

Abstract
The clinical impact of rare loss-of-function variants has yet to be determined for most genes. Integration of DNA sequencing data with electronic health records (EHR) could enhance our understanding of the contribution of rare genetic variation to human disease.1 By leveraging 10,900 whole exome sequences linked to EHR data in the Penn Medicine Biobank (PMBB), we addressed the association of the cumulative effects of rare predicted loss-of-function (pLOF) variants per individual gene on human disease on an exome-wide scale, as assessed using a set of diverse EHR phenotypes. After discovering 97 genes with exome-by-phenome-wide significant phenotype associations (p < 10−6), we replicated 26 of these in PMBB, as well as in three other medical biobanks and the population-based UK Biobank (UKB). Of these 26 genes, five had associations that have been previously reported and represented positive controls, whereas 21 had phenotype associations not previously reported, among which were genes implicated in glaucoma, aortic ectasia, diabetes mellitus, muscular dystrophy, and hearing loss. These findings show the value of aggregating rare pLOF variants into “gene burdens” for identifying new gene-disease associations using EHR phenotypes in a medical biobank. We suggest that application of this approach to even larger numbers of individuals will provide the statistical power required to uncover unexplored relationships between rare genetic variation and disease phenotypes.
A “genome-first” approach, in which genetic variants of interest are identified and then subsequently associated with phenotypes, has the potential to inform the genetic basis of human disease and reveal new insights into gene function and human biology.2 This approach can be applied to “medical” biobanks consisting of healthcare populations with DNA sequence data linked to extensive EHR phenotype data, thus permitting “phenome-wide association studies” (PheWAS) as an agnostic approach to determining the clinical impact of specific genetic variants.3 Genome-first approaches utilizing PheWAS have primarily focused on individual common variants of modest effect.4 Very rare and private coding variants are more likely to have larger effect sizes and are of great interest, but are generally too rare to study in a univariate fashion.5 Aggregation of multiple rare variants in a gene (i.e. “gene burden”) not only increases the statistical power of regression analyses but also enables gene-based association studies to describe the clinical implications of loss of gene function in human disease.6
Previously, we leveraged the Penn Medicine Biobank (PMBB, University of Pennsylvania), a large academic medical biobank with whole-exome sequencing (WES) data linked to EHR data, to show that aggregating rare, loss-of-function variants in a single gene or targeted sets of genes to conduct gene burden PheWAS has the potential to uncover novel pleiotropic relationships between the gene and human disease.7,8 We applied rare pLOF-based gene burden PheWAS on an exome-wide scale, utilizing WES data to conduct exome-by-phenome-wide association studies (ExoPheWAS) to evaluate in detail the clinical phenotypes (i.e. phecodes) associated with rare pLOF variants on a gene-by-gene basis across the human exome, and replicated our top results in several other medical biobanks.
We interrogated a dataset of 10,900 individuals with WES data in PMBB (Table 1) for carriers of rare (MAF ≤ 0.1% in gnomAD) pLOF variants, which include frameshift insertions or deletions, gain or loss of stop codon, and disruption of canonical splice site dinucleotides. The distribution of the number of carriers for rare pLOF variants per gene was on a negative exponential distribution (Extended Data Figure 1). We chose to interrogate genes with at least 25 heterozygous carriers for rare pLOFs (N=1,518 genes), for which we show that statistical power to detect association is sufficient as a function of effect size and the associated phenotype’s number of cases (Extended Data Figure 2). We collapsed rare pLOF variants into gene burdens across these 1518 genes for ExoPheWAS analyses with 1000 binary phecodes with at least 20 cases (Figure 1). Given that p values for gene burden association studies interrogating rare loss-of-function variants may be inflated due to their higher likelihood of increasing disease risk compared to other variants,9 we found that our associations roughly deviated from the fitted expected distribution at an observed p<E-06 (Extended Data Figure 3). We identified 97 gene burdens with phenotype associations at p<E-06 (Figure 2, Table S1). We addressed potential inflation issues regarding small sample sizes by using Firth’s penalized likelihood approach, and found that beta and significance estimates were consistent with exact logistic regression (Table S1).
Table 1. Demographics and disease prevalence of the PMBB discovery cohort.
Demographic information and clinical phenotypic counts for all individuals with WES linked to EHRs in the PMBB. Clinical phenotypes were defined by phecodes (see Methods). Data are represented as count data with percentage prevalence in the population in parentheses, where appropriate. AFR, Africa; AMR, the Americas; EAS, East Asia; EUR, Europe; SAS, South Asia; GERD, gastroesophageal reflux disease.
| Basic demographics | |
| Total population, N | 10900 |
| Female, N (%) | 4432 (40.7) |
| Median Age (at biobank entry), years | 67.0 |
| Genetically informed ancestry | |
| AFR, N (%) | 2172 (19.9) |
| AMR, N (%) | 304 (2.8) |
| EAS, N (%) | 79 (0.7) |
| EUR, N (%) | 8198 (75.2) |
| SAS, N (%) | 114 (1.0) |
| Cardiovascular phenotypes | |
| Essential hypertension, N (%) | 6441 (59.1) |
| Ischemic Heart Disease, N (%) | 5008 (45.9) |
| Myocardial infarction, N (%) | 1640 (15.0) |
| Cardiomyopathy, N (%) | 1976 (18.1) |
| Congestive heart failure; nonhypertensive, N (%) | 3695 (33.9) |
| Heart transplant/surgery, N (%) | 518 (4.8) |
| Cardiac dysrhythmias, N (%) | 5784 (53.1) |
| Atrial fibrillation and flutter, N (%) | 3782 (34.7) |
| Cerebrovascular disease, N (%) | 1706 (15.7) |
| Peripheral vascular disease, N (%) | 954 (8.8) |
| Aortic aneurysm, N (%) | 836 (7.7) |
| Atherosclerosis, N (%) | 539 (4.9) |
| Endocrine/metabolic phenotypes | |
| Type 2 diabetes, N (%) | 2799 (25.7) |
| Overweight, obesity and other hyperalimentation, N (%) | 2275 (20.9) |
| Hyperlipidemia, N (%) | 6231 (57.2) |
| Hypercholesterolemia, N (%) | 2034 (18.7) |
| Hypothyroidism, N (%) | 1314 (12.1) |
| Gout and other crystal arthropathies, N (%) | 811 (7.4) |
| Gastrointenstinal phenotypes | |
| Esophagitis, GERD and related diseases, N (%) | 2526 (23.2) |
| Gastrointestinal hemorrhage, N (%) | 660 (6.1) |
| Diverticulosis and diverticulitis, N (%) | 610 (5.6) |
| Chronic liver disease and cirrhosis, N (%) | 449 (4.1) |
| Renal phenotypes | |
| Chronic renal failure, N (%) | 2135 (19.6) |
| End stage renal disease, N (%) | 510 (4.7) |
| Kidney replaced by transplant, N (%) | 283 (2.6) |
| Neuropsychiatric phenotypes | |
| Mood disorders, N (%) | 1353 (12.4) |
| Anxiety, phobic and dissociative disorders, N (%) | 1322 (12.1) |
| Delirium dementia and amnestic and other cognitive disorders, N (%) | 123 (1.1) |
| Respiratory phenotypes | |
| Chronic airway obstruction, N (%) | 1314 (12.1) |
| Asthma, N (%) | 920 (8.4) |
| Obstructive sleep apnea, N (%) | 1623 (14.9) |
| Respiratory failure, insufficiency, arrest, N (%) | 697 (6.4) |
| Sensory phenotypes | |
| Cataract, N (%) | 796 (7.3) |
| Hearing loss, N (%) | 579 (5.3) |
| Glaucoma, N (%) | 449 (4.1) |
| Congenital phenotypes | |
| Cardiac and circulatory congenital anomalies, N (%) | 780 (7.2) |
| Genitourinary congenital anomalies, N (%) | 151 (1.4) |
| Cystic kidney disease, N (%) | 108 (1.0) |
| Congenital anomalies of great vessels, N (%) | 77 (0.7) |
Figure 1. Flow chart for exome-by-phenome-wide association analysis using electronic health record phenotypes.
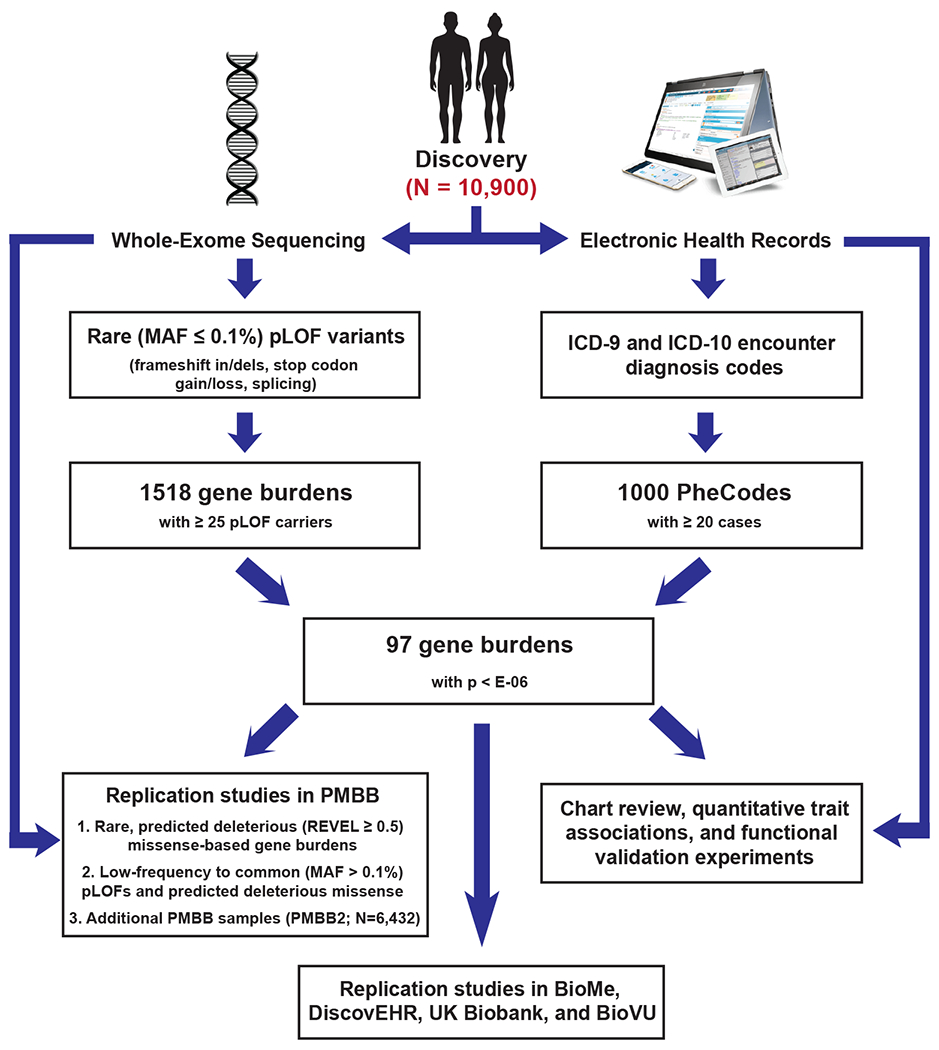
Flowchart diagram outlining the primary methodologies used for conducting the exome-by-phenome-wide association study and for evaluation of the robustness of the associations, indicating that 97 genes had associations at a significance level of p<E-06 via logistic regression. The pathways starting with short descending arrows represent the 'discovery phase', in which predicted loss-of-function (pLOF)-based gene burdens were studied on an exome-by-phenome-wide scale in 10,900 individuals from the Penn Medicine Biobank (PMBB). “Replication studies in PMBB” refers to analyses of gene-phenotype associations using REVEL-informed missense-based gene burdens and univariate analyses within the discovery PMBB cohort, as well as in an independent cohort of African Americans in the PMBB (the PMBB2 cohort; N=6,432). Additional replication studies included analyses of gene-phenotype associations using pLOF-based gene burdens, REVEL-informed missense-based gene burdens, and univariate analyses in BioMe (N=23,989), DiscovEHR (N=85,450), and the UK Biobank (N=32,268), as well as univariate analyses in BioVU (N=66,400).
Figure 2. ExoPheWAS plot exhibits the landscape of gene-phenotype associations across the exome and phenome in the Penn Medicine Biobank.
