

Abstract
Pediatric patients with congenital heart defects (CHD) often present with heart failure from increased load on the right ventricle (RV) due to both surgical methods to treat CHD and the disease itself. Patients with RV failure often require transplantation, which is limited due to lack of donor availability and rejection. Previous studies investigating the development and in vitro assessment of a bioprinted cardiac patch composed of cardiac extracellular matrix (cECM) and human c-kit + progenitor cells (hCPCs) showed that the construct has promise in treating cardiac dysfunction. The current study investigates in vivo cardiac outcomes of patch implantation in a rat model of RV failure. Patch parameters including cECM-inclusion and hCPC-inclusion are investigated. Assessments include hCPC retention, RV function, and tissue remodeling (vascularization, hypertrophy, and fibrosis). Animal model evaluation shows that both cell-free and neonatal hCPC-laden cECM-gelatin methacrylate (GelMA) patches improve RV function and tissue remodeling compared to other patch groups and controls. Inclusion of cECM is the most influential parameter driving therapeutic improvements, with or without cell inclusion. This study paves the way for clinical translation in treating pediatric heart failure using bioprinted GelMA-cECM and hCPC-GelMA-cECM patches.
1. Introduction
Congenital heart defects (CHD) are malformations in the heart during fetal development resulting in life-threatening conditions in pediatric patients.1–4 CHD is the most common congenital defect in the United States with approximately 40 000 new CHD cases annually.4 While therapeutic advances have improved the mortality rates of patients with CHD, complex CHD cases often result in heart failure even with surgical treatments. Specifically, right ventricular (RV) heart failure is a significant clinical problem in the pediatric population.5–9 Increased load on the RV caused by CHD or surgical repair for CHD results in a rapid progression to RV failure due to the limited ability of the RV to respond to changes in afterload.5–9 RV failure is characterized by a variety of factors leading to decreased cardiac output, such as decreased tricuspid annular plane systolic excursion (TAPSE), increased RV dimensions, decreased vascular density, cardiomyocyte hypertrophy, and increased tissue fibrosis.5–12 For patients suffering from RV dysfunction and failure, therapy is limited to transplantation, which is hindered by donor availability and tissue rejection. Reparative therapies to treat RV failure have been explored, mainly using cellular and biomaterial treatment in restoring tissue function or maintaining cardiac output while patients bridge to transplantation.11–13
Injectable cardiac extracellular matrix (cECM) therapy has shown initial safety and feasibility in a phase I clinical trial for treating adult patients post-myocardial infarction.14 Similarly, injectable human c-kit + progenitor cell (hCPCs) therapy has been shown to be safe in small clinical studies and hCPC therapeutic assessment is ongoing in children with hypoplastic left heart syndrome (NCT03406884).15,16 However, there are significant limitations to injectable therapy that may reduce the therapeutic effect overall for treating pediatric patients suffering from CHD and RV dysfunction.17,18 Specifically, injectable cell therapy is limited by early wash-out, high cell death due to the hostile injection environment, and limited cellular functional control.17–19 Additionally, injectable biomaterial therapy is a localized treatment requiring multiple injections, which may not be useful for a global problem such as RV failure, and suffers from limited material control post-injection.20,21
Cardiac patches are solid scaffold devices that overcome limitations in injectable therapy by incorporating biomaterial properties, retaining cells, controlling cellular functionality, and allowing for global treatment with the potential for off-the-shelf therapies.22–24 A previous study investigated the in vitro reparative potential of hCPC-cECM patches that were generated through bioprinting by the incorporation within gelatin methacrylate (GelMA).24 The neonatal hCPC-GelMA-cECM patches showed improved angiogenic potential, improved cell differentiation, device printability, and myocardial-matching mechanical properties compared to patches without cECM. These in vitro results showed that combined hCPC-GelMA-cECM patches could possibly repair the failing RV through combined biomaterial therapy and paracrine factor release. The current study investigates the therapeutic effect of hCPC-GelMA-cECM patches in vivo in a juvenile rat RV heart failure model. Several patch parameters and designs are investigated, including cECM-incorporation and hCPC-inclusion. Cellular retention in patches, functional cardiac outcomes, and tissue-level properties are investigated four weeks after patch implantation to understand and assess the effectiveness of bioprinted hCPC-GelMA-cECM patches for treating pediatric RV failure.
2. Materials and methods
2.1. Materials
Gelatin methacrylate (GelMA) was purchased from CellINK (Gothenburg, Sweden). Triethanolamine (TEOA), 1-vinyl-2-pyrrolidione (NVP), HEPES sodium salt, Triton X-100, sodium dodecyl sulfate, ethanol, pepsin, sucrose, bovine serum albumin (BSA), picrosirius red (direct red 80), aqueous picric acid, and neutral buffered formalin (NBF) 10% were purchased from Sigma-Aldrich (MO, United States). Hams F-12 and penicillin streptomycin (pen–strep) were purchased from Corning Cellgro (NY, United States). Eosin Y sodium salt was purchased from TCI (MA, United States). Nordson EFD 30cc barrels, pistons, and 27-gauge plastic tips were purchased from TEK products (MN, United States). Standard fetal bovine serum (FBS) was purchased from Hyclone (PA, United States). L-Glutamine was purchased from MP Biomedicals (OH, United States). Cell culture dishes were purchased from Cellstar (PA, United States). Dynal magnetic beads, 1,1-dioctadecyl-3,3,3',3'-tetramethylindotricardbocyanine iodide (DiR), and Alexa Fluor 790 carboxylic acid succinimidyl ester (AF790) were purchased from Invitrogen (CA, United States). Phosphate buffered saline (PBS), Tissue-TEK OCT compound, and isopropanol was purchased from Fisher Scientific (NH, United States). Collagenase type 2 (300 U mL−1) was purchased from Worthington (NJ, United States). Anti-c-kit H300 antibodies and Ku86-AF647 antibodies were purchased from Santa Cruz (TX, United States). Bovine fibroblast growth factor (bFGF) was purchased from Stem Cell Technologies (Vancouver, Canada). Fluorescein Griffonia Simplicifolia Lectin I Isolectin B4, Wheat Germ Agglutinate Rhodamine (WGA), and Vectashield antifade mounting solution with DAPI were purchased from Vector Labs (CA, United States). Isoflurane was purchased from Piramal Healthcare (Mumbai, India). Prolene was purchased from Ethicon (NJ, United States). Male adolescent (6–8 weeks old) athymic rats (Crl:NIH-Fox1rnu) were obtained from Charles River Laboratories (Wilmington, MA). Cytoseal mounting medium was purchased from VWR International (Radnor, PA). Histoclear II was purchased from National Diagnostics (Atlanta, GA).
2.2. Human cardiac progenitor cell isolation and culture
All human studies were performed under guidelines set forth by the Declaration of Helsinki and were approved by the Institutional Review Board at Children’s Healthcare of Atlanta and Emory University. As these samples were de-identified and considered waste tissue, the board determined the study was exempt from informed consent. Cells expressing c-kit were isolated from the atrial appendage as previously described.11,24,27,45 In short, right atrial appendage tissue was obtained from pediatric patients aged 0–1 week (neontate) undergoing heart surgeries due to congenital heart diseases. The tissue was enzymatically degraded using collagenase type II (1 mg mL−1) at 37 °C, 5% CO2 for 30 minutes, centrifuged to pellet, and magnetically sorted for c-kit marker. Separated c-kit + cells were expanded, and the expression of c-kit in the cell population was measured by flow cytometry to ensure they were at least 90% positive. Cells from three donors were either pooled at the first passage or cultured separately and combined before printing. hCPCs were grown in T-75 cell culture treated dishes with culture media for expansion, where media consisted of Ham’s F-12 media supplemented with 1× pen– strep, 1% L-glutamine, 10% FBS, and 10 ng mL−1 bFGF.
2.3. Cardiac extracellular matrix isolation and characterization
All tissue is collected after euthanasia for other procedures and is thus exempt from institutional approvals. Decellularized porcine ventricular extracellular matrix (cECM) was generated and processed as previously described.24,29,30,49 Briefly, porcine ventricular tissue was decellularized using a 1% solution of SDS for 4–5 days and milled into a fine powder. Then, the cECM was processed into liquid form by partial digestion with pepsin (1 mg mL−1), pH and salt were adjusted, and the material was aliquoted, lyophilized, and stored at −80 °C before use.
