

Abstract
Enzymes of the TET family are methylcytosine dioxygenases that undergo frequent mutational or functional inactivation in human cancers. Recurrent loss-of-function mutations in TET proteins are frequent in human Diffuse Large B-Cell Lymphoma (DLBCL). Here we investigate the role of TET proteins in B-cell homeostasis and development of B cell lymphomas with features of DLBCL. We show that deletion of Tet2 and Tet3 genes in mature B cells in mice perturbs B-cell homeostasis and results in spontaneous development of germinal center-derived B cell lymphomas with increased G-quadruplexes and R-loops. At a genome-wide level, G-quadruplexes and R-loops were associated with increased DNA double strand breaks at immunoglobulin switch regions. Deletion of the DNA methyltransferase DNMT1 in TET-deficient B cells prevented expansion of germinal center B cells, diminished the accumulation of G-quadruplexes and R-loops, and delayed B lymphoma development, consistent with the opposing functions of DNMT and TET enzymes in DNA methylation and demethylation. CRISPR-mediated depletion of nucleases and helicases that regulate G-quadruplexes and R-loops decreased the viability of TET-deficient B cells. Our studies suggest a molecular mechanism by which TET loss-of-function might predispose to development of B cell malignancies.
Introduction
The three mammalian TET enzymes (TET1, TET2 and TET3) are Fe(II)), O2 and α-ketoglutarate dependent dioxygenases that sequentially oxidize 5-methylcytosine (5mC) to 5-hydroxymethyl- (5hmC), 5-formyl- (5fC) and 5-carboxyl- (5caC) cytosine1–3. TET enzymes regulate enhancer activity and DNA methylation dynamics during development (including B cell development) 4–8, cell differentiation and cell lineage specification (reviewed in9–12). TET2 gene mutations and/or decreased TET activity have been observed in many hematological malignancies and solid cancers, often through impaired regulation of metabolic enzymes that affect TET activity (reviewed in9,13–16). For instance, TET2 is recurrently mutated in ~10% of Diffuse Large B-cell Lymphoma (DLBCL) 17–19, a heterogeneous malignancy originating in mature B cells undergoing activation and differentiation in germinal centers (GCs). TET2 mutations represent an early driver event in DLBCL6; in mouse models, deletion of Tet2 in hematopoietic lineages disrupted GC B cell homeostasis and promoted development of more aggressive lymphomas when the transcription factor BCL6 was constitutively overexpressed6. 5hmC deposition has been observed at sites of DNA double strand breaks (DSBs) in HeLa cells20, and TET2 is associated with degradation of stalled replication forks in BRCA2-deficient mouse cells21, suggesting that TET proteins regulate genomic integrity.
Two non-canonical DNA structures, R-loops and G-quadruplexes, can act as physical impediments to DNA and RNA polymerases during transcription and DNA replication, and have been linked to replication fork stalling and genome instability (reviewed in22–26). R-loops form, mostly at genomic regions with high GC content, when RNA binds to the transcribed strand of DNA, displacing the non-transcribed DNA strand22,23. G-quadruplexes often form on the displaced G-rich strand when four guanines, one from each of four tracts of two or more guanine bases interspersed with variable numbers of random nucleotides (G>2N1-nG>2N1-nG>2N1-nG>2), form square planar structures known as G-quartets, that are stabilized by Hoogsteen hydrogen bonding23,25,27–29. G-quartets stack above each other and the resulting G-quadruplex structure is further stabilized by monovalent cations24–26. G-quadruplexes and R-loops are commonly associated with gene promoters, 5’ untranslated regions, DNA replication origins, telomeres and other regulatory elements in the mammalian genome22,24–26,30–32. The pathological effects of these structures can be mitigated by the concerted actions of diverse nucleases and helicases, among them RNases H1 and H2 which destroy RNA in RNA:DNA hybrids22,23, and several helicases (e.g. ATRX, FANCD2, and Werner’s (WRN) and Bloom (BLM) syndrome Recq-like DNA helicases) that can bind and resolve G-quadruplexes24–26.
Here we show that profound TET loss-of-function, induced by deletion of the Tet2 and Tet3 genes in mature B cells in mice, is associated with the rapid development of DLBCL-like tumors from GC B cells. Like other malignancies associated with TET loss-of-function in primary mouse cells 33–35, Tet2/3-deficient B cells exhibit increased DNA damage, based on increased staining with phosphorylated H2AX (γH2AX). 5hmC deposition has been observed at sites of DNA double strand breaks (DSBs) in HeLa cells20, and while exploring possible mechanisms for increased γH2AX staining, we noticed a marked accumulation of G-quadruplex (G4) structures and R-loops in expanded TET-deficient B, T and myeloid cells. CRISPR-mediated depletion of RNASEH1 or ATRX, FANCD2 and BLM helicases led to a slight increase in DNA damage and apoptosis in TET-deficient compared to control B cells. Genome-wide mapping and high-throughput genome-wide translocation sequencing (HTGTS) showed a strong correlation of increased G-quadruplex and R-loop structures with increased DNA DSBs in switch regions of immunoglobulin genes in TET-deficient B cells. TET-deficient B cells also showed upregulation of the maintenance DNA methyltransferase DNMT1, as well as a slight but significant increase in DNA methylation at regions associated with G-quadruplexes and R-loops. DNMT1 was recently shown to bind G-quadruplex structures36, and deletion of DNMT1 in TET-deficient B cells was associated with a striking reduction in GC B cells, decreased levels of G-quadruplex and R-loop structures in the surviving B cells, and a marked delay in B lymphoma development. Together, our findings suggest molecular mechanisms through which TET loss-of-function could promote oncogenesis and genome instability in multiple TET-deficient cell types.
Results
TET deficiency in mature B cells causes B cell lymphoma
Tet2 and Tet3 are the major TET paralogs expressed in mature B cells4,5,7. To disrupt Tet2 and Tet3 activity specifically in mature B cells, we generated compound transgenic mice bearing floxed alleles of the Tet2 and Tet3 genes (Tet2fl/fl, Tet3fl/fl, here termed double-floxed (Dfl)), the Cre recombinase under control of the CD19 locus (CD19Cre), and the yellow fluorescent protein (YFP) reporter preceded by the loxP-stop-loxP cassette in the Rosa26 locus (Rosa26-YFPLSL) (Fig. 1a). Whereas CD19Cre and Dfl control mice, and mice with individual deletions of Tet2 or Tet3 (CD19 Tet2 KO and CD19 Tet3 KO mice), survived normally for more than a year of age, Tet2fl/fl, Tet3fl/fl, CD19Cre (CD19 DKO) mice with profound TET deficiency showed spontaneous development of lymphoma with complete penetrance and a median survival age of ~20 weeks (Fig. 1b). The disease was marked by lymphadenopathy and splenomegaly as early as 6 weeks of age (Fig. 1c), with disruption of normal splenic architecture (Extended data Fig. 1a), early expansion of activated CD4 and CD8 T cells (CD44+CD62Llow) as well as T follicular helper cells (CD4+PD1+CXCR5+) and progressive expansion of TET-deficient CD19+YFP+ B cells (Figs. 1d; Extended data Figs. 1b–1k). Transfer of the expanded YFP+ B cells from 8-week-old CD19 DKO (CD45.2+) mice into immunocompetent, CD45.1+ congenic host mice recapitulated the lymphoma, whereas transfer of B cells from Dfl control mice did not result in long-term engraftment (Extended data Figs. 1l–1o).
Figure 1. TET deficiency in mature B cells causes B cell lymphoma.
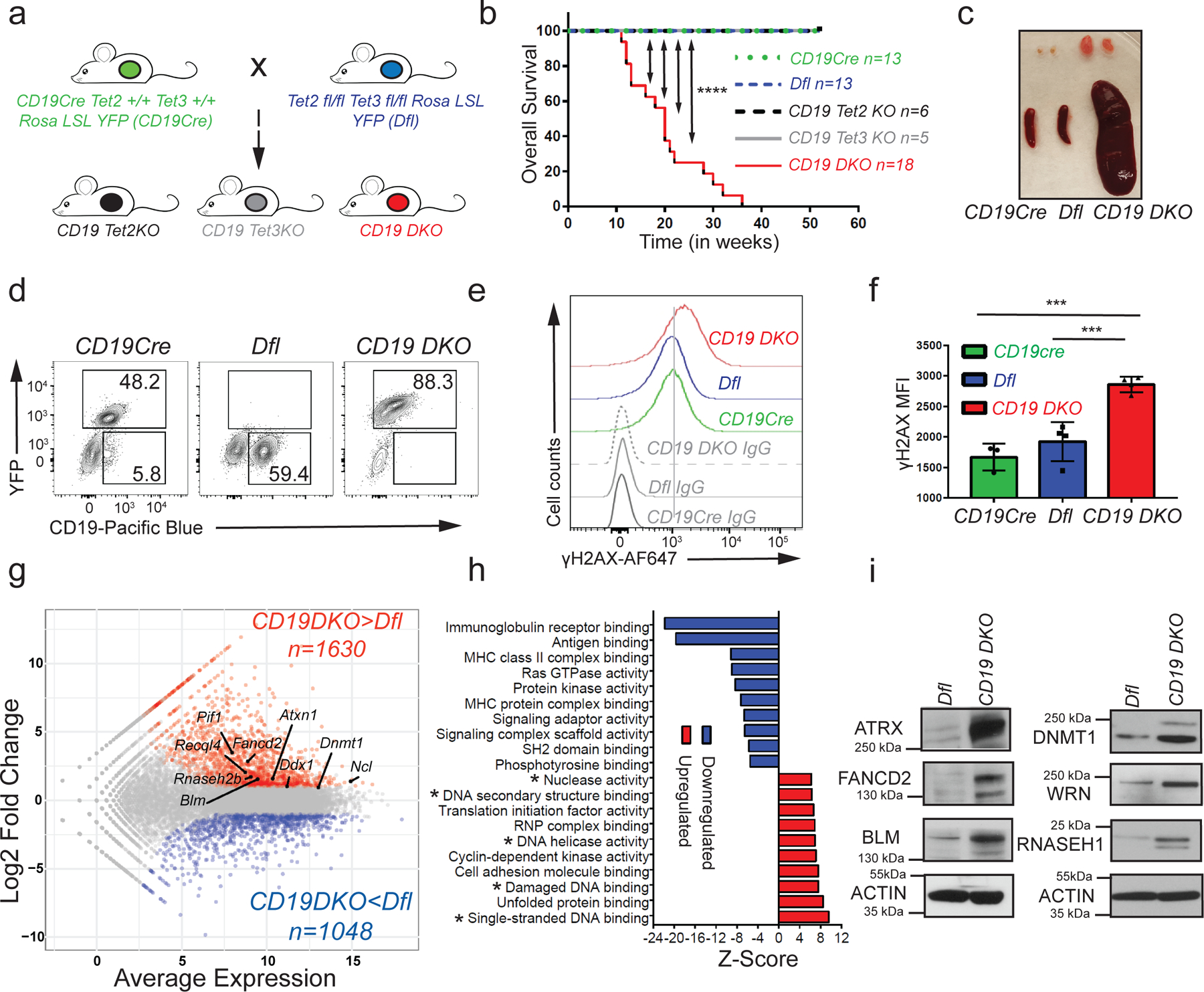
a) Breeding strategy used to generate single Tet2 or Tet3-deficient mice (CD19 Tet2KO or CD19 Tet3KO) and double Tet2, Tet3-deficient mice (CD19 DKO). b) Kaplan-Meir curves displaying overall survival of control Dfl (blue) and CD19 Cre (green) mice, CD19 Tet2KO (black), CD19 Tet3KO (grey) and CD19 DKO (red) mice. Y-axis denotes percent survival and X-axis shows time in weeks. Only the CD19 DKO mice develop B cell lymphoma in the 52-week time period shown. c) Enlarged spleen and lymph nodes in 18 week-old CD19 DKO and control mice. d) Flow cytometry plots of splenocytes from 18-week-old CD19Cre, Dfl and CD19 DKO mice. Numbers show frequencies of B cells among splenocytes. e) Flow cytometry histograms showing γH2AX staining compared to isotype IgG controls in B cells from 8-week-old CD19cre (YFP+), Dfl and CD19 DKO (YFP+) mice. f) Quantification of the median fluorescence intensity (MFI) of γH2AX in CD19Cre (YFP+), Dfl and CD19 DKO (YFP+) B cells from 4 independent experiments. g) MA plot of RNA-sequencing data, displaying changes in gene expression in CD19 DKO B cells compared to Dfl B cells. The highlighted genes represent known G4 and R-loop binders. h) Upregulated (red) and downregulated pathways (blue) in CD19 DKO compared to Dfl B cells. X-axis, Z-score. * Asterisks highlight pathways related to DNA structures and DNA damage. i) Protein levels of RNASE H1 and selected G4-binding helicases and proteins in Dfl and CD19 DKO B cells, assessed by immunoblotting. The data is representative of at least 2 independent experiments. Statistical significance is calculated using the log-rank test (b) or one-way ANOVA (f). Error bars represent mean +/− standard deviation, *** p value ≤0.001, **** p value ≤0.0001.
Transcriptional profiling of CD19 DKO and control Dfl B cells from 8-week-old mice revealed 2,678 differentially expressed genes (DEGs) in CD19 DKO B cells compared with Dfl control B cells. 1630 genes were upregulated and 1048 genes were downregulated (log2 fold change ≥2; FDR ≤0.05; Fig. 1g and Extended data table 2). The transcriptional profile of CD19 DKO B cells resembled that of early germinal center (GC) B cells (Extended data Figs. 2a, 2b). Compared to control Dfl or CD19Cre B cells, a subset of CD19 DKO B cells expressed low IgD and high Ephrin B1 (EFNB1), characteristic of GC B cells (Extended data Fig. 2c). Consistent with their GC origin, the cells showed higher levels of BCL6, activation-induced deaminase (AID; a GC B cell-specific enzyme), and displayed expression of IgG1, IgG2b and IgG3 isotypes, indicating that they were undergoing class switch recombination, albeit without any major changes in germline transcription at the IghM (µ) and IgG1 (γ1) loci (Extended data Figs. 2d–2g). Analysis of variable gene segments of immunoglobulin heavy and kappa (κ) light chains (clonotypes) from Dfl and CD19 DKO B cells showed that the CD19 DKO B cells had undergone oligoclonal expansion compared to Dfl B cells (Extended data Figs. 2h, 2i). Furthermore, CD19 DKO mice showed expansion of GC B cells in Peyer’s patches (Extended data Figs. 2j, 2k). These findings point to a GC origin for expanded B cells from CD19 DKO mice.
Consistent with previous reports in other TET-deficient cell types in mice33–35, CD19 DKO B cells showed increased staining with antibodies to phosphorylated histone H2AX (γH2AX), which marks sites of DNA damage in the genome, compared to B cells isolated from CD19Cre and Dfl control mice (Figs. 1e, 1f). To further characterize the potential mechanisms by which TET deficiency predisposes to increased genomic instability, we scored differentially expressed genes from CD19 DKO B cells (Fig. 1g) for enrichment of annotated molecular pathways. The most downregulated pathways included signaling and scaffolding components of the Ig B cell receptor, the MHC complex and the RAS GTPase complex, which relay signals essential for B cell differentiation, but interestingly, the major upregulated pathways were linked to alterations in secondary DNA structures and DNA damage signaling (Fig. 1h). Compared to control B cells, TET-deficient B cells showed upregulation of mRNAs encoding several helicases and other proteins – FANCD2, BLM, WRN, PIF1, RECQL4, RNASEH2B, nucleolin (Ncl), Ataxin (Atxn1) – that recognize or regulate G-quadruplex (G4) structures and R-loops (Fig. 1g). At the protein level, we confirmed that four known G4-binding helicases ATRX, FANCD2, BLM and WRN25,26, DNMT1 (which also binds G-quadruplex structures36), and RNASEH1 (which like the RNASEH2 complex, specifically degrades the RNA strand of RNA:DNA hybrids in R-loops22,23) showed increased expression in TET-deficient compared to control Dfl B cells (Fig. 1i).
TET deficiency is associated with increased G4s and R-loops
We then tested the effect of TET deficiency on the levels of G-quadruplex structures and R-loops in CD19 DKO B cells. To detect G-quadruplex structures, we used BG4-Ig, a fusion protein of BG4, a single chain variable fragment (scFv) that recognizes G-quadruplex structures37 fused to the mouse IgG1 constant region to improve its valency and sensitivity (Fig. 2a). Treatment of activated primary B cells with a G4-stabilizing ligand, pyridostatin (PDS)38 led to a significant increase in BG4-Ig fluorescence signal, authenticating this approach for G-quadruplex detection (Extended data Figs. 3a, 3b). CD19 DKO B cells from 8 to 10 weeks old mice showed an approximately 1.5- to 2-fold increase in staining with BG4-Ig antibody compared with Dfl or CD19Cre control B cells (Figs. 2b, 2c). We confirmed this result using N-methyl mesoporphyrin IX (NMM), a G4-binding compound that shows a strong increase in fluorescence when it binds G-quadruplex structures39 (Extended data Figs. 3c–e). 38,40CD19 DKO B cells showed a marked increase (~3 fold) in NMM fluorescence signal compared with Dfl and CD19Cre control B cells (Figs. 2d, 2e). Amnis imaging flow cytometry further confirmed increased staining of CD19 DKO B cells with both BG4-Ig and NMM, compared with CD19Cre control B cells (Extended data Figs. 3h–3k).
Figure 2. TET deficiency is associated with increased levels of G-quadruplexes and R-loops.
