–986. [DOI] [PubMed] [Google Scholar]
- 2.Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000; 321: 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leese GP, Wang J, Broomhall J, et al. Frequency of severe hypoglycemia requiring emergency treatment in type 1 and type 2 diabetes: a population-based study of health service resource use. Diabetes Care 2003; 26: 1176–1180. [DOI] [PubMed] [Google Scholar]
- 4.Cryer PE. Perspectives in diabetes: mechanisms of hypoglycemia-associated autonomic failure and its component syndromes in diabetes. Diabetes 2005; 54: 3592–3601. [DOI] [PubMed] [Google Scholar]
- 5.Cryer PE. The barrier of hypoglycemia in diabetes. Diabetes 2008; 57: 3169–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshimatsu H, Oomura Y, Katafuchi T, et al. Effects of hypothalamic stimulation and lesion on adrenal nerve activity. Am J Physiol 1987; 253: R418–R424. [DOI] [PubMed] [Google Scholar]
- 7.Borg WP, During MJ, Sherwin RS, et al. Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J Clin Invest 1994; 93: 1677–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cryer PE, Davis S and Shamoon H. Hypoglycemia in diabetes. Diabetes Care 2003; 26: 1902–1912. [DOI] [PubMed] [Google Scholar]
- 9.Berlin I, Grimaldi A, Landault C, et al. Lack of hypoglycemic symptoms and decreased beta-adrenergic sensitivity in insulin-dependent diabetic patients. J Clin Endocrinol Metab 1988; 66: 273–278. [DOI] [PubMed] [Google Scholar]
- 10.Towler DA, Havlin CE, Craft S, et al. Mechanism of awareness of hypoglycemia: perception of neurogenic (predominantly cholinergic) rather than neuroglycopenic symptoms. Diabetes 1993; 42: 1791–1798. [DOI] [PubMed] [Google Scholar]
- 11.Laitinen T, Huopio H, Vauhkonen I, et al. Effects of euglycaemic and hypoglycaemic hyperinsulinaemia on sympathetic and parasympathetic regulation of haemodynamics in healthy subjects. Clin Sci 2003; 105: 315–322. [DOI] [PubMed] [Google Scholar]
- 12.Marques JL, George E, Peacey SR, et al. Altered ventricular repolarization during hypoglycaemia in patients with diabetes. Diabet Med 1997; 14: 648–654. [DOI] [PubMed] [Google Scholar]
- 13.Frier BM, Schernthaner G and Heller SR. Hypoglycemia and cardiovascular risks. Diabetes Care 2011; 34(Suppl. 2): S132–S137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heller SR and Cryer PE. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes 1991; 40: 223–226. [DOI] [PubMed] [Google Scholar]
- 15.Tkacs NC, Dunn-Meynell AA and Levin BE. Presumed apoptosis and reduced arcuate nucleus neuropeptide Y and pro-opiomelanocortin mRNA in non-coma hypoglycemia. Diabetes 2000; 49: 820–826. [DOI] [PubMed] [Google Scholar]
- 16.Cryer PE. Hypoglycemia-associated autonomic failure in diabetes. Am J Physiol Endocrinol Metab 2001; 281: E1115–E1121. [DOI] [PubMed] [Google Scholar]
- 17.Garg R, Hurwitz S, Turchin A, et al. Hypoglycemia, with or without insulin therapy, is associated with increased mortality among hospitalized patients. Diabetes Care 2013; 36: 1107–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reno CM, Daphna-Iken D, Chen YS, et al. Severe hypoglycemia-induced lethal cardiac arrhythmias are mediated by sympathoadrenal activation. Diabetes 2013; 62: 3570–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flanagan DE, Keshavarz T, Evans ML, et al. Role of corticotrophin-releasing hormone in the impairment of counter-regulatory responses to hypoglycemia. Diabetes 2003; 52: 605–613. [DOI] [PubMed] [Google Scholar]
- 20.McCrimmon RJ, Fan X, Cheng H, et al. Activation of AMP-activated protein kinase within the ventromedial hypothalamus amplifies counterregulatory hormone responses in rats with defective counterregulation. Diabetes 2006; 55: 1755–1760. [DOI] [PubMed] [Google Scholar]
- 21.Song Z and Routh VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose-inhibited neurons in the ventromedial hypothalamus nucleus. Am J Physiol Regul Integr Comp Physiol 2006; 291: R1283–R1287. [DOI] [PubMed] [Google Scholar]
