

Abstract
Background:
Acyl-CoA:cholesterol acyltransferase (ACAT) inhibitors have been considered as potential therapeutic agents to treat several diseases, including Alzheimer’s disease, atherosclerosis, and cancer. While many ACAT inhibitors are readily available, methods to encapsulate them as nanoparticles have not been reported.
New Method:
We report a simple method to encapsulate ACAT inhibitors, using the potent hydrophobic ACAT inhibitor F12511 as an example. By mixing DSPE-PEG2000, egg phosphatidylcholine (PC), and F12511 in ethanol, followed by drying, resuspension and sonication in buffer, we show that F12511 can be encapsulated as stealth liposomes at high concentration.
Results:
We successfully incorporated F12511 into nanoparticles and found that increasing PC in the nanoparticles markedly increased the amount of F12511 incorporated in stealth liposomes. The nanoparticles containing F12511 (Nanoparticle F) exhibit average size of approximately 200nm and are stable at 4°C for at least 6 months. Nanoparticle F is very effective at inhibiting ACAT in human and mouse neuronal and microglial cell lines. Toxicity tests using mouse primary neuronal cells show that F12511 alone or Nanoparticle F added at concentrations from 2–10μM for 24-, 48-, and 72-hours produces minimal, if any, toxicity.
Comparison with Existing Method(s):
Unlike existing methods, the current method is simple, cost effective, and can be expanded to produce tagged liposomes to increase specificity of delivery. This also offers opportunity to embrace water soluble agent(s) within the aqueous compartment of the nanoparticles for potential combinatorial therapy.
Conclusions:
This method shows promise for delivery of hydrophobic ACAT inhibitors at high concentration in vivo.
Keywords: Nanoparticle; ACAT/SOAT; F12511, DSPE-PEG2000; Phosphatidylcholine; Cholesteryl ester
1. Introduction:
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease in the United States and in other countries. The incidence of AD is constantly increasing and is expected to dramatically rise in the next few decades, yet no effective treatment is currently available. With so many failed clinical trials (Cummings et al., 2021), it is important to develop novel targets for AD prevention and treatment. One potential target to consider is acyl-coenzyme A (CoA):cholesterol acyltransferase 1 (ACAT1), also known as sterol O-acyltransferase 1 (SOAT1). ACAT1 is a founding member of the membrane-bound O-acyltransferase (MBOAT) family (Hofmann, 2000; Chang et al., 2011). ACAT1 is the first of 2 ACAT isoenzymes: ACAT1 and ACAT2. ACAT1 (Chang et al., 1993) is expressed ubiquitously in the body and is the main isozyme found in the brain, while ACAT2 is primarily expressed in the intestines and liver (Anderson et al., 1998; Cases et al., 1998; Oelkers et al., 1998; Chang et al., 2000). ACAT2 is also present in various other cell types, but at lower levels than those of ACAT1. ACAT1 and ACAT2 are molecular targets to treat dyslipidemia and atherosclerosis; many small-molecule inhibitors have been developed to inhibit each isozyme (Chang et al., 2009).
The interest in using ACAT inhibitors to treat cardiovascular diseases waned for a few years after several other drugs, including statins, ezetimibe (Davis & Veltri, 2007; Ge et al., 2008), and PCSK9 monoclonal antibody (Rhainds et al., 2012), proved to be more effective than the ACAT inhibitors in reducing serum cholesterol levels. However, recent evidence from several laboratories has cast light on ACAT1 as an important molecular target, again. This time the focus is on using ACAT inhibitors for the treatment of AD (Puglielli et al., 2001; Hutter-Paier et al., 2004; Huttunen et al., 2010; Bryleva et al., 2010; Murphy et al., 2013; Shibuya et al., 2014; Shibuya et al., 2015; van der Kant et al., 2019; Nugent et al., 2020) and for certain forms of cancer (Yue et al., 2014; Yang et al., 2016; Jiang et al., 2019).
Many ACAT inhibitors are readily available since they were originally manufactured as candidate drugs for anti-atherosclerosis treatment. However, to our knowledge, only four of them passed the clinical safety test: CI-1011 (avasimibe) (Insull et al., 2001; Llaverías et al., 2003; Tardif et al., 2004), CS-505 (pactimibe sulfate) (Nissen et al., 2006; Terasaka et al., 2007), K-604 (Ikenoya et al., 2007), and F12511 (eflucimibe) (López-Farré et al., 2008). CI-1011 has been clinically approved, but it is a less potent, isoform-non-specific ACAT inhibitor with half maximal inhibitory concentration (IC50) value of approximately 19μM (Ikenoya et al., 2007). CS-505 is also an isoform-non-specific ACAT inhibitor with an IC50 value of 4.9μM and 3μM for ACAT1 and ACAT2, respectively (Terasaka et al., 2007). K-604 is a high-affinity ACAT1 specific inhibitor; K-604 inhibits ACAT1 with an IC50 value of 0.45μM, which is 229-fold more potent than its IC50 value for ACAT2 (Ikenoya et al., 2007). F12511 is also a high-affinity ACAT1 inhibitor that has been well-studied (Junquero, Bruniquel, et al., 2001; Junquero, Oms, et al., 2001; Junquero, Pilon, et al., 2001). It has a molecular weight of 469.72g/mol and inhibits ACAT1 with an IC50 value of 0.039μM and ACAT2 with a value of 0.11μM (Chang et al., 2000; López-Farré et al., 2008). The IC50 values discussed above are based on ACAT enzymatic assays in vitro (Chang et al., 1998) using extracts of ACAT1 and ACAT2 cells expressing the human form of the respective isozyme.
For AD treatment, none of the ACAT inhibitors have progressed to the preclinical stage. We have begun to search and develop small molecule ACAT inhibitors to treat AD. We chose F12511 to begin our efforts; however, there are two main hurdles. First, F12511 is very hydrophobic, and it is very difficult to increase its solubility (Bougáret et al., 2005). Second, it is unknown if F12511 can cross the blood brain barrier (BBB). The use of nanoparticles to target drugs to the brain and other specific tissues, and to increase the overall bioavailability of the drugs has become increasingly popular over past decades (Kreuter, 2007). The small size of nanoparticles, generally ranging from 10–1000nm, and the relatively simple and cost-effective process to make nanoparticles makes them very attractive vehicles for drug delivery (Yingchoncharoen et al., 2016). Nanoparticles coated with polyethylene glycol (PEG) polymers, often referred to as stealth nanoparticles, show prolonged circulation time, increased half-life, and decreased clearance, and can maintain drug concentrations longer than nanoparticles without PEG, as reviewed in Harris and Chess, 2003. The Food and Drug Administration (FDA) has approved the use of PEG in foods, cosmetics, and pharmaceuticals (Alconcel et al., 2011; Turecek et al., 2016) allowing for its use in various formulations. Gülçür and colleagues used 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy-[poly(ethylene glycol)-2000] (DSPE-PEG2000) to encapsulate curcumin in micelles with a specific tagging peptide to target tumors and reported increased inhibition of tumor formation in vitro (Gülçür et al., 2013). A similar approach was used to successfully target atherosclerotic plaques in mice with DSPE-PEG2000 tagged micelles (Peters et al., 2009). In a separate nanoparticle formulation, PEG tagging was also used to increase bioavailability of nanoparticles with Liver X receptor (LXR) agonist that resulted in anti-inflammatory and anti-atherosclerosis effects (Zhang et al., 2015). Furthermore, phosphatidylcholine (PC) has been used to encapsulate a higher concentration of hydrophobic drugs in DSPE-PEG2000 based nanoparticles (Ashok et al., 2004).
Based on previous work, here we designed a similar PEGylated DSPE nanoparticle with PC included to encapsulate F12511. We also characterized the physical properties of the nanoparticle and its efficacy in inhibiting cholesterol esterification in neuronal and microglial cell lines. In addition, we tested potential toxicity of F12511 alone or F12511 in the nanoparticle (Nanoparticle F) in primary mouse cortical neurons. Overall, our method is simple and reproducible. Nanoparticle F can be easily characterized by several methods including thin layer chromatography (TLC) and dynamic light scattering (DLS), is effective at inhibiting ACAT in intact cells, and does not show toxicity in neuronal cell culture at relevant concentrations.
2. Materials and Methods:
2.1. Lipids, ACAT inhibitors, and solvents:
DSPE-PEG2000 is from Laysan Bio, Inc (mPEG-DSPE, MW 2,000). L-a-Phosphatidylcholine is from Sigma-Aldrich (Catalog No. P-2772). F12511 and K-604 were custom synthesized by WuXi AppTec in China. Based on HPLC/MS and NMR profiles, purity of F12511 was 98% and in stereospecificity; purity of K-604 was 98% in chemical purity. Organic solvents are from Fisher Scientific.
2.2. Cell culture:
Cell lines were kept in a 37°C incubator with 5% CO2. Human neuron-like SH-SY5Y cells were maintained in 1:1 DMEM (Corning, Manassas, VA, USA): Ham’s F-12 (Sigma, St. Louis, MO, USA) medium supplemented with MEM non-essential amino acids (Gibco, Grand Island, NY, USA) and 10% calf serum (R&D Systems, Flowery Branch, GA, USA). Human microglia-like HMC3 cells were maintained in MEM (Corning, Manassas, VA, USA) medium with 10% calf serum. Mouse neuron-like N2a cells were maintained in MEM medium with 10% calf serum. Mouse microglia-like N9 cells were maintained in RPMI (Corning, Manassas, VA, USA) medium with 10% calf serum. HMC3 cells were from ATCC and SH-SY5Y, N2a, and N9 cells were gifts from Drs. Maue, Supattapone, and Hickey at Dartmouth College. For wide field fluorescence and differential interference contrast (DIC) imaging studies, cells were imaged in phenol-free media (Gibco RMPI 1640 (Catalog No. 11-835-030) for N9 cells; Gibco MEM (Catalog No. 51-200-038) for HMC3 and N2a cells; and Gibco DMEM/F-12 (Catalog No. 21–041-025) for SH-SY5Y cells). Primary cortical neurons were dissected from E14.5–16.5 embryos from C57/BL6 mice and plated at 50,000 cells per well in precoated poly-D-lysine (Sigma-Aldrich, St. Louis, MO (Catalog No. P6407)) and laminin-coated (Sigma-Aldrich, St. Louis, MO (Catalog No. L2020)) 96-well tissue culture dishes. Cells were incubated in enriched neurobasal (NB) medium (Thermo Fisher Scientific, Waltham, MA (Catalog No. 21103049)) (500mL NB media, 200mM L-glutamine, B-27 with vitamin A, 1mL PS (1%), and 340mg glucose). Neurons were allowed to grow processes for 4–6 days prior to treatment.
