

Abstract
Qualitative and quantitative mass analysis of antibodies and related macromolecular immune complexes is a prerequisite for determining their identity, binding partners, stoichiometries, and affinities. A plethora of bioanalytical technologies exist to determine such characteristics, typically based on size, interaction with functionalized surfaces, light scattering, or direct mass measurements. While these methods are highly complementary, they also exhibit unique strengths and weaknesses. Here, we benchmark mass photometry (MP), a recently introduced technology for mass measurement, against native mass spectrometry (MS) and size exclusion chromatography multi-angle light scattering (SEC-MALS). We examine samples of variable complexity, namely, IgG4Δhinge dimerizing half-bodies, IgG-RGY hexamers, heterogeneously glycosylated IgG:sEGFR antibody–antigen complexes, and finally megadalton assemblies involved in complement activation. We thereby assess the ability to determine (1) binding affinities and stoichiometries, (2) accurate masses, for extensively glycosylated species, and (3) assembly pathways of large heterogeneous immune complexes. We find that MP provides a sensitive approach for characterizing antibodies and stable assemblies, with dissociation correction enabling us to expand the measurable affinity range. In terms of mass resolution and accuracy, native MS performs the best but is occasionally hampered by artifacts induced by electrospray ionization, and its resolving power diminishes when analyzing extensively glycosylated proteins. In the latter cases, MP performs well, but single-particle charge detection MS can also be useful in this respect, measuring masses of heterogeneous assemblies even more accurately. Both methods perform well compared to SEC-MALS, still being the most established method in biopharma. Together, our data highlight the complementarity of these approaches, each having its unique strengths and weaknesses.
Introduction
With the continued advancement of antibody-based formats as biopharmaceuticals, analytical techniques providing robust and accurate characterization of these products and related macromolecular immune complexes become increasingly important. Antibody functioning strongly depends on noncovalent protein–protein interactions, with their unique structural organization bridging molecular recognition with the recruitment of effector functions.1 Structurally, standard IgG-based antibodies are homo-heterodimers consisting of two heavy chains (HCs) and two light chains (LCs) that are connected through several disulfide bridges. Target recognition is enabled by two variable antigen–binding (Fab) arms, which engage in highly specific epitope–paratope interactions.2,3 Effector functions, on the other hand, are primarily mediated by the constant (Fc) tail, which recruits and directs immune cells by binding to Fc receptor proteins,4,5 but can also initiate humoral immune responses such as the classical complement pathway.6,7 Furthermore, the Fc tail can facilitate the formation of functional oligomers—linked covalently in IgA and IgM or by noncovalent interactions in surface-bound IgGs.8 The ability to accurately characterize antibodies and their interactions with antigens and receptors is thus of crucial importance, both for fundamental research as well as in the optimization of antibody engineering and drug development.
Affinities and kinetics of antibody–antigen interactions are typically assessed by biosensors that quantify interactions with functionalized surfaces. The most prevalent of such approaches is surface plasmon resonance,9,10 but bio-layer interferometry,11 quartz crystal microbalances,12 and Förster resonance energy transfer microscopy13,14 provide accurate readouts down to sub-nanomolar Kd values. However, these techniques are generally limited to binary interactions, preventing their use in studying oligomerization and the formation of larger immune complexes of multiple stoichiometries and compositions.
Low-resolution biophysical methods based on size, charge, diffusion, or light scattering are widely used in academia and industry to study antibody oligomerization and complex formation. Oligomeric distributions and aggregation states can be evaluated using dynamic light scattering.15−17 Analytical ultracentrifugation, on the other hand, can provide quantitative data at a higher resolution,18,19 allowing the technique to be used to study protein–protein interactions as well,20 but arduous experimental procedures make this approach impractical for routine use.21 Moreover, also capillary electrophoresis-related techniques have been applied for the characterization of antibodies and their interactions.22,23
Size exclusion chromatography (SEC) remains the long-standing industry standard for analyzing the quaternary structure of antibody products. SEC utilizes a porous matrix as a stationary phase to enable size-based separation, followed by detection to provide sensitive, highly reproducible, and quantitative data. As SEC alone does not provide an accurate means to assess masses, it often includes average molecular mass determination by coupling to a multi-angle light scattering (MALS) detector, which acts by measuring the light scattering generated by particles, being proportional to their molar mass and concentration. This makes SEC-MALS a very versatile tool for studying antibodies, although it is also hampered by some issues because dilution and shear forces during chromatographic separation can affect equilibria. The technique also requires optimization regarding running conditions, as protein species can adsorb to the matrix.
Direct mass measurement by native mass spectrometry (MS)24,25 represents a relatively newer component of the analytical toolbox for antibody analysis. Compared to the techniques described above, native MS yields higher mass resolution and accuracy that can be used to assess, for instance, microheterogeneity of monoclonal antibodies26,27 and their derivatives,28−30 the formation of antibody–antigen complexes,31−33 and larger megadalton particle immune complexes.34,35 As noncovalent interactions are retained, native MS can also be used to probe binding equilibria of protein–ligand and protein–protein interactions when instrumental parameters are carefully optimized to avoid bias and artifacts.36−40 Direct online coupling of SEC or capillary zone electrophoresis to mass spectrometers can further enable separation and structural characterization of protein assemblies.41,42
Typically, in native MS, mass analysis relies on resolving charge states of the same species in the m/z space of the mass spectrum, which becomes more difficult when the analytes become heavier and more heterogeneous. In such cases, single-particle charge detection-MS (CD-MS)43 may be very useful. This technique, which recently was also demonstrated on commercial Orbitrap-based instruments,35,44 makes it possible to directly assess the mass of single ions by measuring their m/z in parallel with their charge z, as inferred from their single-ion intensity value.
Mass photometry (MP) was recently introduced as a technology that enables mass analysis of proteins and protein complexes under native buffering conditions.45 MP makes use of interferometric scattering microscopy to detect and quantify light scattering caused by single particles.46−50 When particles in solution bind nonspecifically to a glass surface, their scattering signal interferes with the measured reflectivity of the glass/water interface. Because the optical properties and density of proteins are quite uniform, this reflectivity change is proportional to the molecular mass, allowing MP to provide a direct mass measurement for each particle.46,51 MP has already been shown to be able to provide quantitative data, allowing the binding affinities and kinetics of antibody–antigen and antibody–receptor interactions to be explored.45,52,53 However, because typical experimental conditions are limited to low nanomolar range concentrations, the methods outlined in these studies apply only to relatively strong and slowly dissociating protein assemblies.
Here, we compare MP side by side with native MS and SEC-MALS, employing these techniques for the analysis of a variety of antibody formats and heavily glycosylated macromolecular immune complexes. We expand the affinity range of quantitative MP experiments by modeling the dissociation of weaker interactions, and we compare the techniques in terms of their ability to assess a wide range of binding equilibria. We assess the pros and cons of each of these approaches, considering dynamic range, robustness, mass resolution, and mass accuracy, and we highlight their strengths in resolving extensively glycosylated species.
Materials and Methods
More detailed descriptions of the methods are provided in the Supporting Information Methods.
Protein Samples
Anti-EGFR antibodies in IgG4Δhinge, IgG1, and IgG1-RGY format and sEGFR were recombinantly expressed and purified by Genmab.34,40,54,55 Human C1q was obtained from Complement Technology. Samples were buffer-exchanged to the appropriate solution. Protein complexes were assembled by mixing subcomponents at the desired molar ratios, followed by incubation at room temperature for at least 30 min. For quantitative experiments, incubation after preparing a dilution series was proceeded for at least 4 h.
