

Abstract
Recent developments in the SARS-CoV-2 pandemic point to its inevitable transformation into an endemic disease, urging both refinement of diagnostics for emerging variants of concern (VOCs) and design of variant-specific drugs in addition to vaccine adjustments. Exploring the structure and dynamics of the SARS-CoV-2 Spike protein, we argue that the high-mutability characteristic of RNA viruses coupled with the remarkable flexibility and dynamics of viral proteins result in a substantial involvement of allosteric mechanisms. While allosteric effects of mutations should be considered in predictions and diagnostics of new VOCs, allosteric drugs advantageously avoid escape mutations via non-competitive inhibition originating from alternative distal locations. The exhaustive allosteric signaling and probing maps presented herein provide a comprehensive picture of allostery in the spike protein, making it possible to locate potential mutations that could work as new VOC “drivers” and to determine binding patches that may be targeted by newly developed allosteric drugs.
Keywords: allostery, SARS-CoV-2, Spike protein, allosteric drugs, allosteric effects of mutations, mutability, druggability, drug design
Graphical abstract
Tan et al. characterize the dynamics of the SARS-CoV-2 Spike protein and derive allosteric signaling maps for the spike protein, which is of interest for drug development. The analysis of allosteric effects of spike protein mutations allows the prediction of potential drivers in emerging variants of concern.
Introduction
Allostery is a universal property of all proteins and protein machines regardless of their structures, sizes, or functions (Gunasekaran et al., 2004; Mitternacht and Berezovsky, 2011), by which proteins recognize environmental cues in the form of perturbations (Guarnera and Berezovsky, 2019a), such as binding of small ligands (Guarnera and Berezovsky, 2016a), mutations (Guarnera and Berezovsky, 2020; Tee et al., 2019), or post-translational modifications (Berezovsky et al., 2017; Mitternacht and Berezovsky, 2011), and elicit a response at remote locations (Guarnera and Berezovsky, 2019a; Tee et al., 2021). The spike (S) glycoprotein is a large interlocking molecular machine whose ectodomain undergoes extensive conformational rearrangements in order to fuse the viral and cell membranes (Walls et al., 2020) upon attachment of the receptor-binding domain (RBD) to the host receptor (Wrapp et al., 2020). The orchestration and the regulation of these complex processes are facilitated by the modular (Gobeil et al., 2021; Wrapp et al., 2020) and hinged (Turonova et al., 2020) S protein structure, the conformational dynamics of which are regulated through communication between distant subunits, thus hinting at the existence of allosteric communication and signaling between them (Gobeil et al., 2021; Raghuvamsi et al., 2021).
Multiple conformational transitions between states with potentially different epitopes, including immunodominant non-neutralizing ones, further complicated by the possibility of an antigenic drift in the eventually endemic SARS-CoV-2, present challenges for the vaccine design and development of therapeutic approaches against COVID-19 (Cai et al., 2020). The major obstacle in both vaccine and therapeutics development (Gobeil et al., 2021) is the continuous evolution of the RNA virus, with a characteristically high level of emergent mutations (Harvey et al., 2021). While much attention has been placed on the mutations in the RBD, there are ever-increasing data showing that mutations may work indirectly, affecting remote units of the S protein (Grubaugh et al., 2020; Korber et al., 2020; Yurkovetskiy et al., 2020). The “mutability” challenge motivated a high-throughput analysis of the effects of mutations (Li et al., 2020a) with the goal “to predict their effect rather than just to seeing it” (Starr et al., 2020, 2021). Since a number of mutations apparently act indirectly, implicating an allosteric mode of action (Gobeil et al., 2021; Li et al., 2020a; Starr et al., 2020, 2021), our objectives here are to quantify the energetics of the allosteric modulation caused by these mutations and to determine patterns of relevant signaling and locations of potential druggable sites for therapeutic interventions.
In this work, we perform a comprehensive analysis of allosteric signaling that may be involved in the regulation of the S protein activity and its modification upon rapid viral evolution (Harvey et al., 2021). Contrary to previous works discussing circumstantially detected cases of allosteric communication (Gobeil et al., 2021; Raghuvamsi et al., 2021) caused by certain mutation(s), we use our computational framework, which allows us to perform a comprehensive analysis of allosteric signaling on the basis of the structure-based statistical mechanical model of allostery (SBSMMA). The SBSMMA (Guarnera and Berezovsky, 2016b) is an exact model providing the per-residue free energy of allosteric signaling upon ligand/probe binding, mutations, or their combinations, allowing us to build exhaustive allosteric signaling maps (ASMs [Guarnera and Berezovsky, 2019b]) and allosteric probing maps (APMs [Tan et al., 2020]). On the basis of the extensivity of the free energy, we show how high-throughput data from ASMs/APMs can be used, in a generic strategy for predicting new variants of concern (VOCs) driven by the allosterically acting mutations, for determining the rescuing ones (Goodey and Benkovic, 2008; Liu and Nussinov, 2008; Nussinov et al., 2021; Zhang et al., 2020) as well as for finding targets for allosteric drug development that may prevent the emergence of drug resistance in “cocktail” therapeutics (Chi et al., 2020).
Results
Allosteric signaling in the pre-fusion S glycoprotein upon simulated receptor binding
The S glycoprotein from SARS-CoV-2 is a large complex of three interlocking monomers that plays a critical role in viral entry into the host cell by binding to the human ACE2 receptor (Walls et al., 2020; Yan et al., 2020). It consists of the S1 and S2 subunits (Figure 1A), which are involved in the recognition and binding to the host receptors, and the subsequent membrane fusion, respectively (Walls et al., 2020; Wrapp et al., 2020). Binding to host receptors triggers large structural rearrangements in the metastable pre-fusion conformation of the S protein, resulting in the shedding of S1 and, in turn, fusion of the viral envelope with the cell membrane mediated by the S2 core (Li, 2016). Figure 1A shows the S glycoprotein (residues 27–1213) in the open state, in which the RBD of one of the monomers (chain A, orange) is pointed upward to engage a receptor, hereinafter referred to as the “up” conformation. The RBD of chains B and C (green and purple) adopt the “down” conformation inaccessible to receptors. In the closed state, all RBDs adopt the down conformation, capping the top of the S2 subunits (Figure S1). The S1 subunit consists of the RBD and the N-terminal domain (NTD), which are respectively associated with subdomains 1 and 2 (SD1 and SD2) (Figure 1A). SD1 and SD2 together provide an interface between the S1 subunit and the S2 subunit of another monomer. The predominantly α-helical S2 core (Figure 1A) comprises various regions including the fusion peptide (FP), which is inserted into the host-cell membrane after structural rearrangements triggered by receptor binding (Bosch et al., 2003). In this study, we modeled the ectodomain of the pre-fusion S glycoprotein in the open and closed states based primarily on available structures (Walls et al., 2020; Wrapp et al., 2020) obtained from cryo-electron microscopy (cryo-EM). The SBSMMA (Guarnera and Berezovsky, 2016b) allows determining individual residues and sites that undergo positive or negative allosteric modulation as a result of a perturbation, such as mutation(s), binding, glycosylation, or different combinations thereof. A positive allosteric modulation (blue) points to potential conformational changes caused by the work exerted on the residue/site, whereas a negative allosteric modulation (red) reveals a stabilization of modulated residues/sites that dampens their dynamics.
Figure 1.

Allosteric response upon simulated receptor binding
(A) The structure of the spike homotrimer in the open state modeled on a cryo-EM structure (PDB: 6VSB). Chains (A–C) are orange, green, and purple, respectively. Parts considered (Cai et al., 2020; Wrapp et al., 2020): NTD, N-terminal domain; RBD, receptor-binding domain; SD1, subdomain 1; SD2, subdomain 2; RBD hinge, two loops linking RBD and SD1; S1-S2, S1-S2 cleavage site; UH, upstream helix; FP, fusion peptide; FPPR, fusion peptide proximal region; HR1, heptad repeat 1; CH, central helix; BH, beta hairpin; CD, connector domain; HR2, heptad repeat 2.
(B) The allosteric modulation (, kcal/mol) at every residue due to the binding at A.RBM (circled) is depicted using a color-gradient presentation from positive (conformational changes, blue) to negative (stabilization, red) modulation. The complete data on binding to each RBM in the open or closed state are shown in Figures S1B and S1C.
(C) The schematic shows the allosteric communication between RBMs (spheres) of different monomers and from the RBM to the S2 subunit upon ACE2 binding. The bound RBM is indicated by a triangle, and the sign of the modulation in the RBM and S2 is colored in blue, red, or gray for positive, negative, or very weak/absent modulation, respectively.
To explore the allosteric responses initiated by the interactions with and/or binding to a host receptor, we simulated the binding at the receptor-binding motif (RBM, defined in Figure S1A) in the RBD. Figure 1B shows the allosteric modulation of the open S homotrimer upon binding of chain A’s RBM (A.RBM, circled) in the RBD-up conformation. Binding to the A.RBM results in a positive allosteric modulation, initiating a configurational work exerted in several regions of chain A, indicating, thus, their potential conformational changes (Figure 1B): the RBD core, SD1, the loops joining the RBD and SD1 (referred to as RBD hinge), the region linking NTD with SD2, and the lower part of S2, especially in the connector domain (CD). The residues under the strongest allosteric modulation in each of these regions are indicated with the modulation values (Figure 1B, middle structure). The positive configurational work observed upon simulated binding to the A.RBM is largely consistent with the conformational changes revealed in a cryo-EM study, which showed a rigid-body rotation of the bound RBD (up) that shifts its center of mass by approximately 5.5 Å away from the trimer axis, and the movements of NTD, SD1, and SD2 shifting their center of mass by about 1.5–3.0 Å, relative to the unbound S1 subunit in the open state (Benton et al., 2020). In contrast with the domain movement of SD2 revealed in the cryo-EM work, our model suggests that ACE2 binding causes a weak stabilization of A.SD2 (Figure 1B, middle). Specifically, the A.SD1 and A.RBD show the largest positive modulation, indicating that the dissociation of S1 from S2 is likely driven by allosterically induced conformational changes in these regions due to the ACE2-RBM binding at a distance. The large and uniform increase in the configurational work exerted predominantly in the RBD and SD1 compared with elsewhere is further corroborated by the dramatic opening of the spike observed in exascale molecular dynamics simulations (Zimmerman et al., 2021), which is possible only through substantial movements of these two domains.