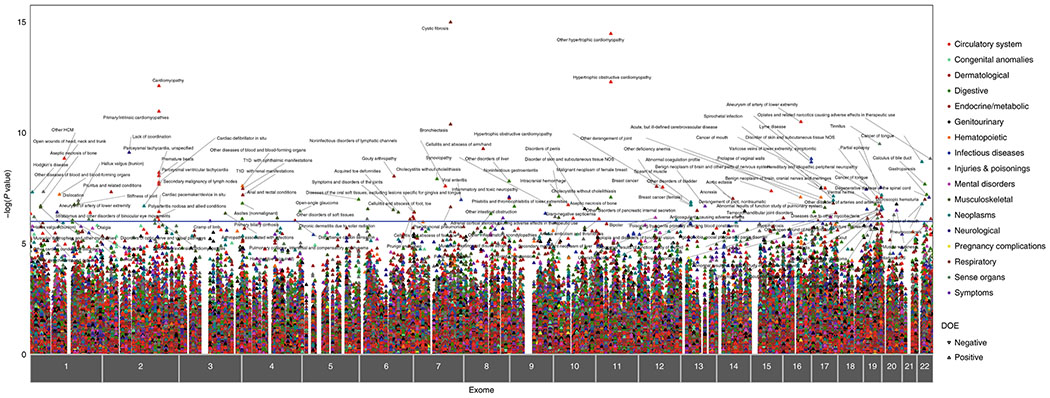
Plot of the results of the exome-by-phenome-wide association study (ExoPheWAS) in the Penn Medicine Biobank for 1518 gene burdens of rare (MAF ≤ 0.1%) predicted loss-of-function (pLOF) variants. The x-axis represents the exome and is organized by chromosomal location. The location of each gene along the x-axis is according to the gene’s genomic location per Genome Reference Consortium Human Build 37 (GRCh37). The association of each gene burden with a set of 1,000 phecodes is plotted vertically above each gene, with the height of each point representing the −log10(p value) of the association between the gene burden and phecode using a logistic regression model. Each phecode point is color-coded according to the phecode group, and the directionality of each triangular point represents the direction of effect (DOE). The blue line represents the significance threshold at p=E-06 to account for multiple hypothesis testing.
We evaluated the robustness of the significant gene-phenotype associations identified via our pLOF-based ExoPheWAS analyses by testing the associations in the same PMBB cohort between a separate group of rare likely deleterious exonic missense variants in the 97 significant genes with the same disease phenotypes that were identified in discovery (Figure 1). We utilized REVEL, an ensemble method for predicting the pathogenicity of missense variants,10 to define predicted deleterious missense variants (REVEL score ≥ 0.5) given the tool’s success in identifying likely pathogenic variants for gene burden association studies.7 First, we separately collapsed rare (MAF ≤ 0.1%), REVEL-informed predicted deleterious missense variants to test discovery-driven associations with their corresponding phenotypes (Table S2). We also interrogated single variants, including both pLOF variants and predicted deleterious missense (REVEL ≥ 0.5) variants, in the 97 genes identified in discovery that were of sufficient frequency (MAF > 0.1%) and therefore were not included in either of the gene burden analyses (Table S3).
We also endeavored to replicate our significant ExoPheWAS discovery analysis associations (Figure 1) using a separate cohort of 6,432 African Americans in PMBB who were exome-sequenced (PMBB2; Table S4–6), as well as two additional medical biobanks with WES linked to EHR phenotypes, namely BioMe (Mount Sinai; Table S7–9) and DiscovEHR (Geisinger Health System; Table S10–12), and the population-based UK Biobank (UKB) (Table S13–15). For each of the 97 significant genes, we interrogated: 1) gene burdens after collapsing rare (MAF ≤ 0.1%) pLOF variants, 2) gene burdens after collapsing non-overlapping rare (MAF ≤ 0.1%) REVEL-predicted deleterious missense variants, and 3) single pLOF or REVEL-predicted deleterious missense variants with MAF > 0.1% for association with their discovery phenotypes. Finally, we further interrogated a targeted list of univariate replications in BioVU (Vanderbilt; Table S16).
We identified a total of 26 robust genes using a Diverse Convergent Evidence (DiCE) approach11 for ranking associations using a combination of the number of significant replications and functional validation (Table 2, Table S17). Five of these genes can be considered positive control gene-disease associations. A gene burden of rare pLOFs in CFTR was significantly associated with cystic fibrosis (CF), a recessive condition caused by biallelic variants in CFTR. This was driven by individuals with a rare pLOF who had a second deleterious CFTR variant—predominantly ∆F508—that was not included in the pLOF gene burden. This association of the CFTR pLOF gene burden with CF was not replicated in other biobanks due to the extremely low case prevalence of CF (Table S18). The CFTR pLOF gene burden was also significantly associated with bronchiectasis independent of a CF diagnosis and occurred in individuals without a second CFTR variant; this finding replicated in all interrogated cohorts. While a predisposition to bronchiectasis due to haploinsufficiency of CFTR has been suggested,12 our finding strengthens this observation. TTN is a known dilated cardiomyopathy gene that replicated convincingly across other cohorts. MYBPC3 is a known hypertrophic cardiomyopathy (HCM) gene that replicated in BioMe and DiscovEHR, but not in UKB, where HCM had a case-control ratio of an order of magnitude lower than the medical biobanks (Table S18). These results indicate that medical biobanks have a different—and sicker—population that enables discovery of associations of human diseases driven by rare genetic variants. A pLOF gene burden in BRCA2 was associated with breast cancer and replicated in all biobanks. BRCA1 was associated with breast cancer in discovery (p=1.29E-04) but due to power did not meet our significance threshold. Finally, CYP2D6 is a P450 enzyme known to metabolize opioids;13 we found that CYP2D6 was significantly associated with adverse effects of therapeutic opiate use.
Table 2. List of robust exome-by-phenome-wide significant gene-phenotype associations.
List of genes among 97 pLOF-based gene burdens with phenotype associations at P < 10−6 in the PMBB discovery cohort that were most robust according to the DiCE approach, which integrates successful replication of the association with clinical and experimental evidence. For replication studies, gene–phenotype associations were evaluated for their robustness by interrogating REVEL-informed missense-based gene burdens and single variants in the same discovery PMBB cohort, and pLOF-based gene burdens, REVEL-informed missense-based gene burdens and single variants in an independent cohort of African Americans in the PMBB (the PMBB2 cohort), as well as in BioMe, DiscovEHR and the UKB. Targeted single variants that showed successful replication in the PMBB, PMBB2 and UKB were additionally analyzed in BioVU. Each gene–phecode association is labeled with the corresponding P value from logistic regression analyses in the discovery phase in the PMBB, as well as the number of total replications and existence of clinical/experimental evidence, fully detailed in Supplementary Table 17. Only associations with at least two total check marks in Supplementary Table 17, where each successful mode of replication in a particular biobank (for example, pLOF burden in BioMe) or the existence of clinical/experimental evidence is labeled with a checkmark, were deemed robust and therefore included here. Previously known associations were considered to represent positive controls. Positive control (above line) and new associations (below line) are each ranked alphabetically by gene name.
| Gene | Phecode Description | Discovery P | Replications (N) | Clinical/Experimental Evidence |
|---|---|---|---|---|
| BRCA2 | Breast cancer | 1.72E-07 | 4 | ✓ |
| CFTR | Bronchiectasis | 2.27E-07 | 10 | ✓ |
| Pseudomonal pneumonia | 4.21E-11 | 5 | ✓ | |
| Cystic fibrosis | 1.05E-15 | 1 | ✓ | |
| CYP2D6 | Opiates and related narcotics causing adverse effects in therapeutic use | 1.50E-09 | 3 | ✓ |
| MYBPC3 | Hypertrophic cardiomyopathy | 3.49E-15 | 5 | ✓ |
| TTN | Cardiomyopathy | 7.83E-13 | 10 | ✓ |
| Cardiac conduction disorders | 6.45E-09 | 10 | ✓ | |
| Cardiac dysrhythmias | 1.77E-08 | 12 | ✓ | |
|
| ||||
| ABCA10 | Benign neoplasm of brain, cranial nerves, meninges | 7.26E-08 | 2 | |
| Abnormal results of function study of pulmonary system | 1.54E-07 | 3 | ||
| BBS10 | Hypertrophic cardiomyopathy | 2.89E-08 | 1 | ✓ |
| CES5A | Abnormal coagulation profile | 8.10E-08 | 5 | |
| CILP | Aortic ectasia | 4.29E-08 | 3 | ✓ |
| CTC1 | Temporomandibular joint disorders | 3.76E-07 | 3 | |
| DNAH6 | Lack of coordination | 7.93E-10 | 2 | |
| DNHD1 | Aseptic necrosis of bone | 2.67E-07 | 4 | |
| EFCAB5 | Prolapse of vaginal walls | 3.19E-08 | 3 | |
| EPPK1 | Phlebitis and thrombophlebitis of lower extremities | 9.19E-08 | 3 | |
| FER1L6 | Muscular wasting and disuse atrophy | 7.18E-07 | 3 | ✓ |
| FLG2 | Stiffness of joint | 1.76E-07 | 2 | |
| MYCBP2 | Spasm of muscle | 2.08E-07 | 2 | ✓ |
| PPP1R13L | Primary open angle glaucoma | 7.29E-07 | 2 | ✓ |
| RGS12 | Type 1 diabetes | 6.48E-08 | 5 | ✓ |
| RTKN2 | Orthostatic hypotension | 7.24E-07 | 5 | |
| SCNN1D | Cardiac conduction disorders | 4.52E-07 | 5 | |
| TGM6 | Lipoma | 2.77E-07 | 4 | |
| TRDN | Acquired toe deformities | 3.90E-07 | 3 | |
| WDR87 | Ventral hernia | 1.70E-07 | 4 | |
| ZNF175 | Tinnitus | 3.24E-10 | 3 | ✓ |
| ZNF334 | Microscopic hematuria | 1.69E-07 | 3 | |
We identified 20 robust genes with novel disease associations that had at least two additional replications beyond the discovery experiment, and one strongly supported by the DiCE analysis (Table 2, Tables S2–S17). Some have prior biological plausibility, and for others we generated additional functional data supporting a biological basis to these associations. For example, a BBS10 gene burden was significantly associated with HCM. BBS10 is one of at least 19 genes implicated in autosomal recessive Bardet-Biedl Syndrome and accounts for ~20% of all cases.14 BBS10 is expressed in the heart15 and cardiac abnormalities have been reported in Bardet-Biedl Syndrome, including hypertrophy of the interventricular septum,16 but cardiac abnormalities due to haploinsufficiency of BBS10 have not been described. We interrogated echocardiography data in carriers of rare pLOF variants in BBS10 in PMBB compared with non-carriers and found increased left ventricular outflow tract (LVOT) stroke volume, consistent with cardiac hypertrophy (Table S19). Rare pLOF variants in SCNN1D, which encodes the delta subunit of the epithelial sodium channel (δENaC), were associated with cardiac conduction disorders and replicated robustly across medical biobanks. SCNN1D is expressed in the heart (unlike epithelial tissue-specific expression for SCNN1A and SCNN1B),17 there is an association between 1p36 deletions (which contains SCNN1D) and congenital heart defects,18 and decreased expression of δENaC may contribute to disrupted Na+ and K+ homeostasis in ischemic heart diseases.19 The association between rare pLOFs in ZNF175 and tinnitus (additionally, hearing loss barely missed our significance threshold), which replicated in BioMe, DiscovEHR, and UKB, is supported by the finding that mice with loss-of-function in Zfp719 (the mouse ortholog) are profoundly deaf and have abnormal Preyer reflex (auditory startle response)20 as well as raised auditory brainstem response thresholds.21 Zfp719 is expressed in inner and outer hair cells of the mouse ear,22 and human ZNF175 has a suggested role in neurotrophin production and neuronal survival.23
Rare pLOFs in FER1L6 were robustly associated with muscular wasting and disuse atrophy. FER1L6 is a member of the ferlin family of genes, and mutations in FER1L1 (dysferlin) are known to cause recessive forms of muscular dystrophy.24 Importantly, loss of the zebrafish ortholog Fer1l6 has been shown to lead to deformation of striated muscle and delayed cardiac development.25 Similarly, pLOFs in MYCBP2, an E3 ubiquitin-protein ligase critical in neuromuscular development in mice,26 Drosophila,27 and C. elegans,28 were associated with muscular spasms and dystrophy. Mice lacking the mouse ortholog Phr1 are lethal at birth without taking a breath due to incomplete innervation of the diaphragm by markedly narrower phrenic nerves that contain fewer axons than controls.26 We found that MYCBP2 showed significantly decreased expression in various lower extremity muscle tissues in tibial muscular dystrophy in humans (Extended Data Figure 4). Our findings suggest that haploinsufficiency in FER1L6 or MYCBP2 increases the risk of developing dystrophic skeletal muscle.