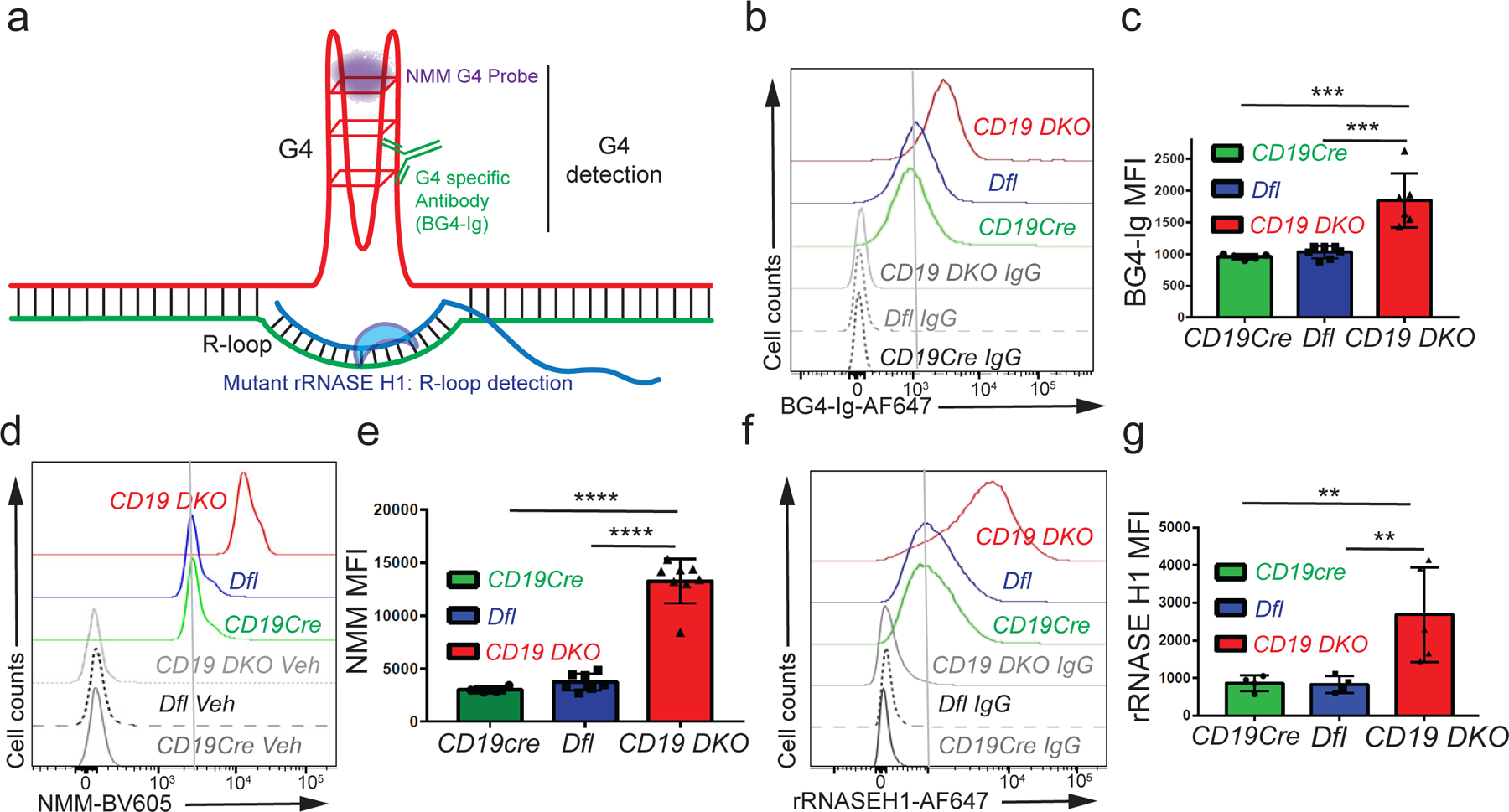
a) Diagrammatic representation of a G-quadruplex with an associated R-loop structure, illustrating the reagents used for detection of G-quadruplexes and R-loops. All experiments were performed in 8 to 10 week-old mice. b) Flow cytometric detection of G-quadruplexes by staining of permeabilized cells with BG4-Ig antibody or isotype IgG controls in B cells from CD19cre (YFP+), Dfl and CD19 DKO (YFP+) mice. c) Quantification of median fluorescence intensity (MFI) of BG4-Ig signal from CD19cre (YFP+), Dfl and CD19 DKO (YFP+) B cells from 4 independent experiments. d) Flow cytometric detection of G-quadruplexes after incubation of cells with NMM or DMSO vehicle controls (Veh) in B cells from CD19Cre (YFP+), Dfl and CD19 DKO (YFP+) mice. e) Quantification of median fluorescence intensity (MFI) of NMM signal from CD19Cre, Dfl and CD19 DKO B cells from 6 independent experiments. f) Flow cytometric detection of R-loops using V5-epitope-tagged recombinant RNASE H1 (rRNASE H1) in B cells from CD19Cre (YFP+), Dfl and CD19 DKO (YFP+) mice. Samples stained with anti-V5 and anti-rabbit secondary antibodies were used as controls (IgG). g) Quantification of median fluorescence intensity (MFI) of R-loops (rRNASE H1) signal from CD19cre (YFP+), Dfl and CD19 DKO (YFP+) B cells from 4 independent experiments. Statistical significance is calculated using one-way ANOVA in c), e) and g). Error bars represent mean +/− standard deviation, ** p value ≤0.01, *** p value≤0.002, **** p value ≤0.0001.
To detect R-loops, we used a recombinant, V5-epitope-tagged version of a catalytically inactive, mutant (D210N) RNASEH1 protein (rRNASEH1)41–43 (Fig. 2a) in preference to the S9.6 antibody against RNA:DNA hybrids44 which gave high background staining. CH12 B cells stained with rRNASEH1 displayed a strong fluorescence signal over background that was significantly diminished upon treatment with catalytically active, RNASE H enzyme (Extended data Figs. 3f, 3g). There was a clear increase (~2-fold) in R-loop levels in CD19 DKO B cells stained with rRNASEH1 compared with Dfl or CD19Cre control B cells (Figs. 2f, 2g; Extended data Figs. 3l, 3m). Together, these studies provide compelling evidence for upregulation of G-quadruplex and R-loop structures in TET-deficient B cells.
The increase in G-quadruplex structures and R-loops occurred early (within 12 days) after deletion of the Tet2 and Tet3 genes in B cells from Cγ1CreTet2fl/flTet3fl/flRosa26-YFPLSL (Cγ1 DKO) mice, in which Cre recombinase expression and TET deletion are induced primarily in GC B cells after immunization. Unlike CD19 DKO mice, unimmunized Cγ1 DKO mice did not show early signs of morbidity and mortality. Immunization with a model antigen, sheep red blood cells (SRBCs), led to a slight increase in spleen cellularity but no apparent change in the frequency of GC B cells in Cγ1 DKO compared with Cγ1Cre or Dfl control mice (Extended data Figs. 4a–4c), but resulted in a significant increase in G4 and R-loop levels in Cγ1 DKO GC B cells compared with Cγ1Cre or Dfl GC B cells (Fig. 3a–3d).
Figure 3. Acute TET deletion is associated with increased levels of G-quadruplexes and R-loops.
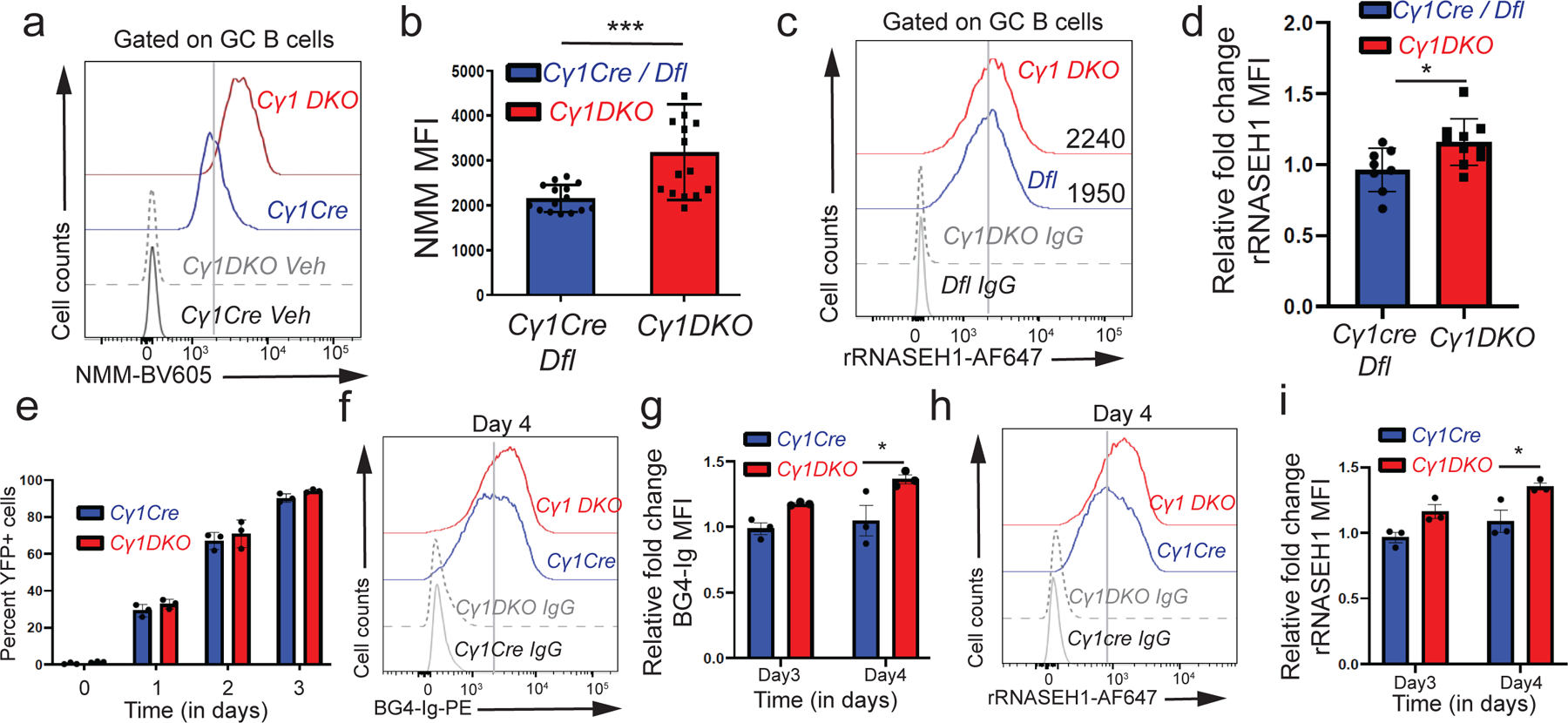
a) Flow cytometric detection of G-quadruplexes with NMM versus DMSO vehicle staining controls (Veh) in GC B cells from Cγ1Cre or Dfl control and Cγ1 DKO mice. b) Quantification of median fluorescence intensity (MFI) of NMM signal from Cγ1Cre or Dfl control and Cγ1 DKO GC B cells from 3 independent experiments. c) Flow cytometric detection of R-loops (rRNASE H1) signal versus IgG staining controls (IgG) in GC B cells from Cγ1cre or Dfl control and Cγ1 DKO mice. d) Fold change in median fluorescence intensity (MFI) signal of R-loops (rRNASE H1) Cγ1 DKO GC B cells relative to Cγ1Cre or Dfl control GC B cells from 2 independent experiments. e) Percent YFP+ B cells from Cγ1Cre control and Cγ1 DKO mice cultured on 40LB media, X-axis denotes time (in days) of culture. f) Flow cytometry detection of G-quadruplexes with BG4-Ig antibody or isotype IgG controls in Cγ1Cre and Cγ1 DKO B cells at day 4 of culture on 40LB feeder cells. g) Quantification of median fluorescence intensity (MFI) of G-quadruplexes (BG4-Ig) signal from Cγ1cre and Cγ1 DKO B cells at day 3 and 4 of culture on 40LB cells from 3 biological replicates. h) Flow cytometry detection of R-loops (rRNASE H1) signal versus anti-rabbit secondary antibody controls (IgG) in Cγ1cre and Cγ1 DKO B cells at day 4 of culture on 40LB cells. i) Quantification of median fluorescence intensity (MFI) of R-loops (rRNASE H1) signal from Cγ1cre and Cγ1 DKO B cells at day 3 and 4 of culture on 40LB cells from 3 biological replicates. Statistical significance is calculated using two-tailed student t-test b) and d) and two-way ANOVA in g) and i). Error bars represent mean +/− standard deviation, * p value ≤0.05, *** p value≤0.002.
We used the 40LB cell culture system, in which B cells are cultured on a mouse fibroblast cell line expressing CD40 ligand and B cell activation factor (BAFF) to induce Cγ1-Cre recombinase activity45, to explore the kinetic relationship between TET loss-of-function and alterations in G-quadruplex and R-loop structures. ~95 to 98% of Cγ1 DKO and Cγ1Cre B cells cultured on 40LB cells underwent genetic recombination (deletion of the LoxP-stop-LoxP cassette), as judged by an increase in YFP expression within 3 days (Fig. 3e). Compared with Cγ1Cre control B cells, Cγ1 DKO B cells displayed a significant increase in G-quadruplex and R-loop levels by 4 days (Figs. 3f–3i). In addition, we deleted Tet genes acutely by tamoxifen treatment of ERT2-Cre Tet2fl/fl Tet3fl/fl Rosa26-YFPLSL (ERT2-Cre DKO) mice; compared to control Dfl mice, these mice show similar B cell development in vivo and similar proliferation of naïve B cells in vitro upon activation with lipopolysaccharide (LPS) and interleukin-4 (IL-4)7. Naïve ERT2-Cre DKO B cells, as well as ERT2-Cre DKO B cells activated in vitro with LPS and IL-4 for 3 days in the presence of tamoxifen (Extended data Fig. 4d), exhibited a significant increase in G-quadruplex levels compared to control Dfl B cells (Extended data Figs. 4e, 4f). Increased levels of G-quadruplexes were also observed in expanded TET-deficient myeloid cells (ERT2-Cre Tet1fl/fl Tet2fl/fl Tet3fl/fl, ERT2creTKO) and iNKT cells (CD4Cre Tet2−/− Tet3fl/fl, CD4Cre DKO) relative to their TET-sufficient counterparts (Extended data Figs. 4g–4j)35,46. In summary, increased levels of G-quadruplexes and R-loops are early events associated with TET deficiency in B cells and other hematopoietic cells.
TET-deficient B cells are sensitive to G4 and R-loop targeting
Since mRNAs encoding G4-resolving helicases and the R-loop resolving enzyme RNase H were upregulated in TET-deficient B cells (Fig. 1g), we asked whether depletion of ATRX, BLM, FANCD2 and RNASEH1 proteins would affect the growth or survival of TET-deficient B cells. Cells were nucleofected with Cas9 ribonucleoprotein complexes (Cas9 RNPs) loaded with the appropriate CRISPR guide RNAs (Extended data Fig. 5a), then cultured in the presence of LPS for 48 hours. This procedure led to efficient depletion of the targeted proteins in CD19 DKO B cells, compared to their levels in cells nucleofected with a control Cas9 RNP directed to the Cd4 locus (Ctrl) (Extended data Fig. 5b). Individual depletion of ATRX, BLM, FANCD2 and RNASEH1 caused a significant increase in apoptosis of CD19 DKO B cells but not Dfl control B cells (measured by flow cytometry for activated caspase 3; Extended data Figs. 5c, 5d), concomitantly with increased DNA damage and increased G-quadruplex levels (assessed by flow cytometry for γH2AX and NMM respectively; Extended data Figs. 5e, 5f). We also used a G4 ligand, pyridostatin (PDS) (Extended data Fig. 5g), to stabilize G-quadruplexes in Dfl and CD19 DKO B cells cultured with LPS with or without PDS for 48 hours. PDS treatment led to an ~2.5-fold increase in apoptosis in CD19 DKO B cells but not in Dfl control B cells (Extended data Figs. 5h, 5k). These studies show that depletion of enzymes that regulate G-quadruplexes and R-loops increases apoptosis in TET-deficient B cells.
Relation of G4s and R-loops to DNA modifications
We mapped the genome-wide distribution of G-quadruplexes and R-loops in control-Dfl and CD19 DKO B cells by chromatin immunoprecipitation followed by sequencing (ChIP-seq) using BG4-Ig47 and MapR42 (a modified CUT&RUN-based method for mapping R-loops which utilizes a recombinant catalytically inactive RNASEH1 protein fused to micrococcal nuclease) respectively. We defined a total of 9722 regions that showed enrichment for G4 and/or R-loop signals over the local background in replicate experiments (Fig. 4a). Consistent with the flow cytometry results, CD19 DKO B cells showed increased G4 and R-loop signals compared with Dfl control B cells (Figs. 4b, 4c). A majority of these (6212 regions; 64%) were present at annotated promoters and 5’UTRs (within +/− 1 kb of transcription start sites (TSS)), and showed enrichment for G-rich DNA sequences predicted to be capable of forming G-quadruplex structures compared to the control regions selected randomly from the genome (Extended data Figs. 6a, 6b). The remaining 3510 regions (36%) were located distant from promoters (median distance ~14 kb from TSS). The G-quadruplex signal was increased at both promoter and non-promoter regions, but only promoter regions showed a significant increase in R-loops signal in CD19 DKO B cells (Extended data Figs. 6c, 6d). The distributions of R-loops and G-quadruplexes at promoter regions were both shifted slightly 5’ of the TSS in CD19 DKO B cells, consistent with the known propensity for G-quadruplex formation on the displaced single DNA strand of R-loops22–26,29. The actual shift in R-loop signal upstream of the TSS might suggest transcriptional pausing at promoter proximal regions, however, the precise reason for the change in the R-loop signal profile in CD19 DKO B cells remains to be determined.
Figure 4. TET-deficient B cells show a genome-wide increase in G-quadruplexes and R-loops and increased translocations to Ig switch regions.