Shifting our focus to modulation occurring in chain B upon binding at A.RBM, we observed a configurational work exerted at B.RBM, likely causing conformational changes in the B.RBD (Figure 1B, right structure), thus yielding allosteric communication between the distant RBMs of chains A and B (Figure 1B). Noteworthy, the S2 subunit of chain B, packed next to A.SD1 and A.SD2, displays opposite allosteric modulations in its upper and lower portions. The N-terminal end of the heptad repeat 1 (HR1) and FP in the lower portion are positively modulated, whereas the C-terminal end of HR1 and parts of the central helix (CH) and upstream helix (UH) located in the upper portion are negatively modulated, preventing its their conformational changes (Figure 1B, inset). The observation that binding at the A.RBM produces a positive configurational work exerted at the N-terminal end of B.HR1 and a concomitant negative modulation at the other end of B.HR1 (Figure 1B, inset) is in good agreement with previous studies on S protein dynamics (Walls et al., 2017). Specifically, it was shown that the HR1 undergoes a jackknife-like movement in the transition from the pre-fusion to the post-fusion state, in which the N-terminal part of HR1 swings upward and is reoriented to form a continuous α-helix with the adjoining central helix (Cai et al., 2020; Walls et al., 2017). Moreover, the FP, a critical element in the fusion machinery, which is also known to undergo large structural rearrangements, as well as the S2′ cleavage site located immediately upstream, yield positive modulation (Figure 1B, right), albeit slightly weaker compared with the other adjacent regions. The C.RBD (excluding the C.RBM) of chain C, C.SD1, and C.SD2 show negative allosteric modulation, and the C.S2 subunit is chiefly positively modulated (Figure 1B, left structure). The S2 coiled-coil stalk formed by the heptad repeat 2 (HR2) region of all three monomers is positively modulated at both ends of the stalk, i.e., next to the CD and near the transmembrane domain.
Summarizing the above observations, one can conclude that binding at the A.RBM induces an allosteric response in several locations of both S1 and S2 subunits in all chains of the S protein, among which the RBD and the adjacent SD1 in S1 exhibit the largest increase in configurational work (Figure 1B), which is likely to result in conformational changes that lead to the dissociation of the monomeric S1 domain upon binding (Benton et al., 2020). While the down conformation of the RBD is commonly deemed inaccessible for receptor binding, from a practical viewpoint some neutralizing antibodies bind to the RBD in the down as well as in the up conformations (Barnes et al., 2020). Therefore, an investigation of the allosteric signaling emanated by the perturbation of down RBDs may have implications for antiviral antibody design (Samsudin et al., 2020), prompting us to simulate binding to the RBMs of both up and down RBDs in the open (1up-2down) and closed (3down) states (Figures S1B and S1C). The modulation modes of allosteric signaling to the other RBMs and the S2 subunit in different scenarios are summarized in Figure 1C. In the open state, binding to A.RBM in the up A.RBD causes positive modulation in B.RBM and the lower part of S2 and negative modulation in the upper part of S2, whereas C.RBM is unaffected, as described above (Figure 1B). Perturbing B.RBM in the down B.RBD leads only to negative modulation in A.RBM and C.RBM. Binding at C.RBM of down C.RBD stabilizes the A.RBM, and, interestingly, leads to extensive allosteric modulation in the S2 subunit (Figures 1C and S1B), similar to that caused by the bound A.RBM. Despite both B.RBD and C.RBD adopting the down conformation, the differences in the signaling from their binding motifs could be attributed to the arrangement of the monomers in the open S structure: in this case, the binding motif in chain B is oriented toward the down C.RBD, whereas the binding motif in chain C faces the up A.RBD. The latter suggests that the allosteric signaling originating from a bound RBM can be affected by the conformation (up/down) and interaction with the RBD in other monomers. In the closed state, in which all three RBDs adopt the down conformation, simulated binding to the RBM of each of the monomers is generally unable to induce allosteric responses in the rest of the S homotrimer (Figure 1C, right structure; Figure S1C).
Allosteric communication between important structural units of the S glycoprotein
In order to obtain details of allosteric communication in the S protein, we derived the ASMs (Guarnera and Berezovsky, 2019b) of the open and closed states of the S glycoprotein, which provide an exhaustive picture of signaling from every protein residue to the rest of the structure regardless of the nature of the original amino acid. In this work, we use ASMs with the allosteric modulation range obtained by modeling each residue’s perturbation in the form of replacement from the smallest (Ala/Gly-like) to the bulkiest (Trp/Phe-like) amino acids, mimicking the residue stabilization as a result of a mutation or of an effector binding (see also STAR Methods for the formal definition of the allosteric modulation range and for a full description of the ASM). The ASMs for both open and closed S structures and the pairwise distance map are shown in Figure S2, and complete, downloadable ASM data are accessible through the AlloMAPS database (Tan et al. 2019) (links to the complete data: http://allomaps.bii.a-star.edu.sg/protView/sarscov2_spike_open, http://allomaps.bii.a-star.edu.sg/protView/sarscov2_spike_closed).
The allosteric communication between functionally important regions of the open and closed forms of the S protein is shown in Figures 2A and S2, respectively. Figure 2B highlights the signaling between the perturbed FP, the fusion peptide proximal region (FPPR [(Cai et al., 2020]), and HR1 in the S2 subunit and the responding RBD, RBM, RBD hinge, and SD1 units in the S1 subunit of the same monomer in the open S structure. The intra-monomer allosteric modulation linking residues in these two sets of regions in S1 and S2 are predominantly positive in both open and closed states (Figures 2B, 2C and S2B). Although the mode and the magnitude of modulations in chains B and C are similar to each other, perturbations in FP, FPPR and HR1 in chain A produce a positive configurational work (Figure 2B, top left) and, potentially, conformational changes in RBD, RBM, RBD hinge, and SD1, and, conversely, perturbations in the latter set of regions lead to prevention of structural changes in the FP, FPPR, and HR1 (Figure 2B, top right). In another example, perturbations in the HR2 of a monomer generally result in negative modulation in RBD, RBM, RBD hinge and SD1 in all monomers of both open and closed forms (here and below marked by bases in illustrations of the S structure) but in positive modulation (marked by arrows) in most of the regions in the S2 subunit such as HR1, BH, and CD (Figures 2A, 2D, and S2C). A notable difference between the signaling patterns observed in the open and closed states is in the modulation of the long S2 stalk (residues 1163–1202, ∼60 Å long): the HR2 regions forming the coiled coil are negatively and positively modulated in the open and closed states, respectively (Figure S2C). In addition to the patterns of signaling that are largely similar across different monomers and conformations (open/closed), we also observed allosteric signals specific only to a certain monomer(s) and conformation. For instance, in the open state, allosteric signaling originating from the S1-S2 cleavage site of chain C (C.S1-S2) promotes conformational changes in all C.S1 regions except C.SD2 while simultaneously precluding conformational changes in the entire C.S2 subunit (Figures 2A, 2E (left structure), S2D). Stabilization of the C.RBM, on the other hand, causes negative modulation in the RBD, RBM, RBD hinge, and SD1 and positive modulation in S2 in all monomers (Figures 2E, right structure, and S2D). In these two cases, the allosteric responses are observed in regions of chain C induced by signals emanating from S1-S2 and RBM in chain C of the open spike structure, whereas perturbations of these regions in other chains do not result in the same pattern of signaling in them.
Figure 2.

Allosteric communication between structural units
(A) ASM containing the average modulation ranges (in kcal/mol) between S protein regions in the open state (the ASM for the closed S protein is shown in Figure S2B).
(B) Parts of the complete ASM in (A) highlighting the intra-monomer communication in chains A–C between two clusters of regions: (i) FP/FPPR/HR1 and (ii) RBD/RBM/RBD hinge/SD1.
(C) Allosteric communication between the two clusters.
(D and E) Example of intra- and inter-monomer signaling observed in (A). The corresponding parts of the ASM are shown in Figures S2C and S2D.
Allosteric signaling to the A.RBM
The comprehensive description of the signaling in the ASMs allows us to explore the locations that may elicit a desired modulation at a site/region of functional importance, hence providing a starting point for allosterically regulated targeting of functional sites. Since chain A undergoes the most significant conformational changes, we focused here on the allosteric communication to A.RBM from distant regions in the open (Figure 3A) and closed (Figure 3C) conformations (see also Figure S3 for the complete data on signaling to A.RBM). The RBM undergoes different modes of modulation: perturbation of closely located C.NTD and B.SD1 causes negative modulation in the A.RBM (Figure 3A), whereas A.FP, A.FPPR, A.HR1, and C.SD1 initiate a positive modulation in A.RBM. Comparison of the responses in the open and closed structures reveals common features: negative modulation (marked by bases) from locations in S1, including C.NTD, C.RBM, and other neighboring regions, and HR2 in the stalk, and positive modulation from A.FP and C.S1-S2 (marked by triangles in Figures 3A and 3C). At the same time, stabilization of B.FPPR, located below A.RBD and A.SD1, leads to a positive modulation in A.RBM in the open state but a negative modulation in the closed state. Negative modulation in A.RBM (Figure 3C) and the down A.RBD (Figure S2B) in the closed state indicates a stabilized down conformation upon introduction of structural order in the B.FPPR, in agreement with the observation that the disordered-to-ordered transition of FPPR facilitates the distal RBD in adopting the down conformation (Cai et al., 2020). In the open state, stabilizing the B.FPPR loop causes positive configurational work in A.RBM (Figure 3A) and the up A.RBD (Figures 2A and 2B), likely leading to conformational changes that shift the RBD from the up to the down form.