2.3. Nanoparticle development and characterization
2.3.1. Nanoparticle formation:
Protocol is modified from Gülçür et al., 2013. Using a clean glass tube (Pyrex no. 9825, 9mL capacity), 60mg of DSPE-PEG2000 is dissolved in 500μL of ethanol (EtOH) for a working concentration of 60mM. Phosphatidylcholine (PC) (dissolved in chloroform) is added at working concentrations ranging from 0 to 12mM to the DSPE-PEG2000 solution while vortexing. F12511 or K-604 is dissolved in 500μL of EtOH at working concentrations of 12–24mM then added to the DSPE-PEG2000/PC mixture while vortexing. The final solution will contain concentrations of 30mM DSPE-PEG2000, 0–6mM PC, and 6–12mM F12511 or K-604. The final solution is then lyophilized under refrigeration overnight to remove organic solvent. After lyophilization, samples can be stored in the dark at −20°C for several weeks until ready to solubilize. To solubilize, the sample is re-suspended in 1mL of 1x phosphate-buffered saline (PBS) and vortexed until sample is in suspension. This step can take approximately 1 hour as the sample needs to rest between repeated vortexing to prevent excessive foaming. Once in fine suspension, the sample is purged with nitrogen, capped, wrapped with parafilm, and bath sonicated (Branson 2510 model) at 4°C for 2–4 rounds at 20 minutes per round. DSPE-PEG2000/PC nanoparticles without ACAT inhibitors should turn clear while F12511/DSPE-PEG2000/PC nanoparticles (Nanoparticle F) and K-604/DSPE-PEG2000/PC nanoparticles (Nanoparticle K) are slightly turbid and may contain some visible precipitate. After sonication, nanoparticles are transferred to sterile Eppendorf tubes and centrifuged at 12,000rpm for 5 minutes to remove unincorporated materials. The supernatant, as well as the pellet, are collected for chemical analysis. In some experiments, as indicated in the figure legend, the nanoparticles were loaded on to a 5mL Sepharose CL-4B column. Sepharose CL-4B contains beads with particle size at 45–165μm. The column (approximately 28mm circumference) was first equilibrated with PBS at room temperature, then loaded with the 1mL of sample, eluted with PBS and fractions of 500μL to 1mL were collected. This method was used to assure that F12511 and K-604 encapsulated in nanoparticles do not appear in the exclusion volume of the Sepharose CL-4B column.
2.3.2. Nanoparticle analysis by thin layer chromatography (TLC):
To confirm the concentration of F12511 or K-604 and DSPE-PEG2000/PC in the nanoparticles, 10μL of each sample component were loaded onto a TLC plate (Miles Scientific Silica gel HL, Catalog #P46911). The solvent systems used were hexanes:ethyl ether:acetic acid (60:40:1) (Bryleva et al., 2010) to detect F12511 and chloroform:methyl acetate:isopropanol:methanol:water (28:25:25:12:10) to detect K-604. The retention factor (Rf) was approximately 0.35 for F12511 and approximately 0.5 for K-604. The content of F12511 or K-604 was determined by extrapolation from a standard curve of a concentration gradient of the respective compound produced in the same TLC plate. The plate was stained with iodine and quantitated by using ImageJ. The TLC/iodine stain method can detect F12511 or K-604, and DSPE-PEG2000/PC separately, thus allowing for encapsulation efficiency determination.
2.3.3. Nanoparticle analysis by fluorescence:
Utilizing F12511’s inherent fluorescence (Mesplet et al., 2005), F12511 concentration was measured using BioTek Synergy Neo2 Hybrid Multi-Mode Plate Reader (excitation λ: 300nm, emission λ: 380nm). A concentration gradient of F12511 (dissolved in EtOH) ranging from 0–13mM was used to create a standard curve. Nanoparticles were diluted in 1x PBS with volume concentrations ranging from 0–90% (i.e., for 90%, 90μL of nanoparticles and 10μL of 1x PBS are mixed). A total of 100μL was loaded per well and samples were measured in triplicate. The amount of F12511 in Nanoparticle F was determined based on the F12511 concentration gradient trendline equation. To address any possible added fluorescence from the nanoparticles themselves, the control nanoparticles of DSPE-PEG2000/PC were also measured and subtracted from corresponding dilution of Nanoparticle F. This method was used to confirm the amount of F12511 encapsulated.
2.3.4. Dynamic light scattering (DLS) measurements:
Size of nanoparticles were measured with Malvern Panalytical Zetasizer Nano-ZS, using 10mm path length disposable cuvettes. Samples were kept at 4°C in Eppendorf tubes wrapped with parafilm and left in the dark whenever possible. Prior to analysis, samples were centrifuged for 5 minutes at 12,000rpm. Most samples required 2-fold dilution to get an accurate reading. Samples were measured in triplicate.
2.4. ACAT activity experiments
2.4.1. Intact cell cholesteryl esterification by 3H-Oleate pulse:
This procedure was according to procedure described in Chang et al., 1986. Monolayers of cells were treated for 2 hours with various concentrations of either control (ethanol or PBS), DSPE-PEG2000/PC nanoparticles, F12511 or K-604 (dissolved in ethanol), or F12511/DSPE-PEG2000/PC (Nanoparticle F) or K-604/DSPE-PEG2000/PC (Nanoparticle K) nanoparticles. Following treatment, cells were pulsed at 37°C for 1–3 hours with 3H-oleate/fatty acid free BSA, then washed with cold PBS, lysed by adding cold 0.2M NaOH at appropriate volume, and placed on orbital shaker for 40 minutes. The solubilized cell slurries were collected into glass tubes, neutralized with 3M HCl and 1M KH2PO4, and lipids were extracted with CHCl3:MeOH (2:1) and water. Samples were vortexed and centrifuged at 500rpm for 10 minutes. The top-phase was removed, and the bottom phase was blown dried by N2 using N-evap apparatus (Organomation Associates, Inc., N-EVAP™ 112). Dried samples were vortexed with ethyl acetate and spotted on TLC plate. The solvent system used was petroleum ether:ethyl ether:acetic acid (90:10:1). The cholesteryl ester band was scraped from the TLC and measured by scintillation counter for radioactivity.
2.4.2. In vitro mixed liposomal ACAT activity assay:
This method was done as described in Chang et al., 1998 and Neumann et al., 2019. Monolayers of cells were treated for 4 hours with either control (dimethyl sulfoxide (DMSO)), or with 0.5μM F12511 in DMSO. Cells were then washed with drug-free media kept at 37°C for various amounts of time. At the indicated times, cells were washed with cold PBS, then lysed in a 2.5% 3-((3-Cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS), 1M KCl in 50mM Tris at pH 7.8 buffer. Lysed cells were collected and aliquoted into pre-chilled tubes containing the mixed liposomal mixture of taurocholate, phosphatidylcholine, and cholesterol. Tubes were vortexed and kept on ice for 5 minutes. Samples were then incubated in a shaking 37°C water bath with 3H-oleoyl CoA added for 10 minutes. The assay was stopped by adding CHCl3:MeOH (2:1). Lipids were extracted and analyzed by the same TLC method described under “2.4.1. Intact cell cholesteryl esterification by 3H-Oleate pulse.”
2.5. Microscopy experiments
2.5.1. Transmission electron microscopy (TEM):
TEM images of Nanoparticle F were taken by adding a small drop (1–2μL) of sample on to a copper grid with a carbon film. Prior to loading the samples, the grid was modified by glow discharge treatment using Quorum GloQube to make the carbon film hydrophilic. The sample was loaded onto the grid, dried, and stained with UranyLess (Electron Microscopy Sciences) and Lead Citrate 3% (Electron Microscopy Sciences). The grids were then imaged using a FEI Tecnai F20 TEM.
2.5.2. Wide-field fluorescence and differential interference contrast (DIC) microscopy:
Mouse and human neuronal (N2a and SH-SY5Y) and microglial (N9 and HMC3) cells were imaged using the 60x objective of Nikon Eclipse Ti-E microscope. Cells were plated in Mattek 12-well glass bottom plates (1.5 glass thickness, 14mm glass diameter, uncoated) for live-cell imaging. Prior to plating, the plates were coated with poly-d-lysine overnight, washed 3 times with sterile water, and washed once with PBS. The evening before imaging, cell media (described under “2.2. Cell culture”) was changed. On the day of imaging, cells were treated for 2 hours with 0.5μM EtOH (control), F12511 (EtOH), Nanoparticle F, or DSPE-PEG2000/PC. The cells were then stained for nuclear visualization with membrane-permeable Hoechst 33258 solution (Sigma-Aldrich, Catalog #94403) for 20–30 minutes and washed 3 times with PBS. Cells were imaged live in phenol-free media (described under “2.2. Cell culture”) at 37°C with 5% CO2 using Tokai Hit stage top incubation system.
2.6. Toxicity experiments
2.6.1. Lactate dehydrogenase (LDH) toxicity assay:
Primary cortical neurons were harvested and maintained as described under “2.2. Cell culture.” Neurons were treated with either F12511 alone (dissolved in EtOH), F12511/DSPE-PEG2000/PC nanoparticles (Nanoparticle F), or DSPE-PEG2000/PC nanoparticles at 2–100μM concentrations for 24-, 48-, or 72-hours. After treatment, the conditioned media from the cells was collected, spun at 12,000rpm for 30 minutes to remove cellular debris, supernatant transferred to new tube, and frozen at −80°C until time of assay. Using Cytotoxicity Detection Kit (LDH) (Roche Catalog No. 11 644 793 001), media and reaction mixture were mixed, incubated for 30 minutes at room temperature protected from light, and the absorbance was measured at 490nm.
2.6.2. Ethical handling of research animals:
Mouse studies were conducted in accordance with the federal ARRIVE guidelines. The Institutional Animal Care and Use Committee (IACUC) at Dartmouth approved all mouse experiments under the protocol “Animal Models of Neurodegenerative Diseases” (#00002117, MCH). C57/BL6 mice used for these studies were purchased from the Jackson Laboratory (#000664, Bar Harbor, ME).
3. Results:
3.1. Nanoparticle development and characterization
3.1.1. A thin-layer chromatographic method to quantitate F12511.
To quantify the F12511 content in nanoparticles, we developed a method by using thin layer chromatography (TLC) followed by iodine staining and ImageJ analysis. The TLC system used clearly separates F12511 or K-604 from DSPE-PEG2000/PC, as shown in Figures 2 and 5. To determine the amount of F12511 in the nanoparticles, a series of F12511 at different concentrations were spotted onto a TLC plate along with the actual sample. After TLC and iodine stain, a trendline, equation, and R-squared value can be calculated to produce a standard curve. Using the extrapolated equation, the concentration of F12511 in the nanoparticles is determined by using ImageJ analysis (Figure 2A).