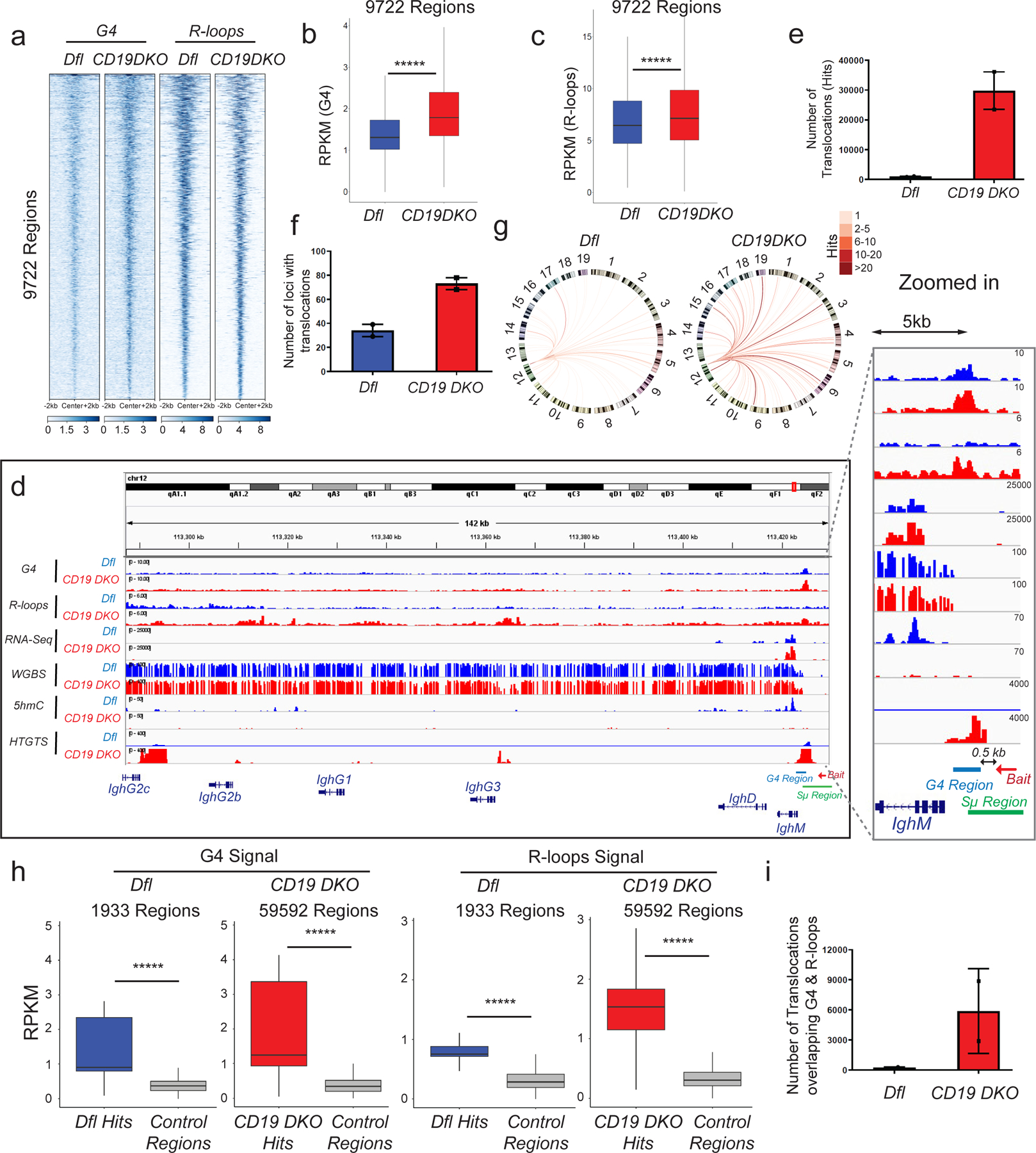
a) Heatmap showing enrichment of G-quadruplex (G4) structures (average of 2 replicates) and R-loops (average of 3 replicates) in Dfl and CD19 DKO B cells. Reads per million (RPM) values in 9722 regions with overlapping high intensity signals for both G-quadruplexes and R-loops are plotted in +/− 2 kb windows from the center of the region. b), c) Box and whisker plots quantifying enrichment (RPKM, reads per kilobase per million) of (b) G-quadruplex structures (2 biological replicates) and (c) R-loops (3 biological replicates) in the 9722 regions from Dfl and CD19 DKO B cells. d) Genome browser view showing data from Dfl (blue tracks) and CD19 DKO (red tracks) B cells. Green and blue bars at the bottom show the location of Sµ and G4 regions. The red arrow indicates the bait sequence located 5’ of the switch µ (Sµ) region used to capture translocations by HTGTS. The zoomed in panel on the right shows the distribution of signals around the IghM region. e)- f) Quantification of (e) total number of translocations (hits), and (f) the number of genomic loci to which translocations occur in CD19 DKO and Dfl B cells from 2 biological replicates. g) Circos plots to visually depict all translocations identified by HTGTS in Dfl and CD19 DKO. Colored lines connect the Sµ bait with the translocation partner regions. Color scale represents the number of translocations identified in a 10 kb window. Translocations from two Dfl and CD19 DKO replicates were concatenated for this representation. h) Box and whisker plots quantifying G-quadruplexes (left panel) and R-loops (right panel) RPKM signal in translocation partner regions of translocations identified in CD19 DKO and Dfl B cells. Regions were extended +/−1 kb from the center of the junctions in the translocation partner regions from 2 biological replicates. i) Quantification of the number of translocations (hits) overlapping G-quadruplexes and R-loops regions in CD19 DKO and Dfl B cells from 2 biological replicates. Statistical significance is calculated using Kruskal-Wallis test and the ad hoc Dunn’s test in b), c) and h). Boxes in box and whisker plots represent median (center) with 25th to 75 th percentile and whiskers represent maxima/minima. Error bars represent mean +/− standard deviation, ***** p value <0.000001.
To relate the distribution of TET-regulated DNA modifications to the distribution of G-quadruplexes and R-loops, we mapped 5hmC by HMCP (5hmC Pull-down), a method similar to hME-Seal48 in which 5hmC is glucosylated and biotinylated prior to DNA precipitation using streptavidin beads. As expected, CD19 DKO B cells showed a strong depletion of 5hmC across the genome (Extended data Fig. 6e). In Dfl B cells, we observed a significant enrichment of 5hmC signal at and near (+/− 1kb) G-quadruplex and R-loop forming regions compared to control random regions in euchromatin (Extended data Fig. 6f). Whole Genome Bisulfite Sequencing (WGBS) analysis identified 6948 differentially-methylated regions (DMRs) genome-wide, of which 5934 (~85%) were hypermethylated and 1014 (~15%) were hypomethylated in CD19 DKO B cells compared with control cells (Extended data Fig. 6g). G4- and R-loop-enriched promoter and non-promoter regions displayed a slight but significant increase in DNA methylation in their flanking regions (+/− 1 to 2 kb) in CD19 DKO compared to Dfl B cells (Extended data Figs. 6c, 6d, 6h, 7a–7c). In contrast, random genomic regions in CD19 DKO B cells were hypomethylated compared with those in control B cells (Extended data Fig. 6i), as we have previously reported for other TET-deficient genomes33,35,46.
In general, more highly expressed genes in both Dfl and CD19 DKO B cells showed greater enrichment for G-quadruplexes and R-loops near the TSS (Extended data Fig. 6j). We also asked whether the presence of G-quadruplex structures and R-loops correlated with differential gene expression. Among all differentially expressed genes (DEGs) in CD19 DKO B cells, a significant proportion (~36%) harbored a G4- and R-loop-enriched region within 1 kb of their TSSs, compared to the proportion (64%) of DEGs without any G4- and R-loop-region and the proportion (22%) of non-DEGs containing a G4- and R-loop enriched region in their promoters (Extended data Fig. 6k, scatter plot and pie chart). However, both up- and down-regulated genes had G-quadruplexes and R-loops at their promoters (Extended data Figs. 6k, 7a–c), consistent with the proposed roles of G4 and R-loop structures in both up- and down-regulation of gene expression22,25.
TET-deficient B cells show genome-wide increase in Ig-translocations
To investigate the potential link between increased G-quadruplexes/R-loop levels and increased DNA breaks in CD19 DKO compared to control Dfl B cells (Figs. 1e, 1f), we performed locus-directed high-throughput genome-wide translocation sequencing (HTGTS49). Using a biotinylated capture oligonucleotide bait located at a well-characterized G4- and R-loop-forming region present within the switch µ region in the IgH locus (Fig. 4d), we observed a striking increase in the absolute numbers of translocations arising from DNA breaks within or up to 500 bases 5’ of this region (Figs. 4d, 4e, 4g; Extended data Fig. 7d), as well as an increase in the number of genomic loci harboring translocations in CD19 DKO compared with Dfl B cells (Figs. 4f, 4g, Extended data Fig. 7d and Extended data table 3). The regions captured as translocation partners of the switch µ region by HTGTS showed significant enrichment of G-quadruplex and R-loop signals compared to control regions in both Dfl and CD19 DKO B cells (Fig. 4h). The absolute number of DNA breaks (translocations) that directly overlap G4- and R-loop-enriched regions were also significantly increased in CD19 DKO B cells compared with Dfl B cells (Fig. 4i). The vast majority (~98%) of DNA breaks were located in the IgH locus associated with IgM as well as other Ig isotypes (Fig. 4d). Compared to control euchromatic regions, a substantial proportion (~85%) of HTGTS hits harbored canonical motifs predicted to form G-quadruplexes (Extended data Fig. 7e) and showed significant enrichment of DNA motifs targeted by the cytidine deaminase AID in B cells (Extended data Fig. 7f). Together, these studies show increased G-quadruplexes and R-loops in CD19 DKO B cells, correlating with an increase in the levels of DNA double-strand breaks at Ig switch regions.
DNMT1 deletion delays oncogenesis in TET-deficient mice
DNMT and TET enzymes have opposing biochemical activities in DNA methylation and demethylation respectively. A recent study showed that binding of DNMT1 to G-quadruplex structures inhibits its catalytic activity36. We confirmed the DNMT1-G-quadruplex interaction (Extended data Fig. 8a, b), then asked whether (given the strong upregulation of DNMT1 in TET-deficient B cells (Fig. 1i)), depleting DNMT1 in TET-deficient B cells affected the pathogenesis of B cell lymphoma. We generated Dnmt1 fl/fl Rosa26-YFPLSL mice expressing Cre recombinase under control of the CD19 locus (CD19Cre Dnmt1 knock out (KO)) and bred them with Tet2fl/fl Tet3fl/fl mice (Triple floxed-Tfl) (Fig. 5a). The CD19 DKO mice generated from these crosses succumbed to B cell lymphoma with a median survival of 20 weeks, whereas CD19 TKO mice (with additional deletion of Dnmt1) showed a substantial increase in survival (median survival 98 weeks) (Fig. 5b). Compared with CD19 DKO mice, Tfl, CD19 Dnmt1 KO and CD19 TKO mice displayed no apparent signs of splenomegaly at 10 weeks of age (Figs. 5c; Extended data Figs. 8c, 8d); a small proportion (4 out of 14 mice) of CD19 TKO mice did develop splenomegaly, albeit with a very long latency (Extended data Fig. 8e).
Figure 5. DNMT1 deletion delays oncogenesis in TET-deficient mice.
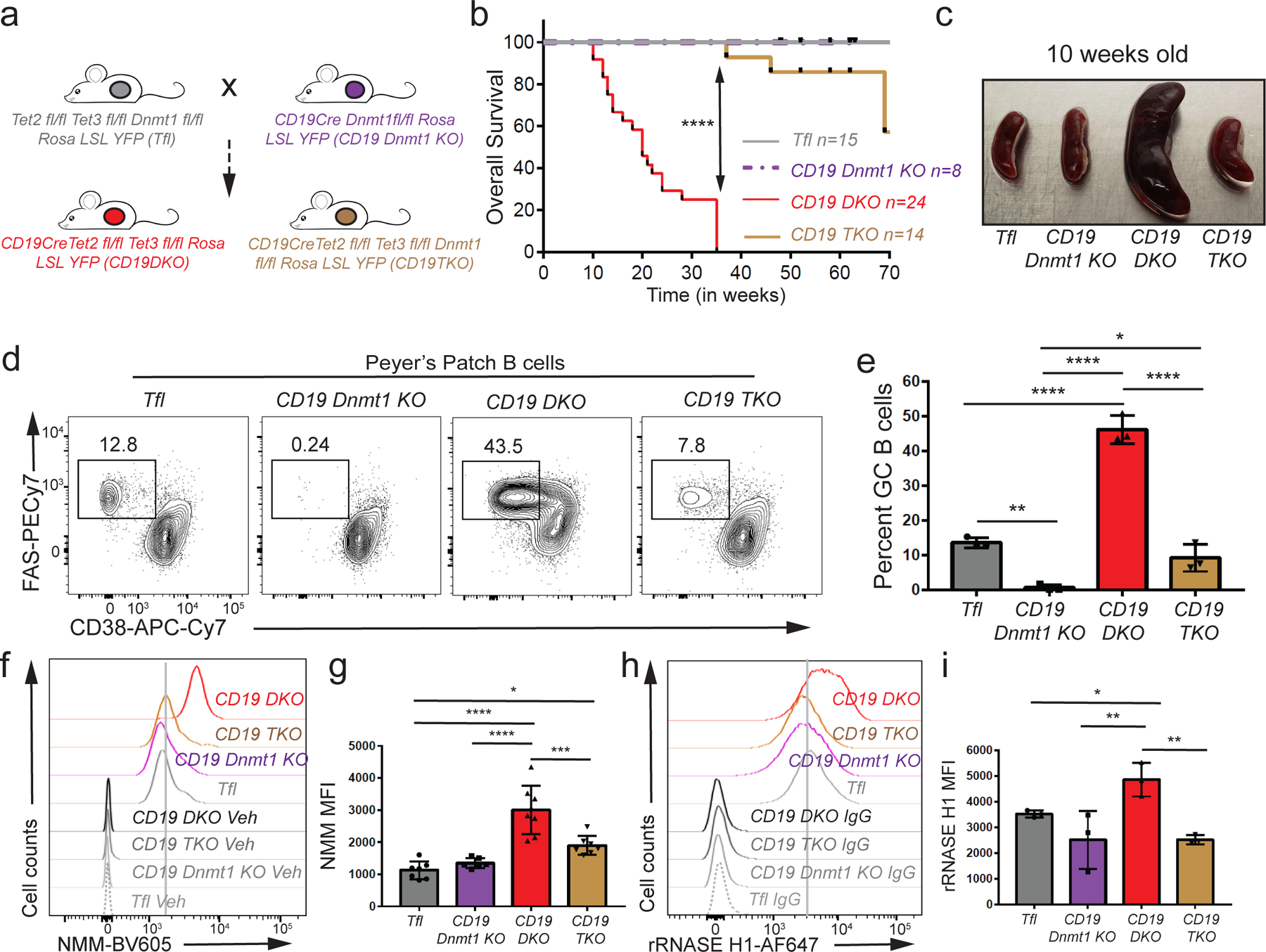
a) Breeding strategy used to generate the triple Dnmt1,Tet2,Tet3 deficient mice(CD19TKO). b) Kaplan-Meir curves displaying the overall survival of Tfl (grey), CD19 Dnmt1 KO (purple), CD19 DKO (red) and CD19 TKO (brown) mice. Y-axis denotes percent survival and X-axis shows time in weeks. c) Spleens from 10 week-old Tfl, CD19 Dnmt1, CD19 DKO and CD19 TKO mice. d) Representative flow cytometry data gated on Peyer’s patch B cells from 10 week-old Tfl, CD19 Dnmt1 KO, CD19 DKO and CD19 TKO mice. Numbers represent frequency of GC B cells, identified as FAS+ (Y-axis) and CD38− (X-axis). e) Quantification of GC B cell frequency in Peyer’s patches of Tfl (YFP−, grey), CD19 Dnmt1 KO (YFP+, purple), CD19 DKO (YFP+, red) and CD19 TKO (YFP+, brown) mice from 3 biological replicates and 2 independent experiments. f) Flow cytometric detection of G-quadruplexes by staining of permeabilized cells with NMM or DMSO vehicle controls (Veh) in B cells from Tfl (YFP−), CD19 Dnmt1 KO (YFP+), CD19 DKO (YFP+) and CD19 TKO (YFP+) mice. g) Quantification of median fluorescence intensity (MFI) of NMM signal from Tfl, CD19 Dnmt1 KO, CD19 DKO and CD19 TKO B cells from 7 biological replicates and 5 independent experiments. h) Flow cytometric detection of R-loops using V5-epitope-tagged recombinant RNASE H1 (rRNASE H1) in B cells from Tfl (YFP−), CD19 Dnmt1 KO (YFP+), CD19 DKO (YFP+) and CD19 TKO (YFP+) mice. Samples stained with anti-V5 and anti-rabbit secondary antibodies were used as controls (IgG). i) Quantification of median fluorescence intensity (MFI) of R-loops (rRNASE H1) signal from Tfl, CD19 Dnmt1KO, CD19 TKO and CD19 DKO B cells from 3 biological replicates and 2 independent experiments. Statistical significance is calculated using one-way ANOVA. Error bars represent mean +/− standard deviation, * p value ≤0.05, ** p value ≤0.01, *** p value ≤0.0005 **** p value <0.0001.
Since GC B cells are the cell-of-origin of the expanded B cells in CD19 DKO mice, we examined the numbers of GC B cells in Peyer’s patches of CD19 DKO versus Dnmt1-Tet2/3 CD19 TKO mice. Deletion of Dnmt1 in B cells completely abrogated GC B cells (Figs. 5d, 5e), similar to previous reports using DNMT1 germline hypomorphs50. CD19 TKO mice displayed GC B cell frequencies considerably lower than the expanded GC B cell population in CD19 DKO mice, but only slightly lower than those in Tfl control mice when gated on YFP+ B cell population (Figs. 5d, 5e). Together, these findings point to a functional interplay between TET and DNMT1 activities in GC B cell development and oncogenic transformation of TET-deficient B cells.
We assessed the levels of G-quadruplexes and R-loops in total splenic B cells and Peyer’s patch GC B cells of 10-week-old mice by flow cytometry. Tet2/3, Dnmt1-deficient CD19 TKO B cells demonstrated a notable decrease in the levels of G4 (using both NMM and BG4-Ig staining) and R-loop (rRNASE H1 staining) structures compared with Tet2/3-deficient CD19 DKO B cells, almost to the levels of control Tfl B cells (Figs. 5f–I; Extended data Figs. 8f, 8g). The decrease in the levels of G4 and R-loop structures in CD19 TKO B cells was also accompanied by decreased levels of γH2AX compared with CD19 DKO B cells (Extended data Fig. 8h). Similar decreases in G-quadruplex and R-loop levels were observed when CD19 TKO GC B cells isolated from Peyer’s patches were compared to CD19 DKO GC B cells (Extended data Figs. 8i–8l). Altogether, these results show that DNMT1 deletion inhibits the development of GC B cells to delay oncogenesis, and is associated with decreased levels of G-quadruplex and R-loop structures (Extended data Fig. 8m).
Discussion
Our studies establish a causal relationship between TET deficiency and the development of mature B cell neoplasms. The fully penetrant progression of B cell lymphomas in CD19 Tet2/3 DKO mice is consistent with the frequent occurrence of TET gene mutations or dysregulation of TET activity in human DLBCL17–19. We show that profound inactivation of TET function, through deletion of both the Tet2 and Tet3 genes, perturbs normal B cell homeostasis, leading to spontaneous expansion of B cells with a GC phenotype. We also document a clear association between TET deficiency, increased G-quadruplexes and R-loops, and increased DNA double strand breaks, particularly at Ig-switch regions in B cells. TET-deficient B cells upregulated mRNAs encoding several proteins that recognize and regulate R-loops and G-quadruplex structures, including RNase H1, Rnaseh2b, DNMT1, Fancd2/FANCD2, ATRX, BLM, WRN, Recql4, and Pif122–26. Genetic disruption of the gene encoding one of the G-quadruplex binders, DNMT1, in CD19 Tet2/3 DKO B cells was associated with decreased levels of the precursor GC B cells, decreased G-quadruplex and R-loop structures and a notable increase in the survival of Tet2/3-deficient mice.
A recent study showed that TET proteins limit the activation of self-reactive B cells in the periphery and serve as critical regulators of B cell tolerance51. While the identity of the antigen(s) driving the expansion of CD19 DKO B cells in our system is not known, it is possible that TET-deficient B cells expand because they are self-reactive. The oligoclonal (rather than polyclonal) nature of the BCRs observed in expanded CD19 DKO B cells implies antigen-driven selection52. This is consistent with the well-documented relationship between the expression of self-reactive B cell receptors and two mature B cell-derived malignancies, chronic lymphocytic leukemia (CLL) and the activated B cell (ABC) subtype of DLBCL, in humans52.