Native MS and CD-MS
For native MS, proteins in 150 mM aqueous ammonium acetate pH 7.5 were measured by direct infusion from a static nano-electrospray ionization (ESI) source. Quantitative experiments were performed on a modified LCT time-of-flight instrument (Waters), measuring samples in triplicate. All other experiments were performed on a Q Exactive Plus UHMR Orbitrap instrument (Thermo Fisher Scientific). For CD-MS, dilute samples were measured at low pressure and high resolution (1 s transient) for accurate determination of both m/z and z of single ions.
SEC-MALS
SEC-MALS experiments were performed on a Waters HPLC with an in-line UV detector (Waters 2487 Dual Absorbance), a MALS detector (MiniDAWN, Wyatt Technology), and an RI detector (Optilab, Wyatt Technology). Proteins were separated on an SRT SEC-500 column (Sepax Technologies) using 100 mM sodium phosphate, 100 mM sodium sulfate, and pH 6.8 as mobile phase at 0.35 mL/min. Data were processed by ASTRA software (Wyatt) based on MALS-RI for antibody mass determination or MALS-UV-RI (Protein Conjugate Analysis) for the analysis of glycan contributions and larger complexes.
Mass Photometry
MP experiments were performed by measuring the samples in PBS on a Refeyn OneMP mass photometer (Refeyn). Triplicate measurements of 12,000 frames were combined into a single mass histogram. When measuring protein complexes, high concentration solutions were jump-diluted to nM range measurement concentrations in approximately 5–30 s. For quantitative experiments, a dilution series was measured in triplicate in recordings of 6000 frames. For these experiments, dissociation upon jump dilution was modeled to infer complex abundance in the original solution.
Results and Discussion
We started our MP analysis by characterizing the monomer–dimer equilibrium of hinge-deleted human IgG4 (IgG4Δhinge) molecules, providing a simple and small one-component system, for which we reported earlier data from SEC and native MS.40 Deletion of the hinge region removes the disulfide bonds that bridge the two HCs, meaning that the two-halves of the antibody interact solely via noncovalent interactions. This results in an equilibrium between antibody half molecules (HLs) and HL dimers (HL)2. Previous work from our group assessed the effects of specific mutations in the CH3 domain on this equilibrium by native MS and SEC,40 providing a panel of highly similar samples, with Kd values spanning 6 orders of magnitude (10–10 to 10–4 M). Because MP experiments are typically performed at concentrations of only a few nM, we used jump dilution to quickly dilute concentrated samples just before starting the measurement. Assuming that the koff of the interaction is low enough, the observed distribution of protein assemblies should then reflect that of the original concentrated sample.
Jump-Diluted IgG4Δhinge Dimers Dissociate during MP Analysis
The distinct light scattering caused by single particles of different masses as measured by MP can be converted into masses and shown in histograms. The mass histograms of “wt” IgG4Δhinge jump-diluted from a 16 μM solution to a measurement concentration of 4 nM reveal two distinct distributions centered at the expected masses, namely, 73 kDa HL and 146 kDa (HL)2 (Figure 1A; see Table S1 for an overview of all measured masses). The particle counts constituted about 74% of (HL)2 dimers (mass abundance of 85%), which is lower than expected for this relatively strong interaction (Kd = 50 nM by native MS40). Upon further inspection, we observed that the abundance of the dimer already decreased during the first seconds of the MP measurement with a pattern resembling exponential decay (Figure 1B). This indicates that the (HL)2 dimer readily starts to dissociate upon jump dilution as the sample re-equilibrates to the measurement concentration, meaning that simply summing the data of the measurement window will lead to an underestimation of the actual dimer abundance.
Figure 1.

Qualitative and quantitative characterization of IgG4Δhinge mutants by MP. (A) Mass histogram showing particle counts of “wt” IgG4Δhinge in PBS, jump-diluted from 16 μM and measured at 4 nM, with normal distributions fitted for HL (bright red, 26%) and (HL)2 (dark red, 74%). This histogram corresponds to the first 80 s after jump dilution. Masses are indicated as the mean of a fitted normal distribution. (B) Monomer–dimer distribution during an extended experiment in triplicate (shades of red) revealed that the abundance of “wt” (HL)2 decreased during the analysis time window. Data were split into bins of 100 events, and an exponential decay function was fitted to the dimer abundance within the bin to determine the koff. (C) Determined koff was used to estimate the ratio between HL and (HL)2 at the instant of jump dilution for a dilution series of the “wt” measured in triplicate, revealing the apparent Kd of each measurement, followed by the calculation of a Kd value for the whole dilution series. (D) Fractional dimer abundances and Kd values resulting from a dilution series of four IgG4Δhinge mutational variants, demonstrating that MP can assess affinities over a broad dynamic range.
Modeling for Dissociation Expands the Affinity Range of IgG4Δhinge Mutants Assessable by MP
To obtain a more accurate representation of the oligomer distributions in solution before jump dilution, we adjusted our data processing approach by modeling (HL)2 dissociation. When the sample concentration is diluted by several orders of magnitude, especially when [HL] ≪ Kd, we can assume that the initial decrease in (HL)2 abundance is driven primarily by koff. Thus, by determining koff of the interaction, we can fit an exponential decay function to the measured [(HL)2] over time to estimate [(HL)2] before jump dilution (see Supporting Information Methods). We evaluated this method using a panel of four IgG4Δhinge mutants spanning a broad affinity range, measuring a dilution series to determine their Kd (Figure 1C,D). Modeling for dissociation allowed us to determine affinities well into the μM range, substantially improving the dynamic range of quantitative MP experiments. Still, a few issues remained. We found that the most consistent results were obtained for relatively strong interactions with low koff values, such as those of “wt” IgG4Δhinge (Kd = 2.3 nM). For the even stronger interactions of the R409K mutant (Kd = 9.4 pM), the equilibrium was still mostly geared toward the dimer at concentrations assessable by MP, reducing the precision of Kd determination for this mutant. Weaker interactions such as those of the D399S (Kd = 7.8 μM) and L368A (Kd = 54 μM) mutants could also be measured, although higher dissociation rates (0.012 and 0.029 s–1) reduced the accuracy of the model. Nonetheless, MP experiments led to the same affinity ranking of the mutants as did native MS, although with some discrepancies between the obtained absolute Kd values (Table S2). An important difference is that native MS is performed with a volatile buffering solution (i.e., aqueous ammonium acetate), while MP enabled the use of PBS as a more physiological buffer. Furthermore, standard native MS is also somewhat restrained to high nM to low μM concentrations, reducing the accuracy by which (sub) nM range Kd values can be assessed. MP and native MS are highly complementary in this sense, as they each have their own distinctive preferred concentration range.