Figure 3.

Allosteric signaling to the RBM
(A) The average modulation ranges at A.RBM residues upon a single mutation in different regions (except the A.RBM itself and A.RBD) in the open state.
(B) Illustration of the effects of signaling from individual residues along with their average modulation ranges in the A.RBM residues in the open state.
(C) Same as (A) for closed state.
(D) Same as (B) for closed state. The complete data are available in Figure S3. The triangles mark perturbed regions that cause the same modulation mode in both states. The illustration at the center summarizes the signaling targeting A.RBM from various locations.
Consideration of signaling at the single-residue level shows that stabilizing perturbations at residues 403, 455, 495, and 501 (mutations occur at these sites in VOCs) in C.RBM prevent conformational changes of A.RBM in the open state (up RBD; Figure 3B). Perturbations of residues 828, 829, and 831 in A.FP induce a positive modulation at about 0.5 kcal/mol, while perturbing residues 844 and 847 in B.FPPR and residues 681 and 682 in C.S1-S2 results in even stronger (∼2-fold) positive modulation. In contrast, residues 838 and 840 in B.FPPR, 1135 and 1140 in A.CD, and, 1170 and 1175 in C.HR2 can elicit strong negative modulation to A.RBM in the closed state upon stabilizing perturbations (Figure 3D).
Glycosylation as a source of allosteric signaling
Viral glycosylation is known to play important roles ranging from protein stability and folding to immune evasion, host tropism, and receptor binding (Vigerust and Shepherd, 2007), acting both orthosterically and giving rise to changes in the structure and/or dynamics at distal protein sites via allosteric signaling (Nussinov et al., 2012). Our analysis shows that intra- and inter-monomer signaling caused by glycosylation varies between the open and closed states as well as between corresponding positions in different monomers in some cases (Figures 4 and S4). For example, residues B.74 and A.234 (Figure 4A, first row) in NTD generally cause positive (arrows) modulation in S1 and negative (bases) modulation in S2 upon glycosylation. The N234 glycan is involved in stabilizing RBD in the up conformation (Casalino et al., 2020), while N74 glycan is located near the NTD antigenic supersite potentially affecting the antibody binding (Cerutti et al., 2021; McCallum et al., 2021). Glycosylated residues C.61 and B.61 (Figure 4A, middle row) result in a positive modulation in both S2 cores (stronger in the closed state), whereas the S1 domains are stabilized. Glycosylation of position 1158 in any of the monomers consistently leads to strong allosteric modulation in the open and closed S glycoprotein (Figure 4B) chiefly similar in all sites (Figure 4A, bottom row) except the HR2 stalk, where negative and positive modulations in the open and closed states, respectively, were observed. Several positions, mostly located in the lower part of the S2 bulk, such as residues 709, 717, 801, 1074, 1098, and 1134 (Figure S4), cause weak modulation to all protein sites upon glycosylation.
Figure 4.

Allosteric signaling from glycosylated positions
(A) Center: glycosylated residues are depicted as spheres on the open spike homotrimer. Sides: examples of allosteric modulation originating from different glycosylation sites. The average modulation range () of residues within every responding site/region is provided in full in Figure S4.
(B) The glycosylated positions that can cause strong modulation (, kcal/mol) in multiple sites/regions (with the average in all sites/regions above 0.2 kcal/mol).
Allosteric effects of mutations: from an analysis of known mutations to the prediction of potential new VOCs
The high mutability and rapid evolution characteristics of RNA viruses (Steinhauer and Holland, 1987) prompt a special interest in investigating effects of mutations in the S protein. Only one out of six amino acid substitutions in the spike of the Alpha variant (B.1.1.7, first reported in the UK) and about one-half of the substitutions of the Beta (B.1.351, South Africa), Gamma (P.1, Brazil), and Delta (B.1.617.2, India) variants occur in the RBD (Gu et al., 2020; Jangra et al., 2021; Rees-Spear et al., 2021; Starr et al., 2020; Wang et al., 2021), thus pointing to a specific ratio between their ortho- and allosteric modes of action and calling for a comprehensive analysis of all mutations. A striking example is the D614G mutation in SD2, which promotes the distant RBD in adopting open conformations (Benton et al., 2021; Korber et al., 2020; Mansbach et al., 2021). Therefore, it is important to investigate the allosteric effects of recurrent mutations in the whole homotrimer and to uncover latent mutations with a potential to impact the spike’s functions allosterically. To this end, we use the concurrent ASMs (see STAR Methods for definition), showing effects of genetic mutations present in all chains A–C and evaluating the modulation range exerted at every residue in both open and closed states. Below, we briefly describe the strongest cases of allosteric modulation of the S homotrimer on the basis of the mutational data obtained from (i) high-frequency mutations in the GISAID database (Elbe and Buckland-Merrett, 2017), (ii) strongly modulating positions identified in the ASMs, and (iii) mutations acquired by the VOCs and the amino acid changes with respect to the bat coronavirus RaTG13 (denoted as Bat)—the closest known relative of SARS-CoV-2 with 98 and 90% sequence identity for the ectodomain and the RBD (Wrobel et al., 2020), respectively.
Analysis of the high-frequency GISAID mutations
High frequencies of some S glycoprotein mutations observed in the ongoing pandemic call for investigating their potential allosteric effects. We identified mutations that occur in more than 5.0% out of a total of 1,261,866 sequences from GISAID (from the update on 19/09/2021) and showed their modulation values in the open state (Figure 5A). These mutations are located throughout the structure and around one-third occur in the RBD. Among 17 high-frequency mutations, 11 of them have been used to define the VOCs analyzed here. Mutations of residues 138, 655, and 1027 (Gamma variant) and 452 (Delta variant) cause very weak modulation, whereas residues 417, 477, 478, 484, 501, 681, 950, and 1176 (all except 477 and 1176 are associated with VOCs) cause relatively stronger modulation in multiple regions. In particular, mutating residues 417, 501, and 681 (marked by star symbols; Figure 5A) causes the strongest modulation throughout the spike ectodomain, as identified by the agnostic analysis described below. The S477N mutation (Hodcroft et al., 2020) at the binding interface with the ACE2 receptor was found to confer resistance to neutralization by multiple monoclonal antibodies (mAbs) (Liu et al., 2021) and to improve binding affinity to ACE2 (Starr et al., 2020). Our results show that mutating residue 477 leads to large negative and positive modulation ranges in the S1 and S2 subunits, respectively (Figure 5A), suggesting that the escape mutation may also prompt structural and/or dynamic changes in the S homotrimer via allosteric signaling. The D614G (96.9%) mutation causes ordering of a segment of a disordered loop (residues 620–640 in SD2) which is, in turn, packed between the NTD and the SD1, and stabilizes the furin-cleaved S1-S2 junction (681–686) near the SD2 region that may prevent premature S1 shedding in the D614G mutants (Zhang et al., 2021). We found that in addition to stabilization of the 620–640 loop and the S1-S2 cleavage site (Figures 5A, 5C; weaker in A.S1-S2 compared with that in chains B and C) caused by simulated stabilizing mutation at residue 614, negative allosteric modulation was observed in various distal regions forming the S2 fusion core of the metastable pre-fusion structure, including the fusion peptide and the adjacent proteolytic site (S2′ site). At the same time, mutating residue 614 causes a positive modulation in all RBDs, especially in the up A.RBD (Figure 5C). The positive configurational work suggests potential conformational changes of the RBDs, which is consistent with experiments showing that the G614 trimers predominantly adopt a range of open conformations with at least one RBD pointed upward, as opposed to the D614 trimer frequently yielding a closed conformation (Benton et al., 2021; Mansbach et al., 2021). The long S2 stalk is also positively modulated, indicating its increased flexibility, which may allow the spike ectodomain to scan the host cell surface more efficiently.
Figure 5.

Allosteric modulation caused by high-frequency mutations and those detected by the agnostic analysis
(A) Left, positions that are frequently substituted by another residue (more than 5.0% out of 1,261,866 sequences). Right, the corresponding rows from the ASM.
(B) Some of the residues identified from the agnostic analysis on the open spike (see Figure S5 for complete data).
(C–F) The modulation ranges resulted from mutating residues 614 (C), 681 (D), 985 (E), and both 681 and 985 (F) in all monomers with the average modulation range in some of the affected regions indicated.