2.4. Bioink preparation, bioprinting, and patch formation
Bioinks formulation and patch fabrication were implemented as previously described.24 GelMA bioinks were prepared by mixing GelMA (final concentration 5% w/v), Eosin Y (100 μM), NVP (0.75% v/v), TEOA (0.3% v/v), and HEPES buffer (15 mM). In the case of GelMA-cECM bioinks, the HEPES in the above formulation was replaced by cECM (final concentration 8 mg mL−1) solution in HEPES (15 mM). Bioink pH was adjusted to pH 7.4. For the inclusion of cells, neonatal hCPCs (passage 6–15, pooled or combined across at least three donors) were removed from cell culture plates and labeled with DiR following manufacturer instructions.11,12 After labeling, cells were mixed with 1 mL solution of the bioink solution, producing a final concentration of 3 million cells per mL. All bioink solutions, with or without cells, underwent similar printing protocols. 1 mL of bioink was deposited into sterilized 30 cc printer barrels joined with sterile 27-gauge plastic needle tips. The barrels were put in the low-temperature head of the bioprinter (EnvisionTEC 3D-bioplotter Developer Series), and the bioink was allowed to polymerize for 10 minutes 14 °C. Patches were printed onto a glass slide platform at room temperature, using a pressure 0.7–1 bar and speed of 10 mm s−1. Patches were 10 mm in diameter and 0.6 μm thick, which was printed in 3 layers, with an infill pattern of 90° grids with 0.5 mm spacing. Eight patches were printed at once. CAD models of the patches printed were generated using SOLIDWORKS and imported to the printing control system through the Bioplotter RP program. Following printing, patches were exposed to white light (Braintree Scientific) at 4 °C for 5 minutes to allow for radical polymerization. The patches were removed from glass slides and placed in cell culture media. All patches were kept in cell culture incubators for several hours before surgery to allow for cECM polymerization after GelMA radical polymerization.
2.5. Rat pulmonary artery banding, patch surgery, and echocardiogram follow-up
All studies were done in accordance with the Guide for the Care and Use of Laboratory Animals and was approved by the Emory Institutional Animal Care and Use Committee. RV dysfunction was induced in male juvenile immunocompromised (nude, athymic) rats by placing a pulmonary artery band (PAB) to increase RV afterload.11,12 This model was created with the assistance of the Emory and Children’s Animal Physiology Core and has been used by our group and others.11,12 Briefly, an 18-gauge needle was placed alongside the pulmonary artery, a suture was tied around the pulmonary artery and needle, and the needle was removed. Pulmonary artery constriction leads to a gradual development of clinically relevant RV heart dysfunction. Serial echocardiography was employed to follow the development of RV dysfunction. RV dysfunction was established by two weeks post-banding and is characterized by significantly reduced tricuspid annular plane systolic excursion (TAPSE) and RV hypertrophy.5,11,12 Following RV failure, surgical treatment patch groups (bare GelMA, bare GelMA-cECM, neonatal hCPC-GelMA, and neonatal hCPC-GelMA-cECM,) were attached to the right ventricular epicardium through a suture. Animals were randomized to treatment groups by employing a random assignment program (via Microsoft Excel) using variables of number of surviving animals and number of target groups needed/remaining. Patch surgery was implemented as previously described. Briefly, rats were anesthetized with 2% isoflurane, intubated, and placed on a ventilator (Hallowell Emc Microvent 1). Following thoracotomy, the pericardial sac was very carefully exposed and pulled back. Patches were attached to the RV using a suture. Sham rats were not given full PAB surgery or any patch surgery. PAB control rats were given PAB surgery and thoracotomy after RV failure development but received no patch. Rats were followed longitudinally with echocardiographic exams performed at 0, 2, and 4 weeks post attachment to monitor cardiac function as previously described.11 Briefly, transthoracic echocardiography was evaluated using a Vevo 2100 digital high-frequency ultrasound system (Fujifilm VisualSonics) equipped with an MS250 probe. TAPSE was measured in the apical four-chamber view in M-mode. RV end-systolic and end-diastolic areas and RA-area were measured in the apical four-chamber view in B-mode. Echocardiographic measurements were used to determine changes in TAPSE, end-diastolic dimensions, end-systolic dimensions, and right aria area. TAPSE data was normalized to pre-PAB TAPSE values within individual rats. A total of 72 rats underwent either Sham or PAB surgery. Following surgery, 36 rats exhibited no change in TAPSE or other cardiac parameters post-PAB (if in the PAB control or treatment groups) or died just before implantation and were excluded from data analysis and subsequent surgeries. All patch surgeries, data acquisition, and raw data analysis were blinded before final grouping and analysis.
2.6. In vivo cell retention imaging and analysis
Rats were imaged for cell retention in vivo by evaluating the fluorescence signal from DiR loaded within cells as previously described.11,12 Rats were imaged at 0, 14, and 28 days after patch implantation using an IVIS Spectrum in vivo imager (Perkin Elmer). DiR fluorescence from the hearts was measured as radiant efficiency and normalized within individual rats as percentage retention (given as 100% on day 0) over time.
2.7. Animal sacrifice and heart tissue fixation/freezing
Rats were sacrificed on day 28 after patch implantation via carbon dioxide. Hearts were removed en bloc, washed with sterile 1× PBS, and fixed with 10% NBF for 4–5 hours. Following fixation, hearts were washed with 1× PBS to remove excess NBF, then submerged in 30% sucrose overnight to ensure cryoprotection.11,12 Following cryoprotection, hearts were washed with 1× PBS to remove excess sucrose and immediately frozen in OCT within tissue blocks using liquid nitrogen chilled isopropanol. Hearts were retained at −80 °C before tissue sectioning and analysis.
2.8. Immunohistochemistry methods and analysis
Cryopreserved heart tissues were sectioned into 10 μm thick sections and mounted on charged slides. Tissue sections are allowed to air dry for 10 minutes, followed by fixation with 10% NBF for 5 minutes. Sections were then washed with a wash buffer composed of 1× PBS and 0.1% Triton X-100 3 times for 5 minutes each. Sections were blocked in blocking buffer composed of 1× PBS, 0.1% Triton X-100, and 5% BSA for 30 minutes in a dark, humidified chamber. Following blocking, sections were incubated with a dye/antibody cocktail overnight at 4 °C or for 90 minutes at room temperature.11,12,33–36 The dye/antibody cocktail was composed of isolectin–fluorescein (1 : 100 dilution, Griffonia simplicifolia lectin I isolectin B4, fluorescein), WGA-rhodamine (1 : 300 dilution, wheat germ agglutinin, rhodamine), and monoclonal Ku86-AF647 (1 : 500 dilution) suspended in blocking buffer.11,12 Sections were then washed with wash buffer three times for 5 minutes each. Vectashield antifade mounting medium with DAPI was added to each slide, and a coverslip was placed over the section, followed by a 10-minute incubation to allow DAPI to interact with cell nuclei. Slides were imaged on an Olympus IX81 FluoView FV1000 confocal microscope. Each tissue section was imaged at multiple locations within the RV for all groups and adjacent to the implanted patch for patch groups. Sequential imaging of different fluorescence channels was employed to reduce nonspecific dye bleed-through. Vessel density was analyzed by isolectin staining of EC tubule formation. The number of vessels per 0.2 mm2 area was counted for each tissue section across multiple replicates and averaged within patch or control groups. For patch-adjacent vessel analysis, the number of vessels in the area adjacent to patches was counted, divided by the evaluated area, converted to vessels per 0.2 mm2 area, and averaged within groups. Adjacent area was defined as an 18–20 mm2 rectangular (2 mm width inwards to tissue, 9–10 mm length across tissue) area of the tissue directly adjacent to patches. Cardiomyocyte hypertrophy was analyzed by WGA staining of cardiomyocyte cell walls. Cardiomyocyte cross-sectional areas were evaluated for each tissue section across multiple replicates and averaged within patch or control groups. Cell retention was analyzed by co-expression of human marker Ku86 and DAPI within cardiac patches.