MP Outperforms Native MS in the Mass Assessment of Heavily Glycosylated Antibody–Antigen Assemblies
We next characterized the interactions between antibodies and their antigen by MP and native MS. Several therapeutic antibodies target glycosylated receptor proteins, some of which are notoriously hard to analyze by native MS due to their high degree of microheterogeneity. Methods that can accurately mass measure and quantify these antigens and their interaction with mAbs are therefore of great use to both fundamental and biopharmaceutical research. Here, we analyze an IgG1 mAb targeting epidermal growth factor receptor (EGFR), whereby we used the soluble domain (sEGFR). This protein with a mass of 69,409 Da in its non-glycosylated form is very heterogeneous, harboring 11 typical N-glycosylation motifs that can be variably occupied.56
Starting with the IgG1 alone, although the average mass obtained by MP was in good agreement with native MS, the latter provided unparalleled mass accuracy and resolution, enabling baseline resolution of individual glycoforms (Figure 2). However, native mass analysis of extensively glycosylated sEGFR alone was difficult, being unable to resolve charge states because of the presence of a plethora of proteoglycoforms. This obstacle was overcome by using charge detection MS (CD-MS), recently developed as a methodology for Orbitrap instruments, which provides an extra dimension of data by measuring the charge of the ions independently. CD-MS measured a mass of 88 kDa for sEGFR, in close agreement with an earlier reported value derived by tandem MS experiments.30 Similarly, MP readily provided a mass of 86 kDa, with SEC-MALS-UV-RI also measuring a mass of 91 kDa for sEGFR (Figure S3). Next, when the anti-sEGFR mAb was incubated together with sEGFR, MP presented further advantages. In native MS, additional ion signals were observed for (IgG1)1:(sEGFR)1 (m/z 7000–8500), although poor resolution hampered mass determination, while the full (IgG1)1:(sEGFR)2 complex (m/z 8500–10,000) could not be resolved at all. However, both CD-MS and jump dilution MP enabled the reliable measurement of the average masses for all co-occurring complexes, clearly revealing the stoichiometry. However, binding occupancy was somewhat lower in CD-MS, likely because of re-equilibration upon dilution before the somewhat longer measurements. SEC-MALS was similarly able to discern the full (IgG1)1:(sEGFR)2 complex, although the resolution was substantially lower (Figure S3). Combining them, these data already show that MP and CD-MS have advantages for mass analysis of heterogeneous antibody-antigen complexes.
Figure 2.

MP and CD-MS may overcome certain limitations of native MS in the mass measurements of highly heterogeneous antibody–antigen complexes. (A) MP provides an average mass for IgG1 (upper panel) and sEGFR (middle) and is not hampered by the high micro-heterogeneity of the latter. When 2 μM IgG1 was incubated with 5 μM of sEGFR to form (IgG1)1:(sEGFR)1 and (IgG1)1:(sEGFR)2 complexes, jump dilution MP could resolve these highly heterogeneous species (lower). (B) Although native MS on samples at the same concentrations provided superior mass resolution and accuracy for free IgG1 (upper), resolving individual glycoforms (zoom), the high microheterogeneity of sEGFR, measured separately (middle) and in antibody–antigen complexes (lower), resulted in unresolved features. In these experiments, overlapping charge states prevented mass measurements of these species. (C) More accurate masses could be obtained by native CD-MS, measuring in two dimensions m/z and z (insets) for sEGFR (upper) and all co-occurring species involving IgG1 and sEGFR (lower). For these experiments, the same native MS samples were diluted 20-fold, leading to re-equilibration and thus a lower binding occupancy.
MP, SEC, and Native MS Analyses of the Monomer–Hexamer Equilibrium of Soluble IgG1-RGY Hexamers Produce Consistent Results
We next evaluated the performance of MP in the characterization of larger and more complex antibody-based systems, involved in immune activation through the complement pathway.8,34 Target-bound IgG can initiate complement activity by forming a hexameric binding platform for recognition of complex C1q. Although these IgG oligomers are thought to only form by clustering on antigenic surfaces in vivo, this process can be mimicked in solution using the engineered IgG-RGY platform, a triple mutant that readily forms hexamers in equilibrium with monomers.8,34,55
MP mass histograms and SEC-MALS chromatograms of IgG1-RGY revealed as expected two species corresponding to the monomer (denoted (IgG1)1) and hexamer ((IgG1)6) (Figure 3A,B). While the MP mass of the monomer was in good agreement with the native MS data (Figure 3C), we noticed that the mass of the hexamer was consistently off by about +70 kDa, possibly due to its non-globular shape as a flat disc. In SEC-MALS, we observed peak trailing for the hexamer, potentially driven by shear force-induced dissociation or re-equilibration during separation. As reported previously,8,34 native mass spectra of IgG1-RGY also showed two well-resolved distributions for (IgG1)1 and (IgG1)6, but uniquely also intermediate oligomers at lower abundance. Possibly, the ESI process could result in partial dissociation of the hexamers, as this process is distinct from gas-phase-based tandem MS (Figure S4).
Figure 3.
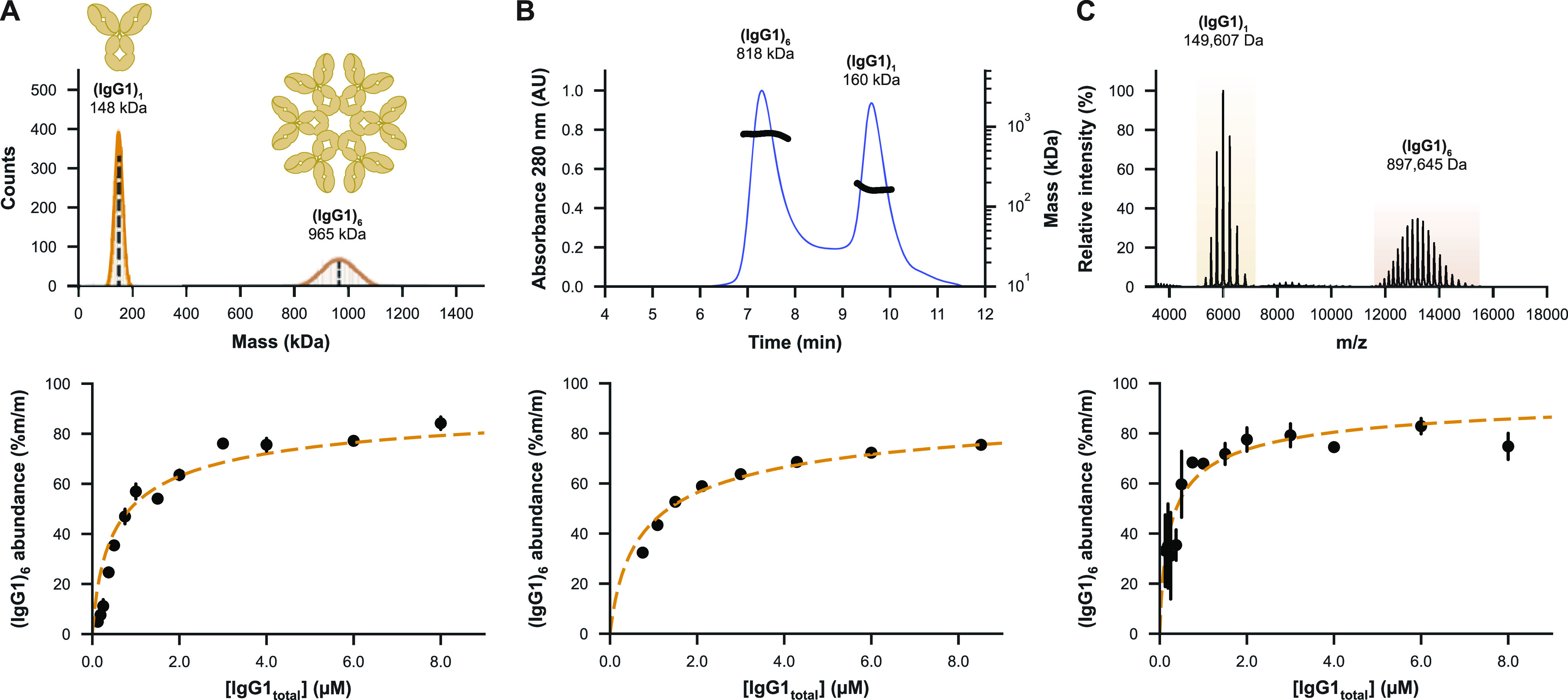
MP enables qualitative and quantitative characterization of the monomer–hexamer equilibrium of IgG1-RGY. (A) MP mass histogram (top) of 2 μM IgG1-RGY in PBS jump-diluted to 10 nM showing monomeric ((IgG1)1) and hexameric ((IgG1)6) species. The mass of the hexamer was consistently measured about 70 kDa too high. The relative abundance of the IgG1-RGY hexamer was measured over a dilution series spanning a concentration range of 0.1 to 8 μM (bottom), with error bars indicating the standard deviation over three technical replicate measurements. (B) SEC-MALS chromatogram of the same 2 μM IgG1-RGY sample (top) and the fractional abundance of the hexamer as measured by SEC-UV over a dilution series (bottom), revealing a similar monomer to hexamer ratio. The quantitative data in the lower panel are adapted from the work of van Kampen et al.58 (C) Native mass spectrum (top) of 2 μM IgG1-RGY measured in 150 mM NH4OAc pH 7.5, revealing two distinct ion series for the monomer and hexamer, with ions originating from intermediate oligomeric states observed at lower abundance. While generally in good agreement with the other methods, hexamer abundances measured by MS (bottom) were less consistent and higher than expected, particularly at the lowest measured concentrations.