Agnostic analysis
We employed the agnostic analysis for identifying residues that can induce the strongest modulation (top 10% in the distributions for positive and negative modulation ranges; see STAR Methods for details) in the entire ectodomain based on its ASM (Tee et al., 2021). The agnostic analysis reveals residues in multiple sites/regions in the spike that can strongly modulate its dynamics, likely leading, thus, to an extensive allosteric response in the structure when mutated. The complete data for both open and closed forms are available in Figure S5. In the open state, almost all of the strongly modulating residues are located in the RBD and S2 stalk (Figure S5). Noteworthy, mutations identified by the agnostic analysis showed a larger than expected overlap with high-frequency mutations in GISAID and with mutations in VOCs: 3 observed cases, compared with 1.7 and 1.8 expected by chance, respectively (see STAR Methods for details). Residues 417, 501, and 681 identified by the agnostic analysis are also mutated in the Alpha, Beta, Gamma, and Delta VOCs (Figure 6A, marked by stars), indicating that a mutation at these residues can induce a large allosteric response in the open S homotrimer (Figures 5B and S5). For instance, stabilizing amino acid substitutions at residue 681 (Alpha and Delta variants) causes large positive modulation at the distant RBD and SD1, which is likely to result in conformational changes in the regions while strongly stabilizing the S2 subunit (Figure 5D). In another example, mutating residue 985, near residues 986 and 987, which are commonly replaced by prolines (2P mutation) (Kirchdoerfer et al., 2018) via protein engineering to stabilize the pre-fusion spike conformation, causes large negative modulation ranges in the RBDs (Figure 5E) and likely prevents conformational changes in the apex of S1. At the same time, positive modulation can be observed in the S2 subunit, suggesting potential conformational changes despite experiments (Kirchdoerfer et al., 2018) showing that 2P mutations (residues 986 and 987) resulted in a structure that is nearly identical in most regions to the wild- type. The opposite allosteric response, caused by stabilizing mutations at residues 681 and 985, suggests that mutating residue 985 might be able to “rescue” the wild-type properties (Goodey and Benkovic, 2008; Liu and Nussinov, 2008; Nussinov et al., 2021; Zhang et al., 2020) in the presence of the 681 mutation, which is widely acquired (16.2%; Figure 5A) by different variants (Alpha/Delta). We simulated mutations at both residues and found that, indeed, mutating residue 985 was able to counteract the modulation due to the 681 mutation at various locations (Figure 5F), especially at the up A.RBD (circled) crucial for ACE2 binding, where the resultant modulation becomes negligible.
Figure 6.

Allosteric effects of mutations from VOCs, Bat, and experiments
(A) Right: allosteric modulation ranges of positions that are mutated in VOCs and/or RaTG13 bat coronavirus. Some experimentally characterized mutations (Li et al., 2020a; Starr et al., 2021) that are not associated with the VOCs/Bat are also included (see Figure S6 for complete data). A mutational effect is denoted by an increase (↑) or decrease (↓) in the infectivity (I) or sensitivity (S) to monoclonal antibodies and/or convalescent sera. Residues identified in the agnostic analysis are marked with a star. Left: the location of the residues in the open spike.
(B–D) Illustration of modulation ranges caused by mutating residues 484 (B), 501 (C), or both (D).
(E) Examples of the allosteric polymorphism for residues 484 and 501. Residues with similar modulation to 484 (cyan) and 501 (yellow) were short-listed if the root-mean-square deviation of per-residue modulation ranges with respect to those from residues 484/501 were below 0.2 kcal/mol and well separated from residues 484/501 (Cα-Cα distance ≥ 11 Å; see Figure S6B for complete data).
VOCs from the perspective of the evolution from the bat coronavirus RaTG13
Considering the evolution of SARS-CoV-2, we analyzed mutations acquired by the Alpha, Beta, Gamma, and Delta variants of concern and/or substituted in the bat coronavirus RaTG13 (VOCs/Bat), the most interesting of which from the perspective of allosteric signaling are exemplified in Figure 6A (open state; complete data for both open and closed states are available in Figure S6A). It appears that some of the mutated residues in VOCs/Bat can induce allosteric responses in the S ectodomain. For example, stabilizing mutations at residues 417 (Beta/Gamma), 484 (Beta/Gamma/Bat), and 501 (all of VOCs/Bat except Delta) in the RBD cause negative and positive modulations in the S1 and S2 subunits, respectively. In particular, residues 417, 501, and 681 can initiate the strongest modulation (marked by a star symbol; Figure 6A, right) upon their substitution (also found by the agnostic analysis above; Figure S5). Noteworthy, mutations K417N/T, E484K, and N501Y confer resistance to antibodies (decreased sensitivity; ↓S symbols in Figure 6A, right) and/or enhance binding affinity to the ACE2 receptor in experimental studies (Gu et al., 2020; Jangra et al., 2021; Rees-Spear et al., 2021; Starr et al., 2020; Wang et al., 2021). Figures 6B and 6C illustrate the per-residue modulation ranges caused by a mutation at residues 484 or 501, respectively. The large positive modulation ranges in S2, which are also observed in the closed state (Figure S6A), indicate conformational changes induced in the distal S2 subunit, including the FP, S1-S2, and S2′ cleavage site and the membrane-proximal end of the S2 stalk, upon a mutation at residue 484/501, likely impacting the spike’s functions via allosteric signaling. Compared with residue 484, mutating residue 501 causes a more extensive negative modulation range across the RBDs and a slightly stronger positive modulation in most parts of the S2 subunit. A double mutation at positions 484 and 501 largely recapitulates the modulation caused by residue 501 alone (Figure 6D). At the time of writing, the Delta variant (B.1.617.2, originating from India), with acquired mutations at residues 478 and 950 in addition to 681 (also present in Alpha) and 452 (present in the Epsilon variant originating from California [Motozono et al., 2021]) is driving a global surge of cases. Although the effects of T478K and D950N in the Delta variant are still unknown, the data presented in Figure 6A show that substitutions of residues 478 (RBD) and 950 (HR1) cause allosteric modulation in both the S1 and the S2 subunits. Moreover, some positions with amino acid substitutions from RaTG13 (Bat) such as residues 372, 459, 490, 493, and 501 can induce a strong allosteric response upon mutation (Figure 6A). Among these positions, the F490S mutation is harbored by the Lambda variant, a variant of interest mainly circulating in South America, while the rest have yet to be reported in emerging strains.
Allosteric polymorphism, new VOCs, and experimentally characterized mutations
Motivated by the obvious allosteric effects of mutations such as 484 and 501, and their apparent involvement in regulation of the S protein function as described above, we tackle here yet another aspect of the allosteric effects of mutations—allosteric polymorphism (Guarnera and Berezovsky, 2020; Tee et al., 2019). According to the concept of allosteric polymorphism, mutations at different locations in a protein can nonetheless induce similar allosteric modulation in a distal region, analogous but not limited to the so-called “latent drivers” in cancerogenesis (Nussinov and Tsai, 2015). Using the exhaustive ASM, we exemplify here analysis of the allosteric polymorphism by identifying residues that can exert per-residue modulation ranges comparable to those from positions 484 and 501. Figure 6E illustrates several residues that exemplify allosteric polymorphism by initiating similar modulation ranges as residues 484 (cyan) and 501 (yellow). For instance, the widespread mutations at residue 478 (9.9%; Figure 5A) acquired by the more recent Delta variant and residue 477 (5.3%) not associated with any VOC, as well as the rarely-reported mutations at residues 369 and 422 (51 and 20 cases on 19/09/2021) at different locations, all result in allosteric modulation almost identical to that induced by mutating residue 484 (11.1%; Beta/Gamma). Moreover, some of these non-VOC mutations have been shown to affect viral fitness in experiments when mutated in experiments (see Figure S6B for complete data). For example, spike mutations at position 476 were shown to escape antibodies REGN10933 and LY-CoV016 (Starr et al., 2021). The Y453F mutation (cyan) has been associated with outbreaks in mink farms and was shown to confer resistance to the REGN10933 antibody (Starr et al., 2021). Additionally, with comparable modulation to that by residue 501, the effects of mutations at residues 404 and 406–409 (yellow) are not yet well described, except those of E406W (Starr et al., 2021) and Q409E (Li et al., 2020a), which respectively decrease and increase the sensitivity to antibodies. Therefore, while the ratio between the ortho- and allosteric components in the effects of mutations should be a topic of future studies, the distal regulatory effects of mutations discussed above coupled with a wide presence of the allosteric polymorphism delineated by the ASM make it rather clear that there might be a number of protein positions that may become major players in newly emerging VOCs.
Finally, we considered allosteric modulation caused by mutations in relation to their experimentally characterized effects on viral fitness. To this end, we used data from two high-throughput mutagenesis experiments (Li et al., 2020a; Starr et al., 2021), which together identified 60 residues causing changes to the infectivity and/or sensitivity to neutralizing mAbs or convalescent sera. The ASM revealed that some of the substitutions (Figures 6A and S6C) that allow the RBD to escape recognition by antibodies (decreased sensitivity, ↓S) also induced strong allosteric responses in distal locations in the spike homotrimer. For instance, mutations at residues 455, 504, and 406, each responsible for the decreased sensitivity to antibodies REGN10933, LY-CoV016 and REGN10933 + REGN10987 cocktail (Starr et al., 2021), respectively, caused large positive modulation in the S2 subunit. Stabilizing mutations of residues in the RBD-ACE2 interface loop (residues 455–504) generally induced an allosteric response in the spike structure (Figure S6C).
Identification of potential allosteric sites
We used the APM (Tan et al., 2020) to develop a combined APM/ASM protocol (Tee et al., 2021), including (i) reverse perturbation of allosteric communication for detecting site-to-site coupling, (ii) targeted analysis for estimating the strength of an allosteric signal, and (iii) agnostic analysis for complementing the signaling to a required value (see STAR Methods for definitions and details).