2.9. Fibrosis staining and analysis
Cryopreserved heart tissues were sectioned into 10 μm thick sections and mounted on charged slides. Tissue sections are allowed to air dry for 10 minutes, followed by fixation with 10% NBF for 5 minutes. Sections were rinsed with deionized water for 3 minutes, rehydrated with 1× PBS for 5 minutes, and stained with a solution of Picrosirius red (1 mg mL−1) in aqueous picric acid for 90 minutes.11,12,37,38 Following staining, slides were rinsed with acidified water for 1 minute, rinsed with deionized water for 3 minutes, and dehydrated with 70% ethanol for 30 seconds. Finally, slides were cleared with Histoclear for 1 minute and mounted with Cytoseal. Slides were imaged on a Hamamatsu NanoZoomer S210 slide scanner (Hamamatsu Photonics). The fibrotic area was quantified using color thresholding on ImagJ (G. Landini software), and % fibrosis was calculated as the fibrotic area/total area of the RV free wall for each slide across replicates and groups.11,12
2.10. Statistics
Numerical data are the mean ± SEM. All data were analyzed using one-way ANOVAs with Tukey’s multiple comparison post-test across all groups. Sample size (n) was 3–7 for all samples (n = 5–7 for cardiac functional data and n = 3–7 for tissue analysis data). In cases where both days and groups were involved, data were compared across groups within the same day and across days within the same group to determine significant differences. All animal-group randomization, patch surgical implantation, raw data acquisition, and raw data analysis were performed blinded before final grouping and data analysis.
3. Results and discussion
3.1. In vivo cell retention
Studies from our laboratory and others have employed a juvenile rat model of RV failure and dysfunction using a pulmonary artery banding (PAB) method to increase RV afterload.11,12 Briefly, juvenile immunocompromised rats (nude, athymic) had a band placed on their pulmonary artery. This method reduced blood flow through the pulmonary artery and caused pressure overload in the RV, resulting in RV hypertrophy and gradual development of RV failure due to the limited ability of the RV to respond to changes in afterload, mimicking the development of RV failure in certain CHD.5,6,11,12 After PAB surgery, rats were followed with echocardiography for two weeks to assess the development of RV dysfunction by a reduction in TAPSE, a measure of global RV function.11,12,25,26 After confirmation of RV dysfunction, patches were randomized across different groups and were surgically attached to hearts directly. Patches were adhered centrally onto the beating RV of each rat using a single suture per patch, covering the majority of the RV wall. For patch mechanical properties and micro-graphical description, reference the previous study by our laboratory on patch design and development.24 Test groups included sham controls, PAB controls that did not receive patches, bare GelMA patches, bare GelMA-cECM patches, neonatal hCPC-GelMA patches, and neonatal hCPC-GelMA-cECM patches. Rats were followed throughout the 4-week therapy timeline with echocardiographic and in vivo fluorescent imaging methods. After four weeks, animals were sacrificed, the hearts were explanted and fixed/frozen en bloc, and the tissue was further analyzed.
Previous studies from our laboratory and others have shown that injected hCPCs are quickly cleared from the tissue, and only 0–30% of hCPCs are retained after 28 days of therapy depending on the method of modulation.11,12,17,27 All cells within patches were labeled with a fluorescent membrane dye, allowing for tracking of cell retention during therapy similar to previous studies.11,12 This methodology has previously shown no detrimental effects on the cells themselves.11,12 The cells were tracked over 28 days (4 weeks) post-implantation with fluorescence, and the radiant efficiency of the fluorescent signal at day 0 after patch implantation was used to normalize signals from days 14 and 28, as seen in Fig. 1. Rats across both GelMA and GelMA-cECM cell-laden patch groups exhibited a clear, strong fluorescent signal at the anatomical location of the heart, as seen by Fig. 1A. Quantification of cell retention, seen in Fig. 1B, showed that fluorescent signal remained consistent throughout 28 days of therapy without any significant differences between days or patch groups. These results show that neonatal hCPCs were retained without significant loss to the circulation or tissue environment throughout therapy. Additionally, there were no differences in retention across material types, indicating that the effectiveness in retention was not an additional confounding variable in the assessment of patch therapeutic benefit across biomaterial groups. Overall, these results showed that cells were retained during therapy. However, the cell tracking method employed in this study may result in residual dye signal originating from tissue if implanted cells do not survive post-implantation. Therefore, in vivo measurements were complemented by tissue analysis to support retention. Explanted tissue sections were analyzed using a human-specific Ku86 marker and nucleus staining with DAPI.11,12,28 As seen in Fig. 1C, fluorescent images focused on neonatal hCPC-GelMA-cECM patches attached to tissue sections showed clear expression of Ku86 (magenta) and nuclei (blue). Cells were retained within patches after 28 days of therapy, reinforcing the in vivo imaging results. While rat imaging data showed a robust fluorescent signal indicating a large cell number, the tissue staining data showed fewer cells. The reduced prevalence of cells at 28 days in the tissue analysis may be due to the direct staining methodology used (as opposed to an amplified immunohistological method), which captures only strongly bound fluorescent antibodies. In addition, small, thin portions of patches captured per tissue section and hCPCs distributed in low cell density along a 3D distribution within patches results in very few cells captured per section, making direct numerical comparison between full rat imaging and tissue staining ineffective. Even with these limitations, human cells were observed to indicate cell retention over 28 days. These studies support the hypothesis that patch therapy retains cells during surgical treatment with minimal loss of cells, compared to injectable therapies.
Fig. 1.
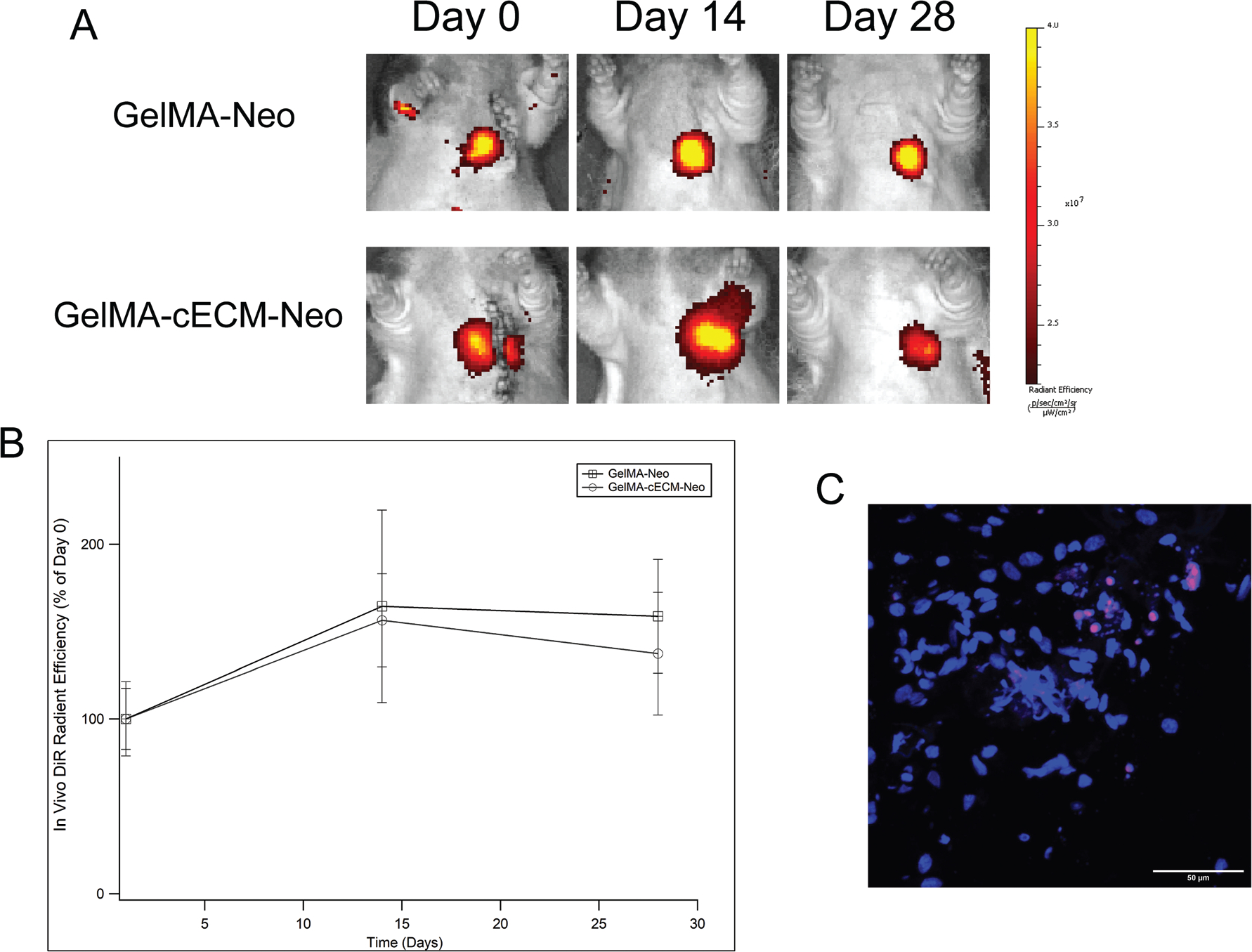
In vivo cell retention. (A) Radiant efficiency signal of rats across neonatal hCPC-patch groups over 28 days of therapy. (B) Quantified fluorescent signal across neonatal hCPC-patch groups over 28 days of therapy, presented as radiant efficiency (% of day 0) normalized to day 0 measurements within rats. n = 3–4 for all time-points. (C) Immunohistological staining of neonatal hCPC-GelMA-ECM patch within tissue sections (image is of patch only), where human marker Ku86 (magenta) and nuclei marker DAPI (blue) is co-localized. Scale bar for stained images is 50 μm.