Rare pLOFs in CES5A were robustly associated with abnormal coagulation. Upon further investigation of EHR lab data in PMBB, we found that carriers of rare pLOF variants in CES5A had increased international normalized ratios (INR; ß=8.2, p=2.13E-02, N=5,275) and partial thromboplastin times (PTT; ß=13.9, p=2.07E-02, N=3,786) compared to non-carriers. Through chart review, we found an enrichment of gastrointestinal bleeding episodes following use of anti-platelet medications among carriers for rare pLOF variants in CES5A. CES5A is part of the family of carboxylesterases, which are known metabolizers of various orally bioavailable drugs, including the anti-platelet medications aspirin and clopidogrel.29 Given its predominant expression in the liver,15 it is thus plausible that haploinsufficiency of CES5A predisposes to adverse effects of anti-platelet medications.
Another novel finding was that rare pLOF variants in PPP1R13L, one of the most evolutionarily conserved inhibitors of p53,30 were associated with primary open angle glaucoma—a disease of the optic nerve head (ONH) that causes progressive vision loss. We interrogated the expression of PPP1R13L in silico using the Ocular Tissue Database (OTDB) and found that it is highly expressed in ocular tissues, with optic nerve and the ONH among the highest (Table S20). Retinal ganglion cells (RGCs) are the primary cells affected by glaucoma, and cells in the ONH such as astroglia, microglia, and endothelial cells mediate RGC degeneration in response to stress such as increased intraocular pressure. We investigated whether Ppp1r13l is differentially expressed in the mouse ONH in glaucoma by comparing microarray gene expression datasets of the ONH.31 We found Ppp1r13l expression to be highest during late-early to moderate stages of glaucoma (Extended Data Figure 5A). Additionally, inhibition of PPP1R13L has been shown to exacerbate retinal ganglion cell (RGC) death following axonal injury.32 We found that the PPP1R13L protein is predominantly localized to the ganglion cell layer in the adult human retina with some expression in the outer and inner plexiform layers, confirming its role in RGC function (Extended Data Figure 5B). Using human induced pluripotent stem cell-derived RGCs (iPSC-RGCs), we found that oxidative stress markedly upregulated PPP1R13L expression (Extended Data Figure 5C) to a much greater extent than even superoxide dismutase 1 (SOD1), which is known to be transcriptionally upregulated in response to oxidative stress. Thus, PPP1R13L is expressed in RGCs, is significantly upregulated by oxidative stress, and may help to prevent RGC death from p53 activation and p53-mediated apoptosis in primary open angle glaucoma.33 Our results are consistent with the concept that haploinsufficiency of PPP1R13L in RGCs increases the visual consequences of primary open angle glaucoma.
Another interesting novel finding was that rare pLOF variants in RGS12 were associated with type 1 diabetes mellitus and its complications. In PMBB, carriers of rare pLOFs in RGS12 had higher median values for random serum glucose than non-carriers (ß=16.9, p=2.91E-02, N=5,389). RGS12, an inhibitor of signal transduction in G protein signaling, contains an N terminus PDZ domain which selectively binds to and represses the macrophage IL-8 receptor CXCR2.34 Activation of macrophage CXCR2 by IL-8 is pro-inflammatory, and its antagonism leads to attenuation of immune cell infiltration and cytokine release as well as a shift of macrophages to the anti-inflammatory M2 state, thereby counteracting inflammatory signal pathways in diabetes.35 To further investigate RGS12 in type 1 diabetes, we generated single-cell RNA-seq data in human pancreatic islets from type 1 diabetes and control subjects collected by the Human Pancreas Analysis Program (HPAP; https://hpap.pmacs.upenn.edu) and interrogated RGS12 expression in distinct functional cells. We found that while RGS12 showed no significant differential expression in pancreatic endocrine or exocrine cells in type 1 diabetes versus control, there was a substantial reduction of expression of RGS12 in peri-islet CD45+ macrophages in type 1 diabetes (Extended Data Figure 6). These results are consistent with a model that RGS12 dampens islet macrophage inflammatory responses and that haploinsufficiency of RGS12 predisposes to greater islet inflammation and higher risk of type 1 diabetes.
Additionally, rare pLOF variants in CILP were associated with aortic ectasia, or dilatation of the aorta often associated with connective tissue disorders. Chart review of CILP pLOF carriers showed an enrichment for ascending thoracic aortic aneurysms. CILP encodes an extracellular matrix protein and is best known for its expression in chondrocytes.36 However, CILP is also expressed in the cardiovascular system,15 and has been shown to be involved in cardiac remodeling in response to pressure overload.37 We performed single-cell RNA-seq of normal mouse aorta and found that Cilp expression was localized mainly to adventitial fibroblasts in the aorta, but showed no significant expression in aortic smooth muscle cells (Extended Data Figure 7A–B). Single-cell RNA-seq of human aorta confirmed that CILP is localized to aortic fibroblasts (Extended Data Figure 7C–D). Importantly, CILP has been reported to modulate TGFB1 signaling and IGF1-induced proliferation,38 and dysregulated TGF-ß signaling has been shown to contribute to the pathogenesis of thoracic aortic aneurysm formation.39 To further interrogate the relationship between CILP and TGFB1 in human fibroblasts, we conducted a meta-analysis of 11 independent microarray and RNA-seq datasets for human fibroblasts from various tissues treated with TGF-ß from the Gene Expression Omnibus (GEO). We found that CILP was in the top 1% of significantly upregulated genes in human fibroblasts when treated with TGF-ß (log2 fold change = 1.964, p = 3.60E-29; Extended Data Figure 7E), confirming its role in a functional feedback loop with TGF-ß as similarly seen in the context of chondrocyte metabolism.36 Furthermore, CILP was differentially co-expressed with IGF1 as well as genes implicated in aortic ectasia including SMAD3, ACTA2, MYH11, and ELN (Extended Data Figure 7E).39 Our findings suggest that haploinsufficiency of CILP predisposes to the risk of developing thoracic aortic dilatation, perhaps through compromising the structural integrity of the aortic wall and contributing to dysregulation of TGF-ß signaling.
There has been a significant gap of knowledge regarding the clinical implications of genetic variants overrepresented among Africans due to the lack of ancestral diversity in the populations that have been studied in previous genetic association studies.40 Our discovery study included 19.9% African ancestry individuals, and three of our replication cohorts included substantial numbers of African-Americans (6,432 in PMBB2, 6,470 in BioMe, and 10,456 in BioVU). Interestingly, we identified 16 rare predicted deleterious single variants which are African ancestry-specific and that replicated associations with the same disease in which a pLOF gene burden was associated in discovery (Table S21). None of these rare variants exist in the GWAS catalog or have been previously mentioned in the published literature. Our findings suggest that larger experiments of this type in ethnically diverse cohorts are imperative for improving our understanding of the contribution of ancestry-specific rare genetic variants to human disease.
A significant challenge in rare variant association studies is the difficulty of performing replication studies. Here we show the value of evaluating the robustness of gene burden associations by interrogating other deleterious variants in the same genes (but in different individuals) in the same biobank cohort. We also performed replication studies in another cohort in PMBB as well as in two other medical biobanks with WES data. These provided more replication than the UKB, which is a population-based biobank and is widely recognized to have a “healthy volunteer selection bias”41 and has lower prevalence of the specific diseases than the medical biobanks (Table S18). This may be one factor explaining the relative lack of novel findings in gene burden studies using UKB for discovery.42,43 Finally, we show that one should not expect a uniform fit for p values when interrogating the cumulative effect of rare pLOF variants, and that the validity of the results is due as much to robust replication in other cohorts as to the determination of a particular significance threshold. To this end, our study emphasizes the value of medical biobanks for discovery of novel gene-disease associations based on rare variants.
In conclusion, we demonstrate the feasibility and value of aggregating rare pLOF variants into gene burdens on an exome-wide scale for association with EHR-derived phenotypes in a medical biobank for discovery of novel gene-disease relationships. Our compelling novel findings based on initial discovery in < 11,000 whole exomes suggest that much larger experiments of this type are likely to be highly informative and will lead to many new insights into the biology of human phenotypes and diseases.
Methods
Setting and study participants
All individuals who were recruited for the Penn Medicine Biobank (PMBB) are patients of clinical practice sites of the University of Pennsylvania Health System. Appropriate consent was obtained from each participant regarding storage of biological specimens, genetic sequencing, access to all available electronic health record (EHR) data, and permission to recontact for future studies. The study was approved by the Institutional Review Board of the University of Pennsylvania and complied with the principles set out in the Declaration of Helsinki.
In addition to our robustness validation analyses within PMBB, replication analyses were conducted using the WES dataset from an additional set of independent African-American individuals in PMBB (PMBB2), BioMe, DiscovEHR, UK Biobank (UKB), as well as imputed genotype data in BioVU, for evaluation of the robustness of gene-phenotype associations identified in PMBB. For replication analyses in BioMe, DiscovEHR, and BioVU, each study was approved by the Institutional Review Board of each respective biobank’s institution. Access to the UK Biobank for this project was from Application 32133.
Genetic sequencing
This PMBB study dataset included a subset of 11,451 individuals in the PMBB who have undergone whole-exome sequencing (WES). For each individual, we extracted DNA from stored buffy coats and then obtained exome sequences generated by the Regeneron Genetics Center (Tarrytown, NY). These sequences were mapped to GRCh37 as previously described.7 Furthermore, for subsequent phenotypic analyses, we removed samples with low exome sequencing coverage (i.e. less than 75% of targeted bases achieving 20x coverage), high missingness (i.e. greater than 5% of targeted bases), high heterozygosity, dissimilar reported and genetically determined sex, genetic evidence of sample duplication, and cryptic relatedness (i.e. closer than 3rd degree relatives), leading to a total of 10,900 individuals.
For replication studies in PMBB2, we interrogated an additional 6,935 individuals of African American ancestry in PMBB who were exome-sequenced by the Regeneron Genetics Center. We focused our replication efforts on 6,432 individuals after removing samples with poor genotype quality, individuals closer than 3rd degree relatives, and those with dissimilar reported and genetically determined sex. These sequences were mapped to GRCh38.
For replication studies in BioMe, we interrogated 6,470 individuals of African ancestry, 8,735 individuals of European ancestry, and 8,784 individuals of Hispanic ancestry with WES data linked to EHR diagnosis phenotypes after removing samples with poor genotype quality, individuals closer than 3rd degree relatives, and those with dissimilar reported and genetically determined sex. These sequences were mapped to GRCh38.
For replication studies in DiscovEHR, we interrogated 70,734 individuals of European ancestry exome-sequenced on the IDT platform and a separate set of 59,133 individuals of European ancestry exome-sequenced on the VCRome platform. We focused our replication efforts on 85,450 individuals (N=48,413 for IDT, N=37,037 for VCRome) after removing samples with poor genotype quality, individuals closer than 3rd degree relatives, those with dissimilar reported and genetically determined sex, and those that self-identified as Hispanic/Latino. These sequences were mapped to GRCh38.
For replication studies in UKB, we interrogated the 34,629 individuals of European ancestry (based on UKB’s reported genetic ancestry grouping) with ICD-10 diagnosis codes available among the 49,960 individuals who had WES data as generated by the Functional Equivalence (FE) pipeline. We focused our replication efforts on 32,268 individuals after removing samples with poor genotype quality, individuals closer than 3rd degree relatives, and those with dissimilar reported and genetically determined sex. The PLINK files for exome sequencing provided by UKB were based on mappings to GRCh38.
For replication studies in BioVU, which has genotype but not large-scale WES data, we focused on a select group of single variants that showed replication in PMBB, PMBB2, and/or UKB. We interrogated these variants for association with specific phecodes in 10,456 individuals of African American ancestry and 55,944 individuals of European ancestry after removing samples with poor genotype quality, individuals closer than 3rd degree relatives, and those with dissimilar reported and genetically determined sex. These sequences were mapped to GRCh37.
Additional information regarding population characteristics, recruitment, and ethical oversight can be found in the Life Sciences Reporting Summary of this study.
Variant annotation and selection for association testing
For all cohorts analyzed, genetic variants were annotated using ANNOVAR (version 2018Apr16)44 as predicted loss-of-function (pLOF) or missense variants according to the NCBI Reference Sequence (RefSeq) database. pLOF variants were defined as frameshift insertions/deletions, gain/loss of stop codon, or disruption of canonical splice site dinucleotides. Predicted deleterious missense variants were defined as those with Rare Exonic Variant Ensemble Learner (REVEL)10 scores ≥ 0.5. Minor allele frequencies for each variant were determined per Non-Finnish European, African, and Latino minor allele frequencies reported by the Genome Aggregation Database (gnomAD) v2.45 pLOF and REVEL-informed missense variants were selected for gene burden testing or univariate association analyses per ancestry group in each cohort according to each ancestry’s corresponding ancestry-specific minor allele frequency thresholds (rare variants with MAF ≤ 0.1% for gene burden testing, single variants with MAF > 0.1%).