Here, we sought to uncover potential allosteric sites targeting different spike regions, using the RBM and RBD hinge in S1, FP, and the adjacent FPPR in S2 and the HR2 region in the S2 stalk as examples. Figure 7 illustrates a sample of 10 potential allosteric sites R1–R10 and the resultant modulation due to ligand binding to each of them (Figure S7 contains APMs of both states). It was found that biliverdin, a heme metabolite, binds to a cleft on one side of the NTD and blocks the conformational rearrangements required for neutralizing antibodies to bind the epitope on the NTD (Rosa et al., 2021). Also, it was found that a polysorbate 80 detergent molecule binds to this cleft (Bangaru et al., 2020). We found that the recruited heme metabolite in the NTD cleft (overlapping with the R1 site) stabilizes its vicinity and the distal B.NTD (likely preventing antibody binding) and the S2 stalk in the closed state (Figure 7), but allosterically promotes conformational changes in the down RBD. The R2 site located between B.RBD and the flanking C.NTD causes even stronger positive modulation at the down B.RBM compared with the nearby R1 site. Hence, binding of natural metabolites to both R1 and R2 sites may also lead to the “opening” of the S1 apex via allostery in addition to the known effect of the R1 site in immune evasion. The modeled R3 site is formed by residues 617–642 in a disordered loop in the close state. We show that binding to the R3 site causes a large negative allosteric modulation in all RBDs, and results in positive configurational work exerted in the entire S2 subunit. Earlier, it was found that a linear epitope (residue 625–642) in the R3 site may elicit neutralizing antibodies with high specificity (Li et al., 2020b), and a potentially druggable hydrophobic pocket underneath the 617–628 loop, overlapping with the R3 site, was also recently discovered (Zuzic et al., 2021), in addition to the allosteric effects of the order-disorder transition at the 620–640 loop in modulating the RBD up-down equilibrium shown in experiments (Zhang et al., 2021). Simulated binding to the R4 site near the junction between the bulk and the stalk of the S2 subunit increases flexibility in the stalk and B.CD while leading to stabilization of the RBD and SD1 of all chains. Ligand binding to the R5 and R6 sites at the coiled-coil region of the HR2 destabilizes the stalk of the closed S glycoprotein and the distant NTD of chains A and B, causing negative modulation from the RBD to SD1 in all monomers.
Figure 7.

Identification of allosteric sites for prospective drug development
Potential allosteric sites (R1–R10, cyan) identified in the open and closed forms of the S protein and effects of signaling (, kcal/mol). Allosteric modulation is in kcal/mol, illustrated by the gradual blue-to-red coloring of structures. The complete list of identified sites is available in the AlloMAPS database (Tan et al., 2019).
In the open state, binding to the R7 site at the hinge of the up A.RBD chiefly increases the work exerted in the A.SD1, as well as weak negative and positive modulations in the S2 bulk and the stalk, respectively. The R8 site consists of residues from A.SD1 and B.NTD. Binding to R8 causes large negative allosteric modulation in the entire B.NTD and slightly weaker modulation in the up RBD, FP, and FPPR in chain A. A linear epitope in A.SD1 (residue 553–564) was discovered to elicit neutralizing antibodies (Li et al., 2020b; Poh et al., 2020). The opposite response can be observed in A.NTD, A.SD1, A.SD2, and B.FPPR. Ligand binding to the R9 site between the NTD and SD2 in chain B negatively modulates the whole B.NTD and the A.RBM, whereas the RBM, FP, FPPR, HR1, and HR2 within the same monomer are positively modulated. We observed different responses upon ligand binding to the HR2 in the open and closed states: binding to the R10 site caused negative modulation in the long S2 stalk in the open-spike form, whereas R5 and R6 caused the opposite response in the closed state; binding to R10 likely leads to conformational changes in most parts of the S2 subunit, unlike R5 and R6, where mixed responses at S2 are observed.
Discussion
The current analysis of the S protein’s allosteric regulation spans all levels of its organization and dynamics from considering major structural units of pre/post-fusion states to communication between functional/regulatory sites and down to individual residues. By first simulating the S protein’s binding to the receptor, we obtained a global picture of the allosteric signaling that facilitates large-scale conformational changes of the protein’s subunits (Figure 1). We found that in the open form the conformational dynamics observed in cryo-EM (Cai et al., 2020) and exascale MD simulations (Zimmerman et al., 2021) are supported by the extensive allosteric signaling between the RBMs of different chains, and RBM and other parts, including FP and the S2 cleavage site, and HR2 in the S2 stalk. At the same time, large-scale allosteric communication is strongly suppressed in the closed state of the S protein. Next, on the basis of a per-residue resolution provided by the ASM, we explored details of the allosteric signaling between critical sites of the S protein (Figure 2), observing a strong communication between the RBMs and, specifically, between elements of RBM, RBD, and RBD hinges, with FP and HR1/HR2 of the S2 subunit. While in some cases communication between the above sites was persistent in all monomers of both the open and the closed states, in others, specific for certain chains in one of the states, we clearly observed an omnipresence and significant signaling between the S1 and S2 parts of the S protein, apparently important for the regulation of its receptor binding and fusion with the cell membrane. Acknowledging a preventive and therapeutic potential of the drug intervention causing RBM conformational changes incompatible with the receptor binding, we specifically analyzed and described in detail perturbations in locations such as RBM (down), S1-S2 cleavage site, FP, FPPR, CD, and HR2 that elicit strong allosteric signals to the RBM in the up conformation (Figure 3). Analysis of allosteric signaling from glycosylated residues revealed several positions in both S1 and S2 that may cause a modulation on RBM, NTD, and a few other units important for function or regulation in both the closed and open states (Figure 4).
The high rate of mutations characteristic of RNA viruses and, as a result, their potential contribution to the emergence of new VOCs, require specific attention to their effects in general and, from the perspective of this work, to its allosteric component. We show here how ASM-based comprehensive analysis combined with available clinical and biophysical data can shed light on the allosteric effects of mutations, allowing us to predict potential drivers of new VOCs (Figures 5 and 6). First, the agnostic analysis identified three strongly modulating positions (residues 417, 501, and 681) that are mutated in the VOCs, frequently mutated based on GISAID data, which is higher than the expected number of overlapping residues between these sets due to chance 0.03 (see STAR Methods for details). Importantly, the agnostic analysis also revealed others causing similarly strong modulation to those mutated positions from the VOCs, providing a list of potential new candidates (Figures 5B and S5). Whereas some mutations in VOCs are located in the RBD and, presumably, work orthosterically, a number of mutations, including recurrent mutations at RBD residues 417, 484, and 501 in several VOCs, could lead to strong allosteric responses in distal functionally important locations in the homotrimer (Figure 6A). Notably, strong allosteric signaling provided by residue 501 explains its role in the Alpha, Beta, and Gamma VOCs, also pointing to other residues that initiate the same signaling as being potential 'drivers' of new emerging VOCs (e.g., residues 403, 455, and 495; Figure 3B) upon mutation and revealing the allosteric polymorphism (see also Figure 6E; Tee et al., 2019).
Using our APM-based (Tan et al., 2020) generic framework for identifying potential allosteric sites (Tee et al., 2021) on the basis of the reverse perturbation (Tee et al., 2018), we predicted a repertoire of candidate sites for modulating remote functional sites/regions in S glycoprotein (Figure 7). We found several binding sites unique in closed and open states, which may serve as targets for ligands and neutralizing antibodies (Samsudin et al., 2020; Cao et al., 2020; Chi et al., 2020; Huo et al., 2020; Wrapp et al., 2020; Zhou et al., 2020). The site’s characteristics may help us to infer the structure of the potential ligand. For example, location of R5, R6, and R10 in the coiled-coil region of the HR2 hints at a possibility of using helical stapled peptides (Kim et al., 2011; Kutchukian et al., 2009) as drug candidates for these sites. Remarkably, whereas the R10 site is found in the open state located in HR2, similar to R5 and R6 detected in the closed state, binding to R10 causes a strong stabilization of the stalk—the opposite effect to that of binding to R5 and R6.
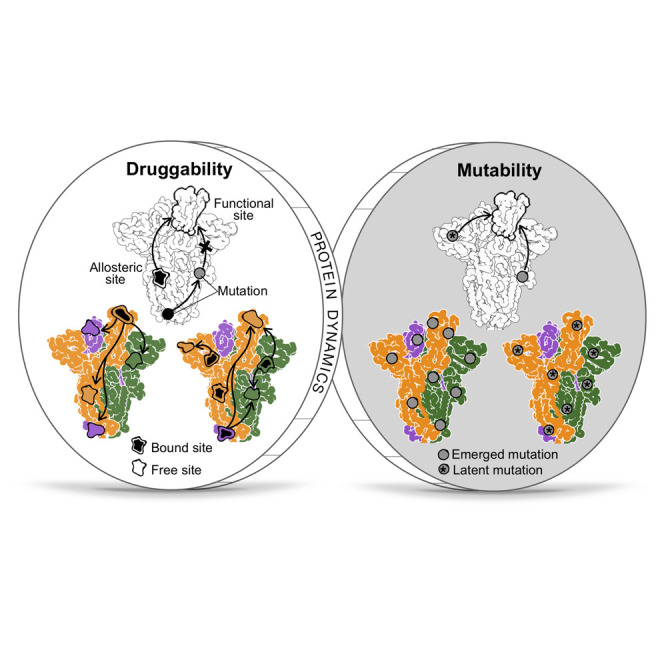
To conclude, although the mutability and the druggability of the S glycoprotein appear to be dichotomous from the perspective of the allosteric regulation of protein functions, both phenomenologies can be viewed as two sides of the same coin, anchored by the cornerstone of protein dynamics (Figure 8 ). On the ‘dark side’, the high mutability of viral proteins is a source of a myriad of mutations, some of them with the potential to modulate the structure and dynamics at another location via allostery, leading to phenotypic differences in the variants such as increased infectivity, virulence, or transmissibility (Figure 8). The allosteric mode of action of some mutations makes their analysis challenging, which is further complicated by the wide presence of allosteric polymorphisms and combined effects of several mutations (Tee et al., 2019) that may result in the emergence of multiple new VOCs. On the ‘bright side’, however, the fundamental property of protein allostery underlined by structural dynamics—remote modulation of protein activity (Berezovsky, 2013; Guarnera and Berezovsky, 2016a)—can be leveraged for identifying druggable regulatory exosites toward modulation of the sites/regions involved in the protein functions with important therapeutic implications (Figure 8). Moreover, allosteric rescue via some emerging mutations (Goodey and Benkovic, 2008; Liu and Nussinov, 2008; Nussinov et al., 2021; Zhang et al., 2020) exemplifies a spontaneous mechanism of neutralization of harmful substitutions. The comprehensive lists of mutations and binding patches provided here on the basis of combined ASM/APM-based analysis of signaling and probe binding can serve as a foundation for future analysis of allosteric signaling and its alteration in the S protein dynamics and for quantification of individual and combined effects of mutations, glycosylation, and binding aimed at the prediction of new VOCs and the design of allosterically acting drugs.