3.2. Cardiac functional assessment
Rats were evaluated throughout therapy by echocardiographic measurements for TAPSE, right ventricular end-diastolic dimension (EDD), right ventricular end-systolic dimensions (ESD), and right atrium area (RA-Area). TAPSE is an indicator of RV global function and has been employed in previous studies as a critical metric to assess therapeutic effects.25,26 In Fig. 2A, TAPSE is presented across 0, 2, and 4 weeks post-patch implantation. Immediately before patch implantation (week 0 values), all groups showed a significantly lower TAPSE than Sham controls, indicating that all rats had developed RV dysfunction post-PAB surgery. Four weeks post-implantation, GelMA-cECM and neonatal hCPC-GelMA-cECM groups showed no significant TAPSE difference compared to Sham controls, indicating similar RV function to healthy controls. Additionally, the neonatal hCPC-GelMA-cECM group showed significant improvements in TAPSE within the patch group between week 0 and week 4. While there were no significant improvements with GelMA patches, both cECM-laden patches were effective in improving global RV function to values that were not statistically different to Sham values.
Fig. 2.
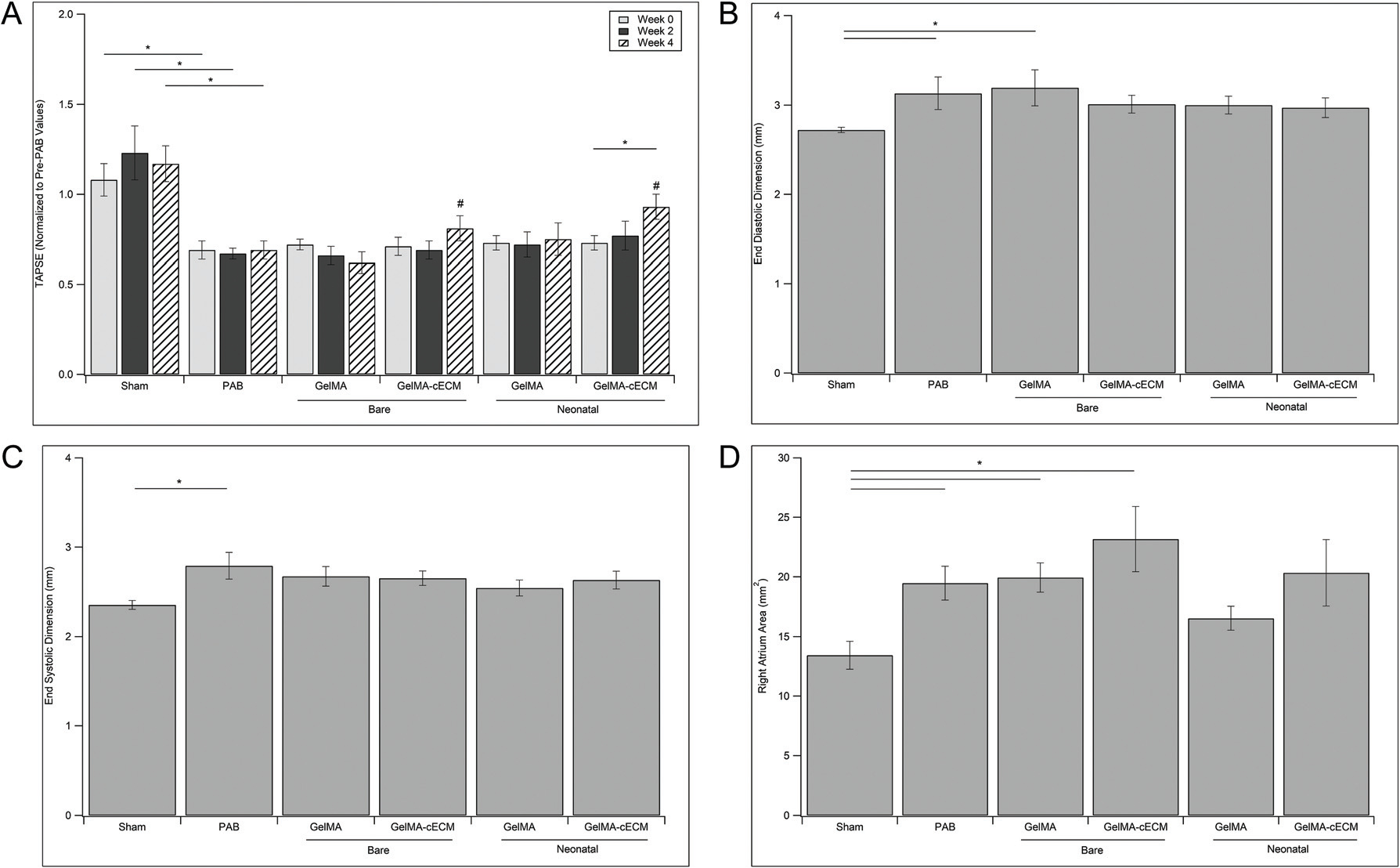
Echocardiographic measurements. (A) TAPSE measurements for control, bare patch, and neonatal patch groups across 0–4 weeks of treatment. TAPSE value is normalized to TAPSE at two weeks before patch implantation (immediately before PAB surgery) within rat. * = p-value <0.05, # = no significant difference from Sham group values within week grouping. (B) End-diastolic dimension (EDD) measurements for control, bare patch, and neonatal patch groups at week 4 post-patch implantation. (C) End-systolic dimension (ESD) measurements for control groups, bare patch groups, and neonatal patch groups at week 4 post-patch implantation. (D) Right atrium area (RA-Area) measurements for control groups, bare patch groups, and neonatal patch groups at week 4 post-patch implantation. In figures B, C, and D, * = p-value <0.05. Statistical values are given by ANOVA with Tukey’s post-test, n = 5–7 for all groups at all time-points.
Evaluation of end-diastolic dimension (EDD), end systolic dimension (ESD), and right atrium area (RA-Area) produced fewer comparable results, where significance between groups was evident only when compared to Sham controls at four weeks post-implantation (Fig. 2B–D). The PAB group showed a significantly larger value for all three additional cardiac metrics compared to the Sham group, indicating control group effectiveness.11,12,25 While no patch group showed differences from PAB controls, cell-laden groups showed no significant differences in cardiac dimensions compared to Sham values across all assessments. In addition, cECM-laden groups showed no differences compared to Sham groups in RV dimensions. Taken together with TAPSE results, these results reinforce the finding that the inclusion of cECM improved the cardiac outcomes during therapy to functional values similar to Sham values, although not different from PAB groups. The bare cECM-laden patches may be effective without cellular-inclusion, where cECM-inclusion was the most important patch parameter. Additionally, GelMA groups were ineffective in facilitating cardiac improvements across all RV metrics and may not by a reparative therapy without cECM-inclusion. In contrast to cECM-laden groups, the only GelMA group that showed any improvements during therapy was neonatal hCPC-GelMA relating to RA-Area, pointing towards cECM potentially being the most impactful patch parameter in treatment. Within cell type, the inclusion of cECM was necessary for improvements in cardiac function over Sham compared to GelMA alone. For biomaterial groups within cell type, both GelMA and cECM groups did not show significant functional improvements with cellular incorporation, although the incorporation of neonatal hCPCs may have boosted this effect for cECM groups. Regardless, neonatal hCPC-GelMA-cECM patches were the only patches that showed improvements in cardiac function to Sham values across all functional measurements, although bare GelMA-cECM patches showed equivalent improvements in most analysis methods. A hypothesis of this work is that incorporating cECM within patches will improve cellular functionality when comparing the GelMA vs. cECM groups laden with the same cell type (neonatal hCPCs).24,29,30 While the neonatal hCPC-GelMA-cECM patch group may have shown the highest TAPSE value across all groups, there were fewer significant differences between neonatal hCPC-GelMA-cECM and neonatal hCPC-GelMA groups directly. These results could indicate that cECM may be the most potent modulator of patch therapeutic potential, and cell inclusion may not be as important.