Clinical data collection
International Classification of Diseases Ninth Revision (ICD-9) and Tenth Revision (ICD-10) disease diagnosis codes and procedural billing codes, medications, and clinical imaging and laboratory measurements were extracted from the patients’ EHR for PMBB. ICD-10 encounter diagnoses were mapped to ICD-9 via the Center for Medicare and Medicaid Services 2017 General Equivalency Mappings (https://www.cms.gov/Medicare/Coding/ICD10/2017-ICD-10-CM-and-GEMs.html) and manual curation. Phenotypes for each individual were then determined by mapping ICD-9 codes to distinct disease entities (i.e. phecodes) via Phecode Map 1.2 using the R package “PheWAS”.46 Patients were determined to have a certain disease phenotype if they had the corresponding ICD diagnosis on two or more dates, while phenotypic controls consisted of individuals who never had the ICD code. Individuals with an ICD diagnosis on only one date as well as individuals under control exclusion criteria based on PheWAS phenotype mapping protocols were not considered in statistical analyses.
All laboratory values measured in the outpatient setting were extracted for participants from the time of enrollment in PMBB until March 20, 2019; all units were converted to their respective clinical Traditional Units. Minimum, median, and maximum measurements of each laboratory measurement were recorded for each individual and used for all association analyses. Inpatient and outpatient echocardiography measurements were extracted if available for participants from January 1, 2010 until September 9, 2016; outliers for each echocardiographic parameter (less than Q1 − 1.5*IQR or greater than Q3 + 1.5*IQR) were removed. Similarly, minimum, median, and maximum values for each parameter were recorded for each patient and used for association analyses.
ICD-9 and ICD-10 codes were similarly mapped to phecodes in PMBB2, BioMe, DiscovEHR, and BioVU for replication studies. For UKB, we used the provided ICD-10 disease diagnosis codes for replication studies, and individuals were determined to have a certain disease phenotype if they had one or more encounters for the corresponding ICD diagnosis given the lack of individuals with more than two encounters per diagnosis, while phenotypic controls consisted of individuals who never had the ICD code. Individuals under control exclusion criteria based on PheWAS phenotype mapping protocols were not considered in statistical analyses.
Association studies
A phenome-wide association study (PheWAS) approach was used to determine the phenotypes associated with rare (MAF ≤ 0.1% in gnomAD) pLOF variants carried by individuals in PMBB for the discovery experiment.47 Each disease phenotype was tested for association with each gene burden or single variant using a logistic regression model adjusted for age, age2, sex, and the first ten principal components (PCs) of genetic ancestry. We used an additive genetic model to collapse variants per gene via the fixed threshold approach.48 Given the high percentage of individuals of African ancestry present in the discovery PMBB cohort, association analyses were performed separately in European (N=8,198) and African (N=2,172) genetic ancestries and combined with inverse variance weighted meta-analysis. Only genes with at least 25 carriers of pLOFs were analyzed in the discovery analysis (N=1,518). Our association analyses considered only disease phenotypes with at least 20 cases, leading to the interrogation of 1,000 total phecodes. All association analyses were completed using R version 3.3.1 (Vienna, Austria). Power analyses were conducted using QUANTO version 1.2.4.49
We further evaluated the robustness of our gene-phenotype associations in the same PMBB discovery cohort by 1) associating the aggregation of rare (MAF ≤ 0.1%) predicted deleterious missense variants in gene burden association tests and 2) testing pLOFs and predicted deleterious missense variants with MAF > 0.1 in univariate association tests. We ensured that individuals were non-overlapping across rare pLOFs, rare deleterious missense, and single variant groups. Rare deleterious missense gene burdens and single variants were analyzed for association with the specific phenotype identified in the pLOF-based gene burden discovery, as well as with related phenotypes in their corresponding phecode families (integer part of phecode). For example, to replicate an association of a gene burden with hypothetical phecode 123.45, we associated other variants in the same gene with phecode 123.45 as well as other related phenotypes under the phecode family 123 (e.g. 123.6). Notably, we checked for the presence of mutual carriers between each gene’s pLOF-based gene burdens and subsequently interrogated missense-based gene burdens or single variants due to linkage disequilibrium and/or rare chance, and only reported replications for which the significant phenotypes’ associations were not being driven by mutual carriers. All association studies in PMBB were based on a logistic regression model adjusted for age, age2, sex, and the first 10 PCs of genetic ancestry.
Additionally, we replicated our findings in PMBB2, BioMe, DiscovEHR, and UKB for genes of interest using pLOF-based gene burden, REVEL-informed missense-based gene burden, and/or univariate association analyses from discovery in PMBB. A specific set of single variants were further replicated in BioVU. Association statistics were calculated similarly to PMBB, such that each disease phenotype was tested for association with each gene burden or single variant using a logistic regression model adjusted for age, age2, sex, and the first 10 PCs of genetic ancestry. In BioMe, the summary statistics obtained from running the logistic regression model separately in individuals of European, African, and Hispanic ancestry were meta-analyzed. In DiscovEHR, the summary statistics obtained from running the logistic regression model separately in individuals of European ancestry on the IDT versus VCRome platforms were meta-analyzed. In BioVU, the summary statistics obtained from running the logistic regression model separately in individuals of European and African ancestry were meta-analyzed. All association analyses for PMBB, PMBB2, BioMe, DiscovEHR, UK Biobank, and BioVU were completed using R version 3.3.1 or later (Vienna, Austria). Further information about association studies in each cohort can be found in the Life Sciences Reporting Summary of this study.
Undercalling of variants in UK Biobank
Given the undercalling of variants largely limited to ~3.25% of the exome target regions in the FE pipeline data, we found that 3 of the 97 genes having associations with p<E-06 from the discovery phase overlap with the undercalled exonic regions, namely CES5A, CYP2D6, and ZC3H3. While all other analyses in this study included variants with less than 5% missingness, we included variants with at least 65% call rate for these three genes, understanding that undercalling per variant is random per individual.
Statistical analyses of clinical measurements
In order to compare available measurements for echocardiographic parameters and serum laboratory values between carriers of predicted deleterious variants and genotypic controls in PMBB, we utilized linear regression adjusted for age, age2, sex, and the first 10 PCs of genetic ancestry in individuals of European ancestry only. These analyses were conducted with the minimum, median, and maximum value as the dependent variable for each echocardiographic parameter and clinical lab measure. All statistical analyses, including PheWAS, were completed using R version 3.3.1 or later (Vienna, Austria).
Chart review to validate robust gene-phenotype associations
To confirm our curated list of robust exome-by-phenome-wide significant associations, we manually chart reviewed the EHR for each carrier of rare pLOF variants in genes that showed at least one mode of replication in any cohort. Importantly, for each gene, we aimed to adjudicate the diagnoses of carriers who were flagged as cases for the relevant associated phenotype. We removed associations for which chart review reduced the prevalence of the diagnosis among carriers and thus changed the association to p > E-06. Furthermore, we removed associations for which chart review could not identify a common underlying etiology among all cases for the diagnosis, paying special attention to phecodes that group “other” diagnoses that do not fit into disease-specific ICD codes (i.e. “other diseases of blood and blood-forming organs”).
We discovered on chart review that individuals who were cases for phecodes “hypertrophic obstructive cardiomyopathy” or “other hypertrophic cardiomyopathy” in PMBB were patients with hypertrophic cardiomyopathy who were being assigned one of the codes due to the lack of a single ICD diagnosis code for hypertrophic cardiomyopathy. Thus, we defined a new phenotype for hypertrophic cardiomyopathy encompassing cases for either phecode, and repeated the association with the pLOF gene burdens of MYBPC3 (positive control) and BBS10 (novel), and confirmed their associations as exome-by-phenome-wide significant (Table S22).
Analysis of publicly available expression datasets from NCBI GEO
We interrogated microarray and RNA-seq data publicly available on the NCBI Gene Expression Omnibus (GEO) platform (https://www.ncbi.nlm.nih.gov/geo/).50 To investigate the novel association between CILP and aortic ectasia, we interrogated 11 different microarray and RNA-seq datasets of human fibroblasts from various tissues treated with TGF-ß (GSE1724, GSE65069, GSE64192, GSE39394, GSE79621, GSE68164, GSE97833, GSE97823, GSE135065, GSE125519, GSE40266). Differential expression for each dataset was interrogated using the GEO2R software via a moderated t-statistic. Meta-analysis of differential expression across the datasets was achieved using the Fisher’s combined probability test. Visualization of the meta-analyzed differential expression was achieved using the R package “MetaVolcanoR 1.0.1”. Identification of the top 1% of differentially expressed genes across all datasets was achieved using the Topconfects method.51
We also analyzed microarray data from muscle biopsies in tibial muscular dystrophy patients versus control (GSE42806) to validate the novel association between MYCBP2 and muscle spasms. Differential expression was interrogated using the GEO2R software via a moderated t-statistic.
In silico analyses for PPP1R13L expression in ocular tissues
To understand the functional relevance of PPP1R13L in the eye, we evaluated its expression in human ocular tissues using the publicly available Ocular Tissue Database (OTDB; https://genome.uiowa.edu/otdb/).52 The OTDB consists of gene expression data for eye tissues from 20 normal human donors, generated using Affymetrix Human Exon 1.0 ST arrays and described as Probe Logarithmic Intensity Error (PLIER) values, where individual gene expression values are normalized with its expression in other tissues.
Gene expression in DBA/2J mouse ocular tissues
We assessed the gene expression of Ppp1r13l in mouse ocular tissues using the publicly available Glaucoma Discovery Platform (http://glaucomadb.jax.org/glaucoma). This platform provides an interactive way to analyze RNA sequencing data obtained from retinal ganglion cells (RGCs) isolated from retina and optic nerve head of a 9-month-old female D2 mouse, which is an age-dependent model of ocular hypertension/glaucoma, and D2-Gpnmb+ mouse that do not develop high IOP/glaucoma.53 For transcriptomic studies, four distinct groups were compared based on axonal degeneration and gene expression patterns. The transcriptome of D2 group 1 is identical to the control strain (D2-Gpnmb+), while D2 groups 2–4 exhibit increasing levels of molecular changes relevant to axonal degeneration when compared to control group. We used the Datgan software to assess the differential expression of Ppp1r13l in the retina.54
Immunolocalization of PPP1R13L in human retina
To study the localization of PPP1R13L protein in different retinal layers of the human eye, we performed immunofluorescence on formalin-fixed paraffin-embedded section (N=3) obtained from normal 68-year old donor’s cadaver eyes with a commercially available antibody, anti-PPP1R13L (Cat# 51141-1-AP, Proteintech, IL, USA). Antigen retrieval was performed in 1X citrate buffer (Life Technologies) warmed to 95°C for 30 minutes. Sections were allowed to cool to room temperature and subsequently blocked in 10% normal goat serum with 1% bovine serum albumin in 1X TBS buffer for one hour. The retinal distribution of PPP1R13L protein was visualized by incubating the retinal section with rabbit polyclonal anti-PPP1R13L antibody at 1:300 dilution overnight at 4°C, followed by chicken anti-rabbit IgG conjugated with Alexa Fluor 594 (Cat# A21442, Life Technologies, Carlsbad, CA) at 1:3000 dilution. Nuclei were stained with the use of Vectashield DAPI in the mounting media. The images were captured using a Zeiss Imager Z1 fluorescence microscope equipped with AxioVS40 software version 4.8.1.0.
Human iPSC-RGC cultures
The human iPSCs were generated from keratinocytes or blood cells via polycistronic lentiviral transduction (Human STEMCCA Cre-Excisable constitutive polycistronic [OKS/L-Myc] Lentivirus Reprogramming Kit, Millipore) and characterized with a hES/iPS cell pluripotency RT-PCR kit.55 The induced pluripotent stem cell-derived retinal ganglion cells (iPSC-RGCs) for our studies were derived using small molecules to inhibit BMP, TGF-beta (SMAD) and Wnt signaling to differentiate retinal ganglion cells (RGCs) from iPSCs. The iPSCs were differentiated into pure iPSC-RGCs with structural and functional features characteristic of native RGC cells based on a previous protocol.56
Evaluating oxidative stress in iPSC-RGCs
Induced pluripotent stem cell-derived retinal ganglion cells (iPSC-RGCs) were incubated with increasing amounts of H2O2 overnight before replacing the cultures with complete media. The cells were collected 24 hours after the H2O2 treatment, and levels of PPP1R13L transcripts were assessed using quantitative RT-PCR and gene expression primers, Fwd-5’- TGCCCCAATTCTGGAGTAGG-3’ and Rev-5’- CGGCACGTGGACACAGATT-3’ following previously established protocols.57 Mean expression levels (±standard error of mean) were calculated by analyzing at least three independent samples with replica reactions and presented on an arbitrary scale that represents the expression over the housekeeping gene ACTB. Relative gene expression was quantified using the comparative Ct method. The relative gene expression was compared against no treatment control to obtain normalized gene expression. A two-tailed unpaired Student’s t test was used for statistical analysis.