G-quadruplex structures have been implicated in initiation of the frequent IgH-BCL2 translocations in follicular lymphoma53,54. We observed an association between increased G-quadruplex and R-loop structures and DNA breaks at the switch regions (primarily switch μ) of Ig isotypes in Tet2/3-deficient B cells. Due to the lack of robust methods for genome-wide mapping of DNA breaks, we relied on HTGTS, a locus-directed method for DNA break mapping at single base resolution49. Since HTGTS maps DNA breaks relative to DNA junctions formed by break ligation (with possible errors due to indels), it is likely that we have underestimated the total number of DNA breaks. Nevertheless, we observed a striking increase in the absolute numbers of DNA DSBs in CD19 DKO B cells when compared with control B cells. The DNA breaks could arise either from conflicts of G-quadruplex and R-loop structures with transcription or DNA replication machineries, or from specific targeting of AID cytidine deaminase, the B cell mutator, to G4 structures and R-loops. In fact, AID possesses a G4-binding activity that is important for its genome-wide targeting55–57. A recent study reported an association between G4 structures and AID activity at commonly mutated genes in B cell lymphoma56; consistent with these findings, we observed an enrichment of AID hotspot motifs within translocation sites with increased levels of G-quadruplexes and R-loops. The detailed mechanisms through which G-quadruplexes and R-loops recruit AID and/or promote genomic instability in B cells remain to be addressed.
TET-dependent methylation changes do not necessarily play a direct biochemical role in the observed increase of R-loop and G quadruplex structures in Tet2/3 deficient B cells. We based this conclusion on the fact that although we observed a slight but significant link between loss of TET activity and increased DNA methylation in the vicinity of R-loops and G-quadruplex structures, we did not observe any dramatic changes in DNA methylation at the R-loop/G-quadruplex-containing regions (median size ~750 bp) themselves. Thus, further studies are needed to fully understand how TET deficiency in multiple cell types results in increased levels of G-quadruplex and R-loop structures.
DNMT1 has been previously shown to be induced in GC B cells50, where it may be needed to maintain the DNA methylation landscape during rapid GC B cell proliferation. Thus, the apparent upregulation of DNMT1 in TET-deficient B cells is likely due to their prominent GC phenotype compared to WT B cells. Conversely, Dnmt1 deletion results in a dramatic decrease of GC B cells, the likely reason for the delayed oncogenesis observed in triple Tet2/3, Dnmt1-deficient B cells. In fact, the late-onset cancers may reflect delayed outgrowth of cells that escaped deletion of one or both Dnmt1 alleles, since in one experiment (unpublished data), PCR analysis of YFP+ total B cells and GC B cells in Peyer’s patches in much older (7-month-old) mice showed almost complete deletion of Tet2 and Tet3 but only ~50% deletion of Dnmt1. DNMT1 is known to be overexpressed in several different hematological and solid cancers58. The potential interplay between TET and DNMT activities in regulating oncogenesis as well as R-loop and G-quadruplex levels in B cells and other cell types remains to be investigated.
Our studies suggest that G-quadruplexes and R-loops could be therapeutic vulnerabilities in cancers with TET loss-of-function. G4-stabilizing ligands were recently shown to decrease cell viability in ATRX-deficient gliomas and BRCA1/2-deficient tumor cells59,60. In our hands, the use of a G4-stabilizing ligand, or depletion of proteins known to regulate G4 or R-loop structures, was associated with increased DNA DSBs and a slight increase in apoptosis in TET-deficient B cells. Furthermore, deletion of DNMT1 in TET-deficient B cells prevented the accumulation of R-loop and G-quadruplex structures in splenic B cells and Peyer’s patch GC B cells and rescued the survival of TET-deficient mice. Follow-up studies in pre-clinical models could test whether a combination of G4-stabilizing agents and DNA methyltransferase inhibitors might synergize to delay the onset and/or progression of B cell lymphomas and other malignancies with TET loss-of function.
Methods
Data Availability.
All genome-wide sequencing datasets have been deposited to Gene Expression Omnibus (GEO) repository, accession number GSE161463. Any data and reagents will also be made available upon request.
Code Availability.
The code used to process the NGS datasets has been deposited in GitHub repository at https://github.com/dsamanie7/Tet2-Tet3_DKO_CD19_cre
Mice.
Tet2fl/fl and Tet3fl/fl mice were generated as previously described61,62. C57BL/6J (000664), CD19 cre (006785), Rosa26-LSL-EYFP (006148), Cγ1Cre (010611), Ubc-CreERT2 (008085; described as ERT2cre) and CD45.1 mice (002014, ptprca) were obtained from Jackson Laboratory. To induce ERT2cre-mediated deletion, Cre-expressing and control mice were intra-peritoneally injected with 2 mg tamoxifen (Sigma) dissolved in 100 µL corn oil (Sigma) daily for 5 days. For transplantation studies, CD45.1 mice were sub-lethally irradiated with 600 rads of X-rays 24 h prior to transfer of 2 million CD19 DKO or Dfl B cells through the retro-orbital sinus. Age and sex matched mice from both sexes were used in the experiments. All mice used were 8 to 16 weeks of age (unless otherwise indicated) and were on a C57BL/6 genetic background, housed in specific-pathogen free animal facility at ambient temperature and humidity with 12h light/12h dark cycle at La Jolla Institute for Immunology. Age and sex matched mice from both sexes were used in the experiments. All studies were performed according to protocols approved by the Institutional Animal Care and Use Committee.
B cell isolation and cell cultures.
Primary B cells were isolated using flow sorting of YFP+ CD19 DKO B cells or YFP- control Dfl B cells for RNA-Seq, WGBS and 5hmC mapping analyses, and using EasySep Mouse Pan B cell isolation kit (#19844 Stem Cell Technology, Canada) for in vitro culture of B cells and CD19 positive selection (for G4 mapping, R-loop mapping and HTGTS studies) from splenocytes. Primary B cells and CH12F3 (CH12) cells were cultured at 37°C, 5% CO2 in RPMI 1640 media supplemented with 10% FBS, 1x MEM non-essential amino acids, 10mM HEPES (pH 7.4), 2mM Glutamax, 1mM sodium pyruvate, 55µM 2-mercaptoethanol (all from Life technologies). B cells (5×105-1×106 cells/mL) were activated with 10 µg/mL LPS (for Cas9 RNP targeting studies) from E. coli O55:B5 (Sigma, St. Louis, MO), 25 µg/mL LPS and 10 ng/mL rmIL-4 for stimulation of Dfl and ERT2cre B cells in the presence of 1 µM 4-hydroxytamoxifen (Tocris). For pyridostatin (PDS) (#SML2690, Sigma-Aldrich) treatment, B cells were activated with 10 µg/mL LPS for 48 h in presence of 10 µM PDS before analysis. All cytokines used above were from Peprotech (Rocky Hill, NJ). CH12F3 cell line was developed in Dr. T. Honjo’s laboratory at Kyoto University and was independently validated using the cell stimulation and Ig class-switching experiments.
For B cell cultures on 40LB feeder cells, 40LB cells were irradiated with 3000 rads of X-rays and plated at a density of 20X104 cells per well on a 12 cm plate and cultured overnight at 37°C, 5% CO2 in DMEM media supplemented with 10% FBS, 10mM HEPES (pH 7.4), 2mM Glutamax, 1mM sodium pyruvate, 55µM 2-mercaptoethanol (all from Life technologies). 40LB cells were obtained from Dr. D. Kitamura’s laboratory (Tokyo University of Sciences) was independently validated by B cell stimulation experiments and periodically tested for mycoplasma contamination. B cells were purified from Cγ1cre and Cγ1 DKO mice using the Mouse B cell isolation kit (#19854 Stem Cell Technology, Canada) and isolated cells were seeded at a density of 2X104 cells per well on 40LB containing 12 cm plate in RPMI 1640 media prepared as above and supplemented with 1ng/ml of rmIL-4. The expanded B cells collected from suspension were analyzed by flow cytometry.
Immunization.
Cγ1Cre, Dfl and Cγ1DKO mice were immunized with sheep red blood cells-SRBCs (#31102, Colorado Serum company, CO, USA) washed two times with PBS and injected in 2 doses, first primed with 200X106 SRBCs followed by a boost at day 5 with 109 SRBCs before analysis of splenocytes at day 12 post first immunization.
Flow cytometry.
Primary cells and in vitro cultured cells were stained in FACS buffer (0.5% bovine serum albumin, 1mM EDTA, and 0.05% sodium azide in PBS) with indicated antibodies for 30 mins on ice. Cells were washed and then fixed with 1% paraformaldehyde (diluted from 4% with PBS; Affymetrix) for 10 min at 25°C before FACS analysis using FACS Celesta and FACS LSR II (BD Biosciences). Antibodies and dyes were from BioLegend, eBioscience, and BD Pharmingen. Data were analyzed with FlowJo (FlowJo LLC, Ashland, OR). The gating strategy for the flow cytometric analysis is displayed in extended data figure 9 and the appropriate gates used in each experiment is described in the corresponding figure legends.
Immunoblotting.
Proteins isolated from cells with NP-40 lysis buffer were resolved using NuPAGE 4–12% Bis-Tris gel (ThermoFisher) and transferred from gel to PVDF membrane using Wet/Tank Blotting Systems (Bio-Rad). Membrane was blocked with 5% non-fat milk in TBSTE buffer (50mM Tris-HCl pH 7.4, 150mM NaCl, 0.05% Tween-20, 1mM EDTA), incubated with indicated primary antibodies, followed by secondary antibodies conjugated with horse-radish peroxidase (HRP) and signal was detected with enhanced chemiluminescence reagents (Invitrogen) and X-ray film. Antibodies against ATRX (1:1000, clone D5), FANCD2 (1:1000, clone Fl17) and BLM (1:1000, clone B4) were purchased from Santa Cruz Biotechnology. Antibodies against RNASE H1 (1:1000, NBP2–20171) from Novus biologicals, and WRN (1:1000, clone 8H3) and β-ACTIN HRP (1:5000, clone 13E5) from Cell signaling and DNMT1 (1:1000, ab19905) from Abcam.
Cas9 RNP targeting.
Alt-R crRNA and Alt-R tracrRNA (from IDT) were reconstituted at concentration of 100 µM in Nuclease-Free Duplex buffer (IDT). RNA duplexes were prepared by mixing oligonucleotides (Alt-R crRNA and Alt-R tracrRNA) at equimolar concentrations in a sterile PCR tube (e.g. 4 µl Alt-R crRNA and 4 µl Alt-R-tracrRNA). Mixed oligonucleotides were annealed by heating at 95C for 5 min in a PCR machine, followed by incubation at 25°C for at least 1 hour. 5.9 µl of crRNA-tracrRNA duplexes (180 pmol) were then mixed with 2.1 µL IDT Cas9 v3 (80 pmol) (together Cas9 RNP) by gentle pipetting and incubated at 25°C for at least 10 min. 35 µl of cell media was aliquoted in a 24 well plate. 2 million B cells were then washed with PBS and resuspended in 15 µl resuspension buffer (4D-Nuclcofector X Kit S, #V4XP-4032; Lonza). Cells in resuspension buffer (12.5µl) were then mixed with Cas9-RNPs (7.5µl) and transferred to the nucleofection cuvette strips and electroporated using the CM-137 program with 4D nucleofector. After electroporation, cells were transferred into 35 µl media in 24 well plate, and incubated for 20 min at 37°C before transferring cells to activation media with 10 µg/ml LPS. The crRNA sequences used are listed in extended data table 1.
RNA extraction, cDNA synthesis, and quantitative RT-PCR.
Total RNA was isolated with RNeasy plus kit (Qiagen, Germany) or with Trizol (ThermoFisher, Waltham, MA) following manufacturer’s instructions. cDNA was synthesized using SuperScript III reverse transcriptase (ThermoFisher) and quantitative RT-PCR was performed using FastStart Universal SYBR Green Master mix (Roche, Germany) on a StepOnePlus real-time PCR system (Applied Biosytems). Gene expression was normalized to Gapdh. Primers are listed in extended data table 1.
G4 detection and mapping.
For flow cytometry-based detection of G4s, cells were stained for cell surface markers according to the flow cytometry staining protocol described above. Following cell surface staining, cells were fixed with 4% paraformaldehyde in PBS for 12 min at 25°C. Using the intracellular transcription factor staining kit (Invitrogen), cells were then permeabilized at 4°C overnight according to manufacturer’s instructions. Cells were then washed with wash buffer and treated with RNASE A (1:50, AM2269; Ambion) for 30 min at 25°C followed by washing and incubation with 1:100 dilutions of BG4-Ig antibody (Ab00174–1.1, 1mg/ml; Absolute Antibody) for 30 min at 25°C or 4°C. This was followed by washing and subsequent staining with anti-mouse IgG1 fluorophore conjugated antibody (BioLegend) to reveal the signal.
The excitation and emission spectra of NMM in presence of G4 DNA, but not in the presence of non-G4 control DNA, closely mimicked that of a fluorophore, Brilliant Violet 605 (BV605). Therefore, we also used NMM to stain for G4. Following the RNASE A treatment cells were incubated with 10 μM NMM (#NMM580; Frontier Scientific) for 30 min at 25°C or 4°C before reading the fluorescence signal in the Brilliant Violet 605 channel. Mouse CH12F3 (CH12) B cells treated with PDS, which binds G-quadruplex structures in a manner distinct from NMM38,40, showed increased NMM fluorescence in flow cytometry (Extended data Fig. 3e), validating NMM as a flow cytometric probe for G-quadruplex structures in cells.
G4 mapping was performed as previously described with minor modifications47. Briefly, ~10 million cells were fixed with 1% formaldehyde at 25°C with nutation for 10 min mins at 1×106 cell/mL in media, quenched with 125 mM glycine, washed twice with ice cold PBS. Cells were pelleted, snap-frozen in liquid nitrogen, and stored at −80°C until use. Cell pellets were then lyzed using 800 μl of ChIP hypotonic solution (Chromatrap) and 150 μl lysis buffer (Chromatrap) supplemented with protease inhibitor cocktail (invitrogen) on ice according to manufacturer’s instructions. The lyzed nuclei suspension was then sonicated using the bioruptor pico sonicator (Diagenode), 16 cycles 30 seconds on / 30 seconds off and the lysates were cleared by centrifugation at >12000g for 10 min. The lysates were diluted 1:10 times in intracellular buffer (25 mM HEPES pH 7.5, 10.5 mM NaCl, 110 mM KCl, 130 nM CaCl2, 1 mM MgCl2) (total 200 μl) then treated with 1mg/ml RNASE A (AM2269; Ambion, 1:100 dilution) for 20 min at 37°C. Chromatin lysates were then incubated with a control IgG or BG4-Ig antibody (Ab00174–1.1, 1mg/ml; Absolute Antibody) (1:100) for 1.5 h at 16 C followed by incubation with protein G magnetic dynabeads (Invitrogen). The beads were then washed 5 times with 1 ml wash buffer (intracellular buffer with 0.05% tween 20). The beads were re-suspended in elution buffer (100 mM NaHCO3, 1% SDS, 1 mg/mL RNaseA; Qiagen) treated with RNASE A (1:100, 100mg/ml) at 37 C for 30 min and proteinase K (1:40, 20mg/ml; Ambion) at 65 C for 2 h to overnight. DNA was purified with Zymo ChIP DNA Clean & Concentrator-Capped Column (Zymo Research, Irvine, CA). The Library was prepared with NEB Ultra II library prep kit (NEB) following manufacturer’s instruction and was sequenced on an Illumina NovaSeq 6000 (50 bp paired-end reads). The control IgG did not pull-down sufficient amount of DNA to prepare libraries, therefore, we prepared libraries from the input DNA and used those as controls. 2 independent experiments were combined for analysis.
NMM oligonucleotide fluorescence enhancement assay.
G4 forming oligonucleotides and control oligonucleotides with mutated G-stretches were ordered from Integrated DNA technologies using standard desalting purification (Extended data table 1). The oligonucleotides were reconstituted at 100μM concentration in 10mM Tris (pH 8.0) and 1mM EDTA (TE buffer) and then diluted to a final concentration of 10μM in buffer containing 20mM HEPES (pH 7.5), 250mM KCl and 1mM DTT. The diluted oligonucleotides were then heated at 95C for 10 min followed by gradual cooling to 25°C (cooled down at 0.1֯C per second ramp rate) in a PCR machine. 50 μl of 10μM was then mixed with 50 μl of 20μM NMM incubated at 25°C for 1 hour and the fluorescence was measured in the Spectramax M2 (Spectral labs) plate reader with excitation at 400nm and emission at 610nm.
Recombinant RNASE H1 purification.
Coding sequence of N-terminal V5-tagged mutant human RNASE H1 (D210N)41 was sub-cloned in frame with Glutathione-S-Transferase (GST) protein separated by a Precission protease cleavage site in the pGEX6p1 vector. BL21 (DE3) cells were transformed with the pGEX6p1 vector with GST-RNASE H1 fusion. Single colonies were picked and grown overnight in 5 ml LB at 37°C before further expanding in 500ml LB (1:100) with shaking at 37°C for 2–3 h until the OD at 600 nm reached 0.5–0.6. 1mM IPTG was then added to induce the protein expression for 3 h at 37°C with shaking. Expression of protein was confirmed by coomassie staining on SDS-PAGE gel by comparing with an uninduced control culture. Cultures were then pelleted at 4500g for 15 min and cell pellets were re-suspended in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl), 15ml for 200ml culture. The cell suspension was then lysed by sonication for a total of 4 min (5 seconds on, 30 seconds off cycle) with a probe sonicator (Missonix) followed by centrifuge at 10,000g for 20min, at 4°C. The supernatant was then mixed with 150µl washed and equilibrated glutathione agarose beads (#16100; Pierce) and rotated for 4 h to overnight. The lysate beads mixture was then washed 3 times with lysis buffer and then re-suspended in 1ml elution buffer (25mM Tris-HCl (pH 7.6), 10% Glycerol, 0.2mM EDTA, 100mM KCl, 1mM DTT) and treated overnight with precission protease (#Z02799; Genescript). The supernatant was then collected and the protein was quantified by absorbance at 280 nm using Nanodrop. The purity of protein was additionally confirmed by coomassie staining of samples on an SDS-PAGE gel.
R-loop detection and mapping (MapR).
For flow cytometry-based detection of R-loops, cells were stained for cell surface marker according to the flow cytometry staining protocol described above. Following cell surface staining, cells were fixed with 4% paraformaldehyde in PBS for 12 min at 25°C. Using the intracellular transcription factor staining kit (Invitrogen), cells were then permeabilized at 4°C overnight according to manufacturer’s instructions. Cells were then washed and incubated with 1:50 dilutions of V5-tagged recombinant mutant RNASE H1 for 30 min at 4°C followed by washing, subsequent staining with anti-V5 tag antibody (1:200, clone D3H8Q; Cell Signaling technologies) and with anti-rabbit secondary antibody (1:1000, #A-21245; Invitrogen) to reveal the signal. For RNASE H digestion and R-loop quantification, CH12 cells were fixed, permeabilized and treated with 20 units of RNASE H (NEB#M0297S) in 100µl of digestion buffer diluted in water for 2 h at 37°C before proceeding to R-loops staining. MapR and immunoprecipitation with S9.6 antibody identify a set of R-loops in common, as well as sets of unique R-loops that depend on differential sequence specifities and/or preferences of S9.6 and RNASE H, thus R-loops identified by MapR may represent a slightly different subset than those identified by other methods63,64.
R-loop mapping was performed by the MapR method, as previously described42. The libraries were sequenced on an Illumina NovaSeq 6000 (50 bp paired-end reads). Three independent experiments were combined for analysis.
Detection of G-quadruplex binding proteins.