3.3. Tissue analysis
While cardiac functional measurements are important parameters in evaluating the therapeutic effect of bioprinted patches, tissue-level effects must also be evaluated to understand how therapies affect cardiac remodeling and function.11,12,31,32 Pediatric RV failure is characterized by progressively increased cardiomyocyte hypertrophy, decreased vascular density, and increased tissue fibrosis.5–8 Quantification of these variables allows for a clear understanding of tissue-level changes due to therapy. Hearts explanted from rats after four weeks of therapy were analyzed using histological methods.11,12,33–38 The first of these assess the vascularization of the RV (Fig. 3), where Fig. 3A shows characteristic images of vessel staining across control, bare patch, and neonatal hCPC-patch groups, and Fig. 3B shows quantified vessel density. PAB and neonatal hCPC-GelMA groups showed significantly lower tissue vascularization compared to Sham groups, indicating limitations in patch therapy towards tissue remodeling when using cell-laden GelMA patches. Alternatively, both GelMA-cECM and neonatal hCPC-GelMA-cECM groups showed improvements in vascularization over PAB controls. These results reinforce the cardiac functional results since only cECM laden groups showed improvements in tissue remodeling and vasculature formation. Previous studies have shown that cECM injection within the myocardium supported infiltration of vasculature in healthy hearts and increased infarct vascularization in damaged hearts, mirroring the results seen here.39–41 Also, both cECM-laden groups (bare and neonatal hCPC patches) showed improved vascularization compared to neonatal hCPC-GelMA groups. These results directly reinforce the in vitro findings of our previous study, where cECM inclusion improved the angiogenic potential of neonatal hCPC-GelMA-cECM patches compared to neonatal hCPC-GelMA patches.24 The effect of cECM on neonatal cell reparative potential may be primarily improving angiogenic effects, such as by releasing pro-vascularization cytokines over other cytokines, supported by both previous in vitro and current in vivo results in this study.24,29
Fig. 3.
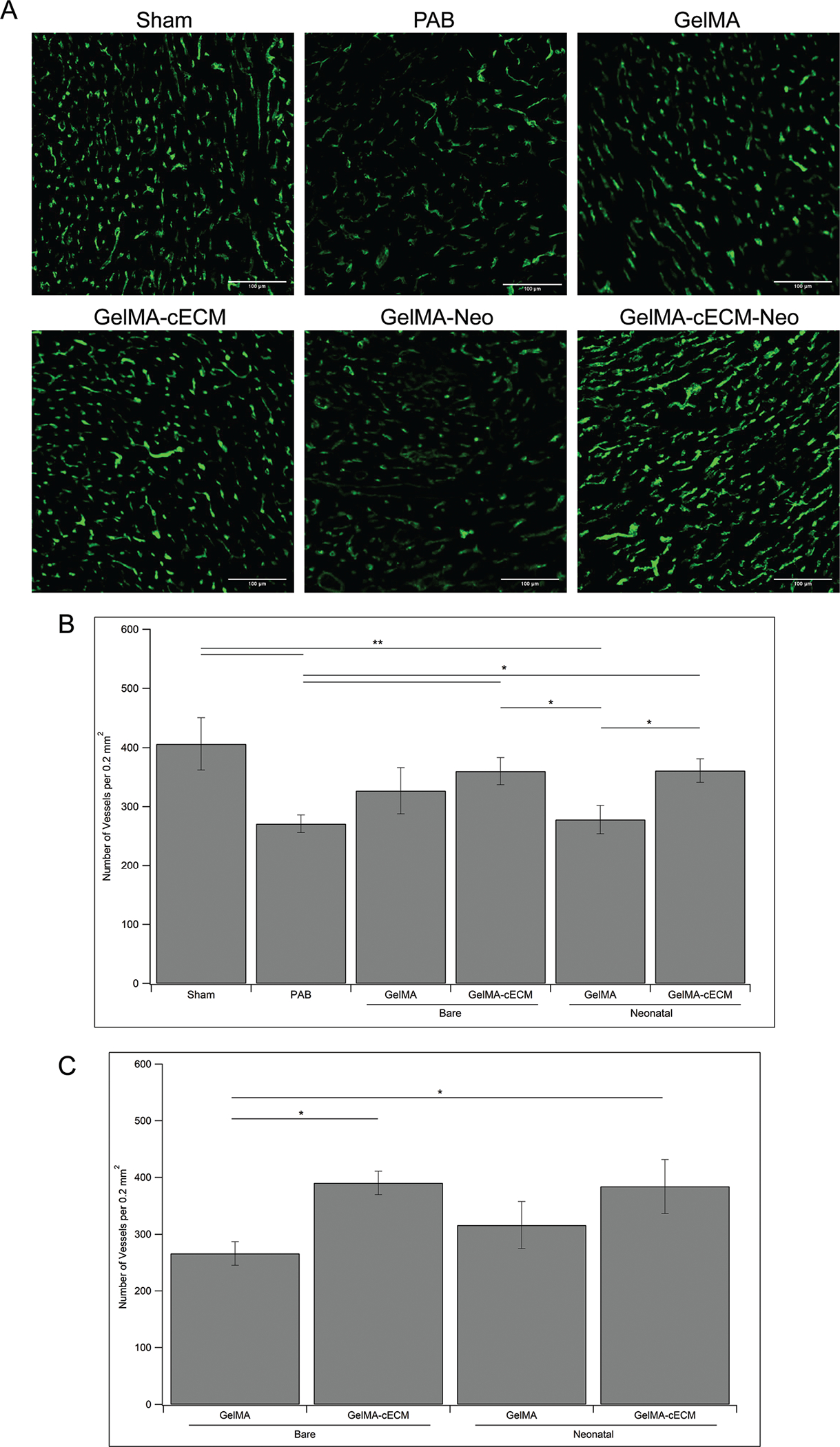
Tissue vascularization. (A) Characteristic images of fluorescent signals from control, bare patch, and neonatal patch tissue sections stained with vessel-specific dye Isolectin B4. Scale bar for all images is 100 μm. (B) Quantified vessel density given as number of vessels per 0.2 mm2 area of the RV overall. (C) Quantified vessel density given as number of vessels per 0.2 mm2 area of the RV adjacent to location of implanted patch. * = p-value <0.05, ** = p-value <0.01, given by ANOVA with Tukey’s post-test, n = 3–7 for all groups at all time-points.
The analysis in Fig. 3B described the vascularization across groups evaluated at multiple sections within the RV. During implantation, patches are placed on the epicardial wall of the RV rather than inside the RV tissue itself, which may mean that there are local changes to tissue vascularization close to the patch implantation site compared to the RV in general.42,43 Cells within patches need blood supply for survival and hCPCs were incorporated within patches to deliver locally released factors to the myocardium.22,44 Taken together, these effects may drive tissue changes at the patch-tissue interface and should be evaluated in addition to overall RV vascularization.45 Vessel density analysis for RV tissue adjacent to patches is presented in Fig. 3C, where only patch groups are considered. Only GelMA-cECM and neonatal hCPC-GelMA-cECM patches showed an improved vessel density adjacent to patches, compared to bare GelMA patches. This trend mirrors the vascularization data seen in Fig. 3B, where only GelMA-cECM and neonatal hCPC-GelMA-cECM patches showed a significantly improved vessel density compared to controls. Unlike RV vascularization overall, there were no significant differences between GelMA-cECM and neonatal hCPC-GelMA-cECM patches considering only patch-adjacent tissue. These results indicate that neonatal cell inclusion in GelMA patches may be producing a local improvement in tissue angiogenesis, although the effects were not sufficiently stronger than the effect of bare GelMA patches. Overall, these results indicate that there may be a local response related to material type only, of which cell-inclusion does not significantly modulate.