Single-cell RNA-seq of human pancreatic islets in type 1 diabetes and control subjects
Pancreatic islets were procured from the HPAP consortium under Human Islet Research Network (https://hirnetwork.org/) with approval from the University of Florida Institutional Review Board (IRB # 201600029) and the United Network for Organ Sharing (UNOS). A legal representative for each donor provided informed consent prior to organ retrieval. For type 1 diabetes (T1D) diagnosis, medical charts were reviewed and C-peptide was measured in accordance with the American Diabetes Association guidelines, leading to five individuals with T1D and six control individuals. T1D individuals were 50% female, and had a median age of 29.5 and median BMI of 21.25. Control individuals were 60% female, and had a median age of 13 and median BMI of 17.3. All individuals were of European ancestry. Organs were recovered and processed as previously described.58 Pancreatic islets were cultured and dissociated into single cells as previously described.59 Total dissociated cells were used for single-cell capture for each of the donors.
The Single Cell 3’ Reagent Kit v2 or v3 was used for generating scRNA-seq data. 3,000 cells were targeted for recovery per donor. All libraries were validated for quality and size distribution using a BioAnalyzer 2100 (Agilent) and quantified using Kapa (Illumina). For samples prepared using The Single Cell 3’ Reagent Kit v2, the following chemistry was performed on an Illumina HiSeq4000: Read 1: 26 cycles, i7 Index: 8 cycles, i5 index: 0 cycles, and Read 2: 98 cycles. For samples prepared using The Single Cell 3’ Reagent Kit v3, the following chemistry was performed on an Illumina HiSeq 4000: Read 1: 28 cycles, i7 Index: 8 cycles, i5 index: 0 cycles, and Read 2: 91 cycles. Cell Ranger 2.1.0 (10x Genomics) was used for bcl2fastq conversion using the command “cellranger mkfastq --id= --run= --csv= --localmem=64 --localcores=30”. Cell Ranger 2.1.0 was used for aligning, filtering, counting, and cell calling with the command “cellranger count --id= --transcriptome= --fastqs= --localmem=64 --localcores=35”. Samples were aggregated using Cell Ranger 2.1.0 using the command “cellranger aggr --id= --csv=”.
Seurat 3.0.2 (http://satijalab.org/seurat/)60 was used for filtering, UMAP generation, and initial clustering. Genes were kept that were in 0.01% of cells (3 cells), resulting in 74% of genes remaining for analysis (24,986 of 33,694 genes). Cells with at least 200 genes were kept; however, all cells had at least 200 genes, so this filtering didn’t eliminate any of the 35,134 cells. nFeature, nCount, percent.mt, nFeature vs nCount, and percent.mt vs nCount plots were generated to ascertain the lenient filtering criteria of 200 > nFeature < 7,500, percent.mt < 30, and nCount <100,000, which led to the filtering out of 66 cells (35,066 cells remaining). Data was then log-normalized, and the top 2,000 variable genes were detected using the “vst” selection method. The data was then linearly transformed, and PCA was carried out on the scaled data, using the 2,000 variable genes as input. To determine the dimensionality of the data (i.e. how many principal components to choose when clustering), we employed two approaches: (1) a Jackstraw-inspired resampling test that compares the distribution of p values of each PC against a null distribution and (2) an elbow plot that displays the standard deviation explained by each principal component. Based on these two approaches, 14 PCs with a resolution of 2 was used to cluster the cells, and non-linear dimensionality reduction (UMAP) was used with 14 PCs to visualize the dataset.
DoubletFinder 2.061 was used to demarcate and remove potential doublets in the data as previously described, with the following details: paramSweep_v3 was used, doubletFinder_v3 was used, 14 PCs were used for pK identification (no ground-truth), and the following parameters were used when running doubletFinder_v3: PCs = 14, pN = 0.25, pK =0.005, nExp = nEx_poi.adj, sct = FALSE. The doublets had higher nCount than the singlets identified using this method, and the 807 doublets were removed from further analyses.
Following doublet removal, the raw data for the remaining 34,259 cells was log normalized, the top 2,000 variable genes were detected, the data underwent linear transformation, and PCA was carried out, as described above. Both the Jackstraw-inspired resampling test and an elbow plot of standard deviation explained by each principal component were used to determine the optimal dimensionality of the data, as described above. Based on these two approaches, 11 PCs with a resolution of 1.2 was used to cluster the cells, and UMAP was used with 11 PCs to visualize the 28 clusters detected.
Garnett was used for initial cell classification as previously described.62 In brief, a cell type marker file with 17 different cell types was compiled using various resources,59,60,63 and this marker file was checked for specificity using the “check_markers” function in Garnett by checking the ambiguity score and the relative number of cells for each cell type. A classifier was then trained using the marker file, with “num_unknown” set to 150, and this classifier was then used to classify cells and cell type assignments were extended to nearby cells, “clustering-extended type” (Louvain clustering).
TooManyCells 2.0.0.0 was then used to cluster and visualize the 34,259 single cells, as previously described.64 Briefly, the raw data from the 34,259 cells were not filtered and were normalized by total count and gene normalization by median count followed by frequency-inverse document frequency (tf-idf) using the flags --normalization “BothNorm and --no-filter. The “clustering-extended type” cell labels from Garnett, as well as the demarcation of canonical cell markers, were used to identify broad classes of cell types found within the pancreas, of which we focused on four: Beta, Stellate, Endothelial, and Immune cells.
Differential genes were found using edgeR 3.24.3 through TooManyCells with the normalization “NoneNorm” to invoke edgeR single cell preprocessing, including normalization and filtering. Briefly, edgeR fits normalized expression data to a negative binomial model and uses an exact test with false discovery rate (FDR) control to determine differential expressed genes.65
Single-cell RNA-seq of mouse aorta
All animal experiments were performed following protocols approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine in accordance with the guidelines of the National Institutes of Health. The Center for Comparative Medicine at Baylor College of Medicine monitors the environmental conditions in the animal husbandry rooms. All mice housed in standard ventilated cages, floor area 65 in2, maximum 4 mice per cage. Room temperatures are maintained at 70°F ± 2°. Normal humidity for animal holding rooms ranges from 30% to 70%. The standard light timer is set on a 14-hour light cycle with the lights coming on at 6 AM and off at 8 PM.
Ascending aortic samples were harvested from Mef2c-Cre ROSA26RmT/mG male mice (N=5) and were pooled in Hanks’ Balanced Salt Solution (HBSS, #14175095, Thermo Fisher Scientific) with 10% fetal bovine serum. Extra aortic tissues were removed and the aortic tissues were cut into small pieces. To digest the aortas, samples were subsequently incubated with an enzyme cocktail (3 mg/ml collagenase type II (LS004176, Worthington); 0.15 mg/ml collagenase type XI (C7657, Sigma-Aldrich); 0.24 mg/ml hyaluronidase type I (H3506, Sigma-Aldrich); 0.1875 mg/ml elastase (LS002290, Worthington); 2.38 mg/ml HEPES (H4034, Sigma-Aldrich)) in Ca/Mg contained-HBSS (#14025092, Thermo Fisher Scientific) for 60 minutes at 37 °C. The cell suspension was filtered through a 40 μm cell strainer (CLS431750-50EA, Sigma-Aldrich), centrifuged at 300 g for 10 minutes, and resuspended using cold HBSS (#14175095) with 5% fetal bovine serum. Cells were stained with DIPI and were sorted to select viable cells (≥ 95% viability) by flow cytometry (FACS Aria III, BD Biosciences).
The cells were dispensed onto the Chromium Controller (10x Genomics) and indexed single cell libraries were constructed by a Chromium Single Cell 3’ v2 Reagent Kit (10x Genomics). cDNA libraries were then sequenced in a pair-end fashion on an Illumina NovaSeq 6000. Raw FASTQ data was aligned by Cell Ranger 3.0 with GRCh38. Mapped unique molecular identifier (UMI) counts were imported into Seurat 3.1.4 and built into Seurat objects using the “CreateSeuratObject” function. Cells expressing less than 200 or more than 5000 genes were filtered out for exclusion of non-cell or cell aggregates. Cells with more than 10% mitochondrial genes were also excluded. Data was then normalized and processed into scaled data. Principal component analysis (PCA) and non-linear dimensional reduction using t-Distributed Stochastic Neighbor Embedding (t-SNE) were performed to create clusters and those visualization. The “FindAllMarkers” function in Seurat was used to identify the conserved marker genes in each cluster.
Single-cell RNA-seq of human aorta
The protocol for collecting human aortic tissue samples for scRNA-seq study was approved by the Institutional Review Board at Baylor College of Medicine. Written informed consent was provided by all participants before enrollment. All experiments conducted with human tissue samples were performed in accordance with the relevant guidelines and regulations. Ascending aortic samples were acquired from 3 controls (2 female and 1 male, heart transplant recipient or lung transplant donor) and 8 individuals with ascending thoracic aortic aneurysm (4 female and 4 male). Additional information can be found in the Life Sciences Reporting Summary of this study. For each sample, a piece of aortic tissue (1-2 cm2) was torn into thin layers and cut into small pieces in Hanks’ balanced salt solution (HBSS, without Ca2+ and Mg2+) (Gibco, Waltham, MA, USA) with 10% fetal bovine serum (FBS). Small pieces of tissue were then moved to enzyme cocktail prepared with 3 mg/ml collagenase type II (LS004176, Worthington Biochemical Corp., Lakewood, NJ, USA), 0.15 mg/ml collagenase type XI (H3506, Sigma Corp., Kanagawa, Japan), 0.25 mg/ml soybean trypsin inhibitor (LS003571, Worthington), 0.1875 mg/ml elastase lyophilized (LS002292, Worthington), 0.24 mg/ml hyaluronidase type I (H3506, Sigma), and 2.38 mg/ml 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, H4034, Sigma) in HBSS (with Ca2+ and Mg2+) (14025092, Thermo Fisher Scientific, Waltham, MA, USA) and were digested in a 37°C water bath for 1 to 2 hours. Tissue dissociation was examined under a microscope. Cell suspensions were collected by using a 40-μm cell strainer (CLS431750-50EA, Corning, Inc., Corning, NY, USA), centrifuged at 300 g for 10 minutes, and resuspended in HBSS (without Ca2+ and Mg2+) (14175095, Thermo Fisher) with 5% FBS, followed with incubation on ice for 30 minutes. Cells were then stained by using a live and dead cell kit (L3224, Thermo Fisher) and were submitted for flow cytometry (BD) for the collection of live singlet cells. The living cell rate was further examined under a microscope by using trypan blue (T8154, Sigma Corp., Kanagawa, Japan) staining.
Single-cell suspensions were submitted to the 10X Genomics Chromium System (10x Genomics, Pleasanton, CA, USA), followed by the construction of 3’ gene expression v3 libraries and sequencing on an Illumina NovaSeq 6000. Raw FASTQ data alignment was processed by using Cell Ranger 3.0, with GRCh38 as a reference. Mapped unique molecular identifier (UMI) counts were loaded to R for further analysis. The single-cell sequencing data were filtered by using Seurat 3.0 with the following criteria: gene count per cell >200 and <4000 (or 5000), percentage of mitochondrial genes <10%, and no HBB gene detected in the cell. Data were then normalized and processed into scale data, linear dimensional reduction, cluster finding, and nonlinear dimensional reduction for visualization according to the Seurat manual. To identify clusters in multiple combined datasets, we performed additional integration after normalization and before scale. The conserved (marker) genes for each cluster were identified by using the function “FindAllMarkers” in Seurat. For reclustering, the UMI count of cells of interest were extracted and analyzed similarly to clusters identified in multiple combined datasets.
Data Availability
All summary statistics for significant gene-phenotype associations from the discovery phase in PMBB as well as significant replications from each replication cohort are fully detailed in the Supplementary Information (Table S1–S16). Data for the individual rare pLOF and missense variants in significant genes that were used for gene burden analyses in the PMBB discovery cohort are also included in the Supplementary Information (Tables S23–S24). In addition, a list of all of the single variants that were used for replication analyses across all the cohorts are provided in the Supplementary Information (Table S25). Each variant in Tables S23–25 is annotated with information regarding genomic location, variant effect, amino acid change, REVEL score (for missense), and minor allele frequency in gnomAD as well as in the PMBB discovery cohort. Additionally, up-to-date summary data for genetic variants captured via whole-exome sequencing in PMBB can be accessed via the Penn Medicine Biobank Genome Browser (https://pmbb.med.upenn.edu/allele-frequency/). Individual-level data are not made publicly available due to research participant privacy concerns; however, requests from accredited researchers for access to individual-level data relevant to this manuscript can be made by contacting the corresponding author. Additionally, public expression datasets were obtained from the Ocular Tissue Database (https://genome.uiowa.edu/otdb/), Glaucoma Discovery Platform (http://glaucomadb.jax.org/glaucoma), and the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/). From NCBI GEO, we interrogated 11 different microarray and RNA-seq datasets of human fibroblasts from various tissues treated with TGF-ß (GSE1724, GSE65069, GSE64192, GSE39394, GSE79621, GSE68164, GSE97833, GSE97823, GSE135065, GSE125519, GSE40266) as well as microarray data from muscle biopsies in tibial muscular dystrophy patients (GSE42806).