Purified naïve B cells were activated in presence of 25 µg/ml LPS and 10ng/ml IL-4 and nuclear extracts from 20 million cells were prepared using Dignam extraction. Cells were resuspended in 10 ml dignam buffer A (10 mM KCl, 1.5 mM MgCl2) supplemented with protease inhibitor cocktail (Thermofisher) and incubated on ice for at least 10 min. The cells were then homogenized and lyzed with a dounce homogenizer (10 strokes) and nuclei were collected by centrifugation at 600 g for 5 min and washed again with buffer A. Cells were then resuspended in 1ml of dignam buffer C (0.2 mM EDTA, 25% glycerol (v/v), 20 mM HEPES-KOH (pH 7.9), 0.42 M NaCl, 1.5 mM MgCl2) supplemented with protease inhibitor cocktail and rotated at 4°C for 1 hour. The lysates were collected and passed through a PD-10 buffer exchange column (GE healthcare) equilibrated with intracellular buffer (25 mM HEPES pH 7.5, 10.5 mM NaCl, 110 mM KCl, 130 nM CaCl2, 1 mM MgCl2). The G4 and non-G4 oligonucleotides were folded in the presence of K+ ions, annealed with a complementary biotinylated oligonucleotide, captured on streptavidin conjugated magnetic beads and incubated with nuclear lysates from B cells activated with LPS and IL-4 to identify G4-binding proteins (Extended data Fig. 8a). We observed preferential binding of the known G4-binders ATRX and BLM as well as the flag-tagged BG4 antibody to the G4 oligonucleotides (Extended data Fig. 8b). The DNMT1 protein also showed preferential binding to G4 forming oligonucleotides compared with non-G4 control oligonucleotides, thus independently validating previous studies (Extended data Fig. 8b). The nuclear lysates collected in intracellular buffer were spike-in with 1 µg flag-tagged BG4 scFv antibody (Millipore), pre-cleared with myone T1 streptavidin-conjugated dynabeads (Life technologies) for one hour and incubated with streptavidin beads conjugated with a mixture of G4 or non-G4 forming oligonucleotides for 4 h (Extended data table 1). Samples were then washed with intracellular buffer with 0.05% tween 20 for a total of 5 washes and denatured using the SDS-PAGE sample buffer for immunoblotting.
RNA-Seq.
RNA from cells was isolated with Trizol (ThermoFisher, Waltham, MA) following manufacturer’s instructions. Isolated RNA was further purified using RNA clean up and concentrator kit (R1013, Zymo research) according to manufacturer’s instructions. 40 ng of purified RNA was then used for preparation of RNA-sequencing libraries with the Nugen ovation RNA-Seq V2 system (now Tecan Genomics) according to the manufacturer recommended protocol. Samples were sequenced on Illumina Hiseq 2500 (single-end 50 bp reads). 2 independent experiments were combined for analysis.
Whole genome bisulfite sequencing (WGBS).
1 μg of genomic DNA was spiked-in with unmethylated lambda-phage DNA (1:200) and sonicated using Bioruptor (9 cycles 30 seconds on, 30 seconds off). The sonicated DNA fragments were end repaired, A-tailed and ligated with methylated adaptors (all using NEB kits). The adaptor ligated fragments were then treated with sodium bisulfite using the Methylcode kit (Invitrogen) for a total of 4 h according to manufacturer’s protocol. The bisulfite converted, adaptor ligated fragments were then amplified with Kapa Hifi Hotstart Uracil+ PCR mix with NEB universal dual indexing primers. The libraries were then sequenced on an Illumina NovaSeq 6000 (150 bp paired-end reads). 2 independent experiments were combined for analysis.
5hmC pull down (HMCP).
5hmC mapping was performed on purified genomic DNA using the HMCP method in collaboration with Cambridge Epigenetix according to the manufacturer’s recommended protocol. The libraries were then sequenced on an Illumina Hiseq 2500 (single-end 50 bp reads). 2 independent experiments were combined for analysis.
Linear Amplification Mediated High Throughput Genome-wide Translocation Capture Sequencing (LAM-HTGTS or HTGTS).
HTGTS protocol was adapted from previously published method49. Briefly, genomic DNA was sonicated with 2 pulses of 5 secs ON and 1 minute off using the Bioruptor pico sonicator (Diagenode). LAM-PCR was performed using Phusion High-Fidelity DNA Polymerase (Thermo Fisher) and Sμ 5’ biotinylated bait (TAGTAAGCGAGGCTCTAAAAAGCA). 4 independent PCRs were run with ~2.5 μg of sonicated DNA each, with 90 cycles of primer elongation. The biotinylated DNA fragments were purified using Dynabeads T1 streptavidin beads (Invitrogen) at 25°C, on a rotisserie, overnight. On bead ligation was performed with T4 DNA ligase (NEB) and annealed bridge adaptors (GCGACTATAGGGCACGCGTGGNNNNNN-NH2 and 5’P-CCACGCGTGCCCTATAGTCGC-NH2) for 1h at 25°C, 2h at 22°C, and overnight at 16°C. 15 cycles of nested PCR were performed on the DNA-bead complexes using an Sμ nested primer (ACACTCTTTCCCTACACGACGCTCTTCCGATCT-GGTAAGCAAAGCTGGGCTTG) and a reverse universal primer I7 (GACTGGAGTTCAGACGTGTGCTCTTCCGATCT-GACTATAGGGCACGCGTGG). The bold regions represent the complementary regions to the genomic sequence in the Sμ nested primer and the bridge adaptor for I7 primer. PCR products were cleaned with the QIAquick Gel Extraction Kit (Qiagen) and the final barcoding/indexing PCR was performed using the P5-I5 and P7-I7 universal dual indexing primers (NEB) for 15 cycles and purified using Ampure beads purification with a cutoff of 200 bp. Library quality was evaluated by Bioanalyzer (Agilent) and sequencing performed using the Illumina Miseq Reagent Kit v2 (600 cycles) following the manufacturer’s instructions. DNA junctions from 2 independent experiments were combined for analyses. The complete list of primers and sequences are included in extended data table 1.
Bioinformatics analyses
The reference genome used was mm10. Heatmaps and profile plots were generated using DeepTools65.
HMCP analysis.
Single-end reads were mapped to the mouse genome mm10 GRCm38 (Dec. 2011) indexed with spiked-in phage lambda, using Bowtie (V 1.1.2) (-S -p 3)66. Reads that mapped to spike-ins were filtered out with samtools view; remaining mapped reads in sam files were sorted and PCR duplicates were removed with Picard (V 2.7.1). Peaks were called with MACS2 (callpeak --keep-dup all -g mm)67. Peaks from the replicates from same condition were merged with mergePeaks from HOMER and kept only those that intersected between the replicates. Intersected peaks from the replicates were merged between the conditions to create the total number of peaks. Tag directories were created and replicates were merged with makeTagDirectory from HOMER68 (-genome mm10); makeMultiWigHub.pl was used to generate tracks.
WGBS analysis.
WGBS reads were mapped with BSMAP (v2.90)69 to both the mouse genome mm10 GRCm38 (Dec. 2011) from UCSC and the lambda genome. Bisulfite conversion efficiency was estimated based on cytosine methylation in all contexts. For all the samples the bisulfite conversion efficiency was higher than 0.9996. Duplicated reads caused by PCR amplification were removed by PICARD’s MarkDuplicates. CpG DNA methylation at both DNA strands was called by methratio.py script, from BSMAP (v2.90) (-g - i “correct” -x CG,CHG,CHH). To identify differentially methylated cytosines and regions (DMCs and DMRs) we used RADmeth methpipe-3.4.2 (adjust -bins 1:100:1 ; merge -p 0.05).
RNA-seq and BCR analysis.
Single-end (50bp) reads were mapped to the mouse genome mm10/GRCm38 using STAR70 (v2.5.3a) (--runThreadN 8 --genomeLoad LoadAndRemove --outFilterMultimapNmax 1 --outFilterType BySJout --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 100000 --outFilterMismatchNmax 0). Counts were obtained with featureCounts (subread-1.4.3-p1) (-g gene_name -s 1). Differentially expressed genes were calculated with DESeq271, filtering out genes that did not have any count in any condition; cut off to define DEGs was an adjusted pvalue 0.05 and a log2 fold change >= +/−1
BCR sequences were retrieved from RNA-Seq data sets, and the frequency of IG chain clonotypes was determined using MiXCR72 (mixcr-1.7–2.1) package with the default parameters “align -c IG -s mmu -p rna-seq -OallowPartialAlignments=true”. Two rounds of contig assembly were performed by employing the “assemblePartial” function; extension of incomplete BCR was done with “extendAlignments” function; assembly and export of the clonotypes was performed using the “assemble” and the “exportClones” --preset min -fraction -targets -vHits -dHits -jHits -vAlignments -dAlignments -jAlignments) functions, respectively.
G4 analysis.
Paired-end (50bp) reads were mapped to the mouse genome mm10 GRCm38 (Dec. 2011) indexed with spiked-in G4 sequences, using Bowtie (V 1.1.2)66. Reads that mapped to spike-ins were filtered out with samtools view; remaining reads in sam format were sorted and PCR duplicates were marked and removed with Picard (V 2.7.1). Peaks were calculated with MACS267 (callpeak –keep-dup all -g mm -p 0.0001), using input as control. Peaks from the replicates from same condition were merged with mergePeaks from HOMER and kept only those that intersected between the replicates. Intersected peaks from the replicates were merged between the conditions to create the total number of peaks. Tag directories were created and replicates were merged with makeTagDirectory from HOMER68 (-genome mm10); makeMultiWigHub.pl was used to generate tracks.
MapR.
Paired-end (50bp) reads were mapped to the mouse genome mm10 GRCm38 (Dec. 2011, using Bowtie (V 1.1.2). Mapped reads were sorted and PCR duplicates were marked and kept with Picard (V 2.7.1). Peaks were calculated with MACS2 (callpeak –keep-dup all -g mm --broad --broad-cutoff 0.1), using input as control. Peaks from the replicates from same condition were merged with mergePeaks from HOMER and kept only those that intersected between the replicates. Intersected peaks from the replicates were merged between the conditions to create the total number of peaks. Tag directories were created and replicates were merged with makeTagDirectory from HOMER68 (-genome mm10); makeMultiWigHub.pl was used to generate tracks. For heatmaps and profiles plots generated with Deeptools, MNase control signal was removed from the RNASEH-MNase signal.
G-quadruplexes and R-loops peaks.
The 9722 G-quadruplex and R-loop regions were obtained from the union of G-quadruplexes and R-loops peaks identified by using “intersectBed” from bedtools73.
HTGTS.
Analysis was done following HTGTS49 pipeline (https://github.com/robinmeyers/transloc_pipeline). Briefly, paired-end reads were trimmed with TranslocPreprocess.pl and full analysis was performed with TranslocWrapper.pl. Post filter analysis was done using TranslocFilter.pl with default parameters. Circos plots were generated using the Circos software74. HTGTS hits were identified in a 10kb window and color scale was assigned based on the number of hits per window.
Potential to form G-quadruplexes analysis.
Regular expressions in ‘awk’ commands were used to categorize the potential to form G-quadruplexes and R-loops peaks from fasta sequences. The loop1–7 is defined as with the following expression: G3+N1–7 G3+N1–7 G3+N1–7G3. The long-loop is defined as sequences with a G4 with any loop of length >7 (up to 12 for any loop and 21 for the middle loop). The simple-bulge is defined as sequences with a G4 with a bulge of 1–7 bases in one G-run or multiple 1-base bulges. And the Two Tetrad/Complex bulge is defined as sequences with G4s with two G-bases per G-run with several bulges of 1–5 bases.
AID motifs hotspots analysis.
Fasta sequences were retrieved from bed files from the G-quadruplexes and R-loops peaks. Library “stringr” and function str_locate_all in R was used to identify the “[A|T][A|G]C[C|T]” or “[ A|G]G[C|T][A|T]” motifs.
Random control regions.
Random regions were retrieved with the function “shuffleBed” from the bedtools15 using as the reference the whole mm10 genome. In particular cases, random regions were retrieved from euchromatin regions defined by the A compartment from Hi-C analysis in naïve and activated B cells75.
Quantification and Statistical Analysis.
Statistical analyses were performed with Graph prism 8 and R version 3.4. The statistical tests used to determine significance in each analysis are described in the figure legends of the corresponding figures. Parametric tests (t-tests, ANOVA) were used in experiments where normal distribution could be assumed, whereas in other cases, where normal distribution could not be assumed, non-parametric tests (Wilcoxon signed-rank test, or Kruskal-Wallis test and the ad hoc Dunn’s test) were used. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications7,35. No data points were excluded from the analysis and appropriate animals/samples for each experiment were chosen randomly. Since our analysis required genotyping of the experimental mouse groups, we were not able to blind ourselves during data collection.
Extended Data
Extended data figure 1. TET deficiency leads to development of mature B cell lymphoma.
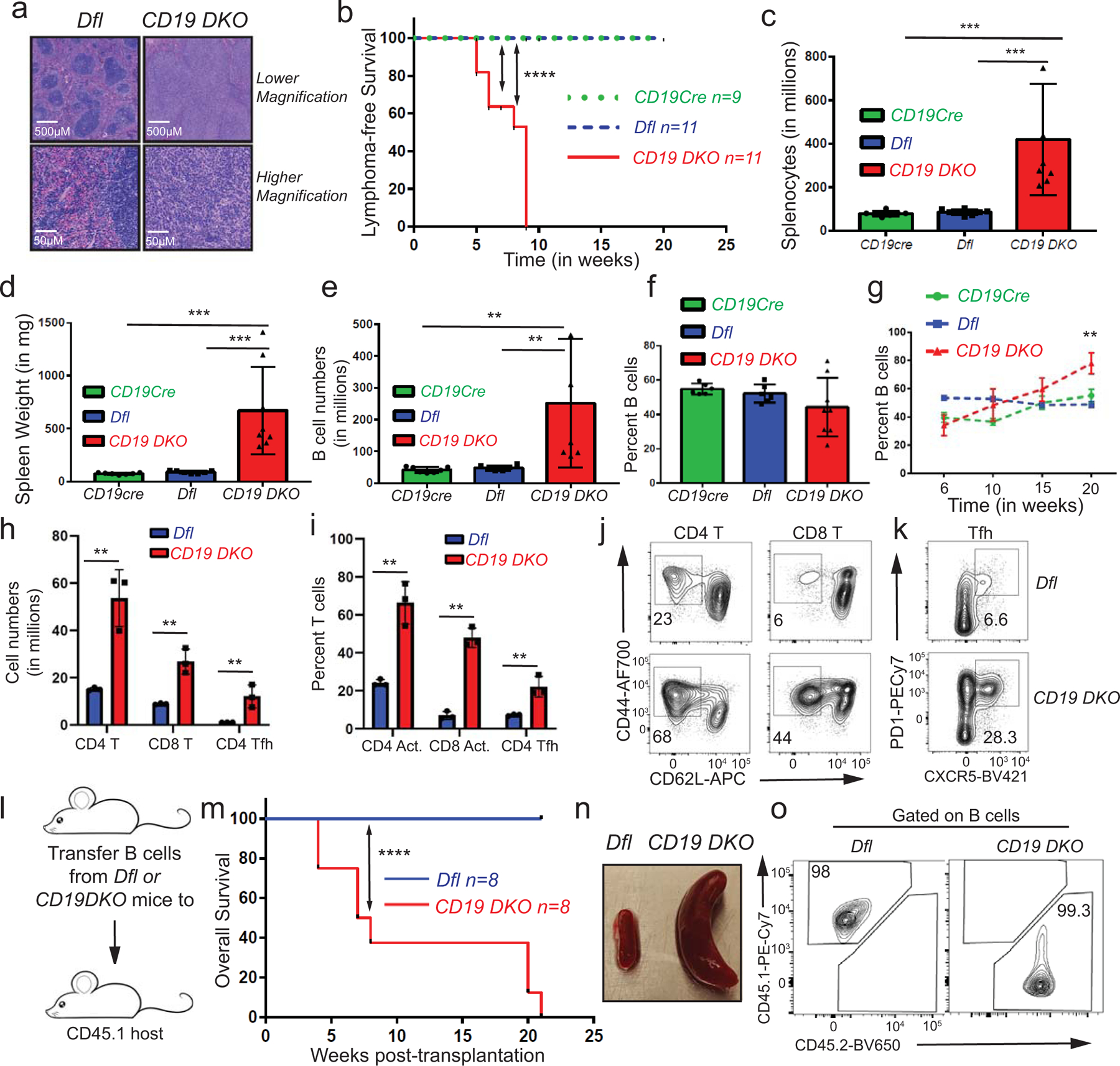
a) H&E staining in spleens from 8 week-old Dfl and CD19 DKO mice The data is representative of 2 independent experiments. b) Kaplan-Meir curves displaying lymphoma-free survival of CD19 DKO (red) and control Dfl (blue) and CD19 Cre (green) mice. Y-axis, percent mice without lymphoma, defined by ≥ 3-fold increase in spleen weight and ≥ 2-fold increase in cellularity. c) – f) Quantification of c) cell numbers, d) spleen weights, e) B cell numbers, f) percent of B cells in spleens of 9 week-old CD19 DKO (red) and control Dfl (blue) and, CD19 Cre (green) mice from at least 5 biological replicates and 4 independent experiments. g) Percent of B cells in spleens of CD19 DKO (red) and control Dfl (blue) and CD19 Cre (green) mice at different ages from 3 independent experiments. h) Quantification of cell numbers of CD4+ T, CD8+ T and CD4+ Tfh cells from spleens of 9 week-old CD19 DKO (red) and control Dfl (blue) mice from 3 biological replicates and 2 independent experiments. i) Percent of activated CD4+ and, CD8+ T cells, and CD4 Tfh cells from spleens of 9 week-old CD19 DKO (red) and control Dfl (blue) mice from 3 biological replicates and 2 independent experiments. j) and k) Flow cytometry plots of j) Activated CD62Llow (X-axis) and CD44+ (Y-axis) CD4+ and CD8+ T cells and k) CD4+ Tfh cells PD1+ (Y-axis) CXCR5+ (X-axis) from spleen of 9 week-old CD19 DKO (red) and control Dfl (blue) mice. Numbers represent frequency of gated populations. l) Scheme of adoptive transfer experiment. B cells from Dfl and CD19 DKO CD45.2 mice were transferred retro-orbitally into sub-lethally irradiated CD45.1 immunocompetent host mice. m) Kaplan-Meir curves displaying overall survival of CD45.1 host mice transplanted with B cells from Dfl or CD19 DKO mice. X-axis denotes time in weeks after transplantation. n) Enlarged spleens in CD45.1 host mice transplanted with CD19 DKO compared to Dfl B cells 8 weeks after adoptive transfer. o) Representative flow cytometry data from the spleens of CD45.1 host mice 8 weeks after transplantation with Dfl and CD19 DKO B cells. Frequencies of the CD45.1+ and CD45.2+ cell populations are shown. Statistical significance is calculated using the log-rank test for b) and m), one-way ANOVA for c)–f) and two-way ANOVA for g)–i). Error bars represent mean +/− standard deviation, ** p value ≤0.01 *** p value ≤0.001, **** p value ≤0.0001.