Quantifying the abundance of the hexamer in a dilution series of IgG1-RGY by all three techniques resulted in highly comparable data, although with some subtle differences. In agreement with earlier studies,57 longer MP recordings showed that hexamers re-equilibrate only very slowly upon jump dilution (Figure S5), meaning that such MP experiments should directly provide an accurate representation of the monomer–hexamer equilibrium. To characterize the equilibrium of IgG1-RGY by MP, we measured a dilution series and compared results to SEC and native MS. MP measurements proved to be quite consistent between replicates and could be performed down to nM range concentrations that cannot be assessed by native MS or SEC (Figure 3A). While SEC also proved to provide very robust data, hexamer abundances were fractionally lower, potentially due to dissociation during separation. Finally, although native MS performed well at higher concentrations, variability increased at lower concentrations. Nonetheless, each of the three techniques revealed that about half of the IgGs are incorporated into hexamers at a concentration of 1 μM, consistent with previously reported data.34
Characterization of Complement Component C1q by MP Exposes Shortcomings of Native MS and SEC-MALS
We next characterized complement component C1q, the recognition complex of the classical complement pathway, revealing striking differences between the three tested techniques. C1q is a 464 kDa 18-membered protein complex that consists of three pairs of triple helices (A2B2C2) that are joined in a stem and end with six globular headpieces.59,60 Although each headpiece has a low affinity for the Fc of IgGs,61,62 clustering into oligomers allows for multivalent binding with increased avidity,63 making the six-armed C1q complex highly compatible with IgM64 and IgG hexamers.8 As demonstrated previously, C1q behaves anomalously in SEC, eluting at a short retention time that suggests a mass of >1 MDa34 (Figure S6A). Because of its open structure, C1q may have a much larger hydrodynamic radius than globular proteins of similar mass, producing a bias in size-based separation. Concordantly, the coupled MALS system revealed that this elution peak did correspond to the free C1q complex with a mass of about 444 kDa. Curiously, the native mass spectra of C1q were consistently marked by the presence of three main ion series corresponding to two-armed, four-armed, and complete six-armed C1q species (Figure S6B), decreasing in abundance with the size of the complex. Partial C1q complexes did not display asymmetrical charge partitioning characteristic for collisional dissociation in the gas phase, suggesting that, similar to intermediate oligomers of IgG-RGY, smaller complexes are formed by the electrospray process. Lastly, MP measurements of C1q revealed predominantly particles of the intact six-armed C1q complex (A6B6C6), with a minor contribution being made by a two-armed A2B2C2 complex (Figure S6C). We thus conclude that MP seems the most unbiased tool for the analysis of C1q.
MP and CD-MS Tackle Mass Heterogeneity When Analyzing Immune Complexes of IgG-RGY Hexamers Bound to Highly Glycosylated Antigens and C1q
Having demonstrated that IgG1-RGY hexamers and C1q can be measured accurately by MP, we next sought to characterize immune activation complexes involving antigen-bound IgG1-RGY hexamers and C1q. When IgG1-RGY was first incubated with C1q, MP revealed the formation of a 1.43 MDa complex corresponding to (IgG1)6:(C1q)1 (Figure 4A). Similar to measurements of the IgG1-RGY hexamers, this mass is about 60 kDa higher than expected, possibly due to the non-globular shapes of both complexes. Next, we assembled larger complement activation complexes associated with highly heterogeneous sEGFR antigens, whereby sEGFR was incubated with preformed (IgG1)6:(C1q)1 complexes (see Figure S7 for the analysis of IgG1-RGY with sEGFR separately). MP revealed the presence of particles with an average mass of 2.35 MDa, likely corresponding to (sEGFR)12:(IgG1)6:(C1q)1. However, as the resulting peak was quite broad, we could not yet exclude the possibility of other (co-occurring) stoichiometries. Similar to MP, SEC-MALS-UV-RI measurement of IgG1-RGY incubated with C1q led to the detection of (IgG1)6:(C1q)1 with a fairly adequate mass of 1.3 MDa (Figure 4B). Although larger complexes of ∼1.9 MDa were observed upon addition of sEGFR, here, the width of this peak and the accuracy of the mass measurement was insufficient for determining the exact stoichiometry, possibly also due to dissociation of sEGFR during separation. In agreement with earlier reports,34 native MS of IgG1-RGY with C1q alone revealed the presence of (IgG1)6:(C1q)1 complexes, for which an accurate mass of 1361 kDa could be established (Figure 4C). Contrary to MP, however, relatively more (IgG1)6 and uniquely also (IgG1)6:(A4B4C4)1 complexes were observed, potentially dissociation products of the full complex formed in the MS source region. While larger but unresolved ions could be detected by native MS after the addition of sEGFR, an inability to resolve charge states prevented mass determination. For these samples, CD-MS enabled confident assignment of a mass of 2.42 MDa, corresponding to the complete (sEGFR)12:(IgG1)6:(C1q)1 assembly (Figure 4D).
Figure 4.