Extended Data
Extended Data Fig. 1. Distribution of number of carriers for rare predicted loss-of-function (pLOF) variants per gene in Penn Medicine Biobank.
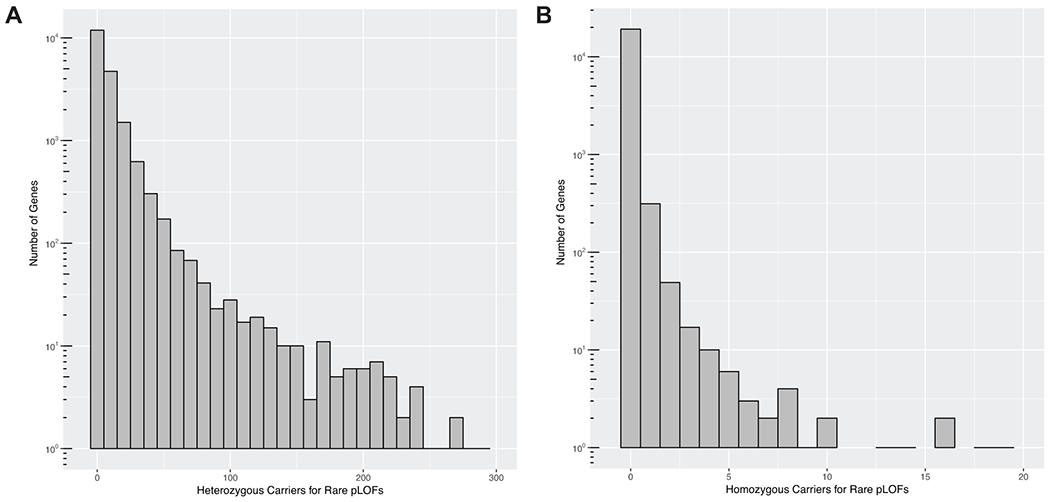
A) Histogram plot for the distribution of number of heterozygous carriers for rare (MAF ≤ 0.1%) pLOF variants per gene in the Penn Medicine Biobank’s (PMBB) exome sequenced cohort. The x-axis represents number of heterozygous pLOF carriers per gene in bin widths of 10, and the log-scaled y-axis represents the number of genes with the x-axis-specified number of heterozygous carriers. B) Histogram plot for the distribution of number of homozygous carriers for rare pLOF variants per gene in PMBB’s exome sequenced cohort. The x-axis represents number of homozygous pLOF carriers per gene in bin widths of one, and the log-scaled y-axis represents the number of genes with the x-axis-specified number of homozygous carriers.
Extended Data Fig. 2. Power analyses for association of gene burdens with at least 25 heterozygous carriers for rare pLOF variants with phenotypes of various case counts.
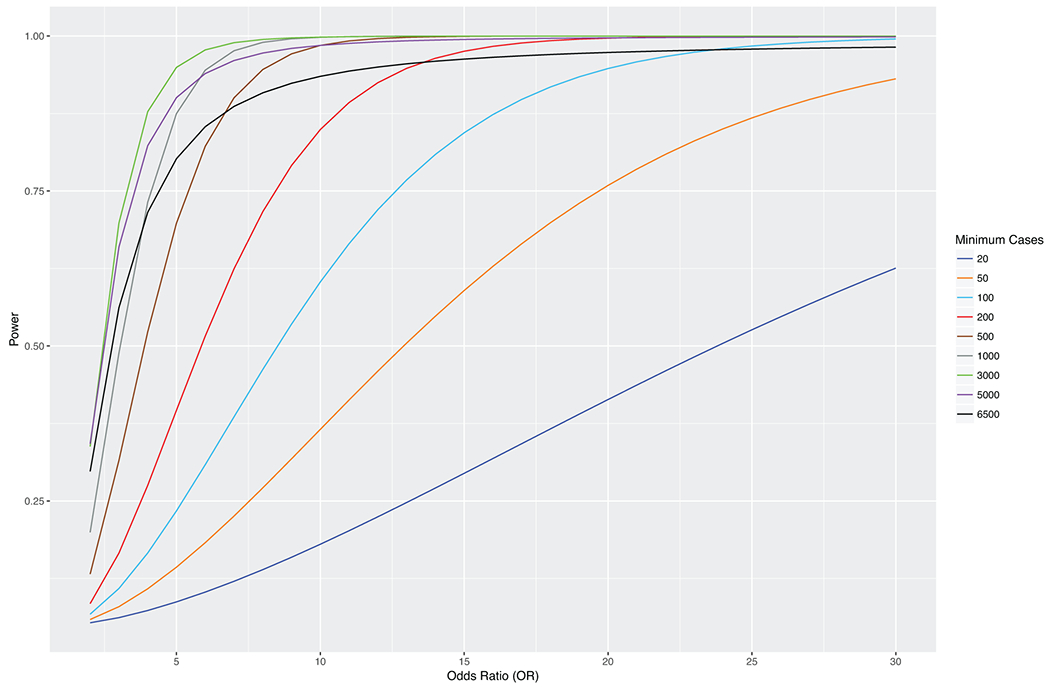
Power analyses for association of gene burdens collapsing rare pLOF variants with 25 heterozygous carriers (i.e. allele frequency = 25/2N ≈ 0.001, where N = 2172 (AFR) + 8198 (EUR)) with phenotypes having various case counts. Phenotype case counts range from 20 to 6500 to reflect the range of case counts for phecodes in the Penn Medicine Biobank discovery cohort, and the power of the gene burden association with each phenotype as a function of odds ratio (OR=exp(beta)) is plotted on separate lines per the plot legend.
Extended Data Fig. 3. Quantile-quantile plot of gene burden testing results from discovery phase of exome-by-phenome-wide association studies in Penn Medicine Biobank.
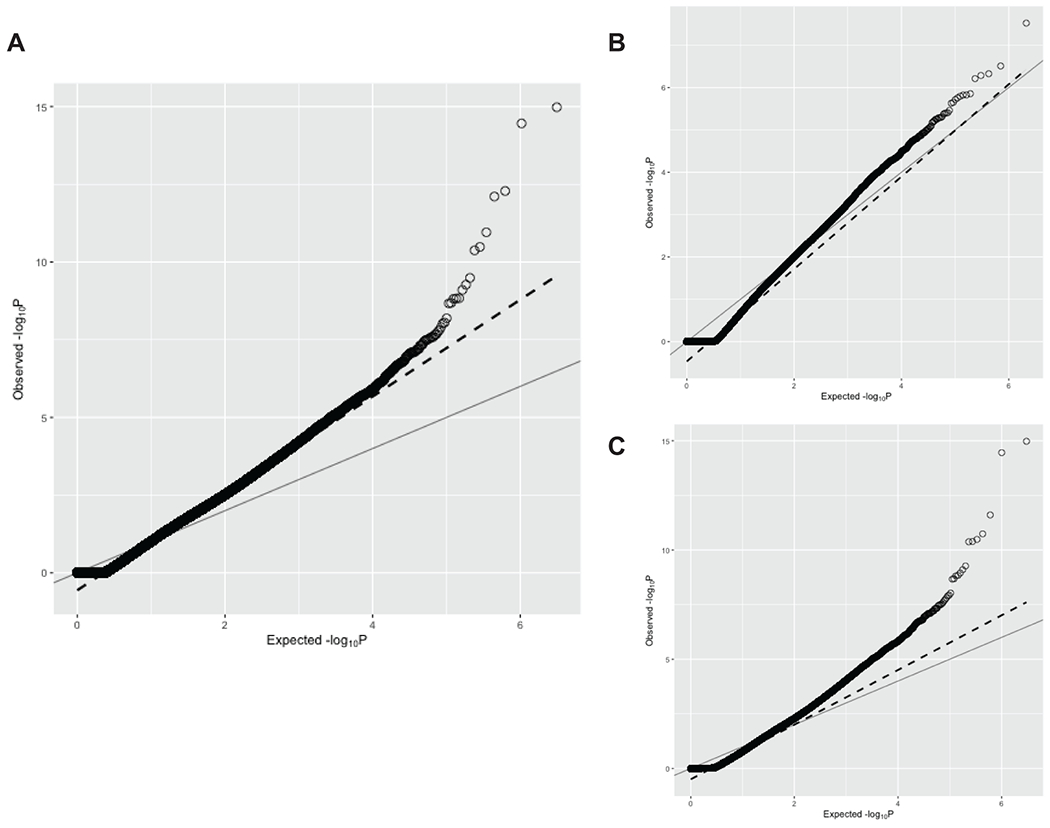
A) Quantile-quantile plot of p values from all exome-by-phenome-wide associations using gene burdens collapsing rare (MAF ≤ 0.1%) predicted loss-of-function (pLOF) variants per gene in the Penn Medicine Biobank (PMBB). The x-axis represents the expected −log10(p value) under the uniform distribution of p values. The y-axis represents the observed −log10(p value) from the discovery phase of the exome-by-phenome-wide gene burden association studies collapsing rare pLOF variants in PMBB. Each point represents an association between one of 1518 gene burdens and one of 1000 phecodes via logistic regression. The solid line shows the relationship between the expected and observed p values under the uniform p value distribution. The dashed line represents the observed fit line between the 50th and 95th percentile of gene burden associations, and the slope of this line is λ∆95 = 1.558. B) AFR-specific QQ plot of p values from all exome-by-phenome-wide associations using gene burdens collapsing rare (MAF ≤ 0.1%) predicted loss-of-function (pLOF) variants per gene in PMBB. Data is presented in a similar manner to panel A. The slope of the fitted line is the AFR-specific λ∆95 = 1.09. C) EUR-specific QQ plot of p values from all exome-by-phenome-wide associations using gene burdens collapsing rare (MAF ≤ 0.1%) predicted loss-of-function (pLOF) variants per gene in PMBB. Data is presented in a similar manner to panel A. The slope of the fitted line is the EUR-specific λ∆95 = 1.251.
Extended Data Fig. 4. MYCBP2 is downregulated in tibial muscular dystrophy.
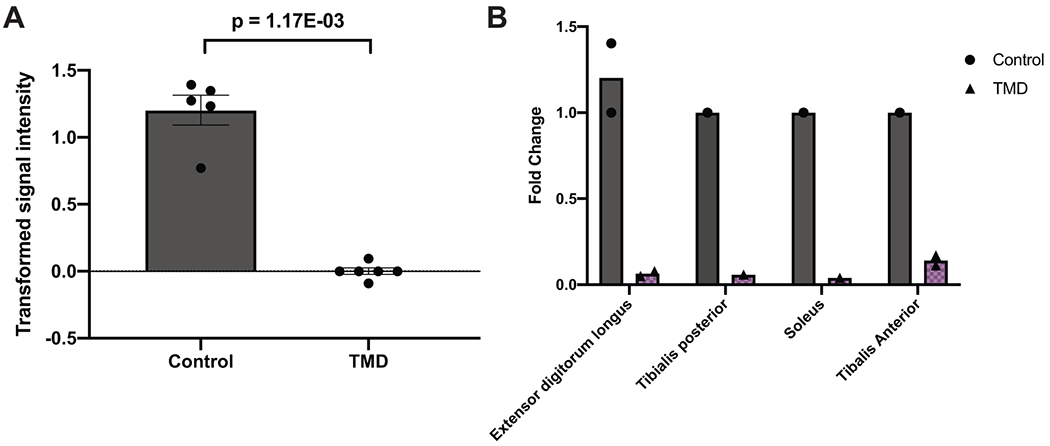
A) Comparison of MYCBP2 expression levels in human distal lower extremity muscles in tibial muscular dystrophy (TMD; N=6 independent muscle samples) versus healthy controls (N=5 muscle samples). Data is presented as mean transformed signal intensity, and error bars denote SEM. Transformed signal intensity values were obtained from GEO Series GSE42806, which are baseline-transformed and MAS5.0-normalized signal intensities, and individual values are plotted overlaying the bar plot. Statistical comparison was based on a moderated t-statistic, and p values were adjusted by Benjamini & Hochberg (FDR) correction. B) Comparison of MYCBP2 expression levels in each distal lower extremity muscle included in the comparison in Extended Data Figure 4A. Data is presented as a bar plot showing mean fold change as compared to a single control sample, and individual values are plotted overlaying the bar plot. Fold changes were calculated based on inverse log-transformed signal intensity values from each lower extremity muscle, including extensor digitorum longus (N=2 independent TMD samples, 2 independent control samples), tibialis posterior (N=1 TMD sample, 1 control sample), soleus (N=1 TMD sample, 1 control sample), and tibialis anterior (N=2 TMD samples, 1 control sample).