Figure 8.

Two sides of the coin: mutability and druggability as a manifestation of the “dark” and “bright” sides of viral protein dynamics
The dark side: Mutations can lead to new VOCs. The bright side: Druggable allosteric sites can be targeted, and rescue mutations can neutralize the effect of the harmful ones.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| ASMs and APMs of the closed form of S protein | This paper | http://allomaps.bii.a-star.edu.sg/protView/sarscov2_spike_open |
| ASMs and APMs of the open form of S protein | This paper | http://allomaps.bii.a-star.edu.sg/protView/sarscov2_spike_closed |
| Cryo-EM structure of S protein with RBD open | RCSB PDB | PDB: 6VSB |
| Cryo-EM structure of S protein ECD in the closed state | RCSB PDB | PDB: 6XR8 |
| NMR structure of SARS-CoV HR2 domain in the prefusion state | RCSB PDB | PDB: 2FXP |
| NMR structure of transmembrane domain of HIV-1 gp41 | RCSB PDB | PDB: 5JYN |
| Software and algorithms | ||
| AlloSigMA | Tan et al. (2020) | http://allosigma.bii.a-star.edu.sg |
| Modeller version 9.21 | Sali and Blundell (1993) | https://salilab.org/modeller/9.21/release.html |
| Other | ||
| Intel Xeon CPU @ 2.20GHz | N/A | N/A |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Igor N. Berezovsky (igorb@bii.a-star.edu.sg).
Materials availability
This study did not generate new materials.
Experimental model and subject details
Building S protein model
Integrative homology modelling was performed to build complete structural models of the S protein in open and closed states using a combination of cryo-EM and NMR structures as templates (details below).
Collection of characterized mutations
A total of 60 residues known to cause phenotypic changes upon non-synonymous substitutions were obtained from the experimental results by Li et al. (Li et al., 2020a) and Starr et al. (Starr et al., 2021) to investigate their potential allosteric modulation. For the latter study, we used only those amino acid positions with a total escape of at least 1.0, which indicates antigenic escape from antibodies.
Method details
Integrative homology modelling
Modeller version 9.21 (Sali and Blundell, 1993) was used to build two full length models of SARS-CoV-2 S protein: i) open state with one RBD in the up conformation and two RBDs in the down conformation; and ii) closed state with all three RBDs in the down conformation. For the open state model, the cryo-EM structure of S protein ECD in the open state (PDB: 6VSB, (Wrapp et al., 2020)) was used as the main template, with missing loops in the NTD and C-terminus of the ECD modelled using the higher resolution cryo-EM structure of S protein ECD in the closed state (PDB: 6XR8, (Cai et al., 2020)). The latter was also used as the main template for the closed state model. The HR2 domain of the S protein stalk was modelled using the NMR structure of SARS-CoV HR2 domain in the prefusion state (PDB: 2FXP, (Hakansson-McReynolds et al., 2006)), which shares 96% sequence identity. The S protein transmembrane (TM) domain was modelled using the NMR structure of HIV-1 gp41 TM domain (PDB: 5JYN, (Dev et al., 2016)), which shares 27% sequence identity. For each state, ten models were built and the three models with the lowest discreet optimized protein energy score (Eramian et al., 2006) were selected for further stereochemical assessment using Ramachandran analysis (Ramachandran et al., 1963). Finally, the best model was chosen as the one with the lowest number of outlier residues.
Structure-based statistical mechanical model of allostery
The structure-based statistical mechanical model of allostery (SBSMMA) introduced and described in previous work (Guarnera and Berezovsky, 2016b; 2019b) is used here to quantify the energetics of the allosteric communication in the spike glycoprotein at the single-residue level. Briefly, the effect of ligands, mutations, their combinations, and probes (in every case it is a perturbation P of the original state 0) is evaluated at the single-residue resolution as a free energy difference . A single crystal structure is used to construct the Cα harmonic model for the unperturbed and perturbed states of the protein. The energy function associated with this perturbed P state is . The first term is the energy function associated with the unperturbed state, where and are the interatomic distances between C atoms in the generic and reference structures, respectively, and are spring constants. The second term , is an additional perturbation harmonic term with its associated perturbation parameter (Guarnera and Berezovsky, 2019a, b; Tee et al., 2021). Three types of perturbations are accounted for within the model: ligand binding, mutations, and the combination of the two. Ligand binding perturbations are defined via the over-stabilization of the interactions between all pairs of residues in the binding site, ligand-probe perturbations are accounted via strengthening the interactions between residues of a three-residue protein chain segment and nine closest residues with the shortest average distance to these three bound by the probe. Two types of mutations, stabilizing (UP) and destabilizing (DOWN), are considered (Guarnera and Berezovsky, 2019a, b; Tee et al., 2021). The set of orthonormal modes , characterizing the configurational ensemble of the original (perturbed) protein state, is calculated from the Hessian matrix . The allosteric potential measures the effect of a perturbation on a particular residue i and is evaluated as the elastic work exerted on residue i as a result of the change of displacement of its neighborhood caused by the low frequency normal modes . For a change of displacement of the residue due to low frequency normal modes, the allosteric potential reads , where and is a set of Gaussian distributed amplitudes with variance . We consider the contribution of the first 10 low frequency modes in the calculation as they capture the relevant conformation changes (Guarnera and Berezovsky, 2016b; 2019b). Integrating over all possible displacements of neighboring residues provides the per-residue partition function and, then, the free energy associated with the transmitted allosteric signal. The free energy difference between two protein states – perturbed and unperturbed, respectively – quantifies the change of the work being exerted by the residue as a result of the perturbation (P), delivered as allosteric signaling: , where the coefficients and are calculated from the normal modes and , respectively. The free energies are the result of a statistical mechanical calculation and they should be a difference of energy ready to perform work.
The work exerted due to purely allosteric effect is called allosteric modulation and is defined as the deviation of the free energy difference from its mean value over all the residues of the protein (protein chain), containing the residue i: . Positive modulation indicates potential conformational changes of residue i whereas a negative modulation may prevent conformational changes. It should be noted that the magnitude of the modulation should be considered in relation to the thermal fluctuations. While per-residue modulations with the magnitude above k B T should be regarded as a strong manifestation of allosteric communication, combinations of low-value modulations in a homogeneously affected region may also result in a significant allosteric modulation. The allosteric modulation on the site can be evaluated by the averaging of per-residue modulations over the residues belonging to this site: .
The allosteric modulation range is also used for estimating the allosteric signalling from any position m regardless of the original residue in the structure to every residue in the protein and for building allosteric signalling maps (ASMs). It is calculated as a strength of the allosteric signal caused by the substitution from the smallest (Ala/Gly-like) to the bulkiest residues (for example, Phe or Trp), and is used as a generic estimate of the allosteric signaling from any protein position m to position i: . This provides a generic estimate of signaling strength from one position to another, regardless of the original residue at the perturbed position. Depending on the task, two types of the ASMs, concurrent and sequential, can be calculated. In the concurrent ASMs the amino acid changes are simulated simultaneously in corresponding positions in all monomers of the oligomer, allowing to consider effects of genetic mutations. The sequential ASMs, in which amino acid changes are simulated in every monomer one after another, are used as a technique for the analysis of the intra- and inter-monomer signaling from a perturbed position in a certain location of the protein oligomeric structure.
To delineate the allosteric communication in this large oligomer of the S protein, we used a top-down approach by first identifying the intra- and inter-monomer communication between protein sites/regions (defined in Figures 1A and S1A), followed by examining the communication at the single-residue resolution. To explore allosteric signalling between different chains of the S oligomer, we used here sequential ASMs where mutations of all residues are made sequentially in every chain. To gain a picture of the allosteric communication between regions, we averaged the modulation range of signalling originated from every residue in a region to each residue in another region and obtained the matrices of allosteric signalling between the S protein regions for the open and closed states (Figures 2A and S2B). Although post-translational modifications are not explicitly modelled in the framework of SBSMMA, covalent linkage of a bulky glycan to a residue would generally stabilize the perturbed residue and its vicinity. The latter can be qualitatively mimicked by the effects of amino acid substitution from a small to a bulkier residue, which we use here to estimate the allosteric modulation caused by the glycosylation at a position in the structure. The complete list of positions that may originate strong allosteric modulation upon mutation is available in the AlloMAPS database ((Tan et al., 2019), links to the complete data: http://allomaps.bii.a-star.edu.sg/protView/sarscov2_spike_open, http://allomaps.bii.a-star.edu.sg/protView/sarscov2_spike_closed).
The Allosteric Probing Map (APM), in which the allosteric modulation on residue is originated by the binding of a small probe to a three-residue segment of the protein modelled sequentially from residue 1 to residue N-2 for a protein chain of N residues are also derived. The allosteric modulation caused by the probe is also evaluated as a background free effect: . The APMs for both states are shown in Figure S7, and the complete list of the binding sites (in addition to those presented in Figure 7 and discussed in the paper) along with the modulation values and the solvent-accessible surface area (SASA) are available on AlloMAPS (Tan et al., 2019).