RV pressure overload drives cardiomyocyte hypertrophy, promotes further cardiomyocyte enlargement, dysfunctional contractility, and eventual RV failure.5–8 Analysis of cardiomyocyte hypertrophy for control, bare patches, and neonatal hCPC patches is seen in Fig. 4, where Fig. 4A shows characteristic images of cardiomyocyte outlines (red) and nuclei (blue), and Fig. 4B shows quantified cardiomyocyte area. PAB and GelMA groups showed significantly higher cardiomyocyte area compared to Sham groups, indicating limitations in patch therapy towards tissue remodeling in line with the vascularization results. Both GelMA-cECM and neonatal hCPC-GelMA groups showed reductions in cardiomyocyte hypertrophy over PAB controls, although they also showed differences from Sham controls, indicating that these patch types may be mildly reduce cardiomyocyte hypertrophy. Neonatal hCPC-GelMA-cECM group showed significant decreases in hypertrophy compared to the PAB and GelMA groups, indicating that the cell-cECMp atchesmay bemost impactful in reducing hypertrophy. These results reinforce the cardiac functional measurements and vessel density analysis, where both cECM-laden groups showed improvements in tissue remodeling and vasculature formation. Also, there was a significant difference in cardiomyocyte area between hCPC-GelMA-cECM and GelMA groups directly. These findings point once again towards cECM-laden patches outperforming pure GelMA patches, in terms of improving tissue remodeling towards repair. Although there was no significant difference between biomaterial types within cell groups in comparing cardiomyocyte area, there was a trend towards reduced cardiomyocyte hypertrophy in the neonatal GelMA-cECM groups compared to the neonatal GelMA groups, similar to trends seen in functional measurements such as EDD and ESD.
Fig. 4.
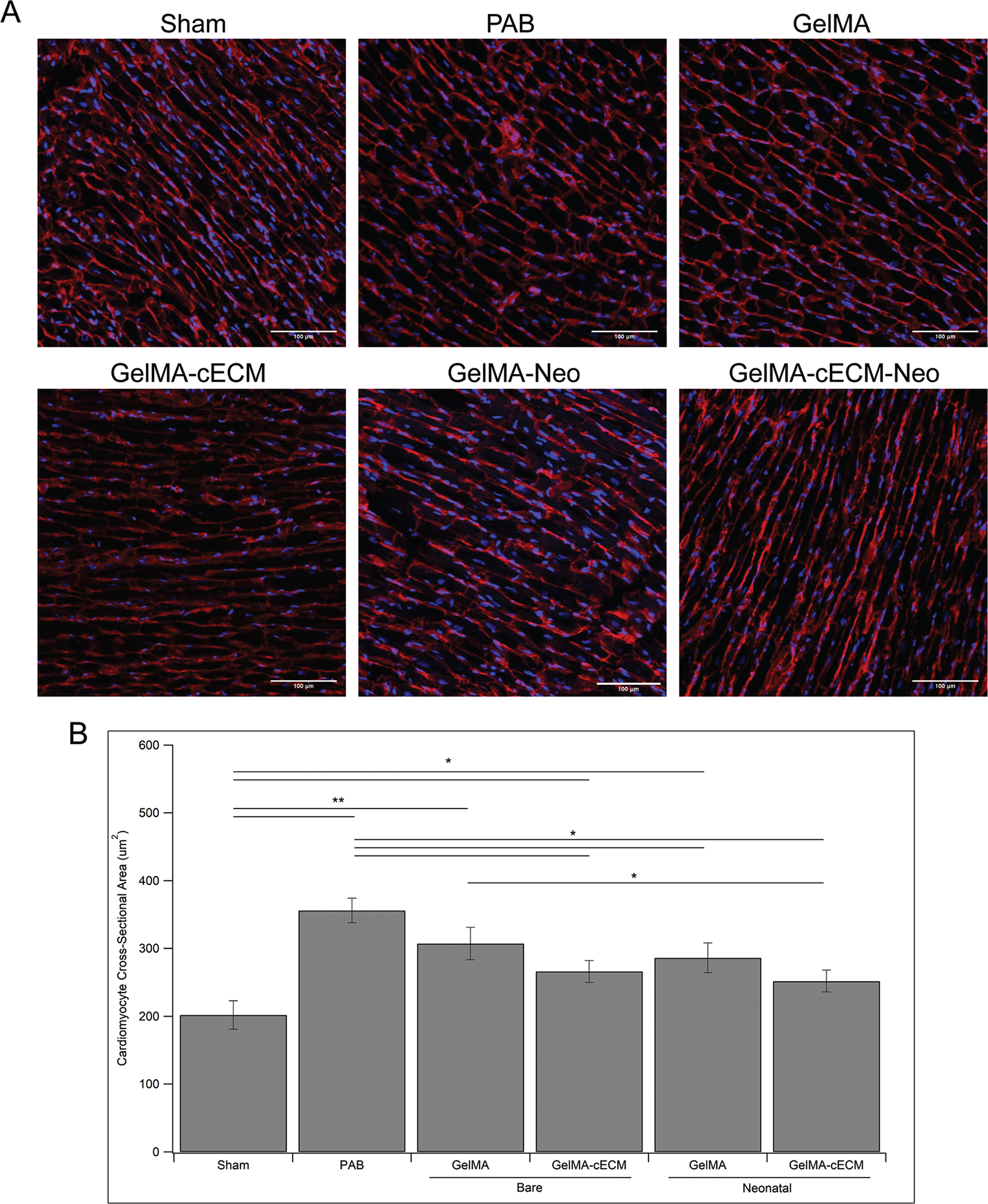
Cardiomyocyte hypertrophy. (A) Characteristic images of fluorescent signals from control, bare patch, and neonatal patch tissue sections stained with cardiomyocyte cell membrane-specific dye Wheat Germ Agglutinate (red) and nuclei marker DAPI (blue). Scale bar for all images is 100 μm. (B) Quantified cardiomyocyte hypertrophy given as average cardiomyocyte cross-sectional area (μm2) in the RV. * = p-value <0.05, ** = p-value <0.01, given by ANOVA with Tukey’s post-test, n = 3–7 for all groups at all time-points.
Finally, RV free wall total fibrosis was evaluated across all groups to understand the therapeutic impact of patches towards tissue remodeling.11,12 Fibrosis in RV dysfunction can result in contractile limitations and myofibroblast recruitment, perpetuating a cycle of tissue damage leading to RV failure.5–8 Analysis of fibrosis for control, bare patch, and neonatal hCPC patch groups is seen in Fig. 5, where Fig. 5A shows characteristic heart section images where darker areas indicate fibrosis, and Fig. 5B shows quantified % fibrosis of the RV free wall across groups. Notably in the GelMA-Neo image, a section of patch is visible on the RV (indicated by an arrow in Fig. 5A), further supporting the findings in Fig. 1 related to patch retention throughout treatment. PAB and neonatal hCPC-GelMA groups showed significantly higher fibrosis compared to Sham group and no groups showed improvements over PAB values, reinforcing the ineffectiveness of GelMA patches in repairing the myocardium. In addition, GelMA-cECM patches showed the lowest Fibrosis values across all groups. These combined results reinforce the findings that the inclusion of cECM is an important factor in developing therapy for treating heart failure, compared to pure GelMA materials. A trend in terms of reduced fibrosis was not seen for neonatal hCPC-GelMA-cECM and neonatal hCPC-GelMA patches, which may indicate that neonatal hCPCs reduce the potential of cECM components to drive fibrosis reduction or collagen remodeling, in contrast with to the improved angiogenic and hypertrophic tissue effects.
Fig. 5.

Tissue fibrosis. (A) Characteristic images of control, bare patch, and neonatal patch tissue sections stained with collagen density/fibrosis-specific dye. Scale bar for all images is set in the Sham image. Scale bar is 4 mm. Arrow indicates retained patch. (B) Quantified tissue fibrosis given as % fibrosis of the RV free wall. * = p-value <0.05, given by ANOVA with Tukey’s post-test, n = 3–7 for all groups at all time-points.