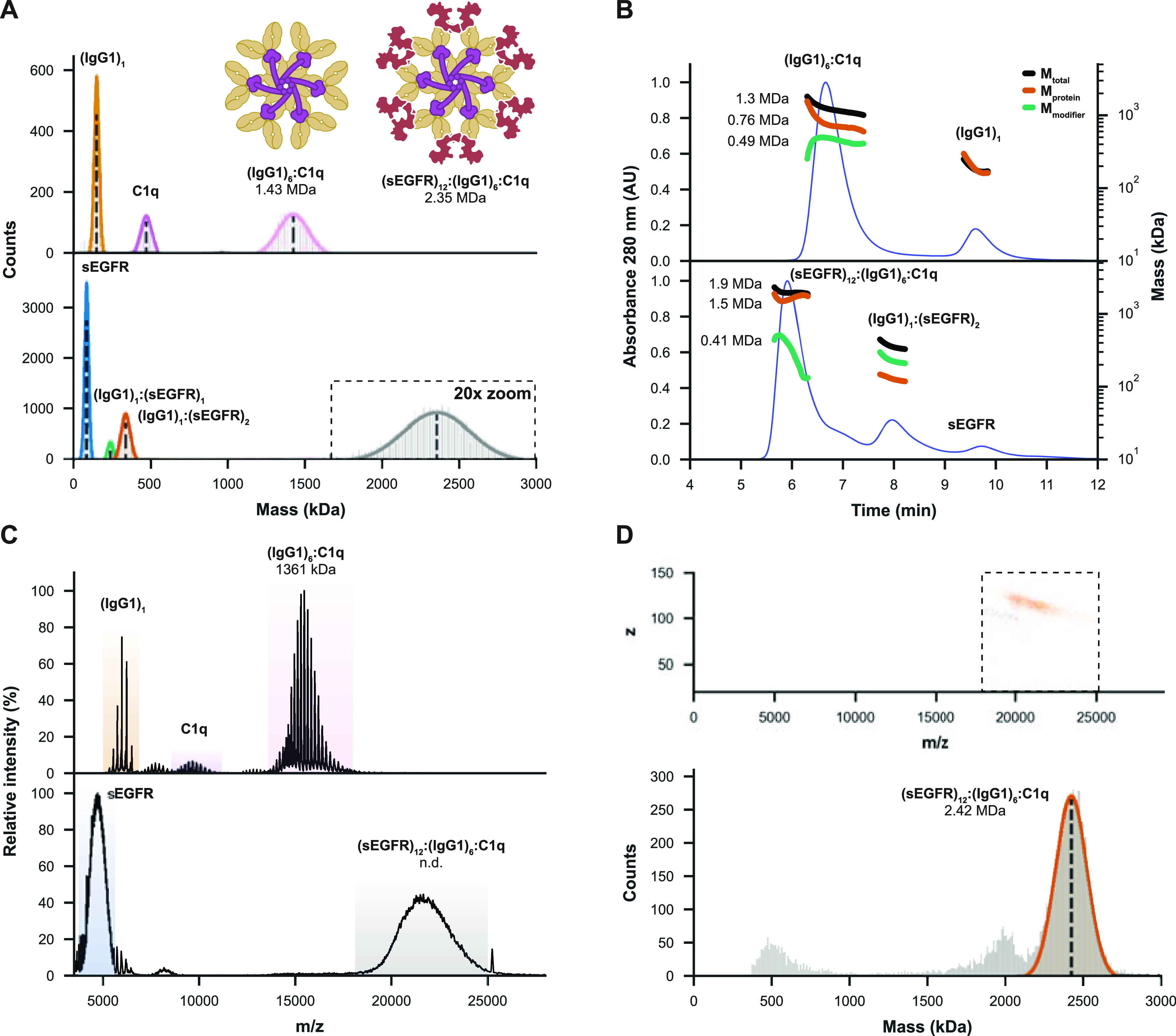
MP and CD-MS successfully determine the mass and stoichiometry of highly heterogeneous (sEGFR)12:(IgG1)6:C1q immune complexes. (A) MP measurements of IgG1-RGY incubated with C1q reveal the formation of (IgG1)6:(C1q)1 complexes, with nearly all IgG hexamers occupied. When incubating C1q with pre-formed (IgG1)6:(sEGFR)12, MP resolves a 2.35 MDa complex, likely corresponding to (sEGFR)12:(IgG1)6:(C1q)1. (B) SEC-MALS-UV-RI analysis similarly reveals the formation of ∼1.3 MDa (IgG1)6:(C1q)1 (with (IgG1)6 measured as the 0.76 MDa “protein” and C1q as a 0.49 MDa “modifier”). When sEGFR was added, SEC-MALS-UV-RI revealed the formation of larger complexes of around 1.9 MDa (1.5 MDa for (sEGFR)12:(IgG1)6 with a 0.41 MDa modifier). (C) Measurement of the same samples by native MS reveals an accurate mass for (IgG1)6:(C1q)1, but the technique struggles with complexes involving sEGFR. Larger ion species were detected in such experiments, but they could not be charge-resolved. (D) Single-particle measurements of the distribution around m/z 21,000 by CD-MS (top) revealed a mass of 2.42 MDa (bottom) corresponding to the expected mass of the full (sEGFR)12:(IgG1)6:(sEGFR)12 complex (bottom).
Comparing Advantages and Disadvantages Reveals That MP, Native MS, and SEC-MALS Are Highly Complementary
Reflecting on the analyses performed in this study on a wide variety of systems, we can compare the advantages and disadvantages of the approaches (see Table S1 for an overview of all the measured masses and Table S3 for a qualitative comparison between the techniques). Analytical SEC-MALS is the most established method, providing reproducible, robust, and quantitative measurements. While accurate for smaller proteins, masses of larger multicomponent systems were underestimated, often by as much as 10–20%, making SEC-MALS suboptimal for large complexes that may dissociate by shear stress or dilution effects during column separation. MP represents a relatively new approach that is a fast and comparatively straightforward technique to measure more accurate masses in native-like buffering solutions, enabling it to tackle multicomponent systems more effectively than SEC-MALS. Still, mass resolution limits the technology mostly to antibody–protein interactions, as mass differences induced by small molecule or peptide binding are mostly too small to resolve. Sample consumption is low, however, and unlike conventional native MS, the technique is not hampered by extensive protein glycosylation. While MP could reliably measure molecular masses and quantify strongly interacting and slowly dissociating protein complexes, jump dilution to nM range concentrations induced dissociation of weaker interactions. Another consideration for using MP is that mass measurements may be affected by the shape of the particles, as we consistently measured a higher-than-expected mass for IgG1-RGY hexamers. Native MS provided superior mass resolution and accuracy, resolving individual proteoglycoforms in samples of moderate complexity and uniquely providing unambiguous stoichiometries for protein complexes. After careful optimization of instrumental parameters, reliable quantification could be achieved when measuring at conventionally used concentrations (around 10–6 M). Certain protein complexes, however, proved to be sensitive to dissociation induced in solution by the ESI process, and performance of conventional native MS deteriorated when analyzing extensively glycosylated proteins. However, when charge states could not be resolved in the m/z domain, this challenge was effectively overcome by CD-MS, providing unparalleled mass accuracy for highly heterogeneous protein assemblies.