Extended Data Fig. 5. Functional validation for the association between PPP1R13L and primary open angle glaucoma.
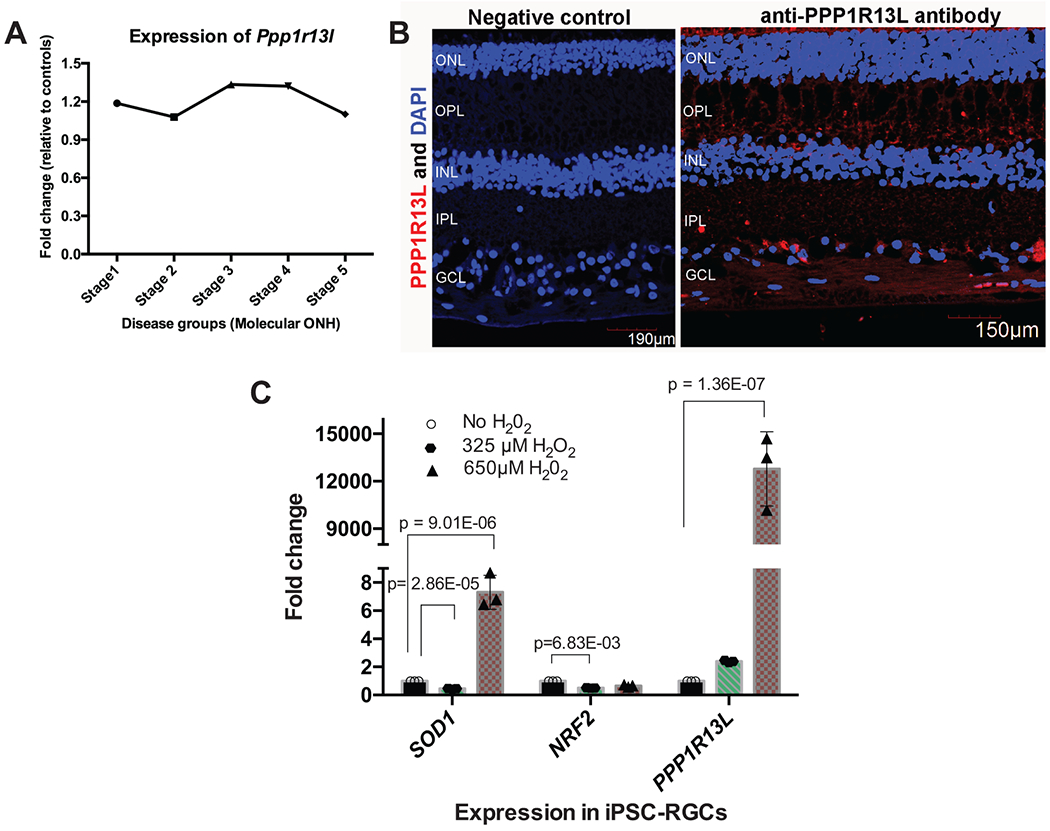
A) Differential expression profile of Ppp1r13l transcript in mouse optic nerve head (ONH) with varying stages of intraocular pressure (IOP)-induced glaucoma. Data represent the fold change in Ppp1r13l expression between different stages of D2 mice (glaucoma, N=50 mice) and D2 Gpnmb+ samples (control, N=10 mice). B) Localization of PPP1R13L protein in the human retina. Shown is the distribution of PPP1R13L by immunohistochemical localization in the retina from normal 68-year-old donor eyes. Overlay of images from DAPI (blue; nuclei) and PPP1R13L (red) in adult human retinal layers are shown on the right. The left represents primary antibody control. Scale bars are shown in each image. The experiment was performed twice independently with consistent results. ONL, outer nuclear layer; OPL, outer plexiform layer; IPL, inner plexiform layer; GCL, ganglion cell layer. C) Relative expression of PPP1R13L transcript in response to oxidative stress in induced pluripotent stem cell-derived retinal ganglion cells (iPSC-RGCs). A two-tailed unpaired Student’s t test was used for statistical analysis, and significant p values are shown. Expression of PPP1R13L in iPSC-RGCs is shown under increasing concentrations of H2O2 treatment (N=3 independent experiments per condition). Plotted are the mean fold changes in comparison to no H2O2, error bars represent standard error of the mean (SEM), and individual values are plotted overlaying the bar plot.
Extended Data Fig. 6. Single-cell RNA-seq of human pancreatic cells shows that RGS12 is not differentially expressed in pancreatic exocrine and endocrine cells, but is reduced in type 1 diabetic peri-islet macrophages.
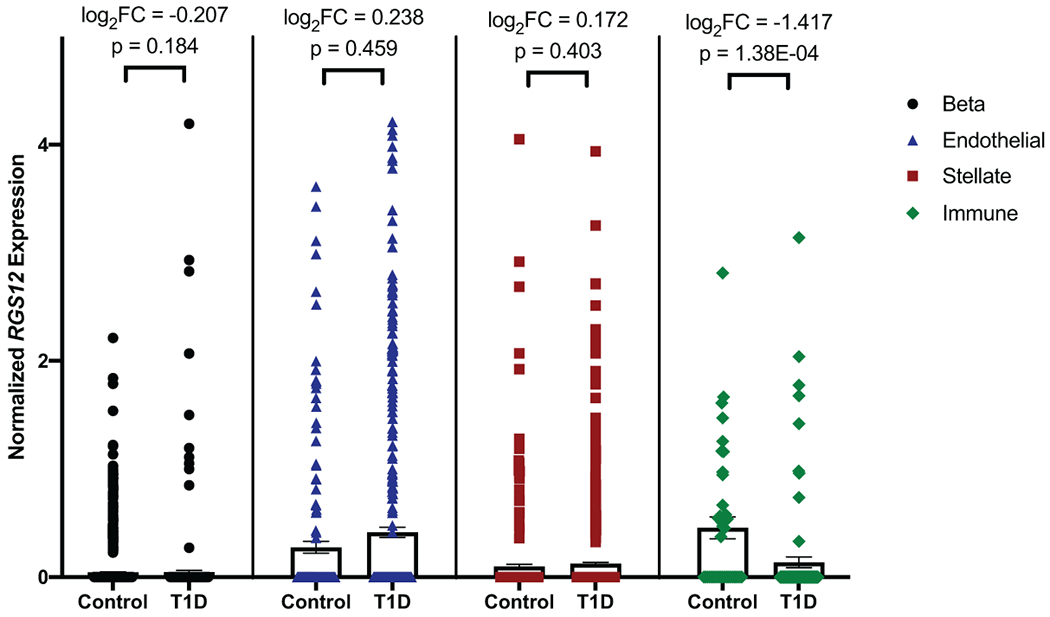
Comparison of RGS12 expression levels in type 1 diabetes (T1D) versus control in pancreatic beta (endocrine; N=2 T1D donors (410 cells), N=6 control donors (1573 cells)), endothelial (N=5 T1D donors (441 cells), N=6 control donors (166 cells)), stellate (exocrine; N=5 T1D donors (910 cells), N=6 control donors (356 cells)), and peri-islet immune (CD45+ macrophages; N=5 T1D donors (95 cells), N=4 control donors (40 cells)) cells based on single-cell RNA-seq. Differential expression of RGS12 in each cell type was determined by edgeR, which fits normalized expression data to a negative binomial model and uses an exact test with false discovery rate (FDR) control to determine differential expressed genes. Data is presented as bars representing mean normalized RGS12 expression and error bars representing standard error of the mean (SEM). Individual points are plotted overlaying their respective bar plots. Differential expression as determined by edgeR are displayed for each cell type as log2 fold change and p values adjusted by FDR correction.
Extended Data Fig. 7. CILP is expressed in aortic adventitial fibroblasts, and is downregulated in human fibroblasts in response to treatment with TGF-ß.
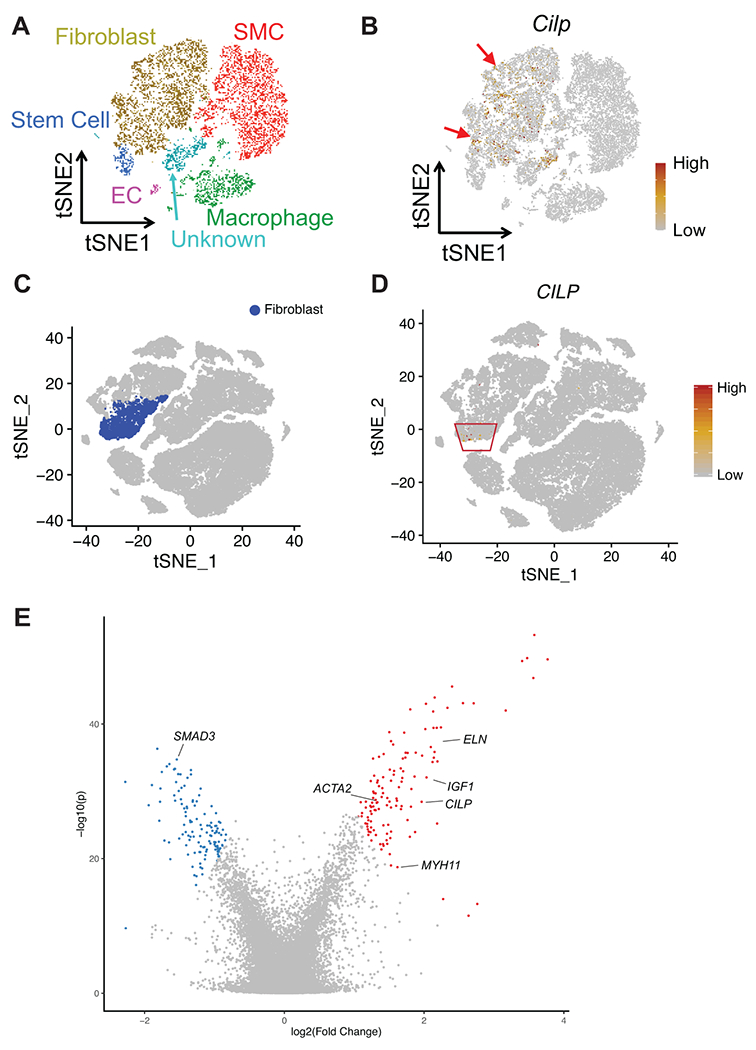
A) t-SNE plot of aortic single cells in mice. Colors denote 6 cell types: smooth muscle cell (SMC), fibroblast, endothelial cell (EC), macrophage, stem cell, unknown. B) Relative expression of Cilp in all cells projected onto a t-SNE plot based on single-cell RNA-seq. The red arrows indicate where Cilp is expressed. C) t-SNE plot of aortic single cells in humans, with fibroblasts highlighted. D) Relative expression of CILP in all cells projected onto a t-SNE plot based on single-cell RNA-seq. The red box indicates where CILP is expressed. E) Volcano plot displaying differential expression of genes from meta-analysis of microarray and RNA-seq data for human fibroblasts treated with TGF-ß (see Methods or Life Sciences Reporting Summary for details about the datasets used). Meta-analysis of differential expression across the datasets was achieved using the Fisher’s combined probability test. The x-axis represents meta-analyzed log2(fold change), and the y-axis represents meta-analyzed −log10(p value). The top 1% of differentially expressed genes across all datasets are labeled in red (upregulation) or blue (downregulation).
Supplementary Material
Acknowledgements
We thank D. Birtwell, H. Williams, P. Baumann, and M. Risman for informatics support regarding the Penn Medicine Biobank. We thank S. Rathi for her help with the real-time PCR experiment on iPSC-RGCs and J. He for her help with iPSC-RGC cultures. We thank S. Dudek for his help with the Penn Medicine Biobank Genome Browser. Research reported in this paper was supported by grants from the National Human Genome Research Institute of the National Institutes of Health under award number F30HG010442 to J.P.; the National Center For Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR001878 to D.J.R; the National Eye Institute of the National Institutes of Health under Award Number R21EY028273-01A1, BrightFocus Foundation grant, Lisa Dean Moseley Foundation Grant, Research to Prevent Blindness, F.M. Kirby Foundation, and The Paul and Evanina Bell Mackall Foundation Trust to V.R.M.C; American Heart Association SFRN in Vascular Disease (18SFRN33960114 and 18SFRN33960163) to S.A.L. and A.D., and National Institutes of Health under award number 1R01HL143359 to Y.H.S. and S.A.L.; Sarnoff Cardiovascular Research Foundation to S.A.; Institute for Translational Medicine and Therapeutics’ (ITMAT) Transdisciplinary Program in Translational Medicine and Therapeutics, and National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR001878 to R.L.K.