Agnostic and targeted analyses of ASM/APM
The agnostic analysis (Tee et al., 2021) of the ASM/APM was carried out to identify perturbations (mutations and probe binding) that can elicit the strongest positive or negative modulation on distal residues (with Cα-Cα distance above 11 Å). We define the average positive regulation due to a perturbation as the average modulation on positively modulated distal residues, and the average negative modulation is defined correspondingly. Perturbations with the strongest 10% in either average positive modulation or average negative modulation are shortlisted. The targeted analysis (Tee et al., 2021) provides a more focused approach, by detecting perturbations that strongly modulate known regions of interest. The modulation of a site/region due to a general perturbation is defined as the mean of the modulation ranges exerted on all residues belonging to the site, . For each targeted site, we ranked perturbations by the allosteric modulation on the given site, and shortlist perturbations in the positive and negative 10% tails for the distribution of .
Reverse perturbation of allosteric communication
We demonstrated previously that simulating ligand binding to a functional site causes large allosteric responses at the residues in the known allosteric site(s), thereby allowing their identification (Tee et al., 2018, 2021). According to the operational definition of allostery (Tee et al., 2018) in the framework of SBSMMA, only a set of distal residues that are beyond a Cα-Cα cutoff distance Å away from the site, denoted as , are considered. For each targeted site, we identified distal residues that are strongly modulated by binding on the site, with a modulation greater than 1.25 times the average magnitude of modulation on distal residues.
Identification of potential allosteric sites
We have previously introduced a generic framework for the inducing and tuning of allosteric signaling via the identification and design of allosteric sites (Tee et al., 2021). The framework comprises independent components including the agnostic and targeted analyses, and the reverse perturbation of allosteric communication. To identify potential allosteric sites that could be a target for drugs, we first considered only the set of residues providing strong allosteric coupling to the target site that are shortlisted in all three approaches. Second, we built patches around residues in this set by including nearby residues with a cutoff distance of 7 Å. The resulting patches form potential allosteric sites that can modulate the dynamics of target sites. The average size of obtained potential allosteric sites is 17 residues, the average relative SASA is 40%, and average absolute SASA is 1160 Å2. The complete data including the binding sites, the resulting modulation and the solvent-accessible surface area calculated by the FreeSASA package (Mitternacht, 2016) are available on AlloMAPS (Tan et al., 2019).
Overlapping mutations in VOCs, GISAID, and agnostic analysis
We computed the overlap between three sets of mutations: (i) 18 mutations in VOCs, (ii) 17 high-frequency mutations in GISAID, and (iii) 119 shortlisted mutations in agnostic analysis of the ASM, limiting this analysis to the residues (numbers 27–1213) where structural information was available. For each set , we defined the probability that a given mutation belongs to a randomized set with the same size : . The number of overlaps expected from random chance between any two sets and is given by the expected overlap in randomized sets of the same sizes: . Similarly, the expected number of random overlaps between three sets , , and is given by .
The number of residues overlapping between the sets from the agnostic analysis (AA set) based on the computed Allosteric Signalling Map, frequently-mutated residues from GISAID (GISAID set), and mutated positions in VOCs (VOC set) are listed below, along with the expected numbers due to chance in parentheses: AA ∩ VOC - 3 (1.8); AA ∩ GISAID - 3 (1.7); VOC ∩ GISAID - 11 (0.3); AA ∩ VOC & GISAID - 3 (0.03).
Quantification and statistical analysis
No statistical analysis was performed.
Additional resources
No additional resources were used.
Acknowledgments
Financial support provided by the Biomedical Research Council via Agency for Science, Technology, and Research (A∗STAR), and partially from grant FY21_CF_HTPO SEED_ID_BII_C211418001 funded by A∗STAR, is greatly appreciated. W.V.T. was also partially supported by the A∗STAR Career Development Fund C210812031.
Authors contributions
Conceptualization, I.N.B.; Methodology, Z.W.T., W.V.T., E.G., and I.N.B.; Investigation, Formal Analysis, Writing – Review & Editing, Z.W.T., W.V.T., Z.W.T., F.S., E.G., P.J.B., and I.N.B.; Software, Z.W.T., W.V.T., and E.G.; Visualization, Z.W.T. and W.V.T.; Writing – Original Draft, W.V.T. and I.N.B.; Supervision, Project Administration, I.N.B.; Funding Acquisition, P.J.B. and I.N.B.
Declaration of interests
The authors declare no competing financial interests.
Published: January 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.12.011.
Supplemental information
Data and code availability
The ASM, APM, and binding sites data have been deposited at the AlloMAPS database ((Tan et al., 2019), see also key resource table above). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Bangaru S., Ozorowski G., Turner H.L., Antanasijevic A., Huang D., Wang X., Torres J.L., Diedrich J.K., Tian J.H., Portnoff A.D., et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science. 2020;370:1089–1094. doi: 10.1126/science.abe1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C.O., Jette C.A., Abernathy M.E., Dam K.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton D.J., Wrobel A.G., Roustan C., Borg A., Xu P., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. The effect of the D614G substitution on the structure of the spike glycoprotein of SARS-CoV-2. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2022586118. e2022586118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton D.J., Wrobel A.G., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature. 2020;588:327–330. doi: 10.1038/s41586-020-2772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezovsky I.N. Thermodynamics of allostery paves a way to allosteric drugs. Biochim. Biophys. Acta. 2013;1834:830–835. doi: 10.1016/j.bbapap.2013.01.024. [DOI] [PubMed] [Google Scholar]
- Berezovsky I.N., Guarnera E., Zheng Z., Eisenhaber B., Eisenhaber F. Protein function machinery: from basic structural units to modulation of activity. Curr. Opin. Struct. Biol. 2017;42:67–74. doi: 10.1016/j.sbi.2016.10.021. [DOI] [PubMed] [Google Scholar]
- Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M., Jr., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369:1586–1592. doi: 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Goreshnik I., Coventry B., Case J.B., Miller L., Kozodoy L., Chen R.E., Carter L., Walls A.C., Park Y.J., et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370:426–431. doi: 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L., Gaieb Z., Goldsmith J.A., Hjorth C.K., Dommer A.C., Harbison A.M., Fogarty C.A., Barros E.P., Taylor B.C., McLellan J.S., et al. Beyond shielding: the roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci. 2020;6:1722–1734. doi: 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti G., Guo Y., Zhou T., Gorman J., Lee M., Rapp M., Reddem E.R., Yu J., Bahna F., Bimela J., et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe. 2021;29:819–833.e7. doi: 10.1016/j.chom.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., Zhang Z., Fan P., Dong Y., Yang Y., et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science. 2020;369:650–655. doi: 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev J., Park D., Fu Q., Chen J., Ha H.J., Ghantous F., Herrmann T., Chang W., Liu Z., Frey G., et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science. 2016;353:172–175. doi: 10.1126/science.aaf7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob. Challenges (Hoboken, NJ) 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramian D., Shen M.Y., Devos D., Melo F., Sali A., Marti-Renom M.A. A composite score for predicting errors in protein structure models. Protein Sci. 2006;15:1653–1666. doi: 10.1110/ps.062095806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S.M., Janowska K., McDowell S., Mansouri K., Parks R., Stalls V., Kopp M.F., Manne K., Li D., Wiehe K., et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science. 2021 doi: 10.1101/2021.03.11.435037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodey N.M., Benkovic S.J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- Grubaugh N.D., Hanage W.P., Rasmussen A.L. Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell. 2020;182:794–795. doi: 10.1016/j.cell.2020.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H., Chen Q., Yang G., He L., Fan H., Deng Y.Q., Wang Y., Teng Y., Zhao Z., Cui Y., et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science. 2020;369:1603–1607. doi: 10.1126/science.abc4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnera E., Berezovsky I.N. Allosteric sites: remote control in regulation of protein activity. Curr. Opin. Struct. Biol. 2016;37:1–8. doi: 10.1016/j.sbi.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Guarnera E., Berezovsky I.N. Structure-based statistical mechanical model accounts for the causality and energetics of allosteric communication. PLoS Comput. Biol. 2016;12:e1004678. doi: 10.1371/journal.pcbi.1004678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnera E., Berezovsky I.N. On the perturbation nature of allostery: sites, mutations, and signal modulation. Curr. Opin. Struct. Biol. 2019;56:18–27. doi: 10.1016/j.sbi.2018.10.008. [DOI] [PubMed] [Google Scholar]
- Guarnera E., Berezovsky I.N. Toward comprehensive allosteric control over protein activity. Structure. 2019;27:866–878.e861. doi: 10.1016/j.str.2019.01.014. [DOI] [PubMed] [Google Scholar]
- Guarnera E., Berezovsky I.N. Allosteric drugs and mutations: chances, challenges, and necessity. Curr. Opin. Struct. Biol. 2020;62:149–157. doi: 10.1016/j.sbi.2020.01.010. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K., Ma B., Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- Hakansson-McReynolds S., Jiang S., Rong L., Caffrey M. Solution structure of the severe acute respiratory syndrome-coronavirus heptad repeat 2 domain in the prefusion state. J. Biol. Chem. 2006;281:11965–11971. doi: 10.1074/jbc.M601174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., Consortium C.-G.U., et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodcroft E.B., Zuber M., Nadeau S., Crawford K.H.D., Bloom J.D., Veesler D., Vaughan T.G., Comas I., Candelas F.G., Stadler T., et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv. 2020 doi: 10.1101/2020.10.25.20219063. [DOI] [Google Scholar]