Conclusions
Here, we compared MP with native MS and SEC-MALS for the qualitative and quantitative analysis of antibodies and related immune complexes. Single-molecule and solution-based MP provides a relatively straightforward way to assess protein complexes and can fill gaps between the two other techniques in terms of mass accuracy and resolution, while also being able to quantitatively assess strong and stable protein–protein interactions. Among the main benefits of MP are its high sensitivity (nM) and ability to measure in-solution using a wide variety of buffer solutions. However, here we show that the mass resolving power of MP is still somewhat limited and that some protein complexes may dissociate due to dilution required for measurements. Among the main benefits of native MS are its comparatively very high mass resolution, but only when charge states can be resolved. When high heterogeneity makes this impossible, single-molecule CD-MS can be used to infer charge states in an alternative manner, providing lower resolving power than conventional native MS, but generally still higher than MP. Native MS is, however, a gas-phase technique that requires volatile buffers and has a low tolerance for salts and detergents. SEC and SEC-MALS are well-established technologies for mass assessment of proteins within the biopharmaceutical laboratories, benefiting from their proven robustness and ease of use. However, for assessing protein assemblies, the resolving power of SEC-MALS is relatively low, and quantification may be somewhat hampered by dilution of the sample during separation. Overall, our data show that the tested approaches are highly complementary, each having its unique preferred use cases. With robust commercial instruments now becoming available, these newer techniques may become more accepted. Furthermore, combining techniques, such as SEC coupled to native MS, may overcome some of their weaknesses, while taking advantage of their strengths.
Acknowledgments
We especially thank Genmab research associates Mandy Blom and Clifford Rodriguez for their excellent work on performing the SEC-MALS experiments and fruitful discussions. We further thank members of the Heck laboratory for general support, especially Arjan Barendregt. This research received funding through the Netherlands Organization for Scientific Research (NWO) TTW-NACTAR project 16442 (A.J.R.H. and M.A.d.B.) and the Spinoza Award SPI.2017.028 to A.J.R.H. We further acknowledge funding for the large-scale proteomics facility, the Netherlands Proteomics Center, through the X-omics Road Map program (project 184.034.019) and the EU Horizon 2020 program Epic-XS (project 823839).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c03656.
Supplementary methods, koff and Kd determination for IgG4Δhinge variants, SEC-MALS of IgG1 and sEGFR, native MS gas-phase dissociation of IgG1 hexamers, IgG1 hexamer dissociation upon jump dilution MP, SEC-MALS, native MS and MP characterization of C1q, SEC-MALS, native (CD) MS and MP characterization of (IgG1)6:(sEGFR)12 antibody–antigen complexes, comparison of masses measured by the assessed techniques, MP-derived kinetic rates and Kd values for IgG4Δhinge variants, comparison of advantages and disadvantages of the assessed techniques, and supplementary references (PDF)
The authors declare the following competing financial interest(s): XX, MDvK and BB are Genmab employees and own Genmab warrants and/or stock.
Supplementary Material
References
- Schroeder H. W. Jr.; Cavacini L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. 10.1016/j.jaci.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H.-P.; Lee K. H.; Jian J.-W.; Yang A.-S. Origins of specificity and affinity in antibody-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E2656–E2665. 10.1073/pnas.1401131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela-Culang I.; Kunik V.; Ofran Y. The Structural Basis of Antibody-Antigen Recognition. Front. Immunol. 2013, 4, 302. 10.3389/fimmu.2013.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes J.; Wormald M.; Rudd P.; Davey G. Fc gamma receptors: glycobiology and therapeutic prospects. J. Inflammation Res. 2016, 9, 209–219. 10.2147/jir.s121233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor J. D.; Brown M. P.; Irving H. R.; Zalcberg J. R.; Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J. Hematol. Oncol. 2013, 6, 1. 10.1186/1756-8722-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugurlar D.; Howes S. C.; de Kreuk B.-J.; de Jong R. N.; Beurskens F. J.; Schuurman J.; Koster A. J.; Sharp T. H.; Parren P. W. H. I.; Gros P. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. 10.1126/science.aao4988. [DOI] [PubMed] [Google Scholar]
- Lu L. L.; Suscovich T. J.; Fortune S. M.; Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018, 18, 46–61. 10.1038/nri.2017.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebolder C. A.; Beurskens F. J.; de Jong R. N.; Koning R. I.; Strumane K.; Lindorfer M. A.; Voorhorst M.; Ugurlar D.; Rosati S.; Heck A. J. R.; van de Winkel J. G. J.; Wilson I. A.; Koster A. J.; Taylor R. P.; Ollmann Saphire E.; Burton D. R.; Schuurman J.; Gros P.; Parren P. W. H. I. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H.; Park J.; Kang S.; Kim M. Surface plasmon resonance: a versatile technique for biosensor applications. Sensors 2015, 15, 10481–10510. 10.3390/s150510481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puiu M.; Bala C. SPR and SPR Imaging: Recent Trends in Developing Nanodevices for Detection and Real-Time Monitoring of Biomolecular Events. Sensors 2016, 16, 870. 10.3390/s16060870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat V.; Rafique A. Designing binding kinetic assay on the bio-layer interferometry (BLI) biosensor to characterize antibody-antigen interactions. Anal. Biochem. 2017, 536, 16–31. 10.1016/j.ab.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Ogi H. Wireless-electrodeless quartz-crystal-microbalance biosensors for studying interactions among biomolecules: a review. Proc. Jpn. Acad., Ser. B 2013, 89, 401–417. 10.2183/pjab.89.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschen-Ohm M. P.; White D. S.; Klenchin V. A.; Chanda B.; Goldsmith R. H. Observing Single-Molecule Dynamics at Millimolar Concentrations. Angew. Chem., Int. Ed. 2017, 56, 2399–2402. 10.1002/anie.201612050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal V.; Ha T. Single-molecule fluorescence microscopy of native macromolecular complexes. Curr. Opin. Struct. Biol. 2016, 41, 225–232. 10.1016/j.sbi.2016.09.006. [DOI] [PubMed] [Google Scholar]
- Nobbmann U.; Connah M.; Fish B.; Varley P.; Gee C.; Mulot S.; Chen J.; Zhou L.; Lu Y.; Sheng F.; Yi J.; Harding S. E. Dynamic light scattering as a relative tool for assessing the molecular integrity and stability of monoclonal antibodies. Biotechnol. Genet. Eng. Rev. 2007, 24, 117–128. 10.1080/02648725.2007.10648095. [DOI] [PubMed] [Google Scholar]