Extended data figure 2. Expanded B cells from CD19 DKO mice have a germinal center (GC) origin.
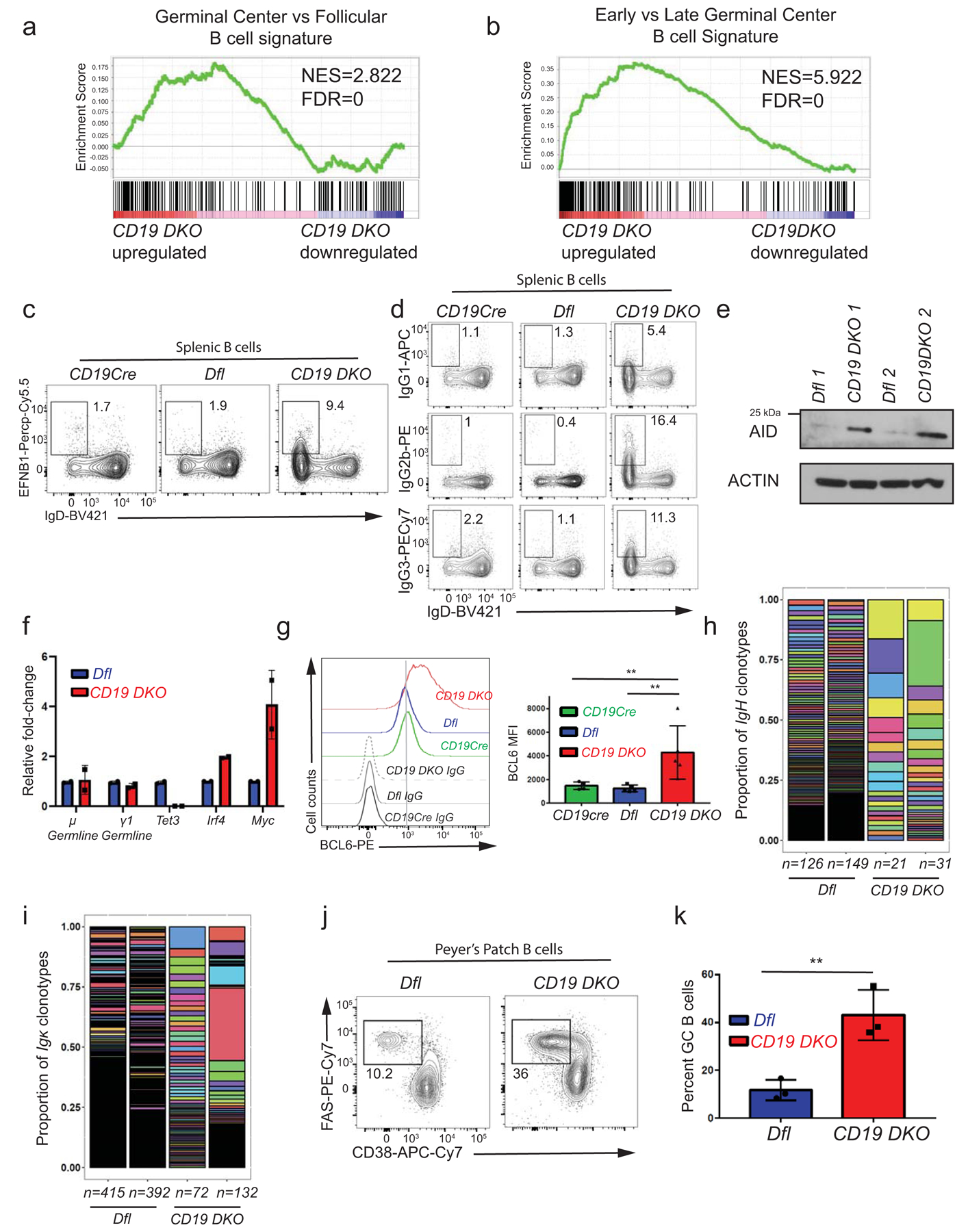
a), b) Gene set enrichment analysis (GSEA) plots showing enrichment for a GC B cell transcriptional signature in the transcriptional profile of CD19 DKO compared to Dfl B cells, using gene sets from a) GC versus follicular B cells and b) early GC versus late GC B cells. Y-axis denotes enrichment score. NES, Normalized enrichment score, FDR, False discovery rate. c) Representative flow cytometry data gated on splenic B cells from 8 week-old Dfl, CD19Cre (YFP+) and CD19 DKO (YFP+) mice. Numbers in the rectangles represent frequencies of GC B cells, identified as EFNB1+ (Y-axis) and IgDlow (X-axis). d) Representative flow cytometry data showing Ig isotype expression, gated on splenic B cells from 8 week-old Dfl, CD19Cre (YFP+) and CD19 DKO (YFP+) mice. Top, IgG1; middle, IgG2b; bottom, IgG3 X-axis shows expression of the default IgD isotype. Numbers represent frequencies of gated cell populations. e) Immunoblot showing AID expression in Dfl and CD19 DKO B cells (2 replicate experiments). Actin is used as a loading control. f) Relative fold-change in expression of μ and γ1 germline, Tet3, Irf4 and Myc transcripts measured by qRT-PCR in Dfl and CD19 DKO B cells from 2 biological replicates. g) Histogram (left panel) and bar-graph (right panel) showing staining with BCL6 antibody compared to isotype IgG controls in B cell from 8 week-old CD19Cre, Dfl and CD19 DKO mice from 3 independent experiments. h) and i) Bar plots displaying the proportions of (h) IgVH and (i) Igκ clonotypes (rearranged variable gene segments) from Dfl (blue) and CD19 DKO (red) B cells, identified from BCR repertoire analysis of RNA-Seq data. Y-axis represents the proportion of each clonotype. Each individual IgVH and Igκ clonotype is displayed using a different color in the bar plots. Numbers at the bottom represent the number of clonotypes identified in two independent replicates of Dfl (blue) and CD19 DKO (red) B cells. j) Representative flow cytometry data gated on Peyer’s patch B cells from 8 week-old Dfl and CD19 DKO mice. Numbers represent frequency of GC B cells, identified as FAS+ (Y-axis) and CD38− (X-axis). k) Quantification of GC B cell frequency in Peyer’s patches of Dfl (blue) and CD19 DKO (red) mice from 3 independent experiments. Statistical significance is calculated using two-tailed student t-test in f), k) and one-way ANOVA in g). Error bars represent mean +/− standard deviation in f), g) and k). ** p value ≤0.01.
Extended data figure 3. TET deficiency is associated with increased levels of G-quadruplexes and R-loops.
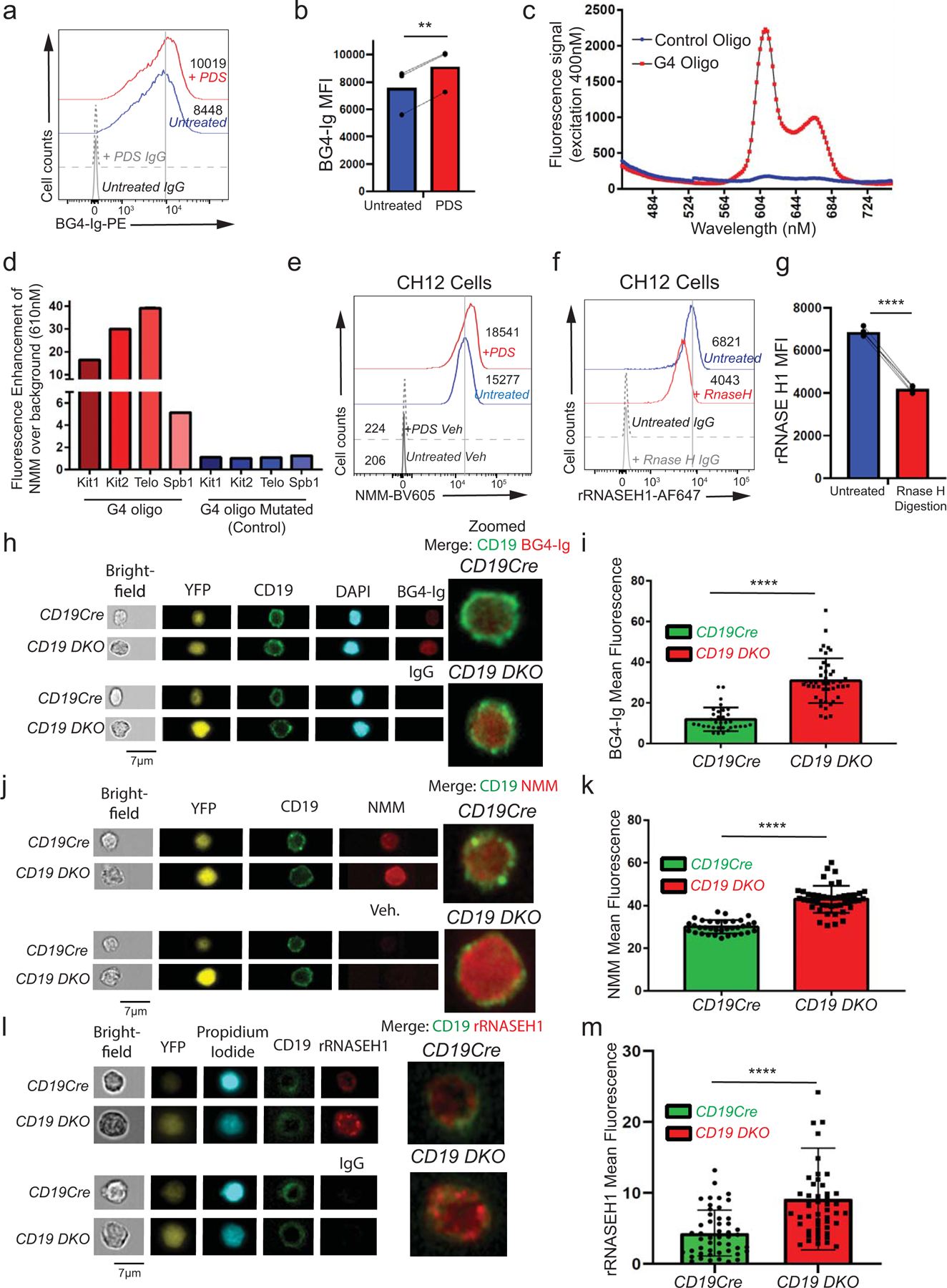
a) Flow cytometric detection of G-quadruplexes with BG4-Ig antibody or isotype IgG controls in primary B cells stimulated with 25 µg/ml LPS for 48 hours and treated with 10 µM pyridostatin (PDS, G4 ligand) for 24 hours. Numbers represent median fluorescence intensity. b) Quantification of median fluorescence intensity (MFI) of BG4-Ig signal from primary B cells treated with (red) or without (blue) PDS. Lines connect paired samples treated with or without PDS from 3 independent experiments. c) Fluorescence emission spectrum of NMM in the presence of a G4-forming oligonucleotide (oligo) from the human c-Kit gene promoter or a control oligo in which guanines in G4-forming regions (G-tracts) were mutated. d) Fluorescence enhancement over background (no oligos) for NMM at 610 nm in presence of known G4-forming oligos (Kit1, Kit2, Spb1) from the c-Kit gene locus or the telomeric repeat (Telo) or their respective mutated versions. e) G-quadruplex levels assessed by NMM or DMSO vehicle control (Veh) staining in untreated CH12 B cells or cells treated with 5 µM pyridostatin (PDS, G4 ligand) for 24 hours. Numbers represent median fluorescence intensity from 3 independent experiments. f) Flow cytometric detection of R-loops using V5-epitope-tagged recombinant RNASE H1 (rRNASE H1) in CH12 cells with or without RNASE H enzyme digestion during staining. Numbers represent median fluorescence intensity. g) Quantification of median fluorescence intensity (MFI) of R-loops (rRNASE H1) signal from CH12 cells with (red) or without (blue) RNASE H enzyme digestion. Lines connect paired samples with or without RNASE H digestion from 3 independent experiments. h) – m) Representative images (h, j, l) and quantification of mean fluorescence signal (i, k, m) of CD19cre and CD19 DKO YFP+ B cells stained with DAPI or propidium iodide and CD19, BG4-Ig (h, i), NMM (j, k) and rRNASE H1 (l, m) or respective controls using the AMNIS imagestream. Data are from two independent experiments. Statistical significance is calculated using paired student t-test in b) and g), two-tailed student t-test i), k) and m). Error bars represent mean +/− standard deviation, ** p value ≤0.01, **** p value ≤0.0001.
Extended data figure 4. TET deficiency in multiple primary cell types is associated with increased DNA G-quadruplex structures.
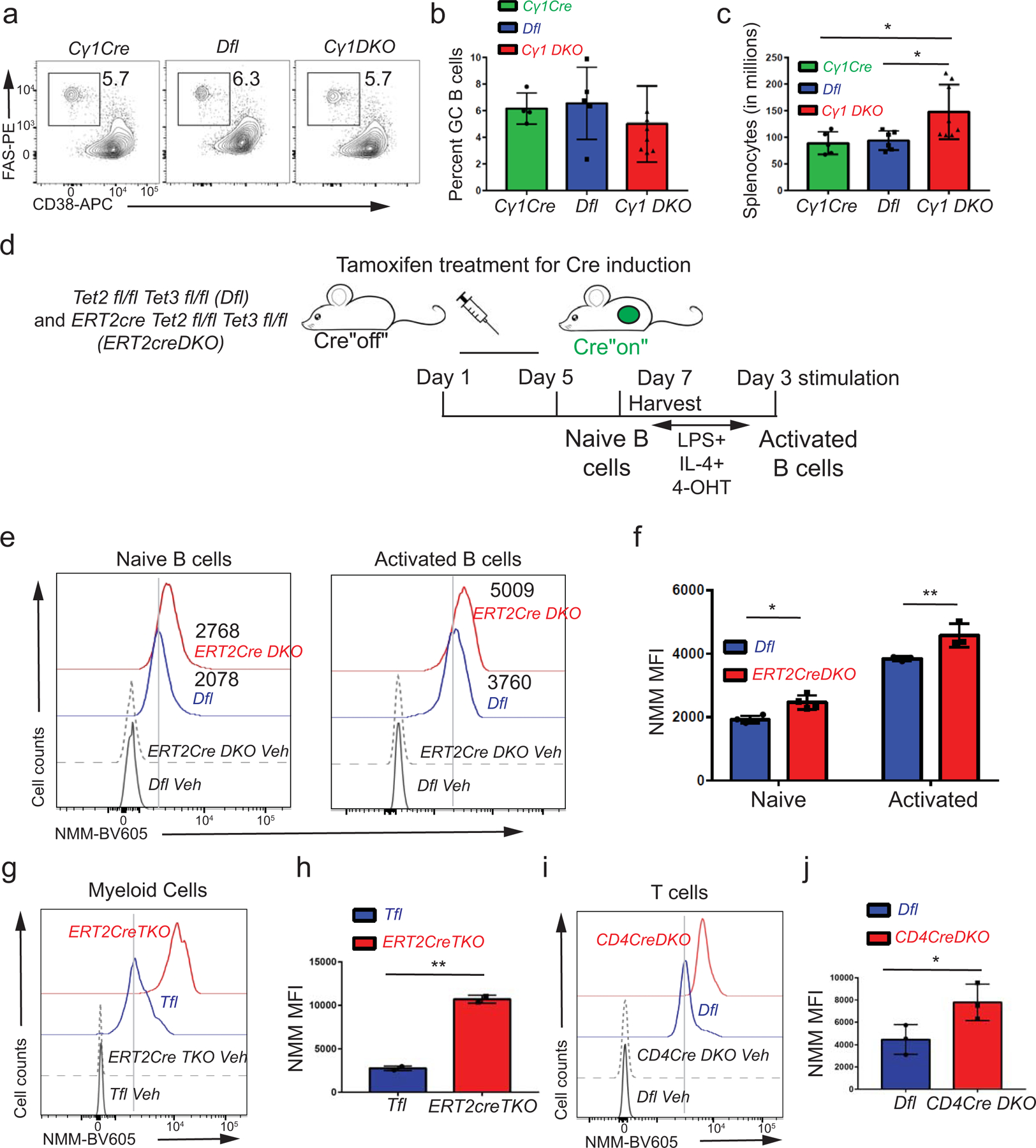
a) Representative flow cytometry data gated on splenic B cells from 8 week-old Cγ1Cre, Dfl and Cγ1 DKO mice 12 days after immunization with SRBCs. GC B cells are identified as FAS+ (Y-axis) and CD38− (X-axis). Numbers represent frequency of GC B cells. b), c) Quantification of (b) GC B cell frequencies and (c) absolute numbers of splenocytes from 8 week-old Cγ1Cre, Dfl and Cγ1 DKO mice 12 days after immunization with SRBCs from 3 independent experiments. d) Experimental design. ERT2Cre DKO or control Dfl mice were injected for 5 consecutive days with tamoxifen to induce Cre expression and TET deletion, then rested for 2 days. Splenic B cells were activated for 72 hours in vitro with LPS and IL-4 in the presence of 4-hydroxytamoxifen (4-OHT). e) G-quadruplex levels in naïve (left panel) and activated (right panel) B cells from tamoxifen-treated ERT2Cre DKO (YFP+) or control Dfl mice. Numbers represent median fluorescence intensity. f) Quantification of median fluorescence intensity (MFI) of NMM signal from naïve and activated B cells from ERT2Cre DKO (YFP+) or control Dfl mice from 3 independent experiments. g), i) G-quadruplex levels assessed by NMM or DMSO vehicle staining (Con) in (g) transferred CD45.2+ myeloid cells from ERT2Cre TKO (YFP+) or control Tfl mice, and (i) transferred CD45.2+ T cells from CD4Cre DKO (YFP+) or control Dfl mice. h), j) Quantification of median fluorescence intensity (MFI) of NMM signal in (h) transferred CD45.2+ myeloid cells from ERT2Cre TKO (YFP+) or control Tfl mice from 2 biological replicates and j) transferred CD45.2+ T cells from CD4Cre DKO (YFP+) or control Dfl mice from 3 biological replicates. Statistical significance is calculated using one-way ANOVA in c), two-way f) and two-tailed student t-test in h) and j). Error bars represent mean +/− standard deviation, * p value ≤0.01, ** p value ≤0.005.
Extended data figure 5. Increased apoptosis and DNA DSBs in TET-deficient B lymphoma cells depleted of enzymes that resolve G-quadruplexes and R-loops.