Figure 2:
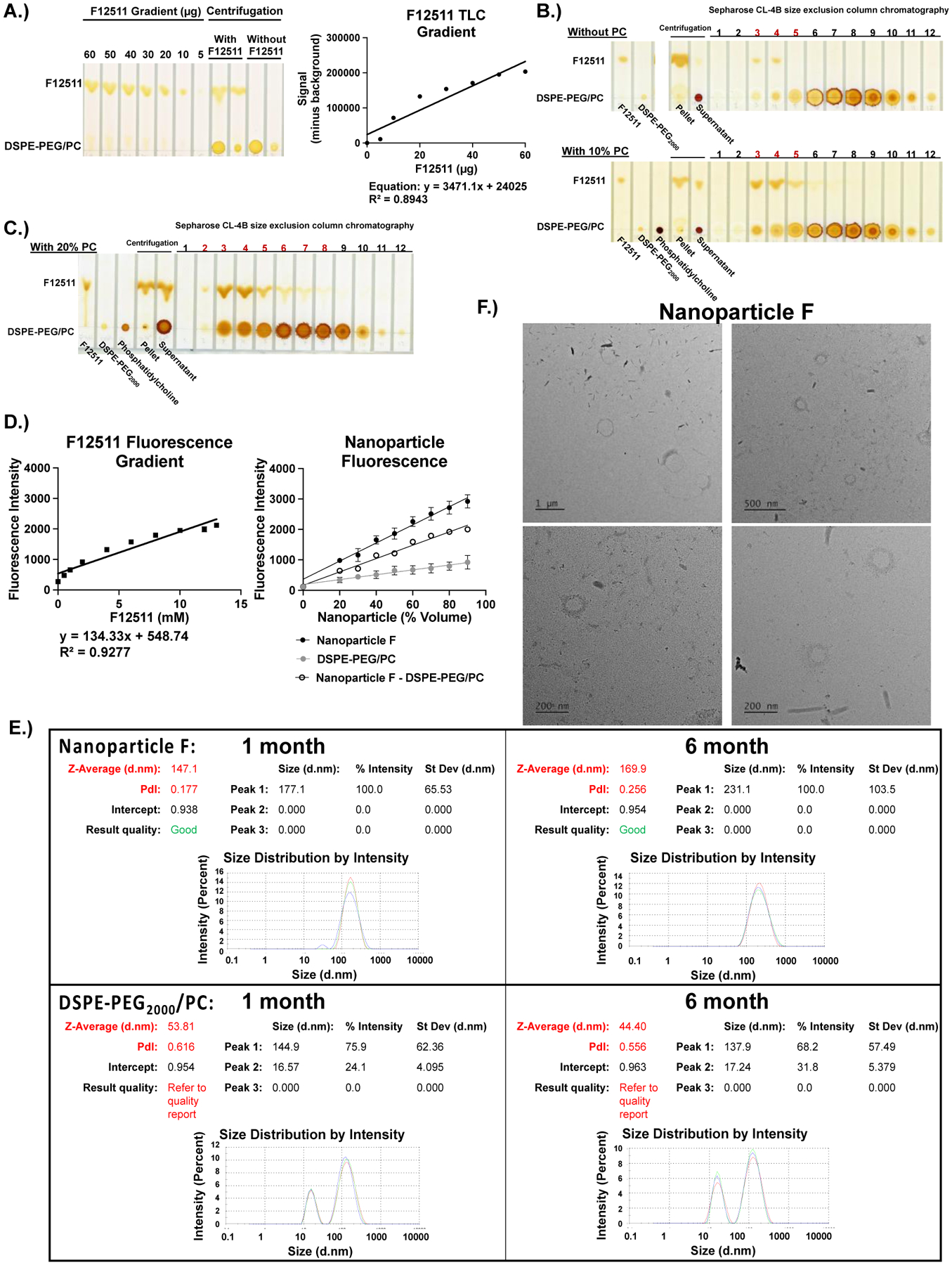
F12511 DSPE-PEG2000/PC nanoparticles (Nanoparticle F) can be characterized qualitatively by TLC and iodine staining methods, as well as quantitatively by using fluorescence, electron microscopy, and Malvern Zeta Sizer methods. A.) A concentration gradient of F12511 (60–5μg) was used to create a standard curve using ImageJ Software. Based on the generated trendline’s equation, the concentration of F12511 was determined. B and C.) Standards (F12511, DSPE-PEG2000, phosphatidylcholine (PC)), pellet (pelleted precipitate), and supernatant are shown in first lanes. The fractions labeled 1–12 refer to the 500μL elution fractions collected from a size exclusion column (Sepharose CL-4B). The red numbers highlight the elution fractions showing encapsulation of F12511 based on the co-elution of F12511 with DSPE-PEG2000 or DSPE-PEG2000/PC. B.) The top TLC plate shows nanoparticles with only DSPE-PEG2000 (no PC). The bottom TLC plate shows Nanoparticle F with DSPE-PEG2000 and 10% PC. C.) The TLC sample shown here came from the nanoparticle system consisting of 40 mol % F12511 (12mM), 20 mol % PC (6mM), and DSPE-PEG2000 (30mM). D.) Fluorescence intensity curves for F12511 alone (left) and nanoparticles (right). E.) Malvern Zeta Sizer “Size Distribution by Intensity” results for 1- and 6-month-old nanoparticles. Each sample was measured in triplicate. F.) Representative TEM images of Nanoparticle F.
Figure 5:
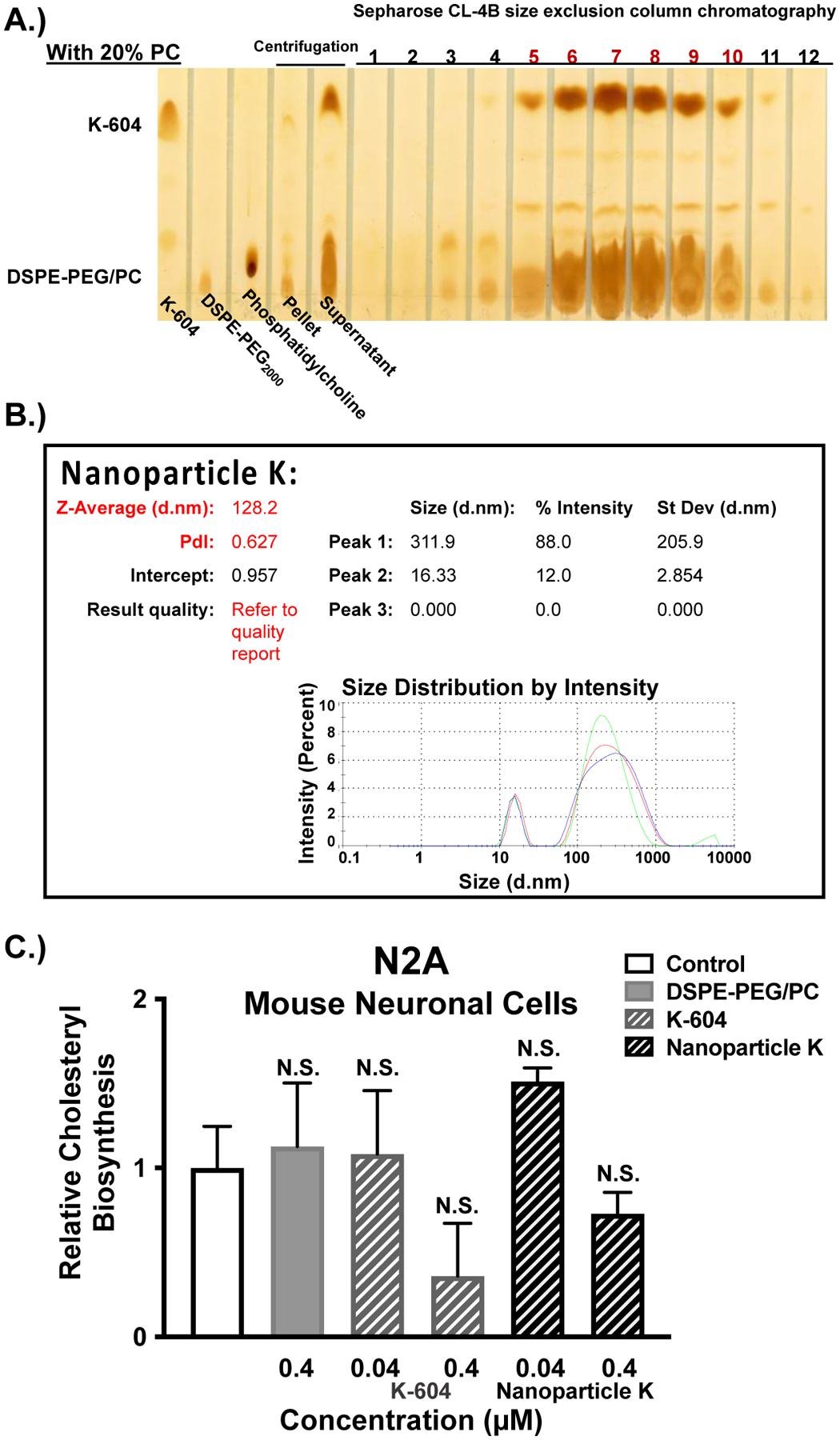
ACAT inhibitor, K-604, is encapsulated at high concentration by using the nanoparticle protocol and displays similar characteristics to Nanoparticle F. A.) Standards (K-604, DSPE-PEG2000, and phosphatidylcholine) are shown in the first 3 lanes followed by the pellet and supernatant samples. The fractions labeled 1–12 refer to the 12 elution fractions, each with 500μL, collected from a size exclusion column (Sepharose CL-4B). The red numbers highlight the elution fractions containing encapsulated nanoparticles of K-604 (Nanoparticle K), based on the co-elution of K-604 with DSPE-PEG2000/PC. The overall Nanoparticle K formulation is 40 mol % K-604 (12mM), 20 mol % PC (6mM), and DSPE-PEG2000 (30mM). B.) Malvern Zeta Sizer “Size Distribution by Intensity” result for freshly solubilized Nanoparticle K. Each sample was measured in triplicate C.) Mouse neuronal (N2a) cells at 80–90% confluency in triplicate (12-well dishes) were either treated for 2 hours with EtOH (control) or with the following agents: 0.4μM DSPE-PEG2000/PC; 0.04μM or 0.4μM of K-604 alone or Nanoparticle K. The cells were then pulsed with 3H-oleate to determine ACAT activity as described in the Methods and Materials section. One-way ANOVA statistical test was completed, and results shown are relative to the control (0.4μM K-604 p-value = 0.08; 0.4μM Nanoparticle K p-value = 0.8). N.S. not significant.