- Huo J., Le Bas A., Ruza R.R., Duyvesteyn H.M.E., Mikolajek H., Malinauskas T., Tan T.K., Rijal P., Dumoux M., Ward P.N., et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020;27:846–854. doi: 10.1038/s41594-020-0469-6. [DOI] [PubMed] [Google Scholar]
- Jangra S., Ye C., Rathnasinghe R., Stadlbauer D., Krammer F., Simon V., Martinez-Sobrido L., García-Sastre A., Schotsaert M. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. The Lancet Microbe. 2021 doi: 10.1016/S2666-5247(21)00068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.W., Grossmann T.N., Verdine G.L. Synthesis of all-hydrocarbon stapled alpha-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011;6:761–771. doi: 10.1038/nprot.2011.324. [DOI] [PubMed] [Google Scholar]
- Kirchdoerfer R.N., Wang N., Pallesen J., Wrapp D., Turner H.L., Cottrell C.A., Corbett K.S., Graham B.S., McLellan J.S., Ward A.B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Scientific Rep. 2018;8:15701. doi: 10.1038/s41598-018-34171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e819. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutchukian P.S., Yang J.S., Verdine G.L., Shakhnovich E.I. All-atom model for stabilization of alpha-helical structure in peptides by hydrocarbon staples. J. Am. Chem. Soc. 2009;131:4622–4627. doi: 10.1021/ja805037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016;3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Wu J., Nie J., Zhang L., Hao H., Liu S., Zhao C., Zhang Q., Liu H., Nie L., et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell. 2020;182:1284–1294.e1289. doi: 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Lai D.Y., Zhang H.N., Jiang H.W., Tian X., Ma M.L., Qi H., Meng Q.F., Guo S.J., Wu Y., et al. Linear epitopes of SARS-CoV-2 spike protein elicit neutralizing antibodies in COVID-19 patients. Cell Mol. Immunol. 2020;17:1095–1097. doi: 10.1038/s41423-020-00523-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Nussinov R. Allosteric effects in the marginally stable von Hippel-Lindau tumor suppressor protein and allostery-based rescue mutant design. Proc. Natl. Acad. Sci. U S A. 2008;105:901–906. doi: 10.1073/pnas.0707401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., VanBlargan L.A., Bloyet L.M., Rothlauf P.W., Chen R.E., Stumpf S., Zhao H., Errico J.M., Theel E.S., Liebeskind M.J., et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29:477–488.e4. doi: 10.1016/j.chom.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansbach R.A., Chakraborty S., Nguyen K., Montefiori D.C., Korber B., Gnanakaran S. The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci. Adv. 2021;7:eabf3671. doi: 10.1126/sciadv.abf3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum M., De Marco A., Lempp F.A., Tortorici M.A., Pinto D., Walls A.C., Beltramello M., Chen A., Liu Z., Zatta F., et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell. 2021;184:2332–2347.e16. doi: 10.1016/j.cell.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitternacht S. FreeSASA: an open source C library for solvent accessible surface area calculations. F1000Res. 2016;5:189. doi: 10.12688/f1000research.7931.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitternacht S., Berezovsky I.N. Coherent conformational degrees of freedom as a structural basis for allosteric communication. PLoS Comput. Biol. 2011;7:e1002301. doi: 10.1371/journal.pcbi.1002301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motozono C., Toyoda M., Zahradnik J., Saito A., Nasser H., Tan T.S., Ngare I., Kimura I., Uriu K., Kosugi Y., et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. 2021;29:1124–1136. doi: 10.1016/j.chom.2021.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R., Tsai C.J. 'Latent drivers' expand the cancer mutational landscape. Curr. Opin. Struct. Biol. 2015;32:25–32. doi: 10.1016/j.sbi.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Nussinov R., Tsai C.J., Xin F., Radivojac P. Allosteric post-translational modification codes. Trends Biochem. Sci. 2012;37:447–455. doi: 10.1016/j.tibs.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Nussinov R., Zhang M., Tsai C.J., Jang H. Phosphorylation and driver mutations in PI3Kalpha and PTEN autoinhibition. Mol. Cancer Res. 2021;19:543–548. doi: 10.1158/1541-7786.MCR-20-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh C.M., Carissimo G., Wang B., Amrun S.N., Lee C.Y., Chee R.S., Fong S.W., Yeo N.K., Lee W.H., Torres-Ruesta A., et al. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020;11:2806. doi: 10.1038/s41467-020-16638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuvamsi P.V., Tulsian N.K., Samsudin F., Qian X., Purushotorman K., Yue G., Kozma M.M., Hwa W.Y., Lescar J., Bond P.J., et al. SARS-CoV-2 S protein:ACE2 interaction reveals novel allosteric targets. Elife. 2021;10:e63646. doi: 10.7554/eLife.63646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran G.N., Ramakrishnan C., Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- Rees-Spear C., Muir L., Griffith S.A., Heaney J., Aldon Y., Snitselaar J.L., Thomas P., Graham C., Seow J., Lee N., et al. The effect of spike mutations on SARS-CoV-2 neutralization. Cell Rep. 2021;34:108890. doi: 10.1016/j.celrep.2021.108890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A., Pye V.E., Graham C., Muir L., Seow J., Ng K.W., Cook N.J., Rees-Spear C., Parker E., Silva Dos Santos M., et al. SARS-CoV-2 can recruit a haem metabolite to evade antibody immunity. Sci. Adv. 2021;7:eabg7607. doi: 10.1126/sciadv.abg7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Samsudin F., Yeo J.Y., Gan S.K., Bond P.J. Not all therapeutic antibody isotypes are equal: the case of IgM versus IgG in Pertuzumab and Trastuzumab. Chem. Sci. 2020;11:2843–2854. doi: 10.1039/c9sc04722k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Addetia A., Hannon W.W., Choudhary M.C., Dingens A.S., Li J.Z., Bloom J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science. 2021;371:850–854. doi: 10.1126/science.abf9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S., Navarro M.J., Bowen J.E., Tortorici M.A., Walls A.C., et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310.e20. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauer D.A., Holland J.J. Rapid evolution of RNA viruses. Annu. Rev. Microbiol. 1987;41:409–433. doi: 10.1146/annurev.mi.41.100187.002205. [DOI] [PubMed] [Google Scholar]
- Tan Z.W., Guarnera E., Tee W.V., Berezovsky I.N. AlloSigMA 2: paving the way to designing allosteric effectors and to exploring allosteric effects of mutations. Nucleic Acids Res. 2020;48:W116–W124. doi: 10.1093/nar/gkaa338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z.W., Tee W.V., Guarnera E., Booth L., Berezovsky I.N. AlloMAPS: allosteric mutation analysis and polymorphism of signaling database. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gky1028. D265–d270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee W.V., Guarnera E., Berezovsky I.N. Reversing allosteric communication: from detecting allosteric sites to inducing and tuning targeted allosteric response. PLoS Comput. Biol. 2018;14:e1006228. doi: 10.1371/journal.pcbi.1006228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee W.V., Guarnera E., Berezovsky I.N. On the allosteric effect of nsSNPs and the emerging importance of allosteric polymorphism. J. Mol. Biol. 2019;431:3933–3942. doi: 10.1016/j.jmb.2019.07.012. [DOI] [PubMed] [Google Scholar]
- Tee W.V., Tan Z.W., Lee K., Guarnera E., Berezovsky I.N. Exploring the allosteric territory of protein function. J. Phys. Chem. B. 2021;125:3763–3780. doi: 10.1021/acs.jpcb.1c00540. [DOI] [PubMed] [Google Scholar]
- Turonova B., Sikora M., Schurmann C., Hagen W.J.H., Welsch S., Blanc F.E.C., von Bulow S., Gecht M., Bagola K., Horner C., et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 2020;370:203–208. doi: 10.1126/science.abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigerust D.J., Shepherd V.L. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 2007;15:211–218. doi: 10.1016/j.tim.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e86. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Tortorici M.A., Snijder J., Xiong X., Bosch B.J., Rey F.A., Veesler D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. U S A. 2017;114:11157–11162. doi: 10.1073/pnas.1708727114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Schmidt F., Weisblum Y., Muecksch F., Barnes C.O., Finkin S., Schaefer-Babajew D., Cipolla M., Gaebler C., Lieberman J.A., et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature. 2021;592:616–622. doi: 10.1038/s41586-021-03324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel A.G., Benton D.J., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020;27:763–767. doi: 10.1038/s41594-020-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkovetskiy L., Wang X., Pascal K.E., Tomkins-Tinch C., Nyalile T.P., Wang Y., Baum A., Diehl W.E., Dauphin A., Carbone C., et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell. 2020;183:739–751. doi: 10.1016/j.cell.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Cai Y., Xiao T., Lu J., Peng H., Sterling S.M., Walsh R.M., Jr., Rits-Volloch S., Zhu H., Woosley A.N., et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science. 2021;372:525–530. doi: 10.1126/science.abf2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Jang H., Nussinov R. PI3K inhibitors: review and new strategies. Chem. Sci. 2020;11:5855–5865. doi: 10.1039/d0sc01676d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D., Duyvesteyn H.M.E., Chen C.P., Huang C.G., Chen T.H., Shih S.R., Lin Y.C., Cheng C.Y., Cheng S.H., Huang Y.C., et al. Structural basis for the neutralization of SARS-CoV-2 by an antibody from a convalescent patient. Nat. Struct. Mol. Biol. 2020;27:950–958. doi: 10.1038/s41594-020-0480-y. [DOI] [PubMed] [Google Scholar]
- Zimmerman M.I., Porter J.R., Ward M.D., Singh S., Vithani N., Meller A., Mallimadugula U.L., Kuhn C.E., Borowsky J.H., Wiewiora R.P., et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021;13:651–659. doi: 10.1038/s41557-021-00707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuzic L., Samsudin F., Shivgan A.T., Raghuvamsi P.V., Marzinek J.K., Boags A., Pedebos C., Tulsian N.K., Warwicker J., MacAry P., et al. Uncovering cryptic pockets in the SARS-CoV-2 spike glycoprotein. bioRxiv. 2021 doi: 10.1101/2021.05.05.442536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ASM, APM, and binding sites data have been deposited at the AlloMAPS database ((Tan et al., 2019), see also key resource table above). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.