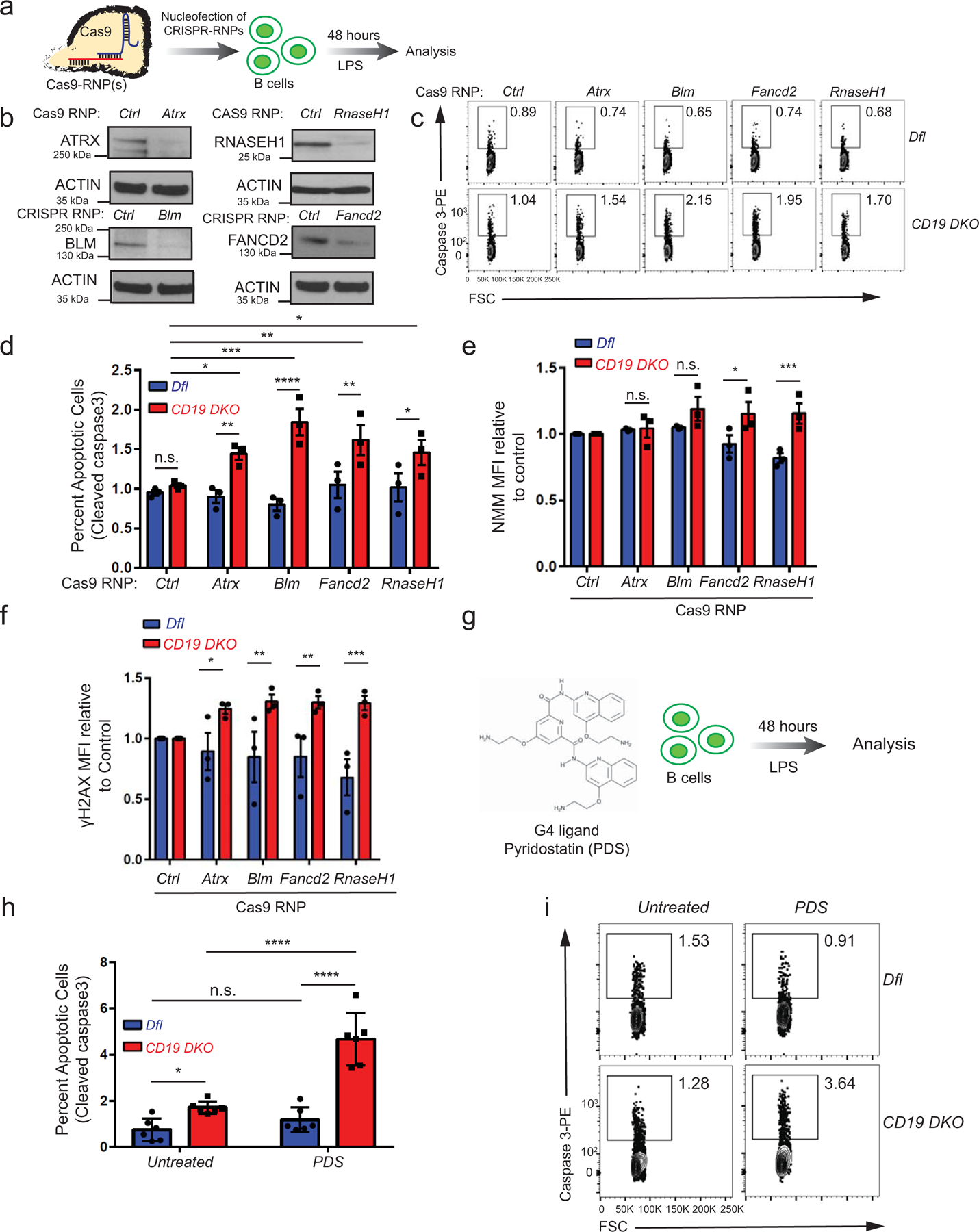
a) Experimental design. Primary B cells from Dfl and CD19 DKO B cells were nucleofected with Cas9 RNPs loaded with sgRNAs against Rnase H1 or the known G4-binding helicases Atrx, Blm and Fancd2, then stimulated with 10 µg/ml LPS for 48 hours before assessing the frequency of apoptotic cells by flow cytometry for cleaved Caspase 3. b) Representative immunoblots showing decreased protein levels of ATRX, BLM, FANCD2 and RNASE H1 in CD19 DKO B cells nucleofected 48 hours earlier with the corresponding or CD4 Cas9 RNPs (Ctrl). The data is representative of at least 2 independent experiments. c) Representative flow cytometry plots quantifying percent apoptotic cells in Dfl and CD19 DKO B cells nucleofected with Cas9 RNPs. Y-axis, staining for cleaved Caspase 3; X-axis, forward scatter (FSC). d) Quantification of apoptosis, measured as percent of cells showing staining for cleaved Caspase 3, in cells nucleofected with Cas9 RNPs to Atrx, Blm, Fancd2, Rnase H from 3 biological replicates. e) Quantification of G-quadruplexes as NMM median fluorescence intensity (MFI) in Dfl and CD19 DKO B cells 48 hours after nucleofection with Cas9 RNPs. The signal is normalized to the NMM MFI of the same biological sample nucleofected with CD4 Cas9 RNPs (Ctrl) from 3 biological replicates. f) Quantification of DNA DSBs, assessed by γH2AX median fluorescence intensity (MFI) in Dfl and CD19 DKO B cells 48 hours after nucleofection with the indicated Cas9 RNPs from 3 biological replicates. The signal is normalized to the γH2AX MFI of the same biological sample nucleofected with control Cas9 RNP loaded with sgRNA against CD4 (Ctrl). g) Experimental design. Dfl and CD19 DKO B cells were treated for 2 days with the G-quadruplex stabilizing compound pyridostatin (PDS) prior to activation for 48 hours with LPS. h) Quantification of apoptosis, measured as percent of cells showing staining for cleaved caspase 3, in Dfl and CD19 DKO B cells cultured without (untreated) or with 10 µM PDS from 5 biological replicates and 3 independent experiments. i) Representative flow-cytometry plots quantifying percent apoptotic cells in Dfl and CD19 DKO B cells with or without PDS treatment. Y-axis, staining for cleaved caspase 3; X-axis, forward scatter (FSC). Statistical significance is calculated using two-way ANOVA. Error bars represent mean +/− standard error, * p value ≤0.05, ** p value ≤0.01, *** p value ≤0.0005 **** p value <0.0001.
Extended data figure 6. TET deficiency is associated with genome-wide accumulation of G-quadruplexes and R-loops.
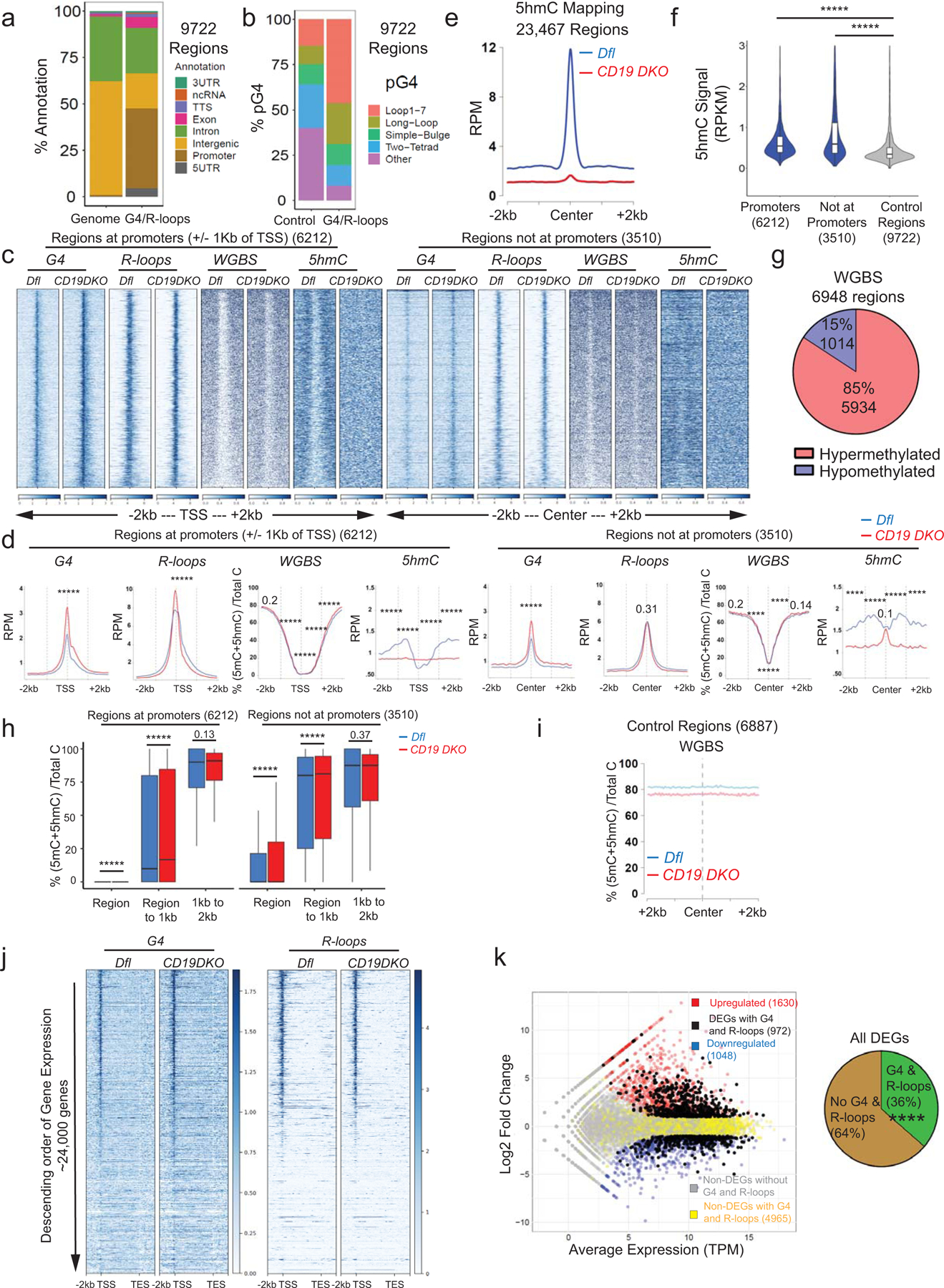
a) Genome annotations of regions enriched for G-quadruplexes (G4) and R-loops (right bar) compared to their representation in the mouse genome (mm10) (left bar). b) Relative representation of different classes of motifs predicted to form G-quadruplexes (pG4) in control regions selected randomly from the genome (left bar) and regions enriched for G-quadruplexes and R-loops (right bar). c) Heat maps showing enrichment (RPM) for G-quadruplexes and R-loops in CD19 DKO and control B cells. The signal is plotted in a +/− 2 kb window from the center of the regions ordered based on decreasing intensity from top to bottom in the entire 4 kb window. R-loop signal is plotted after background subtraction of MNase-alone control. d) Profile histograms showing the signals for G-quadruplexes (G4) (RPM, reads per million), R-loops (RPM), WGBS (percent of 5mC+5hmC/unmodified C) and 5hmC (RPM). The 9722 regions enriched for both G-quadruplexes and R-loops are divided into two categories – 6212 regions overlapping promoters (left panels) and 3510 regions not at promoters (right panels). Dashed grey lines indicate the center of the region and the 1 kb boundaries located on either side of the center. Blue and red lines show data from Dfl and CD19 DKO B cells, respectively. Asterisks represent statistical significance calculated by comparing the signals between Dfl and CD19 DKO B cells, either within the G-quadruplex and R-loop forming regions, the region to +/−1kb window or +/−1kb to 2kb window for respective datasets. e) Profile histograms showing the 5hmC signal in Dfl (blue) and CD19 DKO (red) B cells in 23,467 regions identified as enriched for 5hmC signal. f) Violin plots quantifying enrichment (RPKM) of 5hmC signal in Dfl B cells in the +/−1 kb from G-quadruplex and R-loop forming regions at promoters, non-promoter regions and control regions randomly located in euchromatin (Hi-C A genomic compartment) from 2 biological replicates. g) Pie chart showing the differentially methylated regions (DMRs) in CD19 DKO compared to control Dfl B cells. Of a total of 6948 DMRs identified by WGBS, 1014 (15%) showed reduced DNA methylation (hypomethylation) and 5934 (85%) showed increased DNA methylation (hypermethylation). h) Box and whisker plots quantifying percent of 5mC+5hmC/unmodified C (from WGBS) at and near the G4 and R-loop forming regions overlapping promoters and regions not overlapping promoters in Dfl (blue) and CD19 DKO (red) B cells from 2 biological replicates. The signal is plotted in three windows; window 1, within the G4 and R-loop regions; window 2, from region to +/− 1kb on either side and; window 3, +/−1kb to +/−2kb on either side. i) Percent of 5mC+5hmC/unmodified C (from WGBS) in random genomic regions of Dfl (blue) and CD19 DKO (red) B cells. j) Heatmaps of enrichment (RPM) for G-quadruplexes (left) and R-loops (right) in Dfl and CD19 DKO B cells, ordered in descending order of gene expression. k) MA plot (left) showing differentially expressed genes (DEGs) in CD19 DKO B cells. Red dots, upregulated DEGs; blue dots, downregulated DEGs; black dots, DEGs with G-quadruplexes and R-loops at their promoters (+/−1kb of TSS); yellow dots, non-DEGs with G-quadruplexes and R-loops at their promoters; grey dots, non-DEGs without G-quadruplexes and R-loops. The pie-chart (right) shows the percent of DEGs with (green) and without (brown) G-quadruplexes and R-loops at their promoters. Asterisks indicate statistical significance. Statistical significance is calculated using Kruskal-Wallis test and the ad hoc Dunn’s test in d), f) and h), Chi-square test in j). Boxes in box and whisker plots represent median (center) with 25th to 75 th percentile and whiskers represent maxima/minima. **** p value ≤0.0001 and ***** p value <0.000001.
Extended data figure 7. Genome-wide analysis of TET deficient B cells.
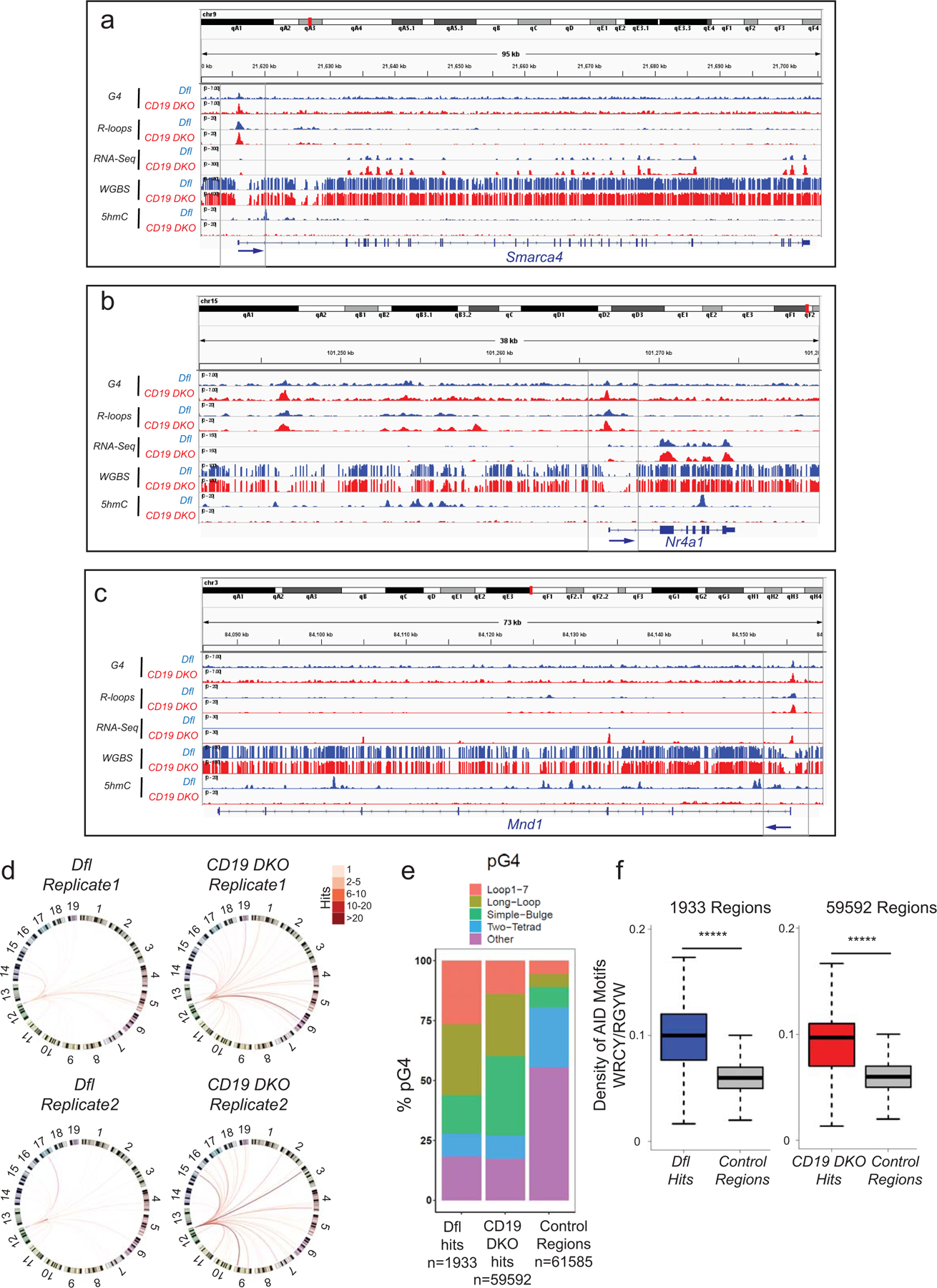
a)–c) Genome browser tracks showing the distribution of G-quadruplexes (G4), R-loops, RNA-Seq, WGBS, and 5hmC datasets for Dfl (blue tracks) and CD19 DKO (red tracks) B cells. Grey boxes indicate regions of interest. The blue arrows at the bottom show the location of the TSS and the direction of transcription. d) Circos plots to visually depict all translocations identified by HTGTS in Dfl and CD19 DKO replicates. Colored lines connect the Sµ bait with the translocation partner regions. Color scale represents the number of translocation partner regions identified in a 10 kb window. Translocations from two Dfl and CD19 DKO replicates are represented separately. e) Relative representation of different classes of motifs predicted to form G-quadruplexes (pG4) in control genomic regions selected randomly (right bar) and +/−300 bp from the center of translocation partner junctions identified from translocations in Dfl and CD19 DKO B cells (right bar). The numbers (n) of Dfl and CD19 DKO hits, and control regions are included in the plots. f) Density of AID motifs (WRCY/RGYW) in +/−300bp from the center of translocation partner junctions identified from translocations in Dfl and CD19 DKO B cells compared to control random regions in euchromatin (Hi-C A compartment) from 2 biological replicates. Statistical significance is calculated using the Wilcoxon signed rank test f), ***** p value <0.000001.
Extended data figure 8. DNMT1 deletion delays oncogenesis in TET-deficient mice.