3.1.2. The addition of phosphatidylcholine to DSPE-PEG2000 nanoparticles considerably improves the encapsulation of F12511.
We prepared the DSPE-PEG2000 nanoparticles to encapsulate F12511 by using the lyophilization/resolubilization method, as described in Gülçür et al., 2013 and Jhaveri and Torchilin, 2014, and analyzed the resultant nanoparticles according to the scheme outlined in Figure 1. The result showed that this method was effective at encapsulating F12511, but at a low encapsulation efficiency of approximately 6 mol % of DSPE-PEG2000. Ashok et al., 2004 suggested that the addition of phosphatidylcholine (PC) may improve drug solubilization in nanoparticles. We tested this possibility by adding various amounts of PC (10% or 20%) to the DSPE-PEG2000 ethanol mixture (Figure 1, step 2), then proceeded with nanoparticle formation and analysis. As shown in the bottom iodine-stained TLC in Figure 2B, there is an increase in detectable F12511 with DSPE-PEG2000 and 10% PC in the supernatant and the corresponding elution fractions from the size exclusion column (seen by comparing the red labeled fractions 3–5 where F12511 and DSPE-PEG2000/PC co-elute). While the addition of PC at 10 mol % of DSPE-PEG2000 was able to incorporate about 20 mol % F12511 (Figure 2B, lower TLC panel), doubling PC and F12511 concentrations led to a notably higher encapsulation (Figure 2C). With 20 mol % (6mM) PC, the nanoparticles were able to encapsulate approximately 40 mol % (12mM) F12511; there is a large amount of F12511 detected in the supernatant, as well as in the corresponding eluted fractions from the column, that co-elute with DSPE-PEG2000/PC (Figure 2C). Based on several preparations of the nanoparticles made, after fractionation by centrifugation, the result of TLC analysis shows that there is approximately 9mM of F12511 encapsulated, rather than the full 12mM present in the unfractionated samples.
Figure 1:
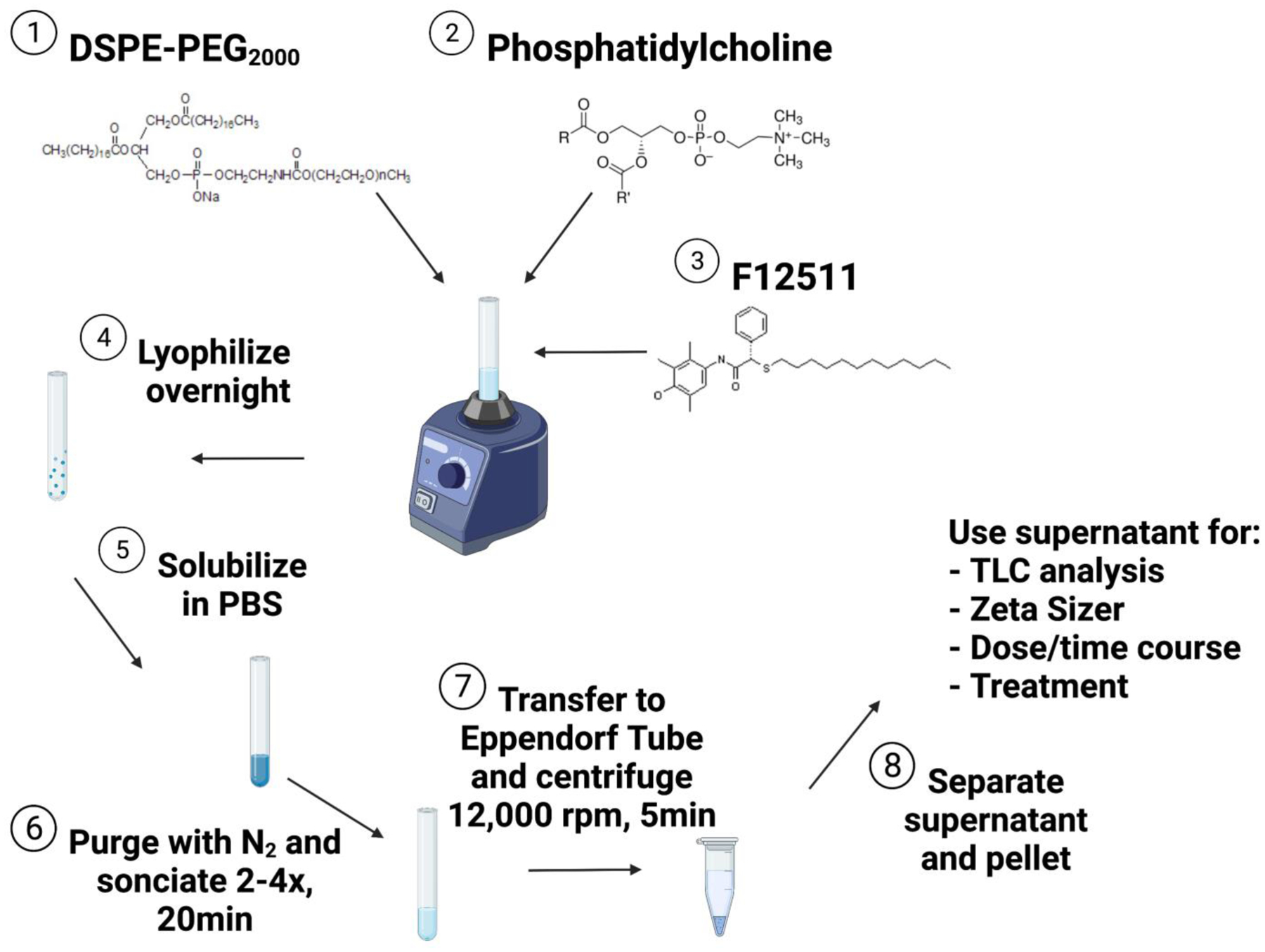
Schematic of nanoparticle protocol. Structures for DSPE-PEG2000, phosphatidylcholine, and F12511 are from Laysan Bio, Inc., Sigma-Aldrich, and López-Farré et al., 2008, respectively. Figure was created with BioRender.com.
The concentration of F12511 in Nanoparticle F was also determined by fluorescence. Following the principle by Mesplet et al., 2005, the inherent fluorescence of F12511 was utilized (as described in Materials and Methods) to confirm the results obtained by using TLC quantitation method. The concentration gradient trendline equation of F12511 (Figure 2D, left) was used to determine F12511 concentration in the nanoparticles. As expected, the nanoparticles show a linear increase in intensity as the concentration of the nanoparticles increases (Figure 2D, right). Interestingly, DSPE-PEG2000/PC nanoparticles without F12511 appear to also have some fluorescence as volume concentration increases; therefore, Nanoparticle F was “standardized” by subtraction of DSPE-PEG2000/PC value for each respective point. Based on this condition and considering the percent volume of nanoparticle at each point, the average of all 8 measurements was approximately 9.9mM. This value is similar to the TLC quantitation of around 9mM suggesting the encapsulation of F12511 in Nanoparticle F is likely around 9–10mM.
3.1.3. F12511 DSPE-PEG2000/PC nanoparticles (Nanoparticle F) are approximately 200nm.
The size of the nanoparticles was characterized quantitatively by using dynamic light scattering (DLS) measurements via Malvern Zeta Sizer. In addition, the same batch of nanoparticles (with or without F12511) were measured monthly to determine stability of the nanoparticles. The results, shown in Figure 2E, show that when the nanoparticles are kept at 4°C and in the dark, their sizes are essentially unaltered for up to 6 months. Since the nanoparticles are PEGylated, this feature increases the stability of the nanoparticles, as previously described by other investigators (Gref et al., 2000; Miteva et al., 2015; Suk et al., 2016). Interestingly, based on testing different batches of nanoparticles, the results show that the nanoparticles containing F12511 have 1 main peak that ranges from 170–250nm, indicative of unilamellar liposomes, while the “empty” nanoparticles containing only DSPE-PEG2000/PC have 2 peaks at approximately 15–20nm and 140–160nm. This is likely due to the DSPE-PEG2000 and PC forming separate, smaller micelles (15–20nm) as well as the unilamellar liposomes with sizes ranging from 140–160nm. The appearance of 2 peaks in DSPE-PEG2000/PC nanoparticles likely affected the quality reading of the sample and accounts for the “Quality Report” results in Figure 2E. The zeta potential for the nanoparticles remained in the low neutral range likely due to the presence of high salt in the PBS used to resolubilize the nanoparticles after lyophilization.
The size and shape of the nanoparticles were confirmed using transmission electron microscope (TEM) methods. The results in Figure 2F suggest that Nanoparticle F is a liposome with a size around 170– 200nm. This is consistent with the DLS measurements; however, the TEM data might suggest a slightly smaller size. When control nanoparticles consisting only of DSPE-PEG2000/PC were prepared, we were unable to recover clearly defined nanoparticles. Again, this seems to reflect the result of the DLS measurements since the quality (“Quality Report” in Figure 2E) for the control nanoparticles was never optimal, despite various dilution methods used. Overall, these results strongly suggest that F12511 stabilizes the nanoparticles to form liposomes with a consistent size range.
3.2. F12511 and Nanoparticle F inhibitory effects
3.2.1. Nanoparticle F significantly inhibits ACAT activity in mouse and human neuronal and microglial cell lines.
To test the efficacy of the nanoparticles at inhibiting ACAT activity, mouse and human neuronal and microglial cell lines were utilized. Intact cells were treated with either control (EtOH), DSPE-PEG2000/PC nanoparticles, F12511 alone (dissolved in EtOH), or F12511 DSPE-PEG2000/PC nanoparticles (Nanoparticle F). The results show that across all cell lines, both F12511 alone and Nanoparticle F tested at 0.04μM and 0.4μM significantly inhibited ACAT in a dose dependent manner (Figure 3A). These results suggest that Nanoparticle F is almost as efficient as F12511 alone in inhibiting ACAT in intact cells, even with a short treatment time of 2 hours. One interesting note is that based on results presented in Figure 3A, F12511 alone or Nanoparticle F is more effective in inhibiting ACAT in human cell lines (SH-SY5Y and HMC3 cells) than in mouse cell lines (N2a and N9 cells).