- 22.Chan O, Cheng H, Herzog R, et al. Increased GABAergic tone in the ventromedial hypothalamus contributes to suppression of counterregulatory responses after antecedent hypoglycemia. Diabetes 2008; 57: 1363–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang L, Sanders NM, Dunn-Meynell AA, et al. Prior hypoglycemia enhances glucose responsiveness in some ventromedial hypothalamic glucosensing neurons. Am J Physiol Regul Integr Comp Physiol 2008; 294: R784–R792. [DOI] [PubMed] [Google Scholar]
- 24.Fioramonti X, Marsollier N, Song Z, et al. Ventromedial hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes 2010; 59: 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fioramonti X, Deak A, Deshpande S, et al. Hypothalamic S-nitrosylation contributes to the counter-regulatory response impairment following recurrent hypoglycemia. PLoS One 2013; 8: e68709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sivitz WI, Herlein JA, Morgan DA, et al. Effect of acute and antecedent hypoglycemia on sympathetic neural activity and catecholamine responsiveness in normal rats. Diabetes 2001; 50: 1119–1125. [DOI] [PubMed] [Google Scholar]
- 27.Herlein JA, Morgan DA, Phillips BG, et al. Antecedent hypoglycemia, catecholamine depletion, and subsequent sympathetic neural responses. Endocrinology 2006; 147: 2781–2788. [DOI] [PubMed] [Google Scholar]
- 28.Fioramonti X, Contie S, Song Z, et al. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in neuropeptide Y and pro-opio melanocortin networks? Diabetes 2007; 56: 1219–1227. [DOI] [PubMed] [Google Scholar]
- 29.Vollmer RR, Balcita-Pedicino JJ, Debnam AJ, et al. Adrenal medullary catecholamine secretion patterns in rats evoked by reflex and direct neural stimulation. Clin Exp Hypertens 2000; 22: 705–715. [DOI] [PubMed] [Google Scholar]
- 30.Wakade AR. Studies on secretion of catecholamines evoked by acetylcholine or transmural stimulation of the rat adrenal gland. J Physiol 1981; 313: 463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levin BE. Reduced paraventricular nucleus norepinephrine responsiveness in obesity-prone rats. Am J Physiol 1996; 270: R456–R461. [DOI] [PubMed] [Google Scholar]
- 32.Anton AH and Sayre DF. A study of the factors affecting the aluminum oxide-trihydroxyindole procedure for the analysis of catecholamines. J Pharmacol Exp Ther 1962; 138: 360–375. [PubMed] [Google Scholar]
- 33.Wilke RA, Riley DA, Lelkes PI, et al. Decreased catecholamine secretion from the adrenal medullae of chronically diabetic BB-Wistar rats. Diabetes 1993; 42: 862–868. [DOI] [PubMed] [Google Scholar]
- 34.Powell AM, Sherwin RS and Shulman GI. Impaired hormonal responses to hypoglycemia in spontaneously diabetic and recurrently hypoglycemic rats. Reversibility and stimulus specificity of the deficits. J Clin Invest 1993; 92: 2667–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shum K, Inouye K, Chan O, et al. Effects of antecedent hypoglycemia, hyperinsulinemia, and excess corticosterone on hypoglycemic counterregulation. Am J Physiol Endocrinol Metab 2001; 281: E455–E465. [DOI] [PubMed] [Google Scholar]
- 36.Carbonaro DA, Mitchell JP, Hall FL, et al. Altered reactivity of the rat adrenal medulla. Brain Res Bull 1988; 21: 451–458. [DOI] [PubMed] [Google Scholar]
- 37.Wilke RA and Hillard CJ. Decreased adrenal medullary catecholamine release in spontaneously diabetic BB-Wistar rats. Diabetes 1994; 43: 724–729. [DOI] [PubMed] [Google Scholar]
- 38.Boksa P and Livett BG. Desensitization to nicotinic cholinergic agonists and K+, agents that stimulate catecholamine secretion, in isolated adrenal chromaffin cells. J Neurochem 1984; 42: 607–617. [DOI] [PubMed] [Google Scholar]
- 39.Wakade AR, Wakade TD and Malhotra RK. Restoration of catecholamine content of previously depleted adrenal medulla in vitro: importance of synthesis in maintaining the catecholamine stores. J Neurochem 1988; 51: 820–829. [DOI] [PubMed] [Google Scholar]
- 40.Davis SN, Mann S, Galassetti P, et al. The effects of differing durations of antecedent hypoglycemia on counterregulatory responses to subsequent hypoglycemia in normal humans. Diabetes 2000; 49: 1897–1903. [DOI] [PubMed] [Google Scholar]
- 41.Morrison SF. Differential control of sympathetic outflow. Am J Physiol Regul Integr Comp Physiol 2001; 281: R683–R698. [DOI] [PubMed] [Google Scholar]