Overall, tissue analysis of rat hearts after four weeks of therapy showed differences between test groups and PAB controls in improvements to vasculature formation, reduction in cardiomyocyte hypertrophy, and reduction in cardiac fibrosis. Of all groups, only bare GelMA-cECM and neonatal hCPC-GelMA-cECM patches showed increased vasculature formation and decreased cardiomyocyte hypertrophy over PAB values. Both bare GelMA-cECM and neonatal hCPC-GelMA-cECM patches showed tissue-level improvements over GelMA groups, such as significance over neonatal hCPC-GelMA in regards to improved vascularization and bare GelMA in regards to reduced myocyte hypertrophy. Additionally, cECM-laden patches were the only patches that showed tissue level improvements over controls compared to the GelMA groups (which showed improvements only through cell-laden patches in reducing hypertrophy), indicating significant repair across all assessment methods (vascularization, cardiomyocyte hypertrophy, and fibrosis). These results point towards cECM inclusion being an important factor for tissue level remodeling in the developed biomaterial-cell patches. There were no significant differences between bare or cell-incorporated patches across vascularization, hypertrophy, or fibrosis analysis within either GelMA or cECM groups specifically.27,46 Alternatively, the only comparison between the neonatal patches in terms of tissue-level remodeling was an improvement in vessel density with the inclusion of cECM, indicating that the neonatal hCPCs may have their primary modulation by cECM related to promoting angiogenesis.27,46–49 Regardless, comparisons between cECM-laden and cECM-free cell-laden patches showed that the inclusion of cECM was required to improve cardiac functionality.
4. Conclusions
The in vivo assessment of bioprinted GelMA-cECM and hCPC-GelMA-cECM patches in a clinically relevant model of pediatric heart failure was investigated in terms of cardiac functional and tissue-level remodeling effects. A juvenile rat model of RV failure generated through PAB was used to establish a clinically relevant heart dysfunction condition.11,12 Patches across different biomaterial types and cell inclusions were implanted onto the failing RV of PAB rats in a randomized and blinded manner. Test groups included Sham controls, PAB controls, bare GelMA and GelMA-cECM patches, and neonatal hCPC-laden GelMA and GelMA-cECM patches. Patches across both cell-laden biomaterial types showed consistent and complete retention of cells during the timeframe of therapy, compared to 0–30% retention seen with injection methods.11,12,17,27 Both cardiac function and tissue-level remodeling were improved with the implantation of neonatal hCPC-GelMA-cECM patches compared to PAB controls. Additionally, bare GelMA-cECM patches showed effectiveness in repairing tissue properties and cardiac functional outcomes. While both neonatal hCPC-GelMA-cECM and bare GelMA-cECM patches may be effective for cardiac repair, bare GelMA-cECM patches do not include primary human cells and represent a reproducible therapeutic technology.14,50,51 Incorporation of cECM into patches was necessary to facilitate cardiac functional and tissue-level repair across all analysis methods and patch types (bare or cell-laden), compared to GelMA groups and PAB controls and reinforces prior in vitro studies.24 A comparison of cell-laden and bare patches showed that the inclusion of cells might not be required for cardiac functional improvements compared to bare patches in the case of GelMA-cECM groups. However, given that neonatal cell inclusion was needed to facilitate improvements for GelMA groups in regards to cardiac functional measurements, the inclusion of cells may improve therapeutic outcomes with pure GelMA patches. Comparisons between neonatal hCPC-laden patch groups showed an improved RV vessel density with cECM-laden patches compared to cECM-free patches.
Altogether, the in vivo analysis of bioprinted hCPC-GelMA-cECM patches shows that acellular GelMA-cECM patches and neonatal hCPC-GelMA-cECM patches are effective in treating RV failure, both in terms of functional and tissue improvements over PAB controls and in comparison to cECM-free GelMA patches, regardless of cell inclusion. Overall, cECM is the most potent modulator of patch therapeutic improvements in a model of RV failure. These findings point towards the use of bioprinted GelMA-cECM patches as effective and reproducible therapies in treating pediatric RV failure, paving the way for larger-scale animal models and human clinical trials using the technology. Further tissue level analysis (such as cardiomyocyte proliferation, myofibroblast activation, and macrophage phenotype and activation) may provide additional information to understand the in vivo effect of bare and cell-laden GelMA-cECM patches.52–54
Future directions include improving cellular function within bioprinted patches, improving patch therapeutic potential by incorporating non-cellular components, and clinical investigations of GelMA-cECM patch therapy overall. The current study has shown that hCPC-incorporation may not provide substantial therapeutic benefit over bare GelMA-cECM patches. Further cellular modulation may improve cell-laden patch therapeutic outcomes by improving cell function, such as by incorporating improved cellular manufacturing methods, computational determination of optimal cell populations, secretory cell types such as mesenchymal stem cells (MSCs), and optimized cell density and number within printed patches.55–58 Alternatively, the effectiveness of cell-free GelMA-cECM patches may be improved by incorporating extracted paracrine factors within patches to further develop an off-the-shelf device. Loading patches with potent emerging therapeutics such as exosomes allows for highly tailorable devices that benefit from combined cECM-exosomes therapy.59,60 In addition, exosome release profiles may be modulated by endogenous ECM-exosome interactions.61
Improved cell sourcing, manufacturing, and optimization, additional cellular modulation, and exosome loading of GelMA-cECM patches may produce substantial improvements to the therapeutic potential of hCPC-GelMA-cECM patches compared to the results seen in this work. Regardless, the effectiveness of cell-free GelMA-cECM patches in treating RV failure was evident and warrants further investigation in immunocompetent rats, large animals, and ultimate human patient assessment.62,63 GelMA-cECM patches may provide effective therapy for decreasing the mortality rate and improving the quality of life for the many pediatric patients suffering from RV dysfunction and failure.
Acknowledgements
This study was supported by the NHLBI to MED and KLC (HL146147). DB was supported by the Cell and Tissue Engineering NIH Biotechnology Training grant (T32 GM008433). JH was supported by the NHLBI training grant (T32HL105373).
Footnotes
Conflicts of interest
Dr Karen L. Christman is a cofounder, board member, consultant, and holds an equity interest in Ventrix, Inc.
Data availability
All raw data from this study are available from the corresponding author by reasonable request.
Notes and references
- 1.Rossano JW, Kim JJ, Decker JA, Price JF, Zafar F, Graves DE, Morales DL, Heinle JS, Bozkurt B and Denfield SW, Circulation, 2010, 122, A1374. [Google Scholar]
- 2.Robbins JM, Bird TM, Tilford JM, Cleves MA, Hobbs CA, Grosse SD and Correa A, Morb. Mortal. Wkly. Rep, 2007, 56, 25. [Google Scholar]
- 3.Bove EL, Pediatr. Cardiol, 1998, 19, 308. [DOI] [PubMed] [Google Scholar]
- 4.Jokerst CE and Cummings KW, Adv. Clin. Radiol, 2020, 2, 37–63. [Google Scholar]
- 5.Mandras SA and Desai S, in StatPearls, StatPearls Publishing, 2017. [PubMed] [Google Scholar]
- 6.Reddy S and Bernstein D, Circulation, 2015, 132, 1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greyson CR, Crit. Care Med, 2008, 36, S57. [DOI] [PubMed] [Google Scholar]
- 8.Ma M, Gauvreau K, Allan CK, Mayer JE Jr. and Jenkins KJ, Ann. Thorac. Surg, 2007, 83, 1438. [DOI] [PubMed] [Google Scholar]
- 9.Alsoufi B, Deshpande S, McCracken C, Kogon B, Vincent R, Mahle W and Kanter K, J. Thorac. Cardiovasc. Surg, 2015, 150, 1455. [DOI] [PubMed] [Google Scholar]
- 10.Cheng JW and Nayar M, Am. J. Geriatr. Pharmacother, 2009, 7, 233. [DOI] [PubMed] [Google Scholar]