Figure 3:
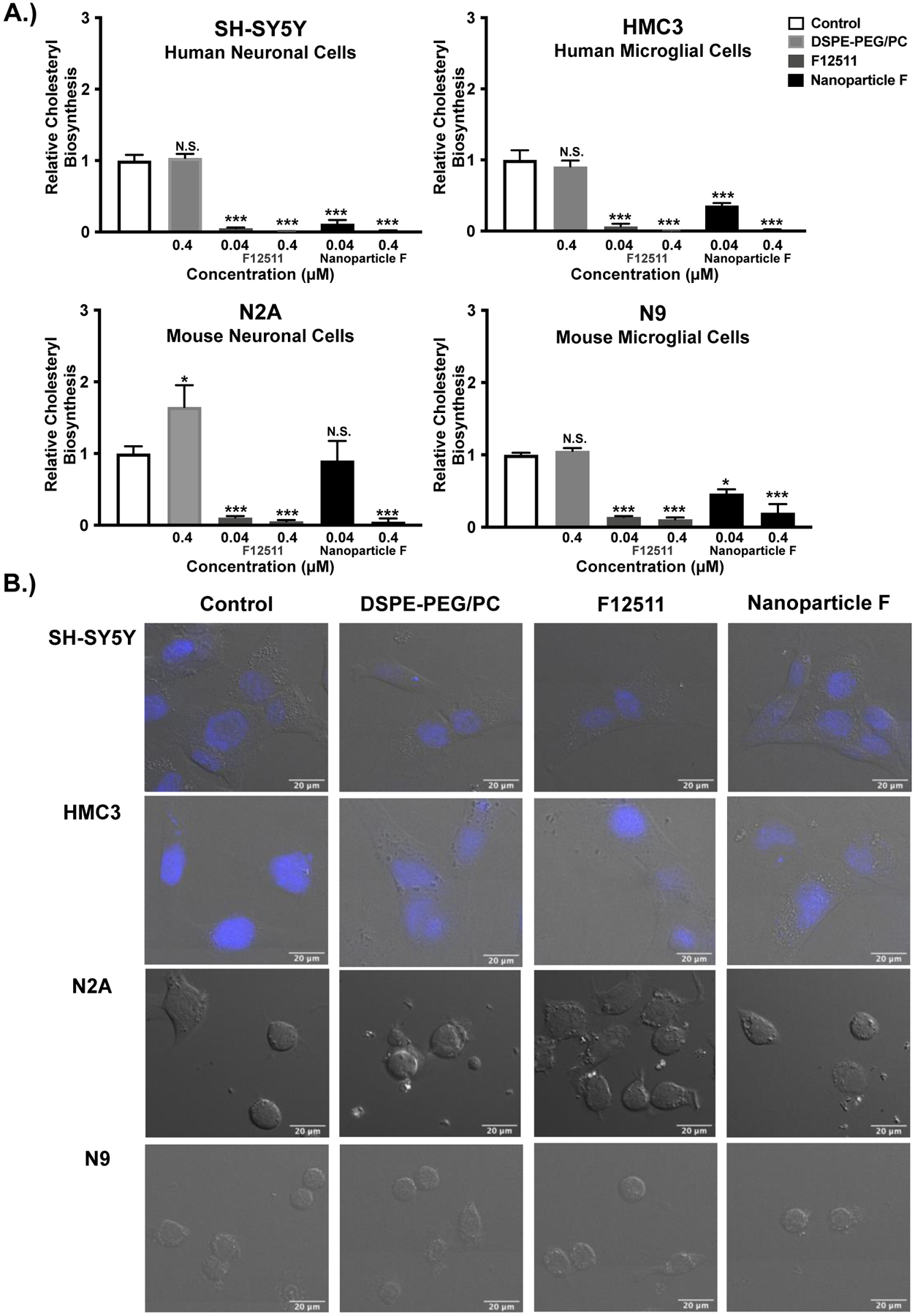
Nanoparticle F decreases ACAT activity in intact cells of human and mouse origin with no obvious effects on cell morphology. Results and images from human neuronal (SH-SY5Y), human microglial (HMC3), mouse neuronal (N2a), and mouse microglial (N9) cells. A.) Cells at 80–90% confluency in triplicate dishes (12- or 6-well dishes) were treated for 2 hours with EtOH (control) or with the following agents: 0.4μM DSPE-PEG2000/PC; 0.04μM or 0.4μM of F12511 alone or of Nanoparticle F. The cells were then pulsed with 3H-oleate to determine ACAT activity as described in the Materials and Methods section. One-way ANOVA statistical test was completed, and results shown are relative to the control. N.S. not significant; p<0.001 ***, p<0.01 **, p<0.05 *. B.) Cells as indicated were treated for 2 hours with 0.5μM EtOH (control), DSPE-PEG2000/PC, F12511 alone (EtOH) or Nanoparticle F and imaged. Representative images of DIC and Hoechst (blue, top 2 rows). Images were analyzed using ImageJ. Scale bar is 20μm.
3.2.2. Acute Nanoparticle F treatment does not produce obvious changes in cell morphology.
To look for possible acute effects of the nanoparticles on morphology in the cell lines used in Figure 3A, cells were treated for 2 hours with 0.5μM of EtOH (control), DSPE-PEG2000/PC nanoparticles, F12511 alone (EtOH), or Nanoparticle F. Live cell differential interference contrast (DIC) microscopy was utilized for viewing to avoid fixing cells which might impact cell morphology. Furthermore, since both SH-SY5Y and HMC3 cell lines are relatively flat and difficult to distinguish with DIC, the images shown for these 2 cell lines also include Hoechst dye (blue) to visualize the nuclear compartment. The results (Figure 3B) suggest that in the 4 cell lines examined, there are no obvious differences in cell morphology between various treatment groups.
3.2.3. F12511 and Nanoparticle F strongly inhibit ACAT even after washout.
F12511 inhibits ACAT1 at high affinity with an IC50 value and inhibitory constant (Ki) of 39nM (Chang et al., 2000). In general, enzyme inhibitors with a low Ki interact with the target enzyme with low dissociation rate. We suspect that once F12511 binds to ACAT1, it may dissociate from the enzyme slowly. We tested this possibility at the cellular level by incubating F12511 alone or Nanoparticle F in neuronal and microglial cell types. After 2 hours of treatment, media containing F12511 was removed, cells were washed twice with drug-free media, and placed in drug-free media for either 0-, 2-, 4-, or 8-hours. Afterwards, the ACAT activity in these cells was measured by performing 3H-oleate pulse. The results (Figure 4A) show that in F12511 treated cells, ACAT activity remained severely inhibited in all cell types examined, even after 8 hours of drug removal from the media. For Nanoparticle F treated cells, ACAT activity also remained severely inhibited for 4–6 hours. At 8 hours, significant inhibition occurred in SH-SY5Y, N2a and N9 cells but not in HMC3 cells. Together, these results strongly suggest that in intact cells, once bound to ACAT1, F12511 slowly dissociates from the enzyme.
Figure 4:
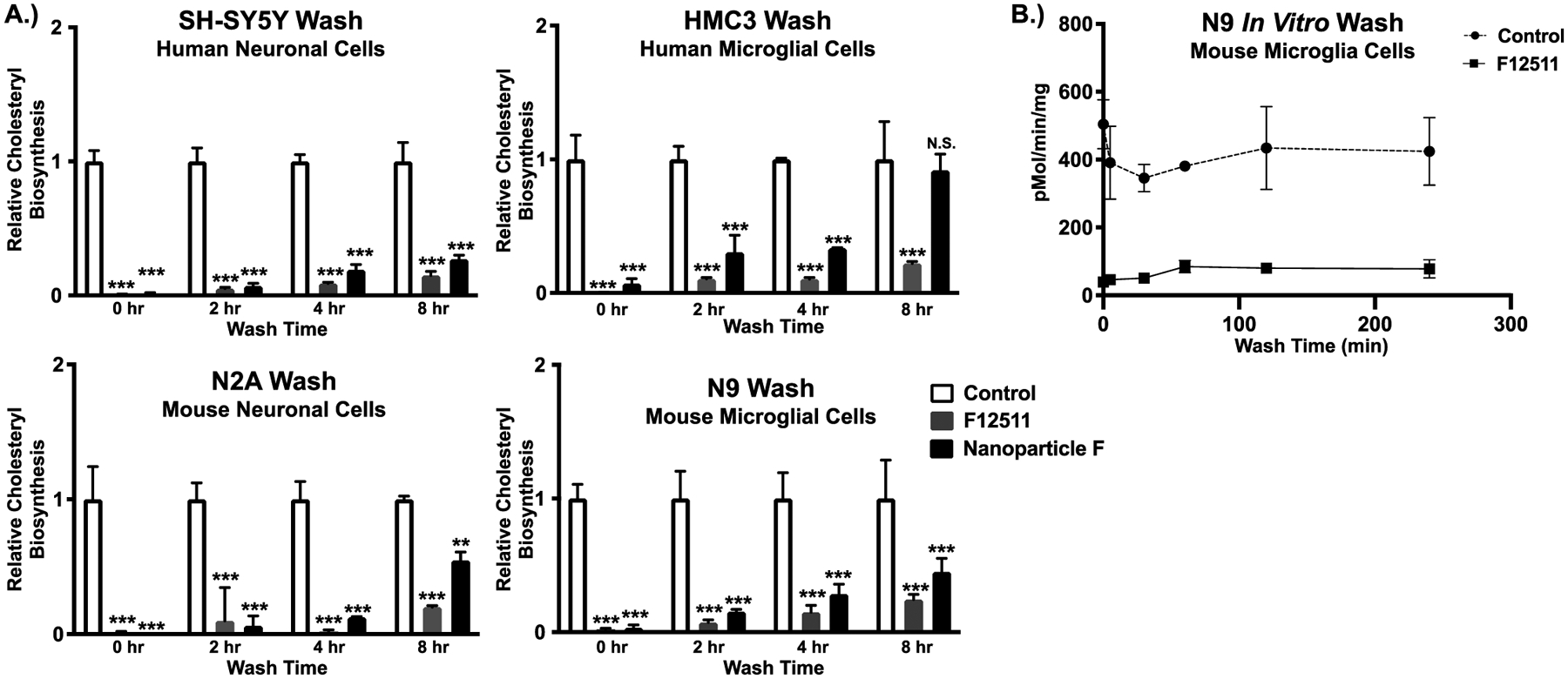
ACAT activity remains severely inhibited in cells treated with F12511 or with Nanoparticle F for several hours after F12511 removal from media. A.) Human neuronal cells (SH-SY5Y), human microglial cells (HMC3), mouse neuronal cells (N2a), and mouse microglial cells (N9) were either treated for 2 hours with 0.5μM EtOH (control), F12511 alone, or Nanoparticle F. After treatment, the media was removed, cells were washed twice with drug-free media kept at 37°C, given fresh drug-free media, and put back in the 37°C incubator with 5% CO2 for 0-, 2-, 4-, and 8-hour(s). The cells were then pulsed with 3H-oleate and ACAT activity was determined by cholesteryl ester formation in intact cells. Two-way ANOVA was completed, and reported results are relative to the control. N.S. not significant; p<0.001 ***, p<0.01 **, p<0.05 *. B.) Mouse microglial cells (N9) were treated with DMSO (control) or with 0.5μM F12511 in DMSO for 4 hours. After treatment, cells were washed twice with drug-free media at 37°C, given fresh drug-free media, and placed back in the 37°C incubator with 5% CO2 for up to 4 hours. At time indicated, cells were harvested, and the mixed liposomal ACAT activity assay in vitro was performed.