- 42.Patrick RL and Kirshner N. Effect of stimulation on the levels of tyrosine hydroxylase, dopamine beta-hydroxylase, and catecholamines in intact and denervated rat adrenal glands. Mol Pharmacol 1971; 7: 87–96. [PubMed] [Google Scholar]
- 43.Marley E and Prout GI. Physiology and pharmacology of the splanchnic-adrenal medullary junction. J Physiol 1965; 180: 483–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimmermann H and Whittaker VP. Effect of electrical stimulation on the yield and composition of synaptic vesicles from the cholinergic synapses of the electric organ of Torpedo: a combined biochemical, electrophysiological and morphological study. J Neurochem 1974; 22: 435–450. [DOI] [PubMed] [Google Scholar]
- 45.Fischer-Colbrie R, Iacangelo A and Eiden LE. Neural and humoral factors separately regulate neuropeptide Y, enkephalin, and chromogranin A and B mRNA levels in rat adrenal medulla. Proc Natl Acad Sci USA 1988; 85: 3240–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jarry H, Dietrich M, Barthel A, et al. In vivo demonstration of a paracrine inhibitory action of met-enkephalin on adrenomedullary catecholamine release in the rat. Endocrinology 1989; 125: 624–629. [DOI] [PubMed] [Google Scholar]
- 47.Richter R, Michael N and Oehme P. Effect of substance P on catecholamine secretion from rat adrenal medulla in situ. Biomed Biochim Acta 1989; 48: 839–842. [PubMed] [Google Scholar]
- 48.Zhou XF and Livett BG. Substance P has biphasic effects on catecholamine secretion evoked by electrical stimulation of perfused rat adrenal glands in vitro. J Auton Nerv Syst 1990; 31: 31–39. [DOI] [PubMed] [Google Scholar]
- 49.Fischer-Colbrie R, Eskay RL, Eiden LE, et al. Transsynaptic regulation of galanin, neurotensin, and substance P in the adrenal medulla: combinatorial control by second-messenger signaling pathways. J Neurochem 1992; 59: 780–783. [DOI] [PubMed] [Google Scholar]
- 50.Antony S, Peeyush Kumar T, Mathew J, et al. Hypoglycemia induced changes in cholinergic receptor expression in the cerebellum of diabetic rats. J Biomed Sci 2010; 17: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sandoval DA, Ping L, Neill AR, et al. Cortisol acts through central mechanisms to blunt counterregulatory responses to hypoglycemia in conscious rats. Diabetes 2003; 52: 2198–2204. [DOI] [PubMed] [Google Scholar]
- 52.Levin BE, Becker TC, Eiki J, et al. Ventromedial hypothalamic glucokinase is an important mediator of the counterregulatory response to insulin-induced hypoglycemia. Diabetes 2008; 57: 1371–1379. [DOI] [PubMed] [Google Scholar]
- 53.Chan O, Paranjape S, Czyzyk D, et al. Increased GABAergic output in the ventromedial hypothalamus contributes to impaired hypoglycemic counterregulation in diabetic rats. Diabetes 2011; 60: 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Vries MG, Arseneau LM, Lawson ME, et al. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes 2003; 52: 2767–2773. [DOI] [PubMed] [Google Scholar]
- 55.Davis SN, Goldstein RE, Price L, et al. The effects of insulin on the counterregulatory response to equivalent hypoglycemia in patients with insulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1993; 77: 1300–1307. [DOI] [PubMed] [Google Scholar]
- 56.Davis SN, Goldstein RE, Jacobs J, et al. The effects of differing insulin levels on the hormonal and metabolic response to equivalent hypoglycemia in normal humans. Diabetes 1993; 42: 263–272. [DOI] [PubMed] [Google Scholar]
- 57.Davis SN, Shavers C, Collins L, et al. Effects of physiological hyperinsulinemia on counterregulatory response to prolonged hypoglycemia in normal humans. Am J Physiol 1994; 267: E402–E410. [DOI] [PubMed] [Google Scholar]
- 58.Morrison SF and Cao WH. Different adrenal sympathetic preganglionic neurons regulate epinephrine and norepinephrine secretion. Am J Physiol Regul Integr Comp Physiol 2000; 279: R1763–R1775. [DOI] [PubMed] [Google Scholar]
- 59.Kvetnansky R, Sabban EL and Palkovits M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol Rev 2009; 89: 535–606. [DOI] [PubMed] [Google Scholar]
- 60.Brindisi MC, Bouillet B, Verges B, et al. Cardiovascular complications in type 1 diabetes mellitus. Diabetes Metab 2010; 36: 341–344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.