- Stetefeld J.; McKenna S. A.; Patel T. R. Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. 10.1007/s12551-016-0218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon A. D.; Larkin M. I.; Reddick R. M. Free-Solution, Label-Free Protein-Protein Interactions Characterized by Dynamic Light Scattering. Biophys. J. 2010, 98, 297–304. 10.1016/j.bpj.2009.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz S. A.; Philo J. S.. Characterizing Biopharmaceuticals using Analytical Ultracentrifugation. In Biophysical Characterization of Proteins in Developing Biopharmaceuticals; Houde D. J., Berkowitz S. A., Eds.; Elsevier: Amsterdam, 2015; pp 211–260. [Google Scholar]
- Uchiyama S.; Noda M.; Krayukhina E. Sedimentation velocity analytical ultracentrifugation for characterization of therapeutic antibodies. Biophys. Rev. 2018, 10, 259–269. 10.1007/s12551-017-0374-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. Analytical Ultracentrifugation as a Tool for Studying Protein Interactions. Biophys. Rev. 2013, 5, 159–171. 10.1007/s12551-013-0106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi A. V.; Pothecary M. R.; Bain D. L.; Carpenter J. F. Some Lessons Learned From a Comparison Between Sedimentation Velocity Analytical Ultracentrifugation and Size Exclusion Chromatography to Characterize and Quantify Protein Aggregates. J. Pharm. Sci. 2017, 106, 2178–2186. 10.1016/j.xphs.2017.04.048. [DOI] [PubMed] [Google Scholar]
- Moser A. C.; Trenhaile S.; Frankenberg K. Studies of antibody-antigen interactions by capillary electrophoresis: A review. Methods 2018, 146, 66–75. 10.1016/j.ymeth.2018.03.006. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Guttman A.; Rathore A. S. Applications of capillary electrophoresis for biopharmaceutical product characterization. Electrophoresis 2021, 10.1002/elps.202100182. [DOI] [PubMed] [Google Scholar]
- Leney A. C.; Heck A. J. R. Native Mass Spectrometry: What is in the Name?. J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. 10.1007/s13361-016-1545-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamara S.; den Boer M. A.; Heck A. J. R. High-Resolution Native Mass Spectrometry. Chem. Rev. 2021, 10.1021/acs.chemrev.1c00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson N. J.; Hendriks L. J.; de Kruif J.; Throsby M.; Heck A. J. Complex mixtures of antibodies generated from a single production qualitatively and quantitatively evaluated by native Orbitrap mass spectrometry. mAbs 2014, 6, 197–203. 10.4161/mabs.27126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Liu F.; Franc V.; Halim L. A.; Schellekens H.; Heck A. J. R. Hybrid mass spectrometry approaches in glycoprotein analysis and their usage in scoring biosimilarity. Nat. Commun. 2016, 7, 13397. 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valliere-Douglass J. F.; McFee W. A.; Salas-Solano O. Native Intact Mass Determination of Antibodies Conjugated with Monomethyl Auristatin E and F at Interchain Cysteine Residues. Anal. Chem. 2012, 84, 2843–2849. 10.1021/ac203346c. [DOI] [PubMed] [Google Scholar]
- Hengel S. M.; Sanderson R.; Valliere-Douglass J.; Nicholas N.; Leiske C.; Alley S. C. Measurement of in Vivo Drug Load Distribution of Cysteine-Linked Antibody–Drug Conjugates Using Microscale Liquid Chromatography Mass Spectrometry. Anal. Chem. 2014, 86, 3420–3425. 10.1021/ac403860c. [DOI] [PubMed] [Google Scholar]
- Dyachenko A.; Wang G.; Belov M.; Makarov A.; de Jong R. N.; van den Bremer E. T. J.; Parren P. W. H. I.; Heck A. J. R. Tandem Native Mass-Spectrometry on Antibody-Drug Conjugates and Submillion Da Antibody-Antigen Protein Assemblies on an Orbitrap EMR Equipped with a High-Mass Quadrupole Mass Selector. Anal. Chem. 2015, 87, 6095–6102. 10.1021/acs.analchem.5b00788. [DOI] [PubMed] [Google Scholar]
- Atmanene C.; Wagner-Rousset E.; Malissard M.; Chol B.; Robert A.; Corvaïa N.; Dorsselaer A. V.; Beck A.; Sanglier-Cianférani S. Extending Mass Spectrometry Contribution to Therapeutic Monoclonal Antibody Lead Optimization: Characterization of Immune Complexes Using Noncovalent ESI-MS. Anal. Chem. 2009, 81, 6364–6373. 10.1021/ac9007557. [DOI] [PubMed] [Google Scholar]
- Debaene F.; Wagner-Rousset E.; Colas O.; Ayoub D.; Corvaïa N.; Van Dorsselaer A.; Beck A.; Cianférani S. Time Resolved Native Ion-Mobility Mass Spectrometry to Monitor Dynamics of IgG4 Fab Arm Exchange and “Bispecific” Monoclonal Antibody Formation. Anal. Chem. 2013, 85, 9785–9792. 10.1021/ac402237v. [DOI] [PubMed] [Google Scholar]
- Wang G.; de Jong R. N.; van den Bremer E. T. J.; Parren P. W. H. I.; Heck A. J. R. Enhancing Accuracy in Molecular Weight Determination of Highly Heterogeneously Glycosylated Proteins by Native Tandem Mass Spectrometry. Anal. Chem. 2017, 89, 4793–4797. 10.1021/acs.analchem.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; de Jong R. N.; van den Bremer E. T. J.; Beurskens F. J.; Labrijn A. F.; Ugurlar D.; Gros P.; Schuurman J.; Parren P. W. H. I.; Heck A. J. R. Molecular Basis of Assembly and Activation of Complement Component C1 in Complex with Immunoglobulin G1 and Antigen. Mol. Cell 2016, 63, 135–145. 10.1016/j.molcel.2016.05.016. [DOI] [PubMed] [Google Scholar]
- Wörner T. P.; Snijder J.; Bennett A.; Agbandje-McKenna M.; Makarov A. A.; Heck A. J. R. Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nat. Methods 2020, 17, 395–398. 10.1038/s41592-020-0770-7. [DOI] [PubMed] [Google Scholar]
- Eschweiler J. D.; Kerr R.; Rabuck-Gibbons J.; Ruotolo B. T. Sizing Up Protein–Ligand Complexes: The Rise of Structural Mass Spectrometry Approaches in the Pharmaceutical Sciences. Annu. Rev. Anal. Chem. 2017, 10, 25–44. 10.1146/annurev-anchem-061516-045414. [DOI] [PubMed] [Google Scholar]
- Mathur S.; Badertscher M.; Scott M.; Zenobi R. Critical evaluation of mass spectrometric measurement of dissociation constants: accuracy and cross-validation against surface plasmon resonance and circular dichroism for the calmodulin–melittin system. Phys. Chem. Chem. Phys. 2007, 9, 6187–6198. 10.1039/b707946j. [DOI] [PubMed] [Google Scholar]
- Kitova E. N.; El-Hawiet A.; Schnier P. D.; Klassen J. S. Reliable Determinations of Protein–Ligand Interactions by Direct ESI-MS Measurements. Are We There Yet?. J. Am. Soc. Mass Spectrom. 2012, 23, 431–441. 10.1007/s13361-011-0311-9. [DOI] [PubMed] [Google Scholar]
- Daniel J. M.; Friess S. D.; Rajagopalan S.; Wendt S.; Zenobi R. Quantitative determination of noncovalent binding interactions using soft ionization mass spectrometry. Int. J. Mass Spectrom. 2002, 216, 1–27. 10.1016/s1387-3806(02)00585-7. [DOI] [Google Scholar]