We tested the validity of this interpretation by using a different method to monitor ACAT enzyme activity. N9 cells were treated with DMSO as control or with F12511 alone for 4 hours, washed with drug-free media, and incubated in drug-free media for various amounts of time up to 4 hours (Figure 4B). Here, the ACAT activity was monitored by using the ACAT enzyme assay in vitro. The in vitro assay monitors the interaction between ACAT and F12511 in the milieu where detergent taurocholate, phospholipid, and cholesterol are present in abundance (Chang et al., 1998). The result in Figure 4B shows that, even under this condition, the ACAT enzyme activity stayed inhibited by F12511 for up to 4 hours after F12511 removal from the media. Together, the results shown in Figure 4A and 4B suggest that once F12511 binds to ACAT, it does not readily dissociate from the enzyme for several hours and the binding between ACAT1 and F12511 is not readily affected by the presence of cholesterol, phospholipids, or detergent.
3.3. DSPE-PEG2000/PC nanoparticles are applicable to other compounds
3.3.1. K-604 is encapsulated in DSPE-PEG2000/PC nanoparticles (Nanoparticle K) and exhibits similar characteristics to Nanoparticle F.
To determine the transferability of this DSPE-PEG2000/PC nanoparticle composition, another ACAT inhibitor, K-604, was investigated. Following the same nanoparticle protocol of 30mM DSPE-PEG2000 and 6mM PC, but with 12mM K-604 rather than 12mM F12511, the results showed effective encapsulation of large concentrations of K-604 (Figure 5A). Using the same nanoparticle preparation for K-604, a large amount of K-604 is detected in the supernatant. The result of the size exclusion column chromatography also shows that most of the K-604 signal co-elutes with DSPE-PEG2000/PC signal and these signals appear in the column-included fractions (Figure 5A).
The size of Nanoparticle K was determined using Malvern Zeta Sizer for DLS measurements. In this case, the nanoparticles were measured within a few days after the nanoparticles were made (rather than testing for stability as in Figure 2E for Nanoparticle F). Compared to Nanoparticle F, Nanoparticle K was larger with a main peak at size of about 310nm (Figure 5B). Interestingly, Nanoparticle K had a similar distribution to control DSPE-PEG2000/PC nanoparticles (Figure 2E) with one large peak of around 300nm as well as a smaller peak of 16nm. Again, suggesting a smaller peak of micelles and a larger peak of liposomes. The zeta potential of Nanoparticle K remained in the low to neutral range, similar to control nanoparticles and Nanoparticle F as discussed above.
Lastly, Nanoparticle K was tested for efficacy in inhibiting ACAT activity in cell culture. To determine the efficacy of Nanoparticle K, mouse neuronal (N2a) cells were treated for 2 hours either with 0.4μM EtOH (control), with 0.4μM DSPE-PEG2000/PC nanoparticles, or with 0.04μM and 0.4μM K-604 alone (EtOH) or Nanoparticle K (Figure 5C). As expected, K-604 and Nanoparticle K do not appear to be as potent as F12511 and Nanoparticle F (Figure 3A), respectively, because, as an ACAT1 inhibitor, F12511 has a lower IC50 value than K-604 (please refer to Introduction). Nevertheless, the results showed that at the high concentration tested (0.4μM), there is a trend for both K-604 and Nanoparticle K for inhibiting ACAT activity by about 60% and 30%, respectively. Overall, the results from Figure 5 show that this nanoparticle system can be utilized to encapsulate high concentrations of other hydrophobic compounds.
3.4. F12511 and Nanoparticle F toxicity measurements
3.4.1. No overt neuronal toxicity is detectable after treatment of F12511 alone or with Nanoparticle F at concentrations of 10μM or lower.
F12511 has been tested in clinical trials for atherosclerosis and passed clinical safety tests; however, its potential toxicity in neurons has not been reported. We examined this issue by testing the potential toxicity of F12511 and Nanoparticle F in primary mouse neuronal cells. Using F12511 concentrations from 2μM to 100μM, cells were treated for 24-, 48- or 72-hours and cellular toxicity was assessed by monitoring the leakage of lactate dehydrogenase (LDH) (assay described in Materials and Methods section). The results (Figure 6) show that for the three different agents tested (F12511 alone, Nanoparticle F, and DSPE-PEG2000/PC vehicle), there is minimal, if any, toxicity after 24 hours of treatment. At 48 hours, there is a slight increase in toxicity for F12511 at very high concentrations (near 100μM). At 72 hours, more toxicity is detected at this high concentration range. Importantly, at 72 hours, only very minimal toxicity occurs after treatment of F12511 alone or with Nanoparticle F at concentrations of 10μM, which is more than two orders of magnitude higher than the Ki for F12511 to inhibit ACAT1/SOAT1 (39nM). Another important note is that the treatment time scale for Figures 3 and 4 is only 2–4 hours, while the time scale in the LDH assays (Figure 6) is significantly longer. This suggests that toxicity was not observed within the treatment paradigm used to measure ACAT inhibition and that longer incubations could be performed with minimal toxicity.
Figure 6:
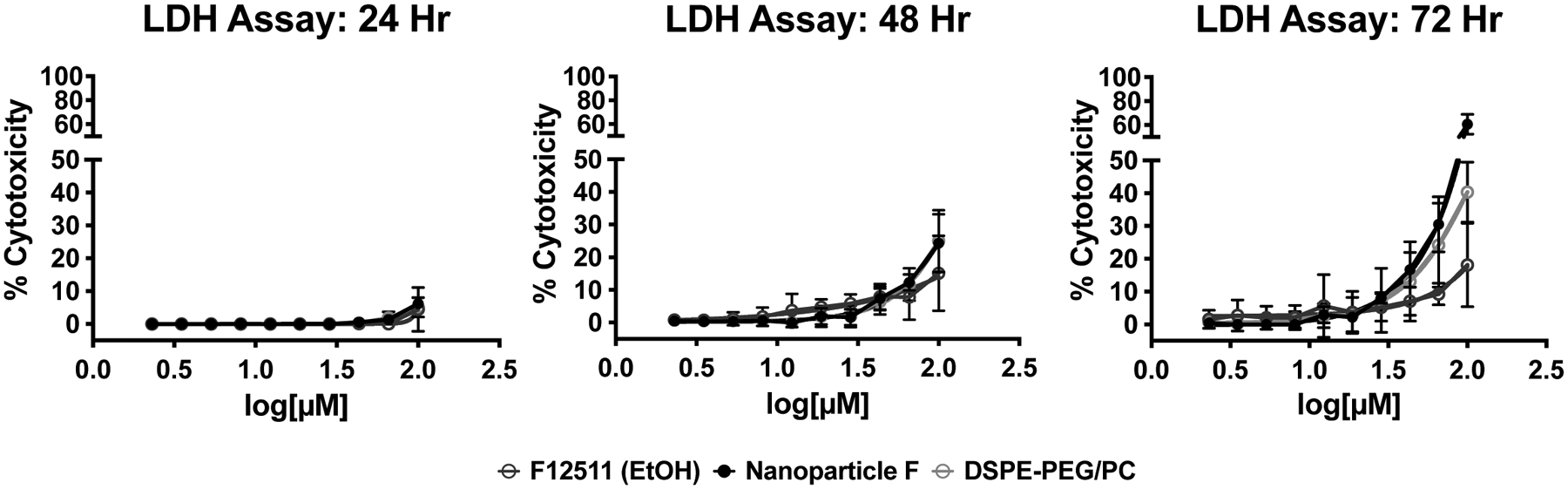
Neither F12511 nor Nanoparticle F appears to have detectable toxicity in primary neuronal cells at relevant concentrations. Toxicity assay is described in detail in the Methods and Materials section. Results are from LDH assays using 2–100μM concentrations of F12511, Nanoparticle F, and DSPE-PEG2000/PC for 24-, 48-, or 72-hours. Conditions were done in duplicate or triplicate. Controls used were media for nanoparticles and EtOH for F12511 alone. Data was transformed, normalized, and best nonfit linear regression determined.
4. Discussion:
Using ACAT inhibitors for AD drug treatment has been of interest for two decades. Since essentially all the ACAT inhibitors are quite hydrophobic, methods to solubilize the ACAT inhibitors and to administer these inhibitors in vivo are key hurdles. The method for delivering the ACAT inhibitors CP-113,818 and CI-1011 in mouse models for AD (Hutter-Paier et al., 2004; Huttunen et al., 2010) was done through the use of a slow injection method that involved a custom made slow-release pellet; the composition of the pellet remained proprietary. A different method to deliver ACAT1 specific inhibitor K-604 intranasally was recently reported (Shibuya et al., 2019). Here, Shibuya and colleagues found that at acidic pH (pH 2.3), K-604 became quite water soluble, and when injected intranasally as a solution at pH 2.3, it significantly inhibited cholesteryl ester content in the mouse brain. However, the long-term safety of this method is unknown. In other reports, Bougáret and colleagues found that F12511 can be solubilized at high concentration by forming a complex with cyclodextrin (Bougáret et al., 2005) and Lee and colleagues reported that CI-1011 can be solubilized in human albumen at high concentration (Lee et al., 2015). While these methods look promising, they do not offer the option for nanoparticle tagging.
Here we report a simple and efficient method to encapsulate a high-affinity ACAT inhibitor F12511 in DSPE-PEG2000/PC nanoparticles. With the addition of PC, the encapsulation of F12511 increased nearly 6-fold, allowing a very high concentration of F12511 to be incorporated. Nanoparticle F exhibits average size of 200nm and is stable at 4°C for several months. Importantly, efficacy tests show that Nanoparticle F is very effective in inhibiting ACAT enzyme activity in human and mouse neuronal and microglial cell lines, and neither F12511 nor Nanoparticle F appear to exhibit detectable toxicity when given to mouse primary neuronal cells at concentrations ranging from 2–10μM for up to 3 days.
The main advantage for this nanoparticle system is its potential impact for in vivo delivery. While F12511 alone (dissolved in DMSO or EtOH) is more potent at inhibiting ACAT in cell culture than Nanoparticle F (as seen in Figures 3 and 4), this would not be feasible for in vivo work. F12511 is very hydrophobic and must be in an emulsion, complexed to something, or as discussed here, encapsulated in nanoparticles for in vivo delivery. We are currently working on delivering Nanoparticle F and Nanoparticle K in vivo.
Another benefit of this method is that it can be expanded to produce tagged nanoparticles to increase the specificity of targeting tissues. It also offers opportunity to incorporate water soluble agent(s) within the aqueous space of the nanoliposomes for potential combinatorial therapy.