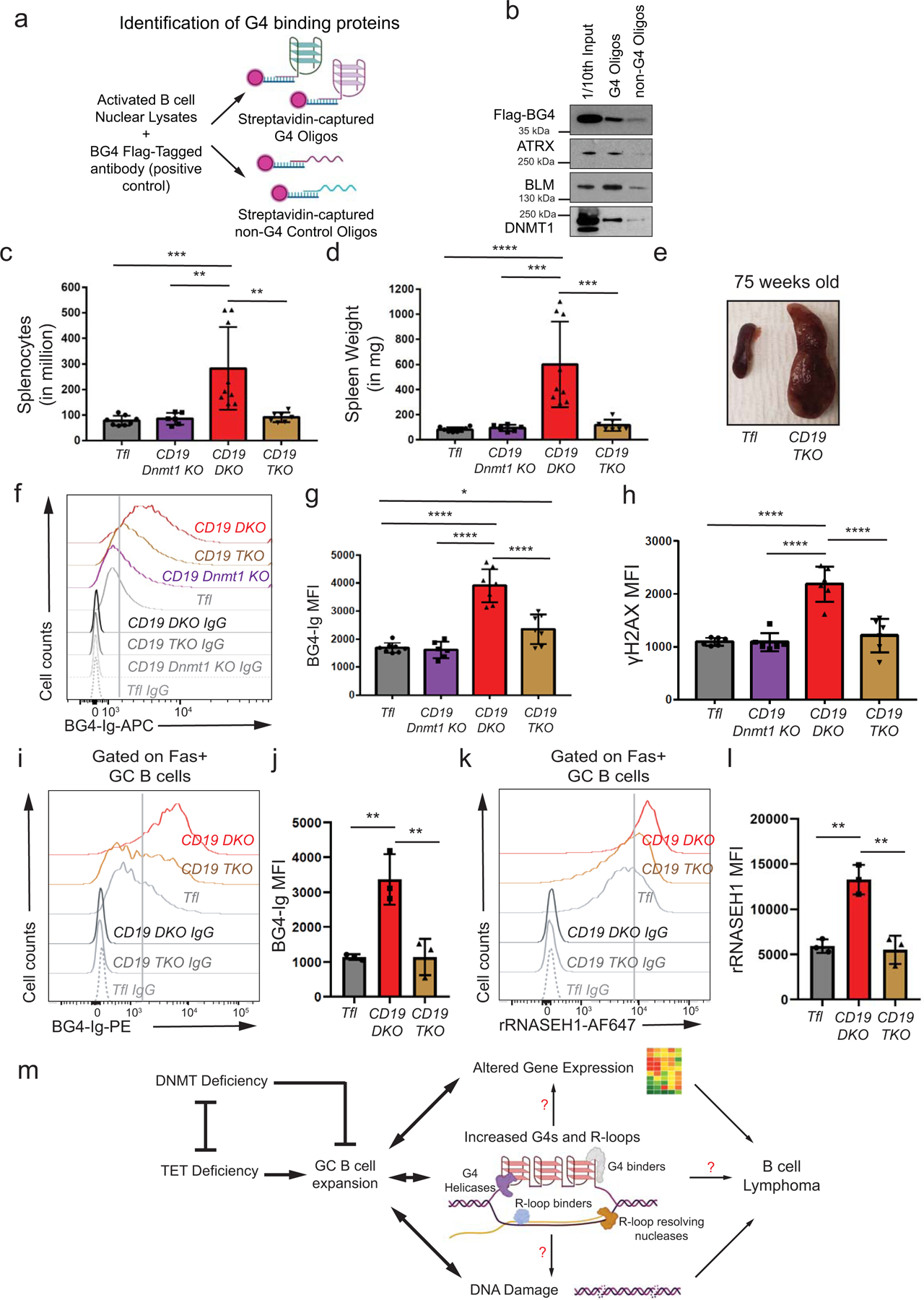
a) Diagrammatic representation of the strategy used to confirm G-quadruplex binding. Nuclear lysates of activated B cells were incubated with biotin-conjugated single stranded G4- or non-G4-forming control oligonucleotides (Oligos) captured using streptavidin beads. b) Immunoblots showing flag-tagged BG4 (positive control), ATRX, BLM and DNMT1 proteins. Left lane, 1/10th input from nuclear lysates (1/10th Input); middle lane, proteins pulled down with G4 forming oligonucleotides; and right lane, proteins pulled down with non-G4 control oligonucleotides. The data is representative of at least 2 independent experiments. c) - d) Quantification of c) cell numbers, d) spleen weights, of 10 week-old Tfl (grey), CD19 Dnmt1 KO (purple), CD19 DKO (red) and CD19 TKO (brown) mice from 5 independent experiments. e) Enlarged spleen of 75-week-old CD19 TKO mice compared with Tfl control mice. f) Flow cytometric detection of G-quadruplexes with BG4-Ig antibody or isotype IgG controls in B cells from Tfl (YFP−, grey), CD19 Dnmt1 KO (YFP+, purple), CD19 DKO (YFP+, red) and CD19 TKO (YFP+, brown) mice. g) – h) Quantification of median fluorescence intensity (MFI) of g) BG4-Ig signal, h) γH2AX signal from Tfl (YFP−), CD19 Dnmt1 KO (YFP+), CD19 DKO (YFP+) and CD19 TKO (YFP+) B cells from 4 independent experiments. i), k) Flow cytometric detection of i) G-quadruplexes with BG4-Ig antibody or isotype IgG controls and k) R-loops using V5-epitope-tagged recombinant RNASE H1 (rRNASE H1) or IgG controls in GC B cells (Fas+) from Tfl (YFP−, grey), CD19 DKO (YFP+, red) and CD19 TKO (YFP+, brown) mice. j), l) Quantification of median fluorescence intensity (MFI) of j) BG4-Ig signal and l) R-loops (rRNASE H1) signal in GC B cells (Fas+) from Tfl (YFP−, grey), CD19 DKO (YFP+, red) and CD19 TKO (YFP+, brown) mice from 3 independent experiments. m) Model proposing functional interplay between TET and DNMT activities to limit GC B cell expansion. TET deficiency in B cells leads to increased G4 and R-loop structures and is associated with altered gene expression, DNA damage and development of B cell lymphoma. Statistical significance is calculated using one-way ANOVA in c), d), g), h), j) and l). Error bars represent mean +/− standard deviation, * p value ≤0.05, ** p value ≤0.01, *** p value ≤0.0005 **** p value <0.0001.
Extended data figure 9. FACS gating strategy and original blots.
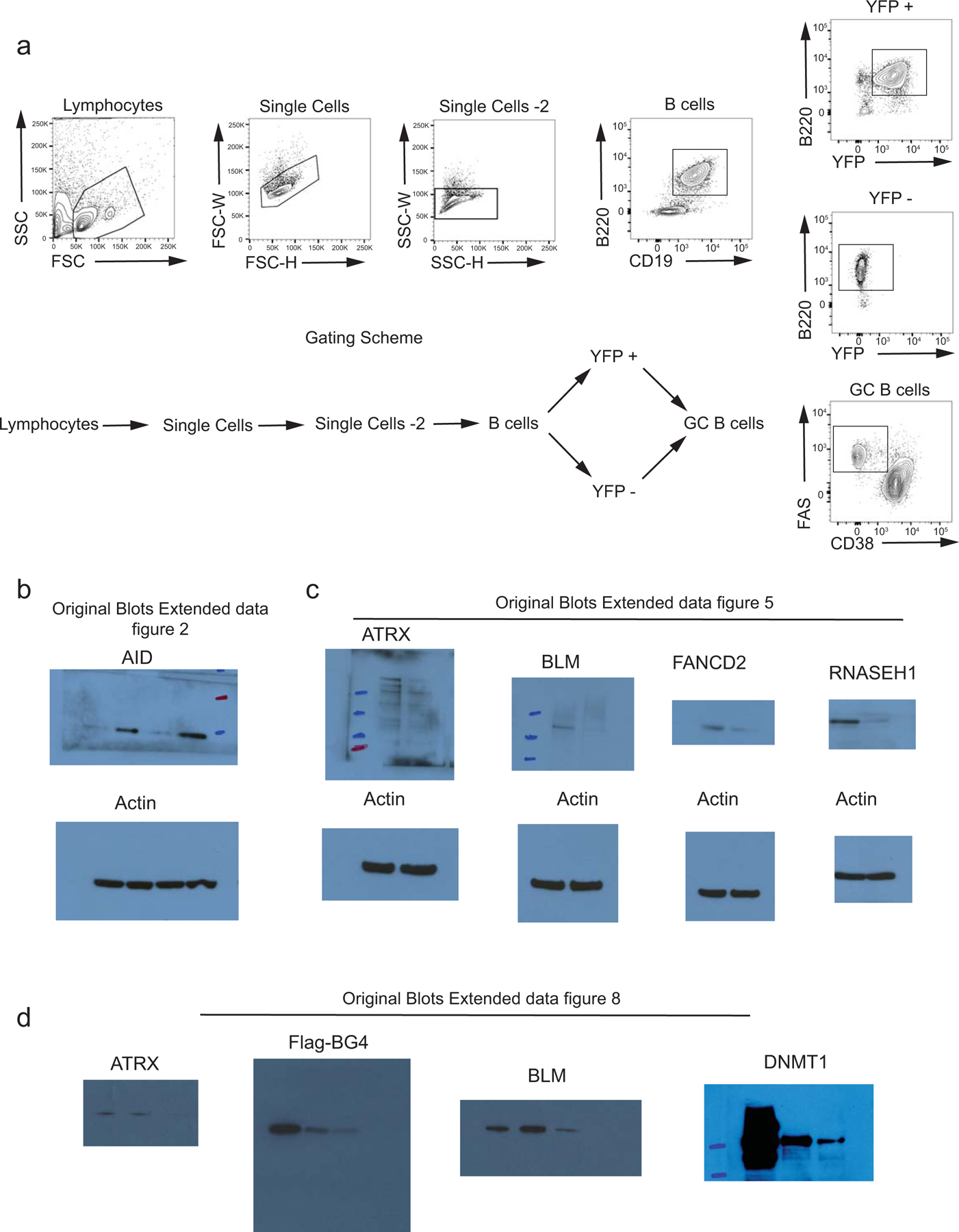
a) Sequential gating strategy used for the flow cytometry analysis. The respective gate names are mentioned in the corresponding figures. b)–e) scanned immunoblots for extended data figures 2 (b), 5 (c) and 8 (d).
Supplementary Material
Extended data table 1: Oligonucleotides and Primers
Extended data table 2: RNA-Seq analysis for differentially expressed genes
Extended data table 3: HTGTS hits
Acknowledgements.
We thank Drs. H. Yuita and I. Lopez-Moyado for generating the ERT2-Cre Tet1fl/fl Tet2fl/fl Tet3fl/fl, ERT2creTKO mice; Dr. D. Kitamura at the Tokyo University of Science for sharing the 40LB cells; Drs. U. Basu and B. Laffleur at Columbia University for help with the HTGTS protocol; our collaborators at Cambridge Epigenetix (UK) for providing the 5hmC mapping kits; the LJI Flow Cytometry Core team: C. Kim, D. Hinz, C. Dillingham, M Haynes, S. Ellis for help with cell sorting; and the LJI Next generation sequencing core members: J. Day, S. Alarcon, H. Dose, K. Tanaguay and A. Hernandez for help with sequencing. BD FACSAria II is supported by NIH (NIH S10OD016262, NIH S10RR027366) and our research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02–05CH11231. The NovaSeq 6000 and the HiSeq 2500 were acquired through the Shared Instrumentation Grant (SIG) Program (S10); NovaSeq 6000 S10OD025052 and HiSeq 2500 S10OD016262. K.S. acknowledges support from National Institutes of Health Grants DP2-NS105576. V.S. was supported by Leukemia and Lymphoma Society Postdoctoral Fellowship (grant ID: 5463–18) and currently by a K99/R00 award from National Cancer Institute (grant ID: CA248835). D.S.C. and E.G.A were supported by University of California Institute for Mexico and the United States and El Consejo Nacional de Ciencia y Tecnología (UCMEXUS/CONACYT) pre-doctoral fellowship Fellowships. This work is supported by the National Institutes of Health (NIH) grants R35 CA210043, R01 AI109842 and AI128589 to A.R and, K99/R00 CA248835, research funds from LLS grant 5463–18 and the Tullie and Rickey families SPARK award from LJI to V.S.
Footnotes
Competing Interests Statement. The Authors declare no competing financial interests.
References
- 1.Tahiliani M et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ko M et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468, 839–843, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito S et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lio CW et al. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orlanski S et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc Natl Acad Sci U S A 113, 5018–5023, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominguez PM et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov 8, 1632–1653, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lio CJ et al. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosikiewicz W et al. TET2 deficiency reprograms the germinal center B cell epigenome and silences genes linked to lymphomagenesis. Sci Adv 6, eaay5872, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lio CJ et al. TET methylcytosine oxidases: new insights from a decade of research. J Biosci 45 (2020). [PMC free article] [PubMed] [Google Scholar]
- 10.Wu H & Zhang Y Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pastor WA, Aravind L & Rao A TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14, 341–356, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmussen KD & Helin K Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30, 733–750, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cimmino L, Abdel-Wahab O, Levine RL & Aifantis I TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell 9, 193–204, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y & Rao A Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet 30, 464–474, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko M, An J & Rao A DNA methylation and hydroxymethylation in hematologic differentiation and transformation. Curr Opin Cell Biol 37, 91–101, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lio CJ, Yuita H & Rao A Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood 134, 1487–1497, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reddy A et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481–494 e415, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitz R et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 378, 1396–1407, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapuy B et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24, 679–690, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kafer GR et al. 5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability. Cell Rep 14, 1283–1292, (2016). [DOI] [PubMed] [Google Scholar]
- 21.Kharat SS et al. Degradation of 5hmC-marked stalled replication forks by APE1 causes genomic instability. Sci Signal 13, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crossley MP, Bocek M & Cimprich KA R-Loops as Cellular Regulators and Genomic Threats. Mol Cell 73, 398–411, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skourti-Stathaki K & Proudfoot NJ A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev 28, 1384–1396, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansel-Hertsch R, Di Antonio M & Balasubramanian S DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat Rev Mol Cell Biol 18, 279–284, (2017). [DOI] [PubMed] [Google Scholar]
- 25.Rhodes D & Lipps HJ G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res 43, 8627–8637, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sauer M & Paeschke K G-quadruplex unwinding helicases and their function in vivo. Biochem Soc Trans 45, 1173–1182, (2017). [DOI] [PubMed] [Google Scholar]
- 27.De Magis A et al. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc Natl Acad Sci U S A 116, 816–825, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray LT, Vallur AC, Eddy J & Maizels N G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat Chem Biol 10, 313–318, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miglietta G, Russo M & Capranico G G-quadruplex-R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res, (2020). [DOI] [PMC free article] [PubMed]
- 30.Chedin F Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet 32, 828–838, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niehrs C & Luke B Regulatory R-loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol 21, 167–178, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanz LA et al. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol Cell 63, 167–178, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.An J et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun 6, 10071, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cimmino L et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol 16, 653–662, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsagaratou A et al. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nat Immunol 18, 45–53, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao SQ et al. DNA G-quadruplex structures mold the DNA methylome. Nat Struct Mol Biol 25, 951–957, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biffi G, Tannahill D, McCafferty J & Balasubramanian S Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5, 182–186, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller S et al. Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells. Org Biomol Chem 10, 6537–6546, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabharwal NC et al. N-methylmesoporphyrin IX fluorescence as a reporter of strand orientation in guanine quadruplexes. FEBS J 281, 1726–1737, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicoludis JM et al. Optimized end-stacking provides specificity of N-methyl mesoporphyrin IX for human telomeric G-quadruplex DNA. J Am Chem Soc 134, 20446–20456, (2012). [DOI] [PubMed] [Google Scholar]
- 41.Chen L et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol Cell 68, 745–757 e745, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan Q, Shields EJ, Bonasio R & Sarma K Mapping Native R-Loops Genome-wide Using a Targeted Nuclease Approach. Cell Rep 29, 1369–1380 e1365, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crossley MP et al. Catalytically inactive, purified RNase H1: A specific and sensitive probe for RNA-DNA hybrid imaging. J Cell Biol 220, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boguslawski SJ et al. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods 89, 123–130, (1986). [DOI] [PubMed] [Google Scholar]
- 45.Nojima T et al. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat Commun 2, 465, (2011). [DOI] [PubMed] [Google Scholar]
- 46.Lopez-Moyado IF et al. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc Natl Acad Sci U S A 116, 16933–16942, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansel-Hertsch R et al. G-quadruplex structures mark human regulatory chromatin. Nat Genet 48, 1267–1272, (2016). [DOI] [PubMed] [Google Scholar]
- 48.Song CX et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol 29, 68–72, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu J et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat Protoc 11, 853–871, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaknovich R et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood 118, 3559–3569, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanaka S et al. Tet2 and Tet3 in B cells are required to repress CD86 and prevent autoimmunity. Nat Immunol 21, 950–961, (2020). [DOI] [PubMed] [Google Scholar]
- 52.Young RM et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A 112, 13447–13454, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nambiar M et al. Formation of a G-quadruplex at the BCL2 major breakpoint region of the t(14;18) translocation in follicular lymphoma. Nucleic Acids Res 39, 936–948, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rabkin CS, Hirt C, Janz S & Dolken G t(14;18) Translocations and risk of follicular lymphoma. J Natl Cancer Inst Monogr, 48–51, (2008). [DOI] [PMC free article] [PubMed]
- 55.Qiao Q et al. AID Recognizes Structured DNA for Class Switch Recombination. Mol Cell 67, 361–373 e364, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu YZ et al. Activation-induced cytidine deaminase localizes to G-quadruplex motifs at mutation hotspots in lymphoma. NAR Cancer 2, zcaa029, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yewdell WT et al. A Hyper-IgM Syndrome Mutation in Activation-Induced Cytidine Deaminase Disrupts G-Quadruplex Binding and Genome-wide Chromatin Localization. Immunity, (2020). [DOI] [PMC free article] [PubMed]
- 58.Zhang W & Xu J DNA methyltransferases and their roles in tumorigenesis. Biomark Res 5, 1, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat Commun 10, 943, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu H et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun 8, 14432, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kang J et al. Simultaneous deletion of the methylcytosine oxidases Tet1 and Tet3 increases transcriptome variability in early embryogenesis. Proc Natl Acad Sci U S A 112, E4236–4245, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ko M et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A 108, 14566–14571, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chedin F, Hartono SR, Sanz LA & Vanoosthuyse V Best practices for the visualization, mapping, and manipulation of R-loops. EMBO J 40, e106394, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konig F, Schubert T & Langst G The monoclonal S9.6 antibody exhibits highly variable binding affinities towards different R-loop sequences. PLoS One 12, e0178875, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xi Y & Li W BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10, 232, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bolotin DA et al. Antigen receptor repertoire profiling from RNA-seq data. Nat Biotechnol 35, 908–911, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krzywinski M et al. Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kieffer-Kwon KR et al. Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Mol Cell 67, 566–578 e510, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended data table 1: Oligonucleotides and Primers
Extended data table 2: RNA-Seq analysis for differentially expressed genes
Extended data table 3: HTGTS hits
Data Availability Statement
All genome-wide sequencing datasets have been deposited to Gene Expression Omnibus (GEO) repository, accession number GSE161463. Any data and reagents will also be made available upon request.