- Rose R. J.; Labrijn A. F.; van den Bremer E. T. J.; Loverix S.; Lasters I.; van Berkel P. H. C.; van de Winkel J. G. J.; Schuurman J.; Parren P. W. H. I.; Heck A. J. R.; Schuurman J.; Parren P. W. H. I.; Heck A. J. R. Quantitative Analysis of the Interaction Strength and Dynamics of Human IgG4 Half Molecules by Native Mass Spectrometry. Structure 2011, 19, 1274–1282. 10.1016/j.str.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Belov A. M.; Viner R.; Santos M. R.; Horn D. M.; Bern M.; Karger B. L.; Ivanov A. R. Analysis of Proteins, Protein Complexes, and Organellar Proteomes Using Sheathless Capillary Zone Electrophoresis - Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 2614–2634. 10.1007/s13361-017-1781-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanAernum Z. L.; Busch F.; Jones B. J.; Jia M.; Chen Z.; Boyken S. E.; Sahasrabuddhe A.; Baker D.; Wysocki V. H. Rapid online buffer exchange for screening of proteins, protein complexes and cell lysates by native mass spectrometry. Nat. Protoc. 2020, 15, 1132–1157. 10.1038/s41596-019-0281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer D. Z.; Jarrold M. F. Single-molecule mass spectrometry. Mass Spectrom. Rev. 2017, 36, 715–733. 10.1002/mas.21495. [DOI] [PubMed] [Google Scholar]
- Kafader J. O.; Melani R. D.; Durbin K. R.; Ikwuagwu B.; Early B. P.; Fellers R. T.; Beu S. C.; Zabrouskov V.; Makarov A. A.; Maze J. T.; Shinholt D. L.; Yip P. F.; Tullman-Ercek D.; Senko M. W.; Compton P. D.; Kelleher N. L. Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nat. Methods 2020, 17, 391–394. 10.1038/s41592-020-0764-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young G.; Hundt N.; Cole D.; Fineberg A.; Andrecka J.; Tyler A.; Olerinyova A.; Ansari A.; Marklund E. G.; Collier M. P.; Chandler S. A.; Tkachenko O.; Allen J.; Crispin M.; Billington N.; Takagi Y.; Sellers J. R.; Eichmann C.; Selenko P.; Frey L.; Riek R.; Galpin M. R.; Struwe W. B.; Benesch J. L. P.; Kukura P. Quantitative mass imaging of single biological macromolecules. Science 2018, 360, 423–427. 10.1126/science.aar5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega Arroyo J.; Andrecka J.; Spillane K. M.; Billington N.; Takagi Y.; Sellers J. R.; Kukura P. Label-Free, All-Optical Detection, Imaging, and Tracking of a Single Protein. Nano Lett. 2014, 14, 2065–2070. 10.1021/nl500234t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piliarik M.; Sandoghdar V. Direct optical sensing of single unlabelled proteins and super-resolution imaging of their binding sites. Nat. Commun. 2014, 5, 4495. 10.1038/ncomms5495. [DOI] [PubMed] [Google Scholar]
- Cole D.; Young G.; Weigel A.; Sebesta A.; Kukura P. Label-Free Single-Molecule Imaging with Numerical-Aperture-Shaped Interferometric Scattering Microscopy. ACS Photonics 2017, 4, 211–216. 10.1021/acsphotonics.6b00912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young G.; Kukura P. Interferometric Scattering Microscopy. Annu. Rev. Phys. Chem. 2019, 70, 301–322. 10.1146/annurev-physchem-050317-021247. [DOI] [PubMed] [Google Scholar]
- Lai S.-H.; Tamara S.; Heck A. J. R. Single-particle mass analysis of intact ribosomes by mass photometry and Orbitrap-based charge detection mass spectrometry. iScience 2021, 24, 103211. 10.1016/j.isci.2021.103211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebel M.; Hugall J. T.; van Hulst N. F. Ultrasensitive Label-Free Nanosensing and High-Speed Tracking of Single Proteins. Nano Lett. 2017, 17, 1277–1281. 10.1021/acs.nanolett.6b05040. [DOI] [PubMed] [Google Scholar]
- Soltermann F.; Foley E. D. B.; Pagnoni V.; Galpin M.; Benesch J. L. P.; Kukura P.; Struwe W. B. Quantifying Protein–Protein Interactions by Molecular Counting with Mass Photometry. Angew. Chem., Int. Ed. 2020, 59, 10774–10779. 10.1002/anie.202001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Piszczek G. Measuring the affinity of protein-protein interactions on a single-molecule level by mass photometry. Anal. Biochem. 2020, 592, 113575. 10.1016/j.ab.2020.113575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleeker W. K.; Lammerts van Bueren J. J.; van Ojik H. H.; Gerritsen A. F.; Pluyter M.; Houtkamp M.; Halk E.; Goldstein J.; Schuurman J.; van Dijk M. A.; van de Winkel J. G. J.; Parren P. W. H. I. Dual Mode of Action of a Human Anti-Epidermal Growth Factor Receptor Monoclonal Antibody for Cancer Therapy. J. Immunol. 2004, 173, 4699–4707. 10.4049/jimmunol.173.7.4699. [DOI] [PubMed] [Google Scholar]
- de Jong R. N.; Beurskens F. J.; Verploegen S.; Strumane K.; van Kampen M. D.; Horstman W.; Engelberts P. J.; Oostindie S. C.; Wang G.; Heck A. J. R.; Schuurman J.; Parren P. W. H. I. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344 10.1371/journal.pbio.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-L.; Taylor A. D.; Lu Q.; Hanash S. M.; Im H.; Snyder M.; Hancock W. S. Identification of Potential Glycan Cancer Markers with Sialic Acid Attached to Sialic Acid and Up-regulated Fucosylated Galactose Structures in Epidermal Growth Factor Receptor Secreted from A431 Cell Line. Mol. Cell. Proteomics 2013, 12, 1239–1249. 10.1074/mcp.m112.024554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz A. R.; Boer M. A. d.; Strasser J.; Zwarthoff S. A.; Beurskens F. J.; de Haas C. J. C.; Aerts P. C.; Wang G.; de Jong R. N.; Bagnoli F.; van Strijp J. A. G.; van Kessel K. P. M.; Schuurman J.; Preiner J.; Heck A. J. R.; Rooijakkers S. H. M. Staphylococcal protein A inhibits complement activation by interfering with IgG hexamer formation. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2016772118 10.1073/pnas.2016772118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kampen M. D.; Kuipers-De Wilt L. H. A. M.; van Egmond M. L.; Reinders-Blankert P.; van den Bremer E. T. J.; Wang G.; Heck A. J. R.; Parren P. W. H. I.; Beurskens F. J.; Schuurman J.; de Jong R. N.. Biophysical Characterization and Stability of IgG1 Variants with Different Hexamerization Propensities; Genmab: Utrecht, the Netherlands, 2021, Unpublished work. [Google Scholar]
- Gaboriaud C.; Thielens N. M.; Gregory L. A.; Rossi V.; Fontecilla-Camps J. C.; Arlaud G. J. Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol. 2004, 25, 368–373. 10.1016/j.it.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Mortensen S. A.; Sander B.; Jensen R. K.; Pedersen J. S.; Golas M. M.; Jensenius J. C.; Hansen A. G.; Thiel S.; Andersen G. R. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 986–991. 10.1073/pnas.1616998114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes-Jones N. C.; Gardner B. Reaction between the isolated globular sub-units of the complement component Clq and IgG-complexes. Mol. Immunol. 1979, 16, 697–701. 10.1016/0161-5890(79)90010-5. [DOI] [PubMed] [Google Scholar]
- Feinstein A.; Richardson N.; Taussig M. I. Immunoglobulin flexibility in complement activation. Immunol. Today 1986, 7, 169–174. 10.1016/0167-5699(86)90168-4. [DOI] [PubMed] [Google Scholar]
- Burton D. R. Immunoglobulin G: Functional sites. Mol. Immunol. 1985, 22, 161–206. 10.1016/0161-5890(85)90151-8. [DOI] [PubMed] [Google Scholar]
- Sharp T. H.; Boyle A. L.; Diebolder C. A.; Kros A.; Koster A. J.; Gros P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 11900–11905. 10.1073/pnas.1901841116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.