Footnotes
Supplementary Information
Supplementary information is available for this paper.
Competing interests
The authors declare no competing interests.
References
- 1.Dewey FE et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 354, doi: 10.1126/science.aaf6814 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Stessman HA, Bernier R & Eichler EE A genotype-first approach to defining the subtypes of a complex disease. Cell 156, 872–877, doi: 10.1016/j.cell.2014.02.002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bush WS, Oetjens MT & Crawford DC Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat Rev Genet 17, 129–145, doi: 10.1038/nrg.2015.36 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Verma A et al. Human-Disease Phenotype Map Derived from PheWAS across 38,682 Individuals. Am J Hum Genet 104, 55–64, doi: 10.1016/j.ajhg.2018.11.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X, Basile AO, Pendergrass SA & Ritchie MD Real world scenarios in rare variant association analysis: the impact of imbalance and sample size on the power in silico. BMC Bioinformatics 20, 46, doi: 10.1186/s12859-018-2591-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S, Abecasis GR, Boehnke M & Lin X Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 95, 5–23, doi: 10.1016/j.ajhg.2014.06.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park J et al. A genome-first approach to aggregating rare genetic variants in LMNA for association with electronic health record phenotypes. Genet Med, doi: 10.1038/s41436-019-0625-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haggerty CM et al. Genomics-First Evaluation of Heart Disease Associated With Titin-Truncating Variants. Circulation 140, 42–54, doi: 10.1161/CIRCULATIONAHA.119.039573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo MH, Plummer L, Chan YM, Hirschhorn JN & Lippincott MF Burden Testing of Rare Variants Identified through Exome Sequencing via Publicly Available Control Data. Am J Hum Genet 103, 522–534, doi: 10.1016/j.ajhg.2018.08.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ioannidis NM et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet 99, 877–885, doi: 10.1016/j.ajhg.2016.08.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciesielski TH et al. Diverse convergent evidence in the genetic analysis of complex disease: coordinating omic, informatic, and experimental evidence to better identify and validate risk factors. BioData Min 7, 10, doi: 10.1186/1756-0381-7-10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casals T et al. Bronchiectasis in adult patients: an expression of heterozygosity for CFTR gene mutations? Clinical genetics 65, 490–495, doi: 10.1111/j.0009-9163.2004.00265.x (2004). [DOI] [PubMed] [Google Scholar]
- 13.Haufroid V & Hantson P CYP2D6 genetic polymorphisms and their relevance for poisoning due to amfetamines, opioid analgesics and antidepressants. Clin Toxicol (Phila) 53, 501–510, doi: 10.3109/15563650.2015.1049355 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Stoetzel C et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat Genet 38, 521–524, doi: 10.1038/ng1771 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Consortium GT The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585, doi: 10.1038/ng.2653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elbedour K, Zucker N, Zalzstein E, Barki Y & Carmi R Cardiac abnormalities in the Bardet-Biedl syndrome: echocardiographic studies of 22 patients. Am J Med Genet 52, 164–169, doi: 10.1002/ajmg.1320520208 (1994). [DOI] [PubMed] [Google Scholar]
- 17.Ji HL et al. delta ENaC: a novel divergent amiloride-inhibitable sodium channel. Am J Physiol Lung Cell Mol Physiol 303, L1013–1026, doi: 10.1152/ajplung.00206.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Battaglia A Del 1p36 syndrome: a newly emerging clinical entity. Brain Dev 27, 358–361, doi: 10.1016/j.braindev.2004.03.011 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Gronich N, Kumar A, Zhang Y, Efimov IR & Soldatov NM Molecular remodeling of ion channels, exchangers and pumps in atrial and ventricular myocytes in ischemic cardiomyopathy. Channels (Austin) 4, 101–107, doi: 10.4161/chan.4.2.10975 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowl MR et al. A large scale hearing loss screen reveals an extensive unexplored genetic landscape for auditory dysfunction. Nat Commun 8, 886, doi: 10.1038/s41467-017-00595-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingham NJ et al. Mouse screen reveals multiple new genes underlying mouse and human hearing loss. PLoS Biol 17, e3000194, doi: 10.1371/journal.pbio.3000194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H et al. Characterization of transcriptomes of cochlear inner and outer hair cells. J Neurosci 34, 11085–11095, doi: 10.1523/JNEUROSCI.1690-14.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilling CE & Carlson KA The effect of OTK18 upregulation in U937 cells on neuronal survival. In Vitro Cell Dev Biol Anim 45, 243–251, doi: 10.1007/s11626-009-9175-8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cacciottolo M et al. Muscular dystrophy with marked Dysferlin deficiency is consistently caused by primary dysferlin gene mutations. Eur J Hum Genet 19, 974–980, doi: 10.1038/ejhg.2011.70 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonventre JA et al. Fer1l6 is essential for the development of vertebrate muscle tissue in zebrafish. Mol Biol Cell 30, 293–301, doi: 10.1091/mbc.E18-06-0401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgess RW et al. Evidence for a conserved function in synapse formation reveals Phr1 as a candidate gene for respiratory failure in newborn mice. Mol Cell Biol 24, 1096–1105, doi: 10.1128/mcb.24.3.1096-1105.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan HI et al. Highwire regulates synaptic growth in Drosophila. Neuron 26, 313–329, doi: 10.1016/s0896-6273(00)81166-6 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Zhen M, Huang X, Bamber B & Jin Y Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26, 331–343, doi: 10.1016/s0896-6273(00)81167-8 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Laizure SC, Herring V, Hu Z, Witbrodt K & Parker RB The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy 33, 210–222, doi: 10.1002/phar.1194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergamaschi D et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 33, 162–167, doi: 10.1038/ng1070 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Howell GR et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. The Journal of clinical investigation 121, 1429–1444, doi: 10.1172/JCI44646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson AM et al. Inhibitor of apoptosis-stimulating protein of p53 (iASPP) is required for neuronal survival after axonal injury. PLoS One 9, e94175, doi: 10.1371/journal.pone.0094175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nickells RW Apoptosis of retinal ganglion cells in glaucoma: an update of the molecular pathways involved in cell death. Surv Ophthalmol 43 Suppl 1, S151–161, doi: 10.1016/s0039-6257(99)00029-6 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Snow BE et al. GTPase activating specificity of RGS12 and binding specificity of an alternatively spliced PDZ (PSD-95/Dlg/ZO-1) domain. J Biol Chem 273, 17749–17755, doi: 10.1074/jbc.273.28.17749 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Cui S et al. The antagonist of CXCR1 and CXCR2 protects db/db mice from metabolic diseases through modulating inflammation. Am J Physiol Endocrinol Metab 317, E1205–E1217, doi: 10.1152/ajpendo.00117.2019 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Mori M et al. Transcriptional regulation of the cartilage intermediate layer protein (CILP) gene. Biochem Biophys Res Commun 341, 121–127, doi: 10.1016/j.bbrc.2005.12.159 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Zhang CL et al. Cartilage intermediate layer protein-1 alleviates pressure overload-induced cardiac fibrosis via interfering TGF-beta1 signaling. J Mol Cell Cardiol 116, 135–144, doi: 10.1016/j.yjmcc.2018.02.006 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Szklarczyk D et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613, doi: 10.1093/nar/gky1131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinard A, Jones GT & Milewicz DM Genetics of Thoracic and Abdominal Aortic Diseases. Circ Res 124, 588–606, doi: 10.1161/CIRCRESAHA.118.312436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sirugo G, Williams SM & Tishkoff SA The Missing Diversity in Human Genetic Studies. Cell 177, 26–31, doi: 10.1016/j.cell.2019.02.048 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fry A et al. Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants With Those of the General Population. Am J Epidemiol 186, 1026–1034, doi: 10.1093/aje/kwx246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cirulli ET et al. Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts. Nat Commun 11, 542, doi: 10.1038/s41467-020-14288-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Z et al. UK Biobank Whole-Exome Sequence Binary Phenome Analysis with Robust Region-Based Rare-Variant Test. Am J Hum Genet 106, 3–12, doi: 10.1016/j.ajhg.2019.11.012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 44.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164, doi: 10.1093/nar/gkq603 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443, doi: 10.1038/s41586-020-2308-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carroll RJ, Bastarache L & Denny JC R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30, 2375–2376, doi: 10.1093/bioinformatics/btu197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denny JC et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 31, 1102–1110, doi: 10.1038/nbt.2749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Price AL et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet 86, 832–838, doi: 10.1016/j.ajhg.2010.04.005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies, http://hydra.usc.edu/gxe (2006).
- 50.Edgar R, Domrachev M & Lash AE Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harrison PF, Pattison AD, Powell DR & Beilharz TH Topconfects: a package for confident effect sizes in differential expression analysis provides a more biologically useful ranked gene list. Genome Biol 20, 67, doi: 10.1186/s13059-019-1674-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagner AH et al. Exon-level expression profiling of ocular tissues. Exp Eye Res 111, 105–111, doi: 10.1016/j.exer.2013.03.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Libby RT et al. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci 22, 637–648, doi: 10.1017/S0952523805225130 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Howell GR, Walton DO, King BL, Libby RT & John SW Datgan, a reusable software system for facile interrogation and visualization of complex transcription profiling data. BMC Genomics 12, 429, doi: 10.1186/1471-2164-12-429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang W et al. Generation of iPSCs as a Pooled Culture Using Magnetic Activated Cell Sorting of Newly Reprogrammed Cells. PLoS One 10, e0134995, doi: 10.1371/journal.pone.0134995 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chavali VRM et al. Dual SMAD inhibition and Wnt inhibition enable efficient and reproducible differentiations of induced pluripotent stem cells into retinal ganglion cells. Sci Rep 10, 11828, doi: 10.1038/s41598-020-68811-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verkuil L et al. SNP located in an AluJb repeat downstream of TMCO1, rs4657473, is protective for POAG in African Americans. Br J Ophthalmol 103, 1530–1536, doi: 10.1136/bjophthalmol-2018-313086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campbell-Thompson M et al. Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev 28, 608–617, doi: 10.1002/dmrr.2316 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang YJ et al. Single-Cell Transcriptomics of the Human Endocrine Pancreas. Diabetes 65, 3028–3038, doi: 10.2337/db16-0405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420, doi: 10.1038/nbt.4096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McGinnis CS, Murrow LM & Gartner ZJ DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324, doi: 10.1016/j.cels.2019.03.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pliner HA, Shendure J & Trapnell C Supervised classification enables rapid annotation of cell atlases. Nat Methods 16, 983–986, doi: 10.1038/s41592-019-0535-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baron M et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst 3, 346–360 e344, doi: 10.1016/j.cels.2016.08.011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwartz GW et al. TooManyCells identifies and visualizes relationships of single-cell clades. Nat Methods 17, 405–413, doi: 10.1038/s41592-020-0748-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang T, Li B, Nelson CE & Nabavi S Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinformatics 20, 40, doi: 10.1186/s12859-019-2599-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All summary statistics for significant gene-phenotype associations from the discovery phase in PMBB as well as significant replications from each replication cohort are fully detailed in the Supplementary Information (Table S1–S16). Data for the individual rare pLOF and missense variants in significant genes that were used for gene burden analyses in the PMBB discovery cohort are also included in the Supplementary Information (Tables S23–S24). In addition, a list of all of the single variants that were used for replication analyses across all the cohorts are provided in the Supplementary Information (Table S25). Each variant in Tables S23–25 is annotated with information regarding genomic location, variant effect, amino acid change, REVEL score (for missense), and minor allele frequency in gnomAD as well as in the PMBB discovery cohort. Additionally, up-to-date summary data for genetic variants captured via whole-exome sequencing in PMBB can be accessed via the Penn Medicine Biobank Genome Browser (https://pmbb.med.upenn.edu/allele-frequency/). Individual-level data are not made publicly available due to research participant privacy concerns; however, requests from accredited researchers for access to individual-level data relevant to this manuscript can be made by contacting the corresponding author. Additionally, public expression datasets were obtained from the Ocular Tissue Database (https://genome.uiowa.edu/otdb/), Glaucoma Discovery Platform (http://glaucomadb.jax.org/glaucoma), and the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/). From NCBI GEO, we interrogated 11 different microarray and RNA-seq datasets of human fibroblasts from various tissues treated with TGF-ß (GSE1724, GSE65069, GSE64192, GSE39394, GSE79621, GSE68164, GSE97833, GSE97823, GSE135065, GSE125519, GSE40266) as well as microarray data from muscle biopsies in tibial muscular dystrophy patients (GSE42806).