5. Conclusion:
One of the key benefits of this method is the ease of reproducing and characterizing the nanoparticles. This new method will give researchers the opportunity to deliver ACAT inhibitors and other hydrophobic compounds/inhibitors at high concentrations as potential therapies to treat various human diseases, including AD. We also anticipate that by using DSPE-PEG2000 and PC based nanoparticles, the bioavailability and half-life of F12511 in vivo will be significantly increased (Allen et al., 1993). We are currently testing the effects of Nanoparticle F and Nanoparticle K in the systemic tissues as well as in the central nervous system (CNS) of mice in vivo.
Highlights:
Encapsulation of ACAT inhibitor F12511 (Nanoparticle F) in stealth liposomes
Nanoparticles are relatively stable when kept at 4°C for several months
Nanoparticle F displays high potency as inhibitor in neuronal and microglial cells
Nanoparticle F has minimal toxicity in primary mouse neurons
This method is applicable to other ACAT inhibitors and hydrophobic compounds
Acknowledgements:
We would like to thank the following funding sources: National Institute of Neurological Disorders and Stroke (NINDS) F31NS110317-02 (ALD); National Institute on Aging (NIA) R01AG063544 (TYC and CC); National Institute of Environmental and Health Sciences (NIEHS) R01 ES024745 (MCH); NIEHS F31ES030982-01 (FLA); and NIH IDeA award for Dartmouth BioMT (P20-GM113132).
We would like to thank Ann Levanway and Zdenek Svindrych, for their help, guidance, and training with the DIC imaging and the F12511 fluorescence work. We thank Michael Ragusa and Maxime Guinel for their generous effort and time with the EM studies. We would also like to thank Dr. Jack Hoopes and Dr. Shan Zhao of Geisel School of Medicine at Dartmouth for providing training, as well as instrument use, of the Malvern Zeta Sizer. We thank the past and present members of the Chang lab for all of their support and feedback. A special thanks to Elaina Melton for suggesting the TLC solvent system for analyzing K-604 nanoparticles.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
Credit Author Statement:
Adrianna L. De La Torre: Conceptualization, Methodology, Validation, Formal Analysis, Investigation, Writing – Original Draft, Visualization. Caleb Smith: Methodology, Validation, Investigation, Writing – Review and editing. Joseph Granger: Conceptualization, Writing – Review and Editing. Faith L. Anderson: Methodology, Investigation, Writing – Review and Editing. Taylor C. Harned: Methodology, Investigation, Writing – Review and Editing. Matthew C. Havrda: Conceptualization, Resources, Writing – Review and Editing. Catherine C. Y. Chang: Conceptualization, Resources, Writing – Original Draft, Supervision, Project Administration, Funding Acquisition. Ta-Yuan Chang: Conceptualization, Resources, Writing – Original Draft, Supervision, Project Administration, Funding Acquisition
References
- Alconcel SNS, Baas AS, & Maynard HD (2011). FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polymer Chemistry, 2(7), 1442–1448. 10.1039/C1PY00034A [DOI] [Google Scholar]
- Allen TM, Hansen CB, & Guo LSS (1993). Subcutaneous administration of liposomes: a comparison with the intravenous and intraperitoneal routes of injection. Biochimica et Biophysica Acta, 1150(1), 9–16. 10.1016/0005-2736(93)90115-G [DOI] [PubMed] [Google Scholar]
- Anderson RA, Joyce C, Davis M, Reagan JW, Clark M, Shelness GS, & Rudel LL (1998). Identification of a Form of Acyl-CoA:Cholesterol Acyltransferase Specific to Liver and Intestine in Nonhuman Primates. Journal of Biological Chemistry, 273(41), 26747–26754. 10.1074/JBC.273.41.26747 [DOI] [PubMed] [Google Scholar]
- Ashok B, Arleth L, Hjelm RP, Rubinstein I, & Önyüksel H (2004). In vitro characterization of PEGylated phospholipid micelles for improved drug solubilization: Effects of PEG chain length and PC incorporation. Journal of Pharmaceutical Sciences, 93(10), 2476–2487. 10.1002/jps.20150 [DOI] [PubMed] [Google Scholar]
- Bougáret J, Leverd E, Ibarra M-D, & Gil A (2005). Anilide and cyclodextrin complexes, their preparation and their use as medicine in particular for treating dyslipidemiae (Patent No. 6,864,246 B2).
- Bryleva* EY, Rogers* MA, Chang CCY, Buen F, Harris BT, Rousselet E, Seidah NG, Oddo S, LaFerla FM, Spencer TA, Hickey WF, & Chang T-Y (2010). ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proceedings of the National Academy of Sciences of the United States of America, 107(7), 3081–3086. 10.1073/pnas.0913828107 [DOI] [PMC free article] [PubMed] [Google Scholar]; *These authors contributed equally to the work
- Cases S, Novak S, Zheng Y-W, Myers HM, Lear SR, Sande E, Welch CB, Lusis AJ, Spencer TA, Krause BR, Erickson SK, & Farese R. v. (1998). ACAT-2, A Second Mammalian Acyl-CoA:Cholesterol Acyltransferase. The Journal of Biological Chemistry, 273(41), 26755–26764. 10.1074/jbc.273.41.26755 [DOI] [PubMed] [Google Scholar]
- Chang CCY, Doolittle GM, & Chang T-Y (1986). Cycloheximide sensitivity in regulation of acyl coenzyme A:cholesterol acyltransferase activity in Chinese hamster ovary cells. 1. Effect of exogenous sterols. Biochemistry, 25(7), 1693–1699. 10.1021/bi00355a038 [DOI] [PubMed] [Google Scholar]
- Chang CCY, Huh HY, Cadigan KM, & Chang T-Y (1993). Molecular Cloning and Functional Expression of Human Acyl- Coenzyme A:Cholesterol Acyltransferase cDNA in Mutant Chinese Hamster Ovary Cells. The Journal of Biological Chemistry, 268(28), 20747–20755. 10.1016/S0021-9258(19)36846-2 [DOI] [PubMed] [Google Scholar]
- Chang CCY, Lee C-YG, Chang ET, Cruz JC, Levesque MC, & Chang T-Y (1998). Recombinant acyl-coA:cholesterol acyltransferase-1 (ACAT-1) purified to essential homogeneity utilizes cholesterol in mixed micelles or in vesicles in a highly cooperative manner. Journal of Biological Chemistry, 273(52), 35132–35141. 10.1074/jbc.273.52.35132 [DOI] [PubMed] [Google Scholar]
- Chang CCY, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, Lin S, Lee C-YG, Strom SC, Kashyap R, Fung J, Farese R. v., Patoiseau JF, Delhon A, & Chang T-Y (2000). Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. Journal of Biological Chemistry, 275(36), 28083–28092. 10.1074/jbc.M003927200 [DOI] [PubMed] [Google Scholar]
- Chang CCY, Sun J, & Chang T-Y (2011). Membrane-bound O-acyltransferases (MBOATs). Frontiers in Biology, 6(3), 177–182. 10.1007/s11515-011-1149-z [DOI] [Google Scholar]
- Chang T-Y, Li B-L, Chang CCY, & Urano Y (2009). Acyl-coenzyme A: cholesterol acyltransferases. American Journal of Physiology Endocrinology and Metabolism, 297, E1–9. 10.1152/ajpendo.90926.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J, Lee G, Zhong K, Fonseca J, & Taghva K (2021). Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 7(1). 10.1002/trc2.12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HR, & Veltri EP (2007). Zetia: Inhibition of Niemann-Pick C1 Like 1 (NPC1L1) to Reduce Intestinal Cholesterol Absorption and Treat Hyperlipidemia. Journal of Atherosclerosis and Thrombosis, 14(3), 99–108. 10.5551/JAT.14.99 [DOI] [PubMed] [Google Scholar]
- Ge L, Wang J, Qi W, Miao H-H, Cao J, Qu Y-X, Li B-L, & Song B-L (2008). The Cholesterol Absorption Inhibitor Ezetimibe Acts by Blocking the Sterol-Induced Internalization of NPC1L1. Cell Metabolism, 7(6), 508–519. 10.1016/J.CMET.2008.04.001 [DOI] [PubMed] [Google Scholar]
- Gref R, Lück M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, & Müller RH (2000). ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids and Surfaces B: Biointerfaces, 18(3–4), 301–313. 10.1016/S0927-7765(99)00156-3 [DOI] [PubMed] [Google Scholar]
- Gülçür E, Thaqi M, Khaja F, Kuzmis A, & Önyüksel H (2013). Curcumin in VIP-targeted sterically stabilized phospholipid nanomicelles: a novel therapeutic approach for breast cancer and breast cancer stem cells. Drug Delivery and Translational Research, 3(6), 562–574. 10.1007/s13346-013-0167-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JM, & Chess RB (2003). Effect of pegylation on pharmaceuticals. Nature Reviews: Drug Discovery, 2, 214–221. 10.1038/nrd1033 [DOI] [PubMed] [Google Scholar]
- Hofmann K (2000). A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochemical Sciences, 25(3), 111–112. 10.1016/S0968-0004(99)01539-X [DOI] [PubMed] [Google Scholar]
- Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, Moir RD, Domnitz SB, Frosch MP, Windisch M, & Kovacs DM (2004). The ACAT Inhibitor CP-113,818 Markedly Reduces Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Neuron, 44, 227–238. 10.1016/j.neuron.2004.08.043 [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Havas D, Peach C, Barren C, Duller S, Xia W, Frosch MP, Hutter-Paier B, Windisch M, & Kovacs DM (2010). The Acyl-Coenzyme A:Cholesterol Acyltransferase Inhibitor CI-1011 Reverses Diffuse Brain Amyloid Pathology in Aged Amyloid Precursor Protein Transgenic Mice. Journal of Neuropathology & Experimental Neurology, 69(8), 777–788. 10.1097/NEN.0b013e3181e77ed9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoya M, Yoshinaka Y, Kobayashi H, Kawamine K, Shibuya K, Sato F, Sawanobori K, Watanabe T, & Miyazaki A (2007). A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis, 191(2), 290–297. 10.1016/j.atherosclerosis.2006.05.048 [DOI] [PubMed] [Google Scholar]
- Insull W, Koren M, Davignon J, Sprecher D, Schrott H, Keilson LM, Brown AS, Dujovne CA, Davidson MH, McLain R, & Heinonen T (2001). Efficacy and short-term safety of a new ACAT inhibitor, avasimibe, on lipids, lipoproteins, and apolipoproteins, in patients with combined hyperlipidemia. Atherosclerosis, 157(1), 137–144. 10.1016/S0021-9150(00)00615-8 [DOI] [PubMed] [Google Scholar]
- Jhaveri AM, & Torchilin VP (2014). Multifunctional polymeric micelles for delivery of drugs and siRNA. Frontiers in Pharmacology, 5(April), 77. 10.3389/fphar.2014.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, Xing B, Sun W, Ren L, Hu B, Li C, Zhang L, Qin G, Zhang M, Chen N, Zhang M, Huang Y, Zhou J, Zhao Y, … He F (2019). Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature, 567(7747), 257–261. 10.1038/s41586-019-0987-8 [DOI] [PubMed] [Google Scholar]
- Junquero D, Bruniquel F, N’Guyen X, Autin J-M, Patoiseau J-F, Degryse A-D, Colpaert FC, & Delhon A (2001). F 12511, a novel ACAT inhibitor, and atorvastatin regulate endogenous hypercholesterolemia in a synergistic manner in New Zealand rabbits fed a casein-enriched diet. Atherosclerosis, 155(1), 131–142. 10.1016/S0021-9150(00)00559-1 [DOI] [PubMed] [Google Scholar]
- Junquero D, Oms P, Carilla-Durand E, Autin J-M, Tarayre J-P, Degryse A-D, Patoiseau J-F, Colpaert FC, & Delhon A (2001). Pharmacological profile of F 12511, (S)-2′,3′,5′-trimethyl-4′-hydroxy-α-dodecylt hioacetanilide a powerful and systemic acylcoenzyme A: cholesterol acyltransferase inhibitor. Biochemical Pharmacology, 61(1), 97–108. 10.1016/S0006-2952(00)00523-2 [DOI] [PubMed] [Google Scholar]
- Junquero D, Pilon A, Carilla-Durand E, Patoiseau J-F, Tarayre J-P, Torpier G, Staels B, Fruchart J-C, Colpaert FC, Clavey V, & Delhon A (2001). Lack of toxic effects of F 12511, a novel potent inhibitor of acyl-coenzyme A: cholesterol O-acyltransferase, on human adrenocortical cells in culture. Biochemical Pharmacology, 61(4), 387–398. 10.1016/S0006-2952(00)00555-4 [DOI] [PubMed] [Google Scholar]
- Kreuter J (2007). Nanoparticles - a historical perspective. International Journal of Pharmaceutics, 331(1), 1–10. 10.1016/j.ijpharm.2006.10.021 [DOI] [PubMed] [Google Scholar]
- Lee SS-Y, Li J, Tai JN, Ratliff TL, Park K, & Cheng J-X (2015). Avasimibe Encapsulated in Human Serum Albumin Blocks Cholesterol Esterification for Selective Cancer Treatment. American Chemical Society Nano, 9(3), 2420–2432. 10.1021/NN504025A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llaverías G, Laguna JC, & Alegret M (2003). Pharmacology of the ACAT Inhibitor Avasimibe (CI-1011). Cardiovascular Drug Reviews, 21(1), 33–50. 10.1111/j.1527-3466.2003.tb00104.x [DOI] [PubMed] [Google Scholar]
- López-Farré AJ, Sacristán D, Zamorano-León JJ, San-Martín N, & Macaya C (2008). Inhibition of acyl-CoA cholesterol acyltransferase by F12511 (Eflucimibe): Could it be a new antiatherosclerotic therapeutic? Cardiovascular Therapeutics, 26(1), 65–74. 10.1111/j.1527-3466.2007.00030.x [DOI] [PubMed] [Google Scholar]
- Mesplet N, Morin P, & Ribet JP (2005). Spectrofluorimetric study of eflucimibe-γ-cyclodextrin inclusion complex. European Journal of Pharmaceutics and Biopharmaceutics, 59(3), 523–526. 10.1016/J.EJPB.2004.09.011 [DOI] [PubMed] [Google Scholar]
- Miteva M, Kirkbride KC, Kilchrist K. v., Werfel TA, Li H, Nelson CE, Gupta MK, Giorgio TD, & Duvall CL (2015). Tuning PEGylation of mixed micelles to overcome intracellular and systemic siRNA delivery barriers. Biomaterials, 38, 97–107. 10.1016/j.biomaterials.2014.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SR, Chang CCY, Dogbevia G, Bryleva EY, Bowen Z, Hasan MT, & Chang T-Y (2013). Acat1 Knockdown Gene Therapy Decreases Amyloid-β in a Mouse Model of Alzheimer’s Disease. Molecular Therapy, 21(8), 1497–1506. 10.1038/mt.2013.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, Chang CCY, & Chang T-Y (2019). Triton X-100 or octyl glucoside inactivates acyl-CoA:cholesterol acyltransferase 1 by dissociating it from a two-fold dimer to a two-fold monomer. Archives of Biochemistry and Biophysics, 671, 103–110. 10.1016/j.abb.2019.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, Schoenhagen P, Waters DD, Pepine CJ, Crowe TD, Davidson MH, Deanfield JE, Wisniewski LM, Hanyok JJ, & Kassalow LM (2006). Effect of ACAT Inhibition on the Progression of Coronary Atherosclerosis. The New England Journal of Medicine, 354, 1253–1263. 10.1056/nejmoa054699 [DOI] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, Lucas A, Baskaran S, Haddick PCG, Lenser M, Earr TK, Shi J, Dugas JC, Andreone BJ, Logan T, … di Paolo G (2020). TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron, 105(5), 837–854.e9. 10.1016/j.neuron.2019.12.007 [DOI] [PubMed] [Google Scholar]
- Oelkers P, Behari A, Cromley D, Billheimer JT, & Sturley SL (1998). Characterization of Two Human Genes Encoding Acyl Coenzyme A: Cholesterol Acyltransferase-related Enzymes. Journal of Biological Chemistry, 273(41), 26765–26771. 10.1074/jbc.273.41.26765 [DOI] [PubMed] [Google Scholar]
- Peters D, Kastantin M, Kotamrajua VR, Karmali PP, Gujraty K, Tirrell M, & Ruoslahti E (2009). Targeting atherosclerosis by using modular, multifunctional micelles. Proceedings of the National Academy of Sciences of the United States of America, 106(24), 9815–9819. 10.1073/pnas.0903369106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli L, Konopka G, Pack-Chung E, Ingano LAM, Berezovska O, Hyman BT, Chang T-Y, Tanzi RE, & Kovacs DM (2001). Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the Amyloid β-peptide. Nature Cell Biology, 3(10), 905–912. 10.1038/ncb1001-905 [DOI] [PubMed] [Google Scholar]
- Rhainds D, Arsenault BJ, & Tardif J-C (2012). PCSK9 inhibition and LDL cholesterol lowering: the biology of an attractive therapeutic target and critical review of the latest clinical trials. Clinical Lipidology, 7(6), 621–640. 10.2217/clp.12.74 [DOI] [Google Scholar]
- Shibuya K, Morikawa S, Miyamoto M, Ogawa S, Tsunenari Y, Urano Y, & Noguchi N (2019). Brain Targeting of Acyl-CoA:Cholesterol O-Acyltransferase-1 Inhibitor K-604 via the Intranasal Route Using a Hydroxycarboxylic Acid Solution. American Chemical Society Omega, 4(16), 16943–16955. 10.1021/ACSOMEGA.9B02307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya Y, Chang CCY, Huang L-H, Bryleva EY, & Chang T-Y (2014). Inhibiting ACAT1/SOAT1 in Microglia Stimulates Autophagy-Mediated Lysosomal Proteolysis and Increases Aβ1–42 Clearance. Journal of Neuroscience, 34(43), 14484–14501. 10.1523/JNEUROSCI.2567-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya* Y, Niu* Z, Bryleva EY, Harris BT, Murphy SR, Kheirollah A, Bowen ZD, Chang CCY, & Chang T-Y (2015). Acyl-CoA:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer’s disease mouse and reduces human P301L-tau content at the pre-symptomatic stage. Neurobiology of Aging, 36(7), 2248–2259. 10.1016/j.neurobiolaging.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]; *These authors contributed equally to the work
- Suk JS, Xu Q, Kim N, Hanes J, & Ensign LM (2016). PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Advanced Drug Delivery Reviews, 99(Part A), 28–51. 10.1016/J.ADDR.2015.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif J-C, Grégoire J, L’Allier PL, Anderson TJ, Bertrand O, Reeves F, Title LM, Alfonso F, Schampaert E, Hassan A, McLain R, Pressler ML, Ibrahim R, Lespérance J, Blue J, Heinonen T, & Rodés-Cabau J (2004). Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation, 110(21), 3372–3377. 10.1161/01.CIR.0000147777.12010.EF [DOI] [PubMed] [Google Scholar]
- Terasaka N, Miyazaki A, Kasanuki N, Ito K, Ubukata N, Koieyama T, Kitayama K, Tanimoto T, Maeda N, & Inaba T (2007). ACAT inhibitor pactimibe sulfate (CS-505) reduces and stabilizes atherosclerotic lesions by cholesterol-lowering and direct effects in apolipoprotein E-deficient mice. Atherosclerosis, 190(2), 239–247. 10.1016/J.ATHEROSCLEROSIS.2006.03.007 [DOI] [PubMed] [Google Scholar]
- Turecek PL, Bossard MJ, Schoetens F, & Ivens IA (2016). PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. Journal of Pharmaceutical Sciences, 105(2), 460–475. 10.1016/J.XPHS.2015.11.015 [DOI] [PubMed] [Google Scholar]
- van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, Steenvoorden E, Rynearson KD, Brouwers JF, Helms JB, Ovaa H, Giera M, Wagner SL, Bang AG, & Goldstein LSB (2019). Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell, 24(3), 363–375.e9. 10.1016/j.stem.2018.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, Yan C, Wang L, Chang CCY, Chang T-Y, Zhang T, Zhou P, Song B-L, Liu W, Sun S-C, … Xu C (2016). Potentiating the antitumour response of CD8 + T cells by modulating cholesterol metabolism. Nature, 531, 651–655. 10.1038/nature17412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingchoncharoen P, Kalinowski DS, & Richardson DR (2016). Lipid-Based Drug Delivery Systems in Cancer Therapy: What Is Available and What Is Yet to Come. Pharmacological Reviews, 68(3), 701–787. https://dx.doi.org/10.1124%2Fpr.115.012070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue S, Li J, Lee S-Y, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, & Cheng J-X (2014). Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metabolism, 19(3), 393–406. 10.1016/J.CMET.2014.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-Q, Even-Or O, Xu X, van Rosmalen M, Lim L, Gadde S, Farokhzad OC, & Fisher EA (2015). Nanoparticles Containing a Liver X Receptor Agonist Inhibit Inflammation and Atherosclerosis. Advanced Healthcare Materials, 4(2), 228–236. 10.1002/ADHM.201400337 [DOI] [PMC free article] [PubMed] [Google Scholar]