- 11.Maxwell JT, Trac D, Shen M, Brown ME, Davis ME, Chao MS, Supapannachart KJ, Zaladonis CA, Baker E and Li ML, Stem Cells, 2019, 37, 1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trac D, Maxwell JT, Brown ME, Xu C and Davis ME, Circ. Res, 2019, 124, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segers VF and Lee RT, Nature, 2008, 451, 937. [DOI] [PubMed] [Google Scholar]
- 14.Traverse JH, Henry TD, Dib N, Patel AN, Pepine C, Schaer GL, DeQuach JA, Kinsey AM, Chamberlin P and Christman KL, J. Am. Coll. Cardiol. Basic Trans. Sci, 2019, 4, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishigami S, Ohtsuki S, Tarui S, Ousaka D, Eitoku T, Kondo M, Okuyama M, Kobayashi J, Baba K and Arai S, Circ. Res, 2014, 116, 653. [DOI] [PubMed] [Google Scholar]
- 16.Ye J and Yeghiazarians Y, J. Cardiovasc. Pharmacol, 2014, 63, 85. [DOI] [PubMed] [Google Scholar]
- 17.Ye L, Zimmermann W-H, Garry DJ and Zhang J, Circ. Res, 2013, 113, 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feyen DA, Gaetani R, Doevendans PA and Sluijter JP, Adv. Drug Delivery Rev, 2016, 106, 104. [DOI] [PubMed] [Google Scholar]
- 19.Gnecchi M, Zhang Z, Ni A and Dzau VJ, Circ. Res, 2008, 103, 1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dawson E, Mapili G, Erickson K, Taqvi S and Roy K, Adv. Drug Delivery Rev, 2008, 60, 215. [DOI] [PubMed] [Google Scholar]
- 21.Pashuck ET and Stevens MM, Sci. Transl. Med, 2012, 4, 160sr4. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Zhu W, Radisic M and Vunjak-Novakovic G, Circ. Res, 2018, 123, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Streeter BW and Davis ME, in Cell Biology and Translational Medicine, Springer, 2018, vol. 5, p. 1. [Google Scholar]
- 24.Bejleri D, Streeter BW, Nachlas AL, Brown ME, Gaetani R, Christman KL and Davis ME, Adv. Healthcare Mater, 2018, 7, 1800672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faber MJ, Dalinghaus M, Lankhuizen IM, Steendijk P, Hop WC, Schoemaker RG, Duncker DJ, Lamers JM and Helbing WA, Am. J. Physiol.-Heart C, 2006, 291, H1580–H1586. [DOI] [PubMed] [Google Scholar]
- 26.Akazawa Y, Okumura K, Ishii R, Slorach C, Hui W, Ide H, Honjo O, Sun M, Kabir G, Connelly K and Friedberg MK, J. Appl. Physiol, 2020, 129, 238–246. [DOI] [PubMed] [Google Scholar]
- 27.Agarwal U, Smith AW, French KM, Boopathy AV, George A, Trac D, Brown ME, Shen M, Jiang R and Fernandez JD, Stem Cells Transl. Med, 2016, 5, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li G, Nelsen C and Hendrickson EA, Proc. Natl. Acad. Sci. U. S. A, 2002, 99, 832–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.French KM, Boopathy AV, DeQuach JA, Chingozha L, Lu H, Christman KL and Davis ME, Acta Biomater, 2012, 8, 4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.French KM, Maxwell JT, Bhutani S, Ghosh-Choudhary S, Fierro MJ, Johnson TD, Christman KL, Taylor WR and Davis ME, Stem Cells Transl. Med, 2016, 8364382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redout EM, Wagner MJ, Zuidwijk MJ, Boer C, Musters RJ, van Hardeveld C, Paulus WJ and Simonides WS, Cardiovasc. Res, 2007, 75, 770–781. [DOI] [PubMed] [Google Scholar]
- 32.Horton KD, Meece RW and Hill JC, J. Am. Soc. Echocardiogr, 2009, 22, 776–792. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Y, Zhang L, Lu Q, Gao Y, Cai Y, Sui A, Su T, Shen X and Xie B, Int. J. Mol. Med, 2017, 40, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore JB, Tang XL, Zhao J, Fischer AG, Wu WJ, Uchida S, Gumpert AM, Stowers H, Wysoczynski M and Bolli R, Basic Res. Cardiol, 2019, 114, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomczyk M, Kraszewska I, Szade K, Bukowska-Strakova K, Meloni M, Jozkowicz A, Dulak J and Jazwa A, Basic Res. Cardiol, 2017, 112, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bageghni SA, Hemmings KE, Zava N, Denton CP, Porter KE, Ainscough JF, Drinkhill MJ and Turner NA, FASEB J, 2018, 32, 4941–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whittaker P, Kloner RA, Boughner DR and Pickering JG, Basic Res. Cardiol, 1994, 89, 397–410. [DOI] [PubMed] [Google Scholar]
- 38.Rich L and Whittaker P, Braz. J. Morphol. Sci, 2005, 22, 97–104. [Google Scholar]
- 39.Singelyn JM, DeQuach JA, Seif-Naraghi SB, Littlefield RB, Schup-Magoffin PJ and Christman KL, Biomaterials, 2009, 30, 5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wassenaar JW, Gaetani R, Garcia JJ, Braden RL, Luo CG, Huang D, DeMaria AN, Omens JH and Christman KL, J. Am. Coll. Cardiol, 2016, 67, 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seif-Naraghi SB, Singelyn JM, Salvatore MA, Osborn KG, Wang JJ, Sampat U, Kwan OL, Strachan GM, Wong J, Schup-Magoffin PJ, Braden RL, Bartels K, DeQuach JA, Preul M, Kinsey AM, DeMaria AN, Dib N and Christman KL, Sci. Transl. Med, 2013, 5, 173ra25–173ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serpooshan V, Zhao M, Metzler SA, Wei K, Wang PA, Mahmoudi M, Malkovskiy AV, Butte MJ and Bernstein D, Biomaterials, 2013, 34, 9048–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang Y, Chen SC, Wei HJ, Wu TJ, Liang HC, Lai PH, Yang HH and Sung HW, J. Thorac. Cardiovasc. Surg, 2005, 130(3), 705–711. [DOI] [PubMed] [Google Scholar]
- 44.Noor N, Shapira A, Edri R, Gal I, Wertheim L and Dvir T, Adv. Sci, 2019, 6, 1900344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell AC, Briquez PS, Hubbell JA and Cochran JR, Acta Biomater, 2016, 30, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agarwal U, George A, Bhutani S, Ghosh-Choudhary S, Maxwell JT, Brown ME, Mehta Y, Platt MO, Liang Y and Sahoo S, Circ. Res, 2017, 120, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gray WD, French KM, Ghosh-Choudhary S, Maxwell JT, Brown ME, Platt MO, Searles CD and Davis ME, Circ. Res, 2015, 116, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amir G, Ma X, Reddy VM, Hanley FL, Reinhartz O, Ramamoorthy C and Riemer RK, Ann. Thorac. Surg, 2008, 86, 1311–1319. [DOI] [PubMed] [Google Scholar]
- 49.Bejleri D and Davis ME, Adv. Healthcare Mater, 2019, 8, 1801217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu B, Lee BW, Nakanishi K, Villasante A, Williamson R, Metz J, Kim J, Kanai M, Bi L, Brown K and Di Paolo G, Nat. Biomed. Eng, 2018, 2, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang K, Ozpinar EW, Su T, Tang J, Shen D, Qiao L, Hu S, Li Z, Liang H, Mathews K, Scharf V, Freytes DO and Cheng K, Sci. Transl. Med, 2020, 12(538), eaat9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morikawa Y, Heallen T, Leach J, Xiao Y and Martin JF, Nature, 2017, 547, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, Lin SCJ, Aronow BJ, Tallquist MD and Molkentin JD, Nat. Commun, 2016, 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang RM, Johnson TD, He J, Rong Z, Wong M, Nigam V, Behfar A, Xu Y and Christman KL, Biomaterials, 2017, 129, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lipsitz YY, Timmins NE and Zandstra PW, Nat. Biotechnol, 2016, 34, 393. [DOI] [PubMed] [Google Scholar]
- 56.Garikipati VNS, Shoja-Taheri F, Davis ME and Kishore R, Circ. Res, 2018, 123, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayourian J, Cashman TJ, Ceholski DK, Johnson BV, Sachs D, Kaji DA, Sahoo S, Hare JM, Hajjar RJ, Sobie EA and Costa KD, Circ. Res, 2017, 121, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu R, Hu X and Wang JA, Stem Cells, 2018, 36, 482–500. [DOI] [PubMed] [Google Scholar]
- 59.Gurunathan S, Kang M-H, Jeyaraj M, Qasim M and Kim J-H, Cells, 2019, 8, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davis ME, Circ. Res, 2016, 119, 1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hernandez MJ, Gaetani R, Pieters VM, Ng NW, Chang AE, Martin TR, van Ingen E, Mol EA, Dzieciatkowska M, Hansen KC, Sluijter JPG and Christman KL, Adv. Ther, 2018, 1, 1800032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wendel JS, Ye L, Zhang P, Tranquillo RT and Zhang JJ, Tissue Eng., Part A, 2014, 20, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao L, Gregorich ZR, Zhu W, Mattapally S, Oduk Y, Lou X, Kannappan R, Borovjagin AV, Walcott GP, Pollard AE and Fast VG, Circulation, 2018, 137, 1712–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw data from this study are available from the corresponding author by